
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrophilically activated nitroalkanes in reaction with aliphatic diamines en route to imidazolines†
Alexander V. Aksenov *a,
Nicolai A. Aksenova,
Nikolai A. Arutiunova,
Vladimir V. Malyugaa,
Sergey N. Ovcharova and
Michael Rubin*ab
*a,
Nicolai A. Aksenova,
Nikolai A. Arutiunova,
Vladimir V. Malyugaa,
Sergey N. Ovcharova and
Michael Rubin*ab
aDepartment of Chemistry, North Caucasus Federal University, 1a Pushkin St., Stavropol 355009, Russian Federation. E-mail: alexaks05@rambler.ru
bDepartment of Chemistry, University of Kansas, 1567 Irving Hill Road, Lawrence, KS 66045-7582, USA. E-mail: mrubin@ku.edu; Tel: +1-785-864-5071
First published on 29th November 2019
Abstract
A novel synthetic methodology for the assembly of imidazolines via an unusual reaction between nitroalkanes and aliphatic 1,2-diamines in the presence of phosphorous acid is described. In contrast to the related highly efficient preparation of benzimidazoles from aromatic amines, this process represents a major synthetic challenge and for a long time was elusive. Analysis of the method limitations is provided.
Introduction
Compounds with a five membered cyclic amidine core isosteric to peptide bond, play a very important role in medicinal chemistry. Targeting adrenergic and imidazoline receptors these small molecules could show regulatory function for the central nervous system, and have found utility in the development of many marketed drugs for the treatment of hypertension, diabetes, neural disorders, etc.1,2 In addition, ligands with a 2-imidozaline structure were shown to target adenosine,3 cholecystokinin,4 cyclooxygenase-2,5 estrogen,6 and histamine7 receptors. Also, molecules of this class were studied as inhibitors of DNA topoisomerase,8 proteasome,9 and cholesterol acyltransferase10 enzymes. Not surprisingly, imidazalines are often regarded nowadays as one of the privileged scaffolds for drug discovery.11,12 Normally, preparation of 2-imidazolines 4 is very straightforward and involves acylation of 1,2-diamines 2 with carbonyl compounds 1 (acyl, thioacyl, carbamoyl halides, esters, carboxylic acids, or nitriles) and subsequent cyclocondensation of the formed β-aminocarboxamide intermediate 3 (Scheme 1).13–16 For number of years we were interested in electrophilic activation of nitroalkanes and their reactions with different nucleophilic species. We pondered if this approach can be employed to access 2-imidazolines. Indeed, in this case, the reaction of diamines 2 with nitroalkanes 5 would provide N-hydroxyamidines 6, which after cyclization and elimination of hydroxylamine should afford the same core 4. Herein we wish to report a full account of this investigation. | ||
| Scheme 1 Formation of imidazolines. | ||
Results and discussion
In recent years, our group has developed an expertise in designing novel acid-mediated multistep cascade transformations of nitroalkenes and nitroalkanes targeting material science and medicinal chemistry applications. In particular, it was shown that nitroalkanes 5 dissolved in polyphosphoric acid (PPA) transform into phosphorylated nitronate 7 showing strong electrophilic properties. This unusual species can be used to design a number of one-pot multi-step transformations involving various carbon-based nucleophiles.17–19 The utilization of nucleophilic amines was also demonstrated.20–24 Mechanistically, the latter process is related to the classical Nef reaction,25–27 employing aniline20–23 or hydrazine24 species 8 instead of water. It was proposed, that this reaction initially provides imidinium ion 9, that can be further employed as a convenient building block for the synthesis of heterocyclic compounds: benzoxazoles 10, benzimidazoles 11, diazines 12–13,20–23 and oxadiazoles 14 (ref. 24) (Scheme 2). Evidently, employment of aliphatic 1,2-diamines in this synthetic scheme would potentially provide 2-imidazolines 4. | ||
| Scheme 2 Activation of nitroalkanes towards nucleophilic attack by amines. | ||
To test this idea, we stirred a mixture of 1-nitropropane (1a) and N-phenyl-1,2-ethylenediamine (2a) in 86% polyphosphoric acid (PPA with 86% P2O5 content, an equivalent of polymeric metaphosphoric acid) under conditions previously optimized for synthesis of benzimidazoles.20 Upsettingly, we failed to observe formation of any imidazolines at 130 °C (Table 1, entry 1), and only traces of the desired product 4aa were detected in the reaction mixture heated at 170 °C. Next we tested reaction mediated by 80% PPA (an equivalent of relatively strong pyrophosphoric acid, H4P2O7), which also failed to provide imidazoline product neat (entry 3) or in the presence of activating additives: PCl3 (entry 4) or EtOH (entry 5). We also failed to observe any reactivity towards formation of 4aa after heating the reagents in the presence of PCl3 in boiling xylene (entry 6). Our attempts on carrying the reaction in orthophosphoric acid, either anhydrous (entry 7) or commercial 86% aqueous (entry 8) proved to be a total fiasco as well. Frustrated with this long series of failures, we started to test the possibility to cyclize the imidazoline core in the presence of other strong Brønsted acids, which ever were used for electrophilic activation of nitroalkanes or nitroalkenes in our laboratories: formic (entry 9), tosic acid in protic (entry 10) or aprotic (entry 11) solvents, and even phosphomolybdic acid (HPA, entry 12), but the stubborn reaction newer worked. We saw the light at the end of the tunnel when one of the test reactions carried out at 170 °C in 86% PPA in the presence of phosphorous acid additive afforded 56% of the desired product (entry 13). Further improvement was achieved after lowering the reaction temperature. The process took longer time for completion, but provided improved yield (entry 14). We also noticed, that reaction is being mediated by H3PO3 namely, while molten PPA serves as polar solvent with slight adverse effect. Indeed, an attempt to reduce the amount of PPA in the mixture led to the further improvement of the reaction outcome (entry 15). Finally, we discovered that the process is best mediated by H3PO3 alone under solvent-free conditions (entry 16).
|
||||
|---|---|---|---|---|
| Medium | Temperature | Time | Yielda | |
| a NMR yield of compound 4aa are shown; test reactions were performed in 1.0 mmol scale. | ||||
| 1 | PPA 86% (2 g) | 130 °C | 7 h | 0% |
| 2 | PPA 86% (2 g) | 170 °C | 7 h | 4% |
| 3 | PPA 80% (2 g) | 170 °C | 7 h | 0% |
| 4 | PPA 80% (2 g), PCl3 (0.5 g) | 130 °C | 7 h | 0% |
| 5 | PPA 80% (0.5 g), EtOH (1 mL) | 90 °C | 7 h | 0% |
| 6 | PCl3 (0.5 g), xylene (1 mL) | 140 °C | 7 h | 0% |
| 7 | H3PO4 100% (2 g) | 170 °C | 7 h | 0% |
| 8 | H3PO4 86% (2 g) | 170 °C | 7 h | 0% |
| 9 | HCOOH (0.5 mL) | 170 °C | 7 h | 0% |
| 10 | AcOH (0.5 mL), TsOH·H2O (190 mg) | 130 °C | 7 h | 0% |
| 11 | TsOH·H2O (95 mg), PhCl (2 mL) | 140 °C | 7 h | 0% |
| 12 | H3PMo12O40 (50 mg), PhCl (2 mL) | 140 °C | 7 h | 0% |
| 13 | PPA 86% (1.5 g), H3PO3 (0.5 g) | 170 °C | 7 h | 56% |
| 14 | PPA 86% (1.5 g), H3PO3 (0.5 g) | 160 °C | 12 h | 63% |
| 15 | PPA 86% (0.5g), H3PO3 (0.5 g) | 160 °C | 6 h | 73% |
| 16 | H3PO3 (0.5 g) | 160 °C | 6 h | 78% |
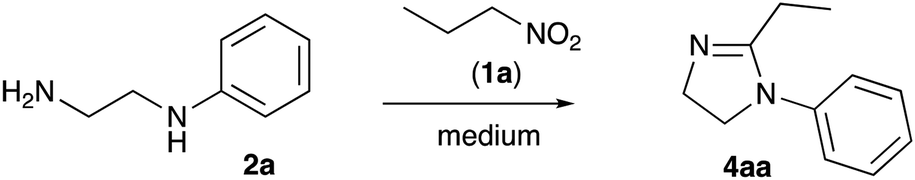
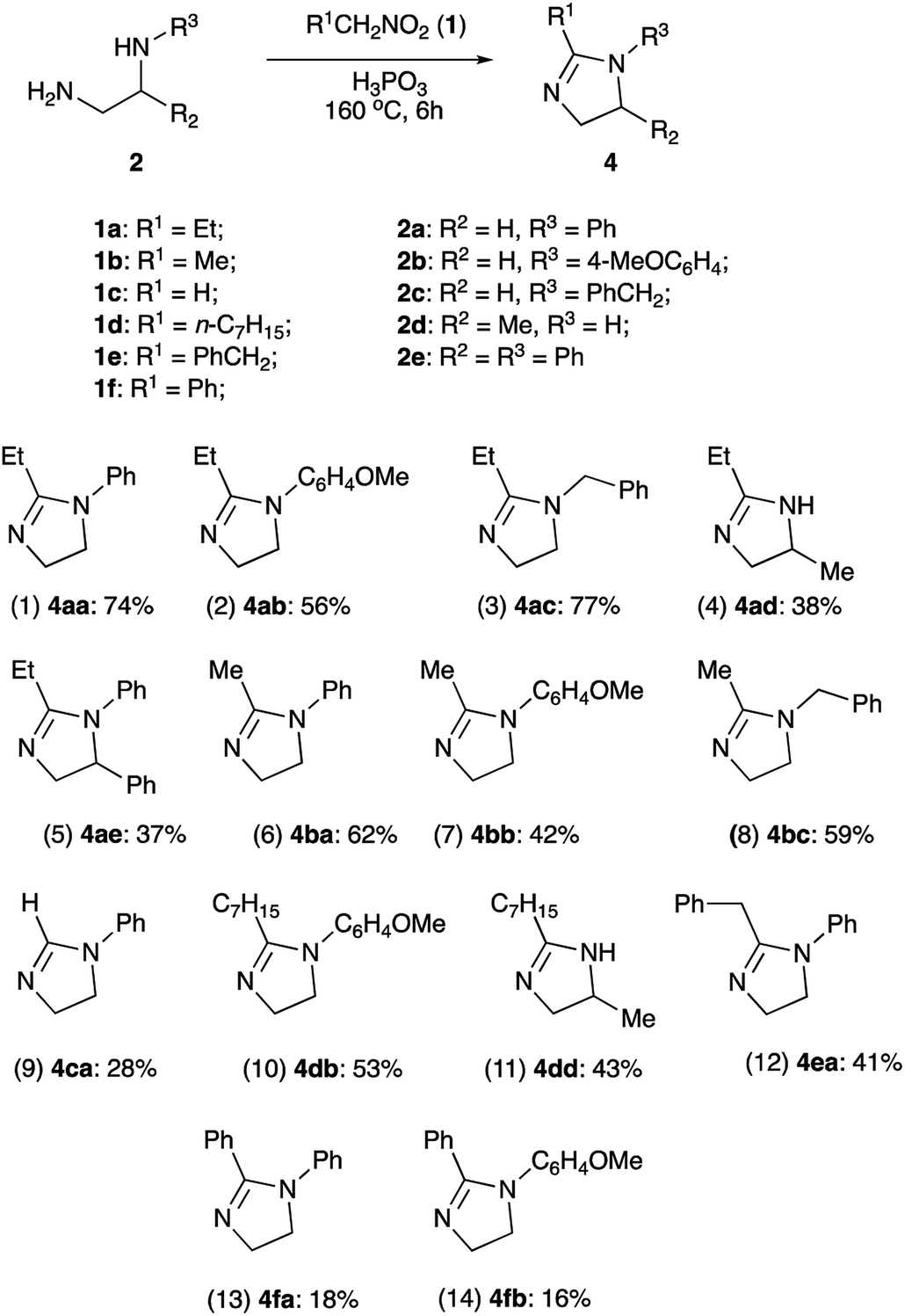
With optimized conditions in hands, we proceeded with evaluation of preparative value of this reaction. After standard aqueous quench and neutralization of the reaction mixture with aqueous ammonia, crude 4aa could be routinely extracted and purified by preparative column chromatography to afford pure product in 74% yield (Scheme 3, entry 1). Ethylenediamines 2b and 2c bearing N-p-anisyl and N-benzyl substituents, respectively, were also tested in reaction with 1-nitropropane (1a) to provide the corresponding products 4ab and 4ac in comparable yields (entries 2, 3). An attempt to use 1,2-propylenediamine (2d) possessing two primary amino groups proved more challenging. In this case yield of product 4ad was quite marginal (entry 4), probably due to significant losses of quite volatile starting material at elevated temperatures despite efficient reflux condenser employed. Another factor affecting the yield could be a sensitivity of the reaction to the steric hindrance at C-1. Indeed, in the reaction of non-volatile diamine 2e bearing even bulkier Ph group at C-1, the yield was also quite low (entry 5).
 | ||
| Scheme 3 Synthesis of imidazolines. | ||
It should be pointed out that volatility of the starting materials escaping the reaction vessel seems to be a major challenge to be addressed in the design of this process. Although this is expected to become a less of an issue upon scale up, in 1.0 mmol scale this has an adverse effect on the reaction performance. Indeed, all reactions involving nitroethane (1b, bp 115 °C) provided consistently lower yields than those carried out in the presence of 1-nitropropane (1a, bp 132 °C) (Scheme 3, entries 6–8). This problem becomes a nuisance when trying to synthesize imidazoline 4ca non-substituted at C-2 from nitromethane (1c, bp 100–102 °C). The yield obtained in this reaction was extremely poor even when we attempted to offset the loss by loading large excess (5 equiv.) of the reagent (entry 9). In contrast, reaction of high boiling nitroalkanes 1d and 1e proceeded smoothly, providing the corresponding cyclic products 4db, 4dd, and 4ea in moderate yields (entries 10–12).
Unexpectedly, we met a major challenge trying to employ α-nitrotoluene (1f) as an electrophilic component of the featured transformation. The reaction was very sluggish, did not proceed to completion, and afforded very poor yields (Scheme 3, entries 13–14). An attempt to carry out this process under more forcing conditions did not lead to improvement, as the products notably decomposed at elevated temperatures. Evidently, in this case, steric issues become greatly prohibitive.
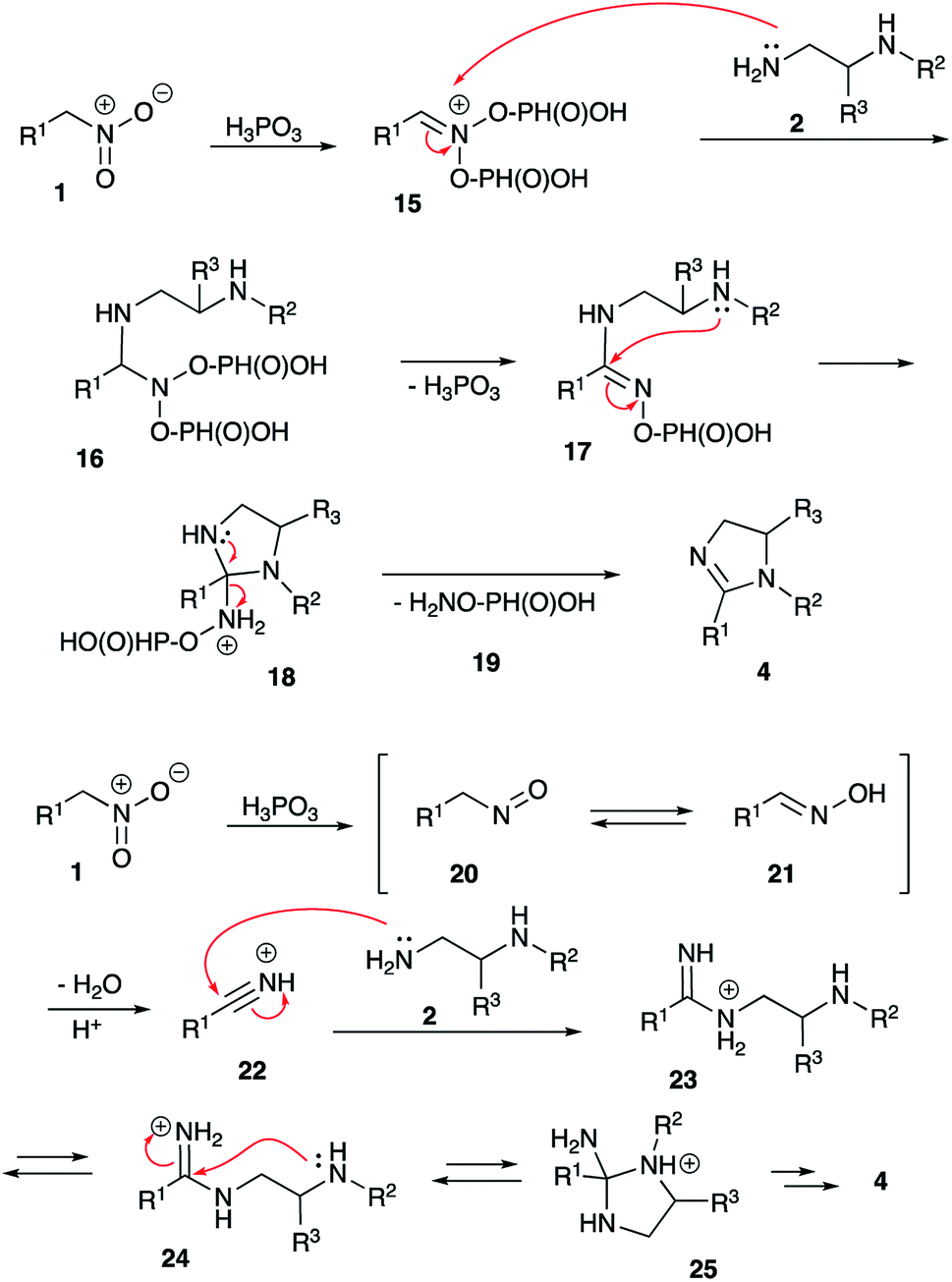
The mechanistic rationale of the described transformation is shown in Scheme 4. Initially, under acidic conditions nitroalkane 1 produces an aci form, which could be stabilized in phosphorylated nitronate form 15. As shown previously, both PPA and H3PO3 enable this step, but diphosphite 15 is less sterically hindered and usually more reactive than diphosphate analog derived from PPA.28 Nucleophilic attack by NH2-moiety of diamine 2 at C-1 of aci-form would first afford aminal 16, which after subsequent elimination of phosphorus acid molecule would be transformed into amidine species 17. The latter would experience 5-exo-trig nucleophilic attack by secondary amine moiety to produce intermediate triaminomethane 18. This step is expected to be quite sensitive to steric factors, which explains poor results obtained in reactions involving reagents with relatively bulky substituents R,1 R2 and R3. Finally, acid-assisted elimination of hydroxylamine (in O-phosphorylated form 19) would yield imidazoline 4.
 | ||
| Scheme 4 Mechanistic rationales. | ||
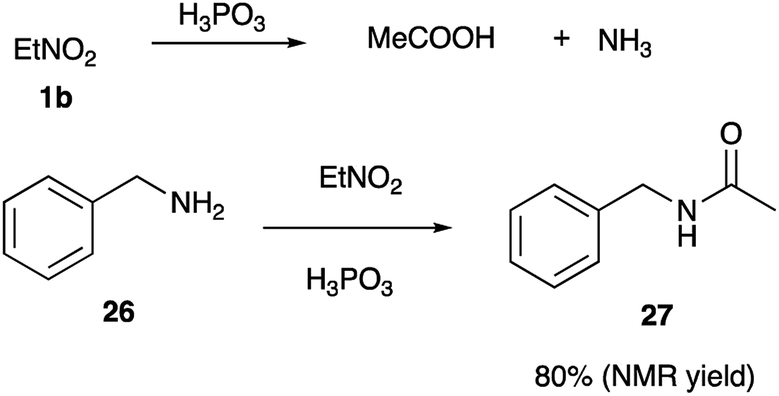
This rationale, however, cannot explain why orthophosphoric acid or PPA, which also are known to stabilize nitronate form via phosphorylation, did not mediate this cyclization. Taking into account, that phosphorous acid has distinct reducing properties, we pondered if this reagent could enable reduction of nitroalkanes into nitroso-compounds 20 (Scheme 4). Once formed, the later should tautomerize into oxime 21, which at elevated temperatures in the presence of acid would loose water to form nitrile (in highly electrophilic protonated form 22). Subsequent nucleophilic attack by primary amine group of diamine 2 shall produce amidinium ion, which exists in equilibrium between forms 23 and 24. Following 5-exo-trig cyclization 24 would afford cyclic triaminomethane species 25, which after elimination of ammonium molecule would yield imidazoline 4 (Scheme 4). It should be pointed out, that H3PO3 indeed seems to be able to reduce nitroalkanes under the featured reaction conditions. Thus, nitroethane was converted into acetonitrile, which in the absence of nucleophilic amines was further hydrolyzed into acetic acid and ammonia (both detected by mass spectroscopy). Interception of nitrile species the presence of benzylamine (26) afforded acetamide 27 in 80% (NMR yield) (Scheme 5).
 | ||
| Scheme 5 Reduction of nitroalkanes with H3PO3. | ||
Next, we attempted to synthesize 2-acyl-substituted imidazolines employing the featured methodology of electrophilic activation of nitroalkanes. To this end, α-nitroacetophenone (1g) was subjected to the reaction with ethylenediamines 2a and 2b under optimized reaction conditions. We were surprised to find that imidazolines 4fa and 4fb were obtained in these reactions as sole isolable products (Scheme 6). The yields of these products were quite moderate, but still significantly better than in attempted reactions involving electrophilically activated α-nitrotoluene (1f) (Scheme 3, entries 13, 14). Such outcome can be explained as follows (Scheme 6). Although, formation of phosphorylated aci-form of nitroalkanes is thermodynamically favored process, it usually requires quite a long time. With electrophilic carbonyl group already present in the substrate 1g, it easily outcompetes the nitronate site, thus, affording hemiaminal 28. The latter may undergo extrusion of nitromethane to yield carboxamide species 29, which can undergo subsequent 5-exo-trig cyclization via “normal” route, affording cyclic intermediate 30. After elimination of water this shall provide imidazoline product 4, missing carbonyl group in the substituent at C-2. It should be pointed out, that this pathway, provides an alternative route for installation of bulky substituents at C-2, although it might be less efficient than “normal route”, involving direct condensation of diamines with carboxylic acid derivatives. Interestingly, reaction of N-benzyl-1,2-ethylenediamine (2c) with α-nitroacetophenone 1g carried at 160 °C was accompanied by competing debenzylation of the resulting imidazoline. After 12 h this process was complete affording heterocycle 4ff as sole product, albeit in modest yield (Scheme 5). Benzyl group is removed in a form of benzyl alcohol (as detected by GC/MS analysis of the crude reaction mixture), which advocates for SN2-mechanism of this process.
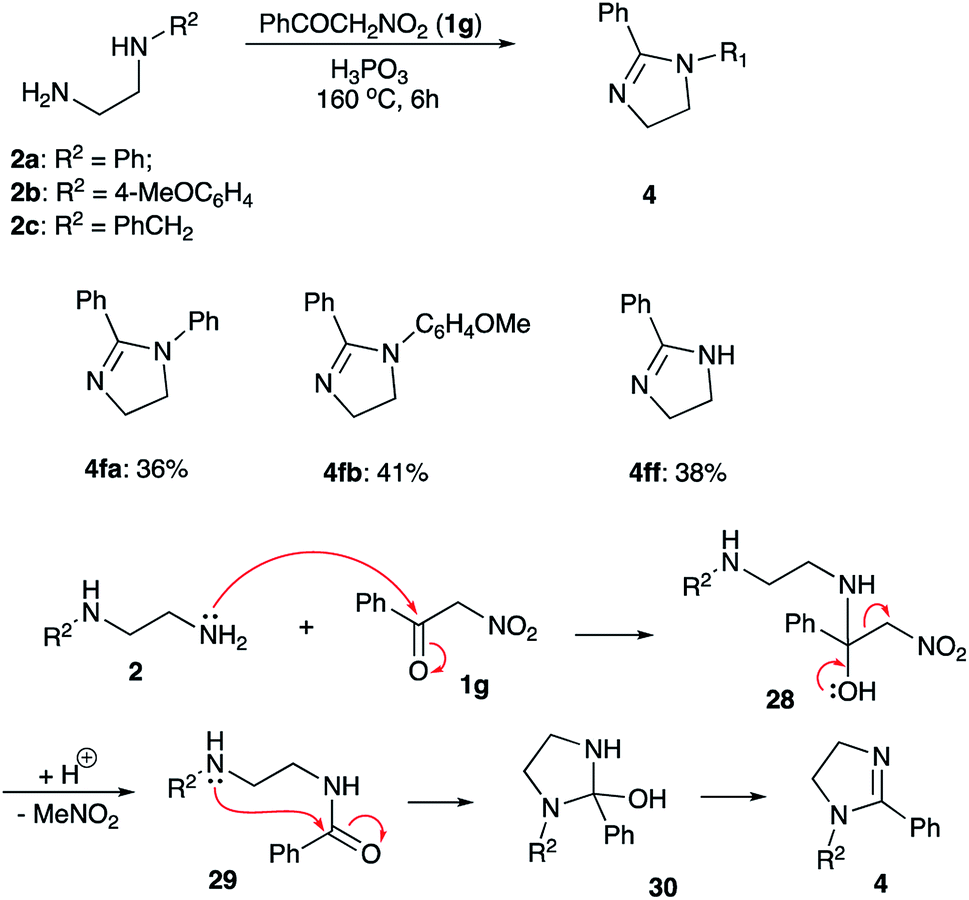 | ||
| Scheme 6 Synthesis of imidazolines from α-nitroacetophenone. | ||
Conclusion
In conclusion, a new protocol was developed, allowing for an assembly of imidazolines from aliphatic 1,2-diamines and electrophilically activated nitroalkanes. Although preparation of benzimidazoles from aromatic analogs can be performed routinely, reaction of aliphatic bis-nucleophiles required careful optimization of the reaction conditions and switching to quite unusual H3PO3 reaction medium. Reaction provides quite moderate yields and is very sensitive to steric factors. Several side processes were studied and the plausible mechanistic rationales explaining the observed limitations was suggested.Experimental part
General information
1H and 13C NMR spectra were recorded on a Bruker Avance-III spectrometer (400 or 100 MHz, respectively) equipped with a BBO probe in CDCl3 using TMS as an internal standard. High-resolution mass spectra were registered with a Bruker Maxis spectrometer (electrospray ionization, in MeCN solution, using HCO2Na–HCO2H for calibration). Melting points were measured with a Stuart SMP30 apparatus. All reactions were performed in oven-dried 5 mL round-bottomed flasks equipped with reflux condensers. The reaction progress and purity of isolated compounds were controlled by TLC on Silufol UV-254 plates, with EtOAc as eluent. Polyphosphoric acids (86% and 80%) were obtained by dissolving of precise amount of P2O5 in 85% orthophosphoric acid according to the published protocols.29,30 All reagents and solvents were purchased from commercial vendors and used as received.2-Ethyl-1-phenyl-4,5-dihydro-1H-imidazole (4aa)
Typical procedure: round bottomed flask (5 mL) equipped with reflux condenser and magnetic bar was charged with N-phenyl-1,2-ethylenediamine (2a) (129 μL, 136 mg, 1.00 mmol), 1-nitropropane (1a) (179 μL, 178 mg, 2.00 mmol), and phosphorous acid (500 mg). The reaction vessel was placed in the oil bath pre-heated to 160 °C and the mixture was stirred for 6 h. Then the mixture was cooled down, quenched with water (10 mL), and basified with 20% aqueous ammonia (10 mL). Crude organic material was extracted with dichloromethane (4 × 15 mL), concentrated in vacuum and purified with preparative column chromatography on silica gel eluting with acetone/EtOAc/Et3N (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4:0.3). The titled compound was obtained as a yellowish oil, Rf 0.40 (acetone/EtOAc/Et3N, 4:4:0.5), Rf 0.37 (EtOH/EtOAc/Et3N, 50:30:3). Yield 174 mg (0.74 mmol, 74%). 1H NMR (400 MHz, CDCl3): δ = 7.31 (t, J = 7.9 Hz, 2H), 7.14–7.03 (m, 3H), 3.78 (s, 4H), 2.28 (q, J = 7.4 Hz, 2H), 1.11 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, CDCl3): δ = 166.1, 141.7, 129.3 (2C), 124.6, 123.3 (2C), 52.9, 52.1, 22.2, 11.0. IR (film, NaCl, cm−1): 3311, 2982, 2938, 1655, 1607, 1503, 1222, 1069; HRMS (ES TOF) calculated for C11H15N2 (M + H)+ 175.1230, found 175.1232 (−1.2 ppm).
:4:0.3). The titled compound was obtained as a yellowish oil, Rf 0.40 (acetone/EtOAc/Et3N, 4:4:0.5), Rf 0.37 (EtOH/EtOAc/Et3N, 50:30:3). Yield 174 mg (0.74 mmol, 74%). 1H NMR (400 MHz, CDCl3): δ = 7.31 (t, J = 7.9 Hz, 2H), 7.14–7.03 (m, 3H), 3.78 (s, 4H), 2.28 (q, J = 7.4 Hz, 2H), 1.11 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, CDCl3): δ = 166.1, 141.7, 129.3 (2C), 124.6, 123.3 (2C), 52.9, 52.1, 22.2, 11.0. IR (film, NaCl, cm−1): 3311, 2982, 2938, 1655, 1607, 1503, 1222, 1069; HRMS (ES TOF) calculated for C11H15N2 (M + H)+ 175.1230, found 175.1232 (−1.2 ppm).
2-Ethyl-1-(4-methoxyphenyl)-4,5-dihydro-1H-imidazole (4ab)
This compound was prepared according to the typical procedure employing N-(4-methoxyphenyl)ethylenediamine (2b) (153 μL, 166 mg, 1.00 mmol), 1-nitropropane (1a) (179 μL, 178 mg, 2.00 mmol), and phosphorous acid (500 mg). The crude material was purified by preparative column chromatography on silica gel eluting with acetone/EtOAc/Et3N (4:1:0.3). The titled compound was isolated as a yellowish oil, Rf 0.41 (acetone/EtOAc/Et3N, 4:1:0.5). Yield 114 mg (0.56 mmol, 56%). 1H NMR (400 MHz, CDCl3): δ = 7.05 (d, J = 8.9 Hz, 2H), 6.88 (d, J = 8.9 Hz, 2H), 3.80 (s, 3H), 3.86–3.70 (m, 4H), 2.17 (q, J = 7.4 Hz, 2H), 1.10 (t, J = 7.5 Hz, 3H). 13C NMR (101 MHz, CDCl3): δ = 167.6, 157.7, 134.7, 126.5 (2C), 114.7 (2C), 55.6, 54.1, 51.9, 21.7, 10.9. IR (film, NaCl, cm−1): 2985, 2880, 2836, 2368, 1650, 1515, 1240, 1034; HRMS (ES TOF) calculated for C12H17N2O1 (M + H)+ 205.1335, found 205.1340 (−2.2 ppm).
1-Benzyl-2-ethyl-4,5-dihydro-1H-imidazole (4ac)
This compound was prepared according to the typical procedure employing N-benzyl-1,2-ethylenediamine (2c) (151 μL, 150 mg, 1.00 mmol), 1-nitropropane (1a) (179 μL, 178 mg, 2.00 mmol), and phosphorous acid (500 mg). The crude material was purified by preparative column chromatography on silica gel eluting with acetone/EtOAc/Et3N, 4:1:0.3. The titled compound was isolated as a yellowish oil, Rf 0.29 (acetone/EtOAc/Et3N, 4:1:1). Yield 145 mg (0.77 mmol, 77%). 1H NMR (400 MHz, CDCl3): δ = 7.34 (t, J = 7.3 Hz, 2H), 7.30–7.19 (m, 3H), 4.28 (s, 2H), 3.69 (t, J = 9.6 Hz, 2H), 3.21 (t, J = 9.7 Hz, 2H), 2.31 (q, J = 7.3 Hz, 2H), 1.23 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, CDCl3): δ = 168.3, 137.8, 128.8 (2C), 127.5, 127.3 (2C), 52.1, 50.6, 50.4, 21.2, 10.9. IR (film, NaCl, cm−1): 3307, 2974, 2938, 2882, 1743, 1655, 1563, 1515, 1459, 1382, 1246; HRMS (ES TOF) calculated for C12H17N2 (M + H)+ 189.1386, found 189.1384 (1.3 ppm).
2-Ethyl-5-methyl-4,5-dihydro-1H-imidazole (4ad)
This compound was prepared according to the typical procedure employing (±)-1,2-diaminopropane (2d) (82 μL, 74 mg, 1.00 mmol), 1-nitropropane (1a) (179 μL, 178 mg, 2.00 mmol), and phosphorous acid (500 mg). The crude material was purified by preparative column chromatography on silica gel eluting first with acetone/EtOAc/Et3N (40:17:3) and acetone/Et3N (3:1) to remove impurities following by acetone/20% aq. ammonia to collect the target molecule. The titled compound was isolated as a yellowish oil, Rf 0.55 (acetone/aq. (20%) NH4OH, 10:2). Yield 42 mg (0.38 mmol, 38%). 1H NMR (400 MHz, CDCl3): δ = 4.14–3.98 (m, 1H), 3.78 (t, J = 10.7 Hz, 1H), 3.24 (dd, J = 11.3, 7.5 Hz, 1H), 2.37 (q, J = 7.6 Hz, 2H), 1.24 (d, J = 6.3 Hz, 3H), 1.19 (t, J = 7.6 Hz, 3H). 13C NMR (101 MHz, CDCl3): δ = 171.4, 53.5, 52.2, 21.4, 20.4, 10.7. IR (film, NaCl, cm−1): 3239, 2974, 2934, 2886, 1747, 1659, 1563, 1402, 1286; HRMS (ES TOF) calculated for C6H13N2 (M + H)+ 113.1073, found 113.1074 (−0.3 ppm).
2-Ethyl-1,5-diphenyl-4,5-dihydro-1H-imidazole (4ae)
This compound was prepared according to the typical procedure employing N-1,1-diphenyl-1,2-ethanediamine (2e), (190 μL, 212 mg, 1.00 mmol), 1-nitropropane (1a) (179 μL, 178 mg, 2.00 mmol), and phosphorous acid (500 mg). The crude material was purified by preparative column chromatography on silica gel eluting with EtOAc/hexanes (1:1). The titled compound was isolated as a yellowish oil, Rf 0.33 (EtOAc/hexanes, 1:1). Yield 93 mg (0.37 mmol, 37%). 1H NMR (400 MHz, CDCl3): δ = 7.34–7.19 (m, 7H), 7.13 (t, J = 7.4 Hz, 1H), 6.98 (d, J = 7.6 Hz, 2H), 5.08 (t, J = 10.2 Hz, 1H), 4.36 (dd, J = 13.9, 10.9 Hz, 1H), 3.76 (dd, J = 14.0, 9.7 Hz, 1H), 2.45–2.23 (m, 2H), 1.19 (t, J = 7.5 Hz, 3H). 13C NMR (101 MHz, CDCl3): δ = 167.5, 141.6, 140.2, 129.4 (2C), 128.9 (2C), 128.0, 127.2 (2C), 126.4, 125.9 (2C), 69.3, 61.7, 22.2, 11.0. IR (film, NaCl, cm−1): 3375, 2934, 2365, 1603, 1493, 1390, 1222, 1029; HRMS (ES TOF) calculated for C17H19N2 (M + H)+ 251.1543, found 251.1550 (−2.8 ppm).
2-Methyl-1-phenyl-4,5-dihydro-1H-imidazole (4ba)
This compound was prepared according to the typical procedure employing N-phenyl-1,2-ethylenediamine (2a) (129 μL, 136 mg, 1.00 mmol), nitroethane (1b) (144 μL, 150 mg, 2.00 mmol), and phosphorous acid (500 mg). The crude material was purified by preparative column chromatography on silica gel eluting with acetone/EtOAc/Et3N, (4:4:0.3). The titled compound was isolated as a yellowish oil, Rf 0.14 (acetone/EtOAc/Et3N, 4:4:0.5), Rf 0.37 (acetone/EtOAc/Et3N, 4:1:1). Yield 99 mg (0.62 mmol, 62%). 1H NMR (400 MHz, CDCl3): δ = 7.30 (t, J = 7.8 Hz, 2H), 7.06 (dd, J = 17.1, 7.7 Hz, 3H), 3.85–3.67 (m, 4H), 2.00 (s, 3H). 13C NMR (101 MHz, CDCl3): δ = 161.5, 141.5, 129.2 (2C), 124.1, 122.2 (2C), 52.1, 51.9, 16.0. IR (film, NaCl, cm−1): 3291, 3042, 2938, 2862, 1650, 1599, 1402, 1318, 1258, 1041; HRMS (ES TOF) calculated for C10H13N2 (M + H)+ 161.1073, found 161.1077 (−2.4 ppm).
1-(4-Methoxyphenyl)-2-methyl-4,5-dihydro-1H-imidazole (4bb)
This compound was prepared according to the typical procedure employing N-(4-methoxyphenyl)ethylenediamine (2b) (153 μL, 166 mg, 1.00 mmol), nitroethane (1b) (144 μL, 150 mg, 2.00 mmol), and phosphorous acid (500 mg). The crude material was purified by preparative column chromatography on silica gel eluting with acetone/EtOAc/Et3N (4:1:0.3). The titled compound was isolated as a yellowish oil, Rf 0.24 (acetone/EtOAc/Et3N, 4:1:0.5). Yield 80 mg (0.42 mmol, 42%). 1H NMR (400 MHz, CDCl3): δ = 7.06 (d, J = 8.9 Hz, 2H), 6.95–6.86 (m, 2H), 3.81 (s, 3H), 3.81–3.72 (m, 4H), 1.91 (s, 3H). 13C NMR (101 MHz, CDCl3): δ = 163.0, 157.4, 134.6, 125.7 (2C), 114.6 (2C), 55.6, 53.4, 51.8, 15.2. IR (film, NaCl, cm−1): 3059, 2989, 2836, 2363, 1615, 1506, 1401, 1244, 1183; HRMS (ES TOF) calculated for C11H15N2O1 (M + H)+ 191.1179, found 191.1174 (2.6 ppm).
1-Benzyl-2-methyl-4,5-dihydro-1H-imidazole (4bc)
This compound was prepared according to the typical procedure employing N-benzyl-1,2-ethylenediamine (2c) (151 μL, 150 mg, 1.00 mmol), nitroethane (1b) (144 μL, 150 mg, 2.00 mmol), and phosphorous acid (500 mg). The crude material was purified by preparative column chromatography on silica gel eluting with EtOH/EtOAc/Et3N (5:3:1). The titled compound was isolated as a yellowish oil, Rf 0.43 (EtOAc/Et3N, 10:1). Yield 103 mg (0.59 mmol, 59%). 1H NMR (400 MHz, CDCl3): δ = 7.43–7.27 (m, 3H), 7.22 (d, J = 7.0 Hz, 2H), 4.33 (s, 2H), 3.72 (t, J = 9.8 Hz, 2H), 3.30 (t, J = 9.7 Hz, 2H), 2.10 (s, 3H). 13C NMR (101 MHz, CDCl3): δ = 165.1, 136.6, 129.1 (2C), 128.0, 127.4 (2C), 50.7, 50.0, 49.8, 14.0. IR (film, NaCl, cm−1): 3030, 2870, 2834, 2361, 1619, 1495, 1418, 1274, 1250, 1029; HRMS (ES TOF) calculated for C11H15N2 (M + H)+ 175.1230, found 175.1232 (−1.2 ppm).
1-Phenyl-4,5-dihydro-1H-imidazole (4ca)
This compound was prepared according to the typical procedure employing N-phenyl-1,2-ethylenediamine (2a) (129 μL, 136 mg, 1.00 mmol), nitromethane (1c) (269 μL, 305 mg, 5.00 mmol), and phosphorous acid (500 mg). The crude material was purified by preparative column chromatography on silica gel eluting with acetone/EtOAc/Et3N (1:6:0.3). The titled compound was isolated as a yellowish oil, Rf 0.36 (acetone/EtOAc/Et3N, 1:6:0.5). Yield 41 mg (0.28 mmol, 28%). 1H NMR (400 MHz, CDCl3): δ = 8.22 (s, 1H), 7.19 (t, J = 7.9 Hz, 2H), 6.73 (t, J = 7.3 Hz, 1H), 6.63 (d, J = 7.7 Hz, 2H), 3.56 (dd, J = 11.6, 5.9 Hz, 2H), 3.33 (t, J = 5.8 Hz, 2H). 13C NMR (101 MHz, CDCl3): δ = 161.9, 147.9, 129.5 (2C), 118.1, 113.0 (2C), 43.9, 37.9. IR (film, NaCl, cm−1): 3351, 3054, 2874, 1663, 1603, 1507, 1386, 1326, 1254, 877, 753; HRMS (ES TOF) calculated for C9H11N2 (M + H)+ 147.0917, found 147.0920 (−2.4 ppm).
2-Heptyl-1-(4-methoxyphenyl)-4,5-dihydro-1H-imidazole (4db)
This compound was prepared according to the typical procedure employing N-(4-methoxyphenyl)ethylenediamine (2b) (153 μL, 166 mg, 1.00 mmol), 1-nitrooctane (1d) (346 μL, 318 mg, 2.00 mmol), and phosphorous acid (500 mg). The crude material was purified by preparative column chromatography on silica gel eluting with hexanes/EtOAc/EtOH/Et3N, 10:1:1:1. The titled compound was isolated as a yellowish oil, Rf 0.68 (hexanes/EtOAc/EtOH/Et3N, 6:1:1:1). Yield 145 mg (0.53 mmol, 53%). 1H NMR (400 MHz, CDCl3): δ = 7.06 (d, J = 8.9 Hz, 2H), 6.90 (d, J = 8.8 Hz, 2H), 3.92–3.70 (m, 7H), 2.24–2.15 (m, 2H), 1.59–1.49 (m, 2H), 1.30–1.11 (m, 8H), 0.83 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CDCl3): δ = 167.4, 158.1, 133.8, 126.7 (2C), 114.8 (2C), 55.6, 54.06, 50.6, 31.7, 29.4, 28.9, 27.9, 26.6, 22.7, 14.2. IR (film, NaCl, cm−1): 2954, 2871, 2840, 1616, 1511, 1458, 1380, 1244, 1043; HRMS (ES TOF) calculated for C17H27N2O1 (M + H)+ 275.2118, found 285.2113 (2.0 ppm).
2-Heptyl-5-methyl-4,5-dihydro-1H-imidazole (4dd)
This compound was prepared according to the typical procedure employing (±)-1,2-diaminopropane (2d) (82 μL, 74 mg, 1.00 mmol), 1-nitrooctane (1d) (346 μL, 318 mg, 2.00 mmol), and phosphorous acid (500 mg). The crude material was purified by preparative column chromatography on silica gel eluting first with acetone/Et3N (9:1) to remove impurities following by acetone/20% aq. ammonia (6:1) to collect the target molecule. The titled compound was isolated as a yellowish oil, Rf 0.37 (acetone/Et3N, 10:1). Yield 78 mg (0.43 mmol, 43%). 1H NMR (400 MHz, CDCl3): δ = 4.01–3.84 (m, 1H), 3.70 (t, J = 10.5 Hz, 1H), 3.15 (dd, J = 11.2, 7.3 Hz, 1H), 2.22 (t, J = 7.8 Hz, 2H), 1.57 (dd, J = 14.6, 7.1 Hz, 2H), 1.33–1.21 (m, 8H), 1.18 (d, J = 6.3 Hz, 3H), 0.84 (t, J = 6.4 Hz, 3H). 13C NMR (101 MHz, CDCl3): δ = 167.3, 56.7, 56.2, 31.8, 29.4, 29.3, 29.1, 26.8, 22.7, 21.9, 14.2. IR (film, NaCl, cm−1): 2938, 2862, 1711, 1559, 1402, 1250, 1009; HRMS (ES TOF) calculated for C11H23N2 (M + H)+ 183.1856, found 183.1854 (0.8 ppm).
2-Benzyl-1-phenyl-4,5-dihydro-1H-imidazole (4ea)
This compound was prepared according to the typical procedure employing N-phenyl-1,2-ethylenediamine (2a) (129 μL, 136 mg, 1.00 mmol), 1-phenyl-2-nitroethane (1e) (269 μL, 302 mg, 2.00 mmol), and phosphorous acid (500 mg). The crude material was purified by preparative column chromatography on silica gel eluting with EtOAc/hexanes/Et3N (4:4:0.5). The titled compound was isolated as a yellowish oil, Rf 0.28 (EtOAc/hexanes/Et3N, 4:4:0.5). Yield 97 mg (0.41 mmol, 41%). 1H NMR (400 MHz, CDCl3): δ = 7.31 (t, J = 7.8 Hz, 2H), 7.25–7.13 (m, 4H), 7.11 (d, J = 6.7 Hz, 2H), 7.06–6.99 (m, 2H), 3.93–3.79 (m, 4H), 3.66 (s, 2H). 13C NMR (101 MHz, CDCl3): δ = 164.2, 141.2, 136.0, 129.4 (2C), 128.9 (2C), 128.5 (2C), 126.8, 125.6, 124.4 (2C), 53.3, 51.8, 34.6. IR (film, NaCl, cm−1): 3323, 3042, 2942, 1651, 1603, 1499, 1262; HRMS (ES TOF) calculated for C16H17N2 (M + H)+ 237.1386, found 237.1391 (−2.2 ppm).
1,2-Diphenyl-4,5-dihydro-1H-imidazole (4fa)
This compound was prepared according to the typical procedure employing N-phenyl-1,2-ethylenediamine (2a) (129 μL, 136 mg, 1.00 mmol), α-nitrotoluene (1f) (274 mg, 2.00 mmol), and phosphorous acid (500 mg). The crude material was purified by preparative column chromatography on silica gel eluting with acetone/EtOAc/Et3N (1:8:0.3). The titled compound was isolated as a yellowish oil, Rf 0.41 (acetone/EtOAc/Et3N, 1:8:0.3). Yield 40 mg (0.18 mmol, 18%). 1H NMR (400 MHz, CDCl3): δ = 7.51–7.45 (m, 2H), 7.38–7.32 (m, 1H), 7.30–7.24 (m, 2H), 7.16 (t, J = 7.9 Hz, 2H), 6.98 (t, J = 7.4 Hz, 1H), 6.79 (d, J = 7.6 Hz, 2H), 4.04 (s, 4H). 13C NMR (101 MHz, CDCl3): δ = 162.7, 143.1, 131.1, 130.1, 128.8 (2C), 128.8 (2C), 128.3 (2C), 123.5, 122.7 (2C), 54.1, 53.0. IR (film, NaCl, cm−1): 3299, 3050, 2946, 2862, 1599, 1495, 1386, 1270; HRMS (ES TOF) calculated for C15H15N2 (M + H)+ 223.1230, found 223.1233 (−1.2 ppm).
The same material was obtained employing α-nitroacetophenone (1g) (330 mg, 2.00 mmol) instead of α-nitrotoluene (1f). Yield 80 mg (0.36 mmol, 36%).
1-(4-Methoxyphenyl)-2-phenyl-4,5-dihydro-1H-imidazole (4fb)
This compound was prepared according to the typical procedure employing N-(4-methoxyphenyl)ethylenediamine (2b) (153 μL, 166 mg, 1.00 mmol), α-nitrotoluene (1f) (274 mg, 2.00 mmol), and phosphorous acid (500 mg). The crude material was purified by preparative column chromatography on silica gel eluting with acetone/EtOAc/Et3N (1:8:0.3). The titled compound was isolated as a yellowish oil, Rf 0.35 (acetone/EtOAc/Et3N, 1:8:0.3); Rf 0.25 (EtOAc/EtOH 1:1); Rf 0.57 (acetone/EtOAc/Et3N, 4:4:0.5). Yield 40 mg (0.16 mmol, 16%). 1H NMR (400 MHz, CDCl3): δ = 7.50 (d, J = 7.3 Hz, 2H), 7.37 (t, J = 7.4 Hz, 1H), 7.29 (d, J = 7.7 Hz, 2H), 6.87 (d, J = 8.9 Hz, 2H), 6.76 (d, J = 8.9 Hz, 2H), 4.14–4.05 (m, 4H), 3.75 (s, 3H). 13C NMR (101 MHz, CDCl3): δ = 164.1, 157.4, 134.9, 131.1, 129.2 (2C), 128.53, 128.47 (2C), 125.8 (2C), 114.6 (2C), 55.6, 55.2, 50.4. IR (film, NaCl, cm−1): 3002, 2840, 2363, 1615, 1511, 1388, 1248, 1179, 1039; HRMS (ES TOF) calculated for C16H17N2O1 (M + H)+ 253.1335, found 253.1339 (−1.5 ppm).
The same material was obtained employing α-nitroacetophenone (1g) (330 mg, 2.00 mmol) instead of α-nitrotoluene (1f). Yield 103 mg (0.41 mmol, 41%).
2-Phenyl-4,5-dihydro-1H-imidazole (4ff)
This compound was prepared according to the typical procedure employing N-benzyl-1,2-ethylenediamine (2c) (151 μL, 150 mg, 1.00 mmol), α-nitroacetophenone (1g) (330 mg, 2.00 mmol), and phosphorous acid (500 mg). The crude material was purified by preparative column chromatography on silica gel eluting with acetone/EtOAc/Et3N (4:4:0.3). The titled compound was isolated as a yellowish oil, Rf 0.35 (acetone/EtOAc/Et3N, 4:4:0.5). Yield 55 mg (0.38 mmol, 38%). 1H NMR (400 MHz, CDCl3): δ = 7.78 (d, J = 7.1 Hz, 2H), 7.40 (dt, J = 23.1, 7.2 Hz, 3H), 3.75 (s, 4H). 13C NMR (101 MHz, CDCl3): δ = 165.0, 130.9, 130.3, 128.5 (2C), 127.2 (2C), 50.1 (2C). IR (film, NaCl, cm−1): 3051, 2875, 2368, 1685, 1598, 1572, 1493, 1380, 1275; HRMS (ES TOF) calculated for C9H11N2 (M + H)+ 147.0917, found 147.0919 (−1.5 ppm).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Russian Foundation for Basic Research (grant #19-03-00308a) and grants from the Ministry of Education and Science of the Russian Federation #4.1196.2017/4.6 and #4.4589.2017/6.7.Notes and references
- D. J. Reis and S. Regunathan, J. Auton. Nerv. Syst., 1998, 72, 80–85 CrossRef CAS PubMed.
- I. F. Musgrave and R. A. Hughes, Ann. N. Y. Acad. Sci., 1999, 881, 301–312 CrossRef CAS PubMed.
- V. Ozola, M. Thorand, M. Diekmann, R. Qurishi, B. Schumacher, K. A. Jacobson and C. E. Muller, Bioorg. Med. Chem., 2003, 11, 347–356 CrossRef CAS.
- I. H. Gilbert, D. C. Rees, A. K. Crockett and R. C. F. Jones, Tetrahedron, 1995, 51, 6315–6336 CrossRef CAS.
- I. K. Khanna, R. M. Weier, Y. Yu, X. D. Xu, F. J. Koszyk, P. W. Collins, C. M. Koboldt, A. W. Veenhuizen, W. E. Perkins, J. J. Casler, J. L. Masferrer, Y. Y. Zhang, S. A. Gregory, K. Seibert and P. C. Isakson, J. Med. Chem., 1997, 40, 1634–1647 CrossRef CAS PubMed.
- R. Gust, R. Keilitz and K. Schmidt, J. Med. Chem., 2001, 44, 1963–1970 CrossRef CAS.
- M. Mor, F. Bordi, C. Silva, S. Rivara, V. Zuliani, F. Vacondio, G. Morini, E. Barocelli, V. Ballabeni, M. Impicciatore and P. V. Plazzi, Farmaco, 2000, 55, 27–34 CrossRef CAS.
- V. Sharma, T. A. Lansdell, S. Peddibhotla and J. J. Tepe, Chem. Biol., 2004, 11, 1689–1699 CrossRef CAS PubMed.
- L. M. Azevedo, T. A. Lansdell, J. R. Ludwig, R. A. Mosey, D. K. Woloch, D. P. Cogan, G. P. Patten, M. R. Kuszpit, J. S. Fisk and J. J. Tepe, J. Med. Chem., 2013, 56, 5974–5978 CrossRef CAS PubMed.
- G. A. Boswell, H.-Y. Li, I. Delucca, J. T. Billheimer, S. Drummond, P. J. Gillies and C. Robinson, Bioorg. Med. Chem. Lett., 1996, 6, 885–888 CrossRef CAS.
- M. Krasavin, Eur. J. Med. Chem., 2015, 97, 525–537 CrossRef CAS PubMed.
- M. Krasavin, Chem. Heterocycl. Compd., 2017, 53, 240–255 CrossRef CAS.
- Y. V. Popov, V. M. Mokhov and I. I. Kalitina, Russ. J. Gen. Chem., 2016, 86, 281–285 CrossRef CAS.
- Y. Liu, H. He, B.-F. Yang, Q. Zhang and G.-Y. Yang, J. Cluster Sci., 2015, 26, 1537–1545 CrossRef CAS.
- X. Sun, J. Qiu, S. A. Strong, L. S. Green, J. W. F. Wasley, J. P. Blonder, D. B. Colagiovanni, S. C. Mutka, A. M. Stout, J. P. Richards and G. J. Rosenthal, Bioorg. Med. Chem. Lett., 2011, 21, 5849–5853 CrossRef CAS PubMed.
- J. H. Forsberg, V. T. Spaziano, T. M. Balasubramanian, G. K. Liu, S. A. Kinsley, C. A. Duckworth, J. J. Poteruca, P. S. Brown and J. L. Miller, J. Org. Chem., 1987, 52, 1017–1021 CrossRef CAS.
- A. V. Aksenov, N. A. Aksenov, O. N. Nadein and I. V. Aksenova, Synlett, 2010, 2628–2630 CrossRef CAS.
- N. A. Aksenov, A. V. Aksenov, O. N. Nadein, D. A. Aksenov, A. N. Smirnov and M. Rubin, RSC Adv., 2015, 5, 71620–71626 RSC.
- A. V. Aksenov, N. A. Aksenov, N. A. Orazova, D. A. Aksenov, M. V. Dmitriev and M. Rubin, RSC Adv., 2015, 5, 84849–84855 RSC.
- A. V. Aksenov, A. N. Smirnov, N. A. Aksenov, A. S. Bijieva, I. V. Aksenova and M. Rubin, Org. Biomol. Chem., 2015, 13, 4289–4295 RSC.
- A. V. Aksenov, N. A. Aksenov, D. S. Ovcharov, D. A. Aksenov, G. Griaznov, L. G. Voskressensky and M. Rubin, RSC Adv., 2016, 6, 82425–82431 RSC.
- A. V. Aksenov, D. S. Ovcharov, N. A. Aksenov, D. A. Aksenov, O. N. Nadein and M. Rubin, RSC Adv., 2017, 7, 29927–29932 RSC.
- A. V. Aksenov, N. A. Aksenov, D. S. Ovcharov, S. V. Shcherbakov, A. N. Smirnova, I. V. Aksenova, V. I. Goncharov and M. A. Rubin, Russ. J. Org. Chem., 2017, 53, 1081–1084 CrossRef CAS.
- A. V. Aksenov, V. Khamraev, N. A. Aksenov, N. K. Kirilov, D. A. Domenyuk, V. A. Zelensky and M. Rubin, RSC Adv., 2019, 9, 6636–6642 RSC.
- J. U. Nef, Justus Liebigs Ann. Chem., 1894, 280, 263–291 CrossRef CAS.
- R. Ballini and M. Petrini, Adv. Synth. Catal., 2015, 357, 2371–2402 CrossRef CAS.
- R. Ballini and M. Petrini, Tetrahedron, 2004, 60, 1017–1047 CrossRef CAS.
- A. V. Aksenov, N. A. Aksenov, D. A. Aksenov, V. F. Khamraev and M. Rubin, Chem. Commun., 2018, 54, 13260–13263 RSC.
- A. L. Huhti and P. A. Gartaganis, Can. J. Chem., 1956, 34, 785–797 CrossRef CAS.
- F. Uhlig, Angew. Chem., 1954, 66, 435–436 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra08630g |
| This journal is © The Royal Society of Chemistry 2019 |