DOI:
10.1039/C9RA08585H
(Paper)
RSC Adv., 2019,
9, 40424-40436
Electrocatalytic water oxidation by a Ni(II) salophen-type complex†
Received
20th October 2019
, Accepted 28th November 2019
First published on 6th December 2019
Abstract
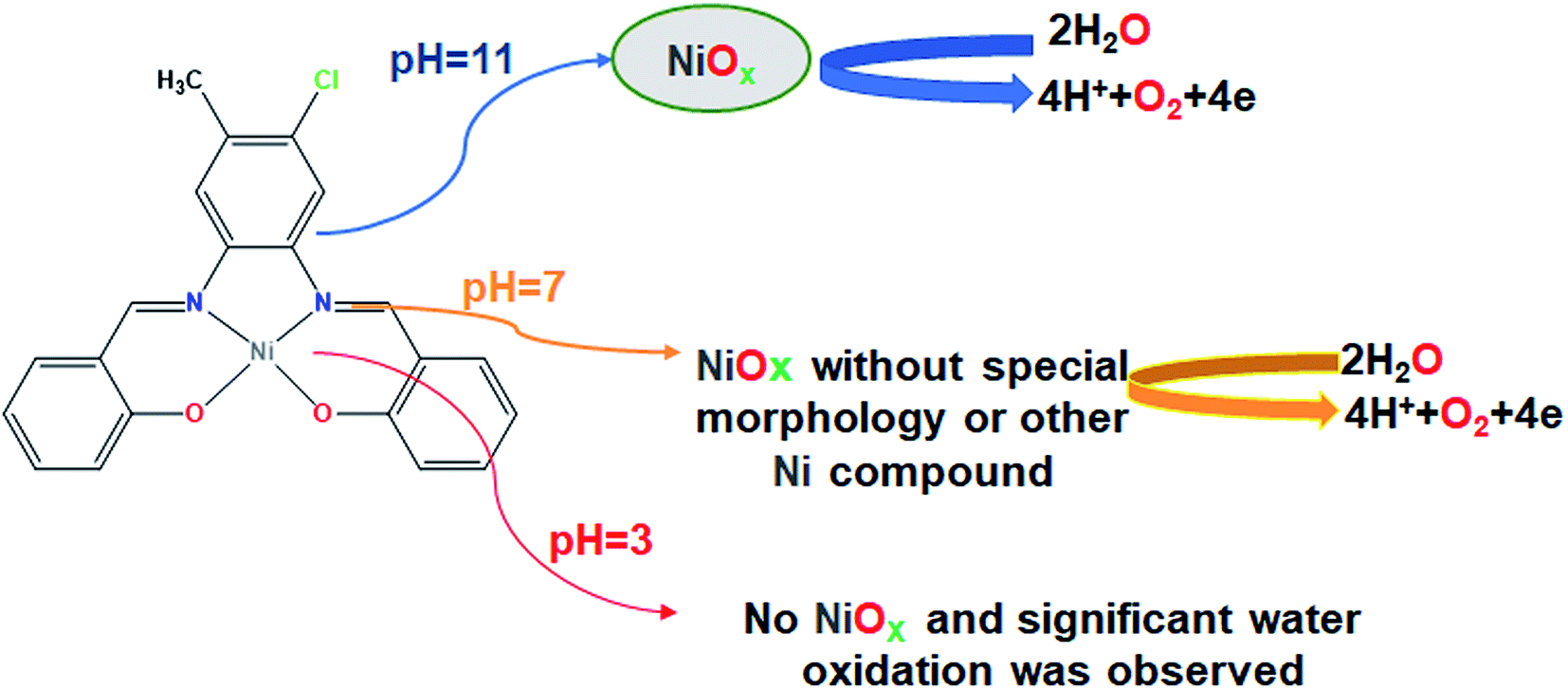
A new mononuclear Ni(II) complex, NiL (1), was synthesized from the reaction of Ni(OAc)2·4H2O and salophen-type N2O2-donor ligand, H2L (where H2L = 2,2′-((1E,1′E)-((4-chloro-5-methyl-1,2-phenylene)bis(azanylylidene))bis(methanylylidene))diphenol), in ethanol. The obtained complex was characterized by elemental analysis, spectroscopic techniques and single crystal X-ray analysis. The complex was studied as a water oxidizing catalyst and its electrocatalytic activity in the water oxidation reaction was tested in 0.5 M of borate buffer at pH = 3, 7 and 11 in a typical three-electrode setup with a carbon paste electrode modified by complex 1 as a working electrode. The linear sweep voltammetry (LSV) curves indicated that complex 1 has a much superior activity and only needs 21 mV vs. Ag/AgCl overvoltage to reach a geometrical catalytic current density of 2.0 mA cm−2 at pH = 11. The onset potential decreased from 1.15 V to 0.67 V vs. Ag/AgCl with an increase of pH from 3 to 13 under a constant current density of 1.0 mA cm−2. Then, to determine the true catalyst for the water oxidation reaction in the presence of complex 1 at pH = 3, 7 and 11, cyclic voltammetry was also performed. The continuous CVs for complex 1 at neutral and alkaline solutions showed significant progress for the water oxidation reaction. In addition, the amperometry tests exhibited excellent stability and high constant current density for water oxidation by CPE-complex 1 under electrochemical conditions at pH = 11 and 7. Although X-ray powder diffraction analysis did not show a pure and crystalline structure for NiOx, the scanning electron microscopy images showed that nickel oxide at pH = 11 and nickel oxide or other Ni-based compounds at pH = 7 are true water oxidizing catalysts on the surface of a CPE electrode. Moreover at pH = 3, no clear water oxidation or NiOx formation was observed.
Introduction
The lack of fossil fuels and many environmental problems such as increasing greenhouse gases, release of pollutants etc., which are produced in the burning of these fuels, reveals the need for new and renewable energy resources.1 For this reason, hydrogen is considered as one of the most important carriers of energy because of its renewable, cheap and clean nature. In recent decades, many efforts have been made to explore reliable methods for the production of hydrogen and oxygen by oxygen evolution and hydrogen evolution reactions (OER and HER) through water splitting by various types of homogenous and heterogeneous catalysts.2 Homogenous water oxidation catalysts are useful in the examination of reaction mechanisms and identification of the active intermediates which will give an opportunity for the precise designing of catalysts, while water insoluble catalysts can allow for easier post-reaction separation and recyclability.3
Water splitting can be divided two half-reactions: proton reduction (2H+ + 2e → H2) and water oxidation (2H2O → 4H+ + O2 + 4e). Water oxidation is a slow electrochemical reaction with a high potential of about 1.23 eV. The oxidation of water to oxygen is a 4e/4H+ process which requires a large overpotential for the O–O bond formation. So, water oxidation has high activation energy and this matter is considered as a limiting step in water splitting.4 On the other hand, at this high voltage the remaining chemical compounds in water, including some ions such as chloride and sulphate, can also be oxidized. Therefore, the design of suitable catalysts to reduce water oxidation potential seems essential. Due to this, the development of stable and cheap water oxidation catalysts is an important challenge for the design of water splitting systems.5 Recently, molecular water oxidation catalysts based on first row transition metals have attracted considerable attention because of their low cost, high abundance and low environmental effects.6 Previous studies indicate that the coordination compounds containing manganese,7 iron,8 nickel,9 cobalt10 and copper ions11 can oxidize water under different conditions. Among water oxidation catalysts based on 3d transition metals, Ni-containing materials have attracted particular attention due to their good water oxidation potential and high oxidation power.12 In fact, Ni-containing mixed-metal oxides or hydroxides are one of the efficient water oxidation catalysts.13 Ni complexes are often investigated as heterogeneous water oxidation catalysts and few reports are available about nickel complexes as homogenous catalysts. For example, Ding groups have been reported mononuclear Ni(II) complexes containing tetradentate N4−, N3O− or N2O-donor ligands (C12H10N4NiO4·4H2O, C11H7N3NiO5·5H2O and C10H4N2NiO6·6H2O). They found that C12H10N4NiO4·4H2O can act as a homogenous molecular catalyst, but two other complexes decompose to NiOx nanoparticles and in fact act as precatalyst for electrochemical water oxidation.14 Najafpour and co-workers presented a tetranuclear Ni(II) complex with bis-[(E)-N,N′-(1-(pyridin-2-yl)ethylidene)]carbohydrazide which catalyzes the oxidation of water. They realized that under water oxidation condition in the presence of this complex, nickel oxide or free Ni(II) ions oxidize water.15 Feizi et al.16 investigated the activity of N,N′-bis-(salicylidene)ethylenediamino nickel(II) at three different pH. It was revealed that the complex is very active for water oxidation at a high pH. Zhang et al.17 designed a Ni(II) complex [Ni(meso-L)] (ClO4)2 (L = 5,5,7,12,12,14-hexamethyl-1,4,8,11-tetraazacyclotetradecane) as a homogenous electrocatalyst for water oxidation which could act at neutral pH and low overpotential in phosphate buffer. Barros et al.18 investigated water oxidation activity of a family of Ni(II) complexes containing oxamidate anionic type of ligands in basic pH. They found that during the water oxidation process, the degradation of the molecular species generates a NiOx layer which is stable and active as electrocatalyst. Han et al.19 reported the water soluble Ni(II) complex of meso-tetrakis(4-N-methylpyridyl)porphyrin which could electrocatalyze water oxidation in neutral aqueous solution with the onset of the catalytic wave appearing at ∼1.0 V (vs. NHE). Sun groups designed a Ni-Py5 [Py5 = 2,6-bis(1,1-bis(2-pyridyl)ethyl)pyridine)] complex which could act as a homogenous electrocatalyst for oxidizing water in aqueous phosphate buffer solutions.20 Hill's group presented a nickel-containing polyoxometalate [Ni5(OH)6(H2O)3(Si2W18O66)]12− which oxidized water in the presence of [Ru(bpy)3]3+. The evidences showed that this catalyst remains molecular throughout and does not form a nickel hydroxide or nickel oxide under the conditions of catalytic water oxidation reaction.21 According to the literature,15,16,22 free Ni(II) ions or Ni oxides can catalyze the water oxidation reaction at low concentration and high pH. There are always some key questions in this regard: “do the Ni complexes molecularly catalyze the oxidation of water?” or “whether the complexes decompose under water oxidation or convert to Ni oxide which catalyzes the water oxidation reaction?” and most importantly, “which is the true catalyst for water oxidation? Metal complex, impurity or decomposed complex?” Determination of this is not easy, and thus leads to a great deal of encouragement by researchers for the preparation of new electrocatalysts of nickel complexes. Here, we reported synthesis and crystal structure of a new Ni(II) complex with salophen-type ligand, (H2L = (2,2′-((1E,1′E)-((4-chloro-5-methyl-1,2-phenylene)bis(azanylylidene))bis(methanylylidene))diphenol). We also investigated the application of this complex for electrochemical water oxidation at pH = 3, 7 and 11. By using various methods we showed that the designed Ni(II) complex is an efficient electrocatalyst for water oxidation in neutral and basic solutions. Researches show that nickel complexes exhibit high activity for water oxidation reaction in basic or high pH conditions in phosphate and borate buffers.18,23 There are few reports of nickel complexes which act as active electrocatalyst for the oxidation of water in neutral pH, and most of them are homogeneous catalysts of nickel complexes in the presence of phosphate buffer.17,19,24 A unique feature of this work compared to previous works is the design of a new heterogeneous electrocatalyst from nickel complexes that can catalyze water oxidation reaction in neutral solutions as well as basic solutions.
Results and discussion
Synthesis and characterization
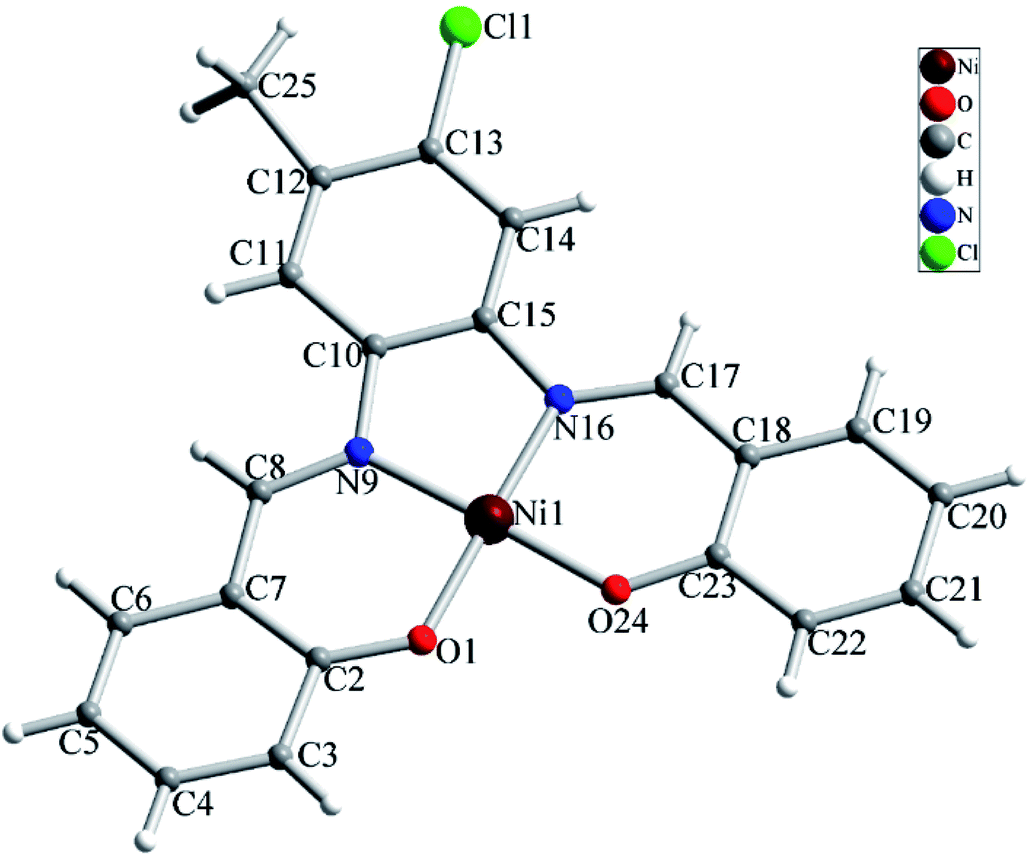
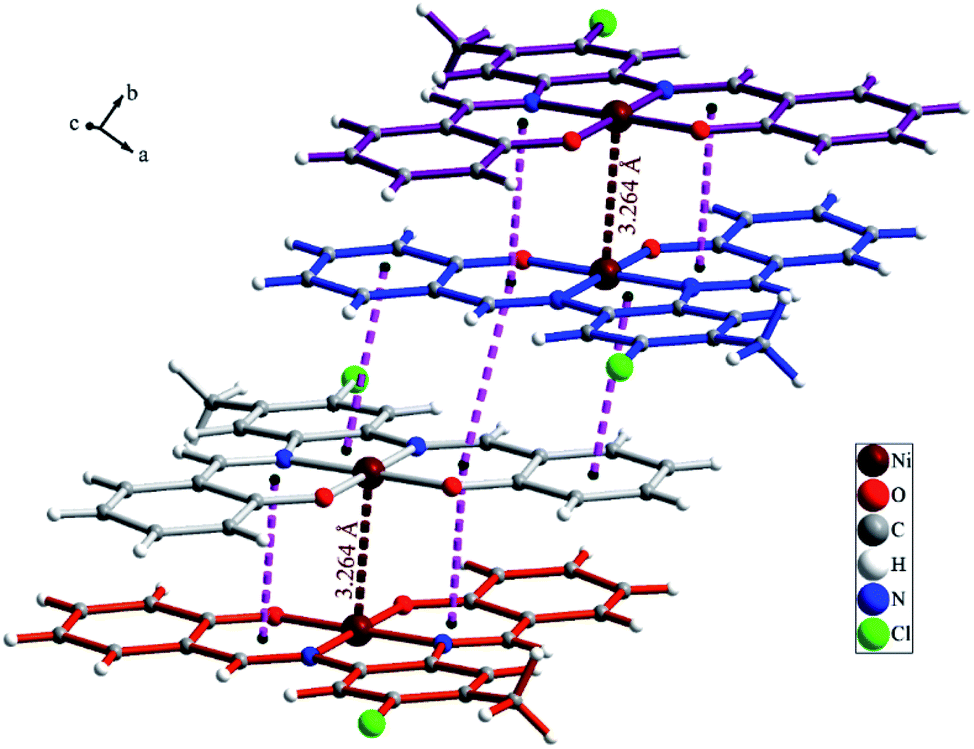
The NiL (1) complex is prepared by the reaction of the salophen-type Schiff-base ligand, H2L (2,2′-((1E,1′E)-((4-chloro-5-methyl-1,2-phenylene)bis (azanylylidene))bis(methanylylidene))diphenol) with Ni(OAc)2·4H2O and characterized by single crystal X-ray analysis and spectroscopic techniques as well as elemental analysis. The molecular structure of complex 1 with atom numbering scheme is shown in Fig. 1 and selected bond lengths and angles around the Ni atom are summarized in Table 1. Structural studies indicated that 1 is a mononuclear Ni(II) complex which crystallizes in P21/c space group of monoclinic system. Complex 1 is a neutral coordination compound and the Ni(II) ion has a square planar cis-NiN2O2 coordination environment. This coordination environment is generated by coordination of two phenolate oxygen and two azomethine nitrogen atoms from the salophen-type N2O2-donor ligand. The Ni–N and Ni–O bond lengths are in the normal range observed for Ni(II) complexes. Due to the coordination of ligand to metal ion a five membered ring (NiN2C2) and two six membered heterocyclic rings containing Ni, N and O atoms (NiC3NO) have been generated in the structure of complex. There is a strong Ni⋯Ni interaction in the crystal structure of complex 1 with Ni⋯Ni distance of 3.264(2) Å (see Fig. 2). Moreover, there are several π⋯π stacking interactions between the phenyl ring and heterocyclic rings containing Ni(II) ion which are shown in Fig. 3. The crystal structure of complex is further stabilized by intermolecular C–H⋯O and C–H⋯Cl interactions (see Fig. S1 and Table S1†).
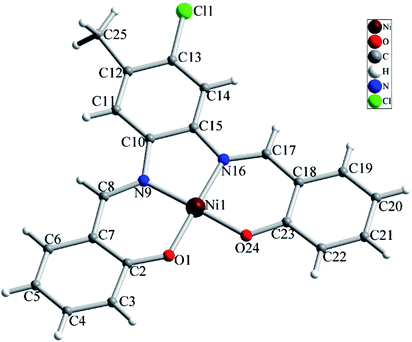 |
| | Fig. 1 Molecular structure of complex 1 with atom numbering scheme. | |
Table 1 Selected bond lengths and angles in the crystal structure of complex 1
| Bond |
Length/Å |
Bond |
Angle/° |
| Ni1–O24 |
1.817(8) |
O24–Ni1–O1 |
83.7(3) |
| Ni1–O1 |
1.834(7) |
O24–Ni1–N9 |
178.2(4) |
| Ni1–N9 |
1.837(9) |
O1–Ni1–N9 |
94.5(4) |
| Ni1–N16 |
1.849(9) |
O24–Ni1–N16 |
95.6(4) |
| C8–N9 |
1.298(14) |
O1–Ni1–N16 |
178.9(4) |
| N16–C17 |
1.302(14) |
N9–Ni1–N16 |
86.1(4) |
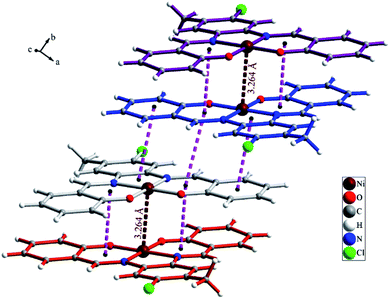 |
| | Fig. 2 Ni⋯Ni and π⋯π interactions in the crystal structure of complex 1. | |
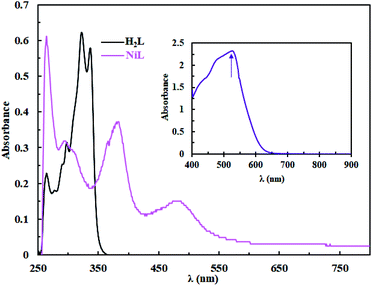 |
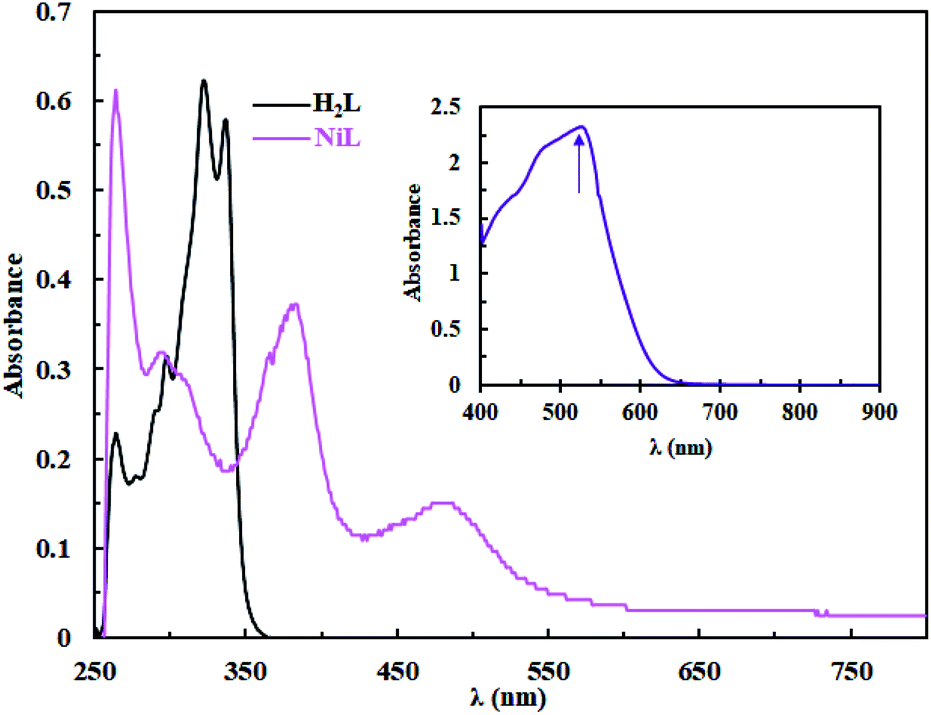
| | Fig. 3 UV/Vis spectra of H2L and complex 1 in DMSO (3 × 10−5 M); inset: d → d transitions of complex 1 (10−3 M). | |
Ni complex 1 is further characterized by FT-IR and 1H NMR spectroscopy. FT-IR spectroscopy data shows that after complexation with nickel, the absorption of imine vibration shifts from 1614 cm−1 for the ligand to shorter wavenumbers about 6 cm−1 for the complex. This suggests the coordination of the electron pair of nitrogen atoms of imine groups to the metal ion center.25,26 Also, the disappearance of the broad absorption band at 3447 cm−1 indicates that the phenolic hydroxyl groups are deprotonated when bound to the Ni(II) ion. In additional, the ν(M–O) and ν(M–N) are identified in the range of 449–595 cm−1. Finally, the medium peaks about 752 cm−1 are related to chlorine (C–Cl) (Fig. S2†).
In the 1H NMR spectrum of the complex, the singlet resonances corresponding to the none-equivalent –OH protons, which are observed at 12.70 ppm and 12.77 ppm for the ligand, are disappear. This indicates that the phenolic hydroxyl groups are deprotonated when bound to a Ni(II) ion. Also, in comparison with the ligand, the imine proton resonances shift to downfield and appear at 9.02 ppm and 8.97 ppm which confirm the coordination of nitrogen atoms of imine groups to the Ni(II) center (in the 1H NMR spectrum of the ligand, the two none-equivalent imine proton resonances are observed at 8.95 ppm and 8.99 ppm).27,28 Moreover, the aromatic protons are observed at 6.73–8.32 ppm and the protons of –CH3 group appear at 2.14 ppm (Fig. S3†).
The UV/Vis spectra of H2L and complex 1 in DMSO solution (3 × 10−5 M) are shown in Fig. 3. The absorption spectra exhibit intense bands in the 258–337 nm region, which is attributed to the π → π* and n → π* intra-ligand transitions.26 The intensity of n → π* transition decreases after complex formation which can be attributed to the coordination of the azomethine groups to the metal ion. For complex 1, the absorption bands at 385 nm and 483 nm can be assigned to the L → M charge transfers transitions (LMCT).26,29 It should be noted that d → d transition appears as a low intensity board absorption band at 531 nm. In addition, the thermal stability of the complex was investigated by thermogravimetric analysis (TGA) under N2 atmosphere. The TGA plot of Ni(II) complex shows that the complex is stable up to 400 °C with a DTGmax = 410 °C and the highest weight loss occurs in the range of 590–800 °C which corresponds to the decomposition of the residue aromatic groups and the formation of metal oxide lattice (see Fig. S4†).
Electrochemistry and water oxidation
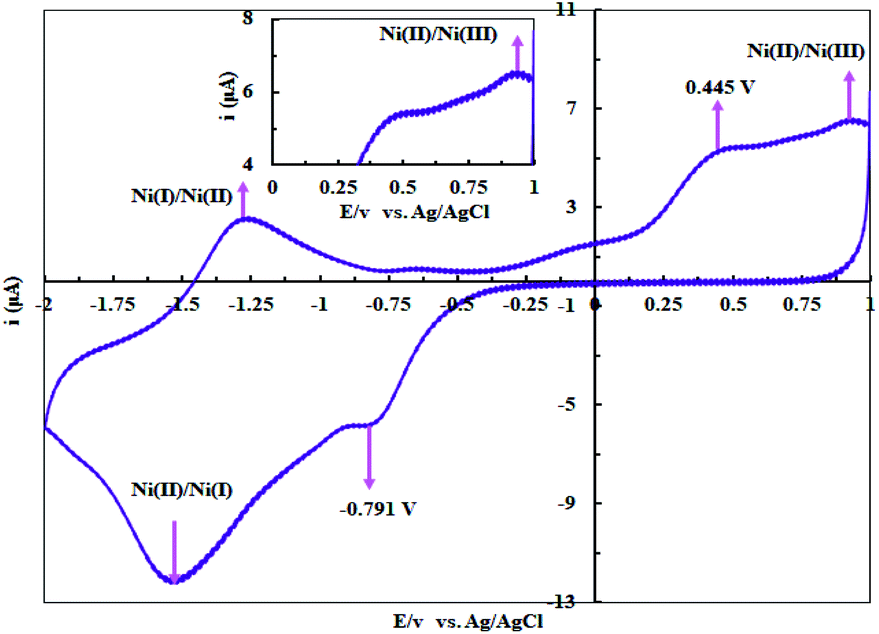
The +2 oxidation state is a common oxidation state for Ni complexes while the other oxidation states (like +1, +3 and +4) are rarely reported.30 According to the literature,31 Ni(II) salen complex (salen = N,N′-bis-(salicylidene)-1,2-ethyleneamine) shows Ni(II)/Ni(III) oxidation at +0.71 V vs. SCE and Ni(I)/Ni(II) oxidation at −1.61 V vs. SCE in DMSO. The Ni(II) complexes with salophen ligand (salophen = N,N′-bis-(salicylidene)-1,2-phenylenediamine) and its phenyl-substituted counterparts display chemically reversible Ni(II)/Ni(III) oxidation. Fig. 4 shows the voltammogram of complex 1 in DMSO solution (10−3 M) containing 0.1 M LiClO4 as a supporting electrolyte at room temperature in a potential range of +1.00 to −2.00 V vs. Ag/AgCl at 50 mV s−1 potential scan rate (the glassy carbon, the Pt wire and the Ag/AgCl electrodes were used as working, counter and reference electrodes, respectively). Complex 1 exhibits a one electron oxidation at 0.934 V vs. Ag/AgCl which can be assigned to the Ni(II) → Ni(III) oxidation process. The anodic peak at −1.25 V and the cathodic peak at −1.51 V are attributed to the redox couple of Ni(I)/Ni(II) with half wave potential of −1.38 V vs. Ag/AgCl. Moreover, the anodic peak at 0.445 V vs. Ag/AgCl and the cathodic peak at −0.791 V vs. Ag/AgCl can be related to the irreversible oxidation and reduction of the ligand.
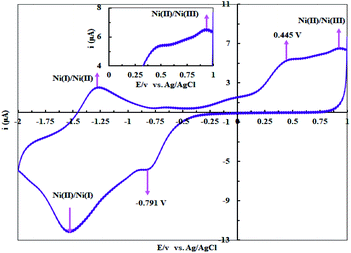 |
| | Fig. 4 The cyclic voltammogram of complex 1 (10−3 M) in DMSO solution containing 0.1 M LiClO4 as a supporting electrolyte at room temperature in a potential rang of +1.0 to −2.0 V vs. Ag/AgCl at 50 mV s−1 potential scan rate. | |
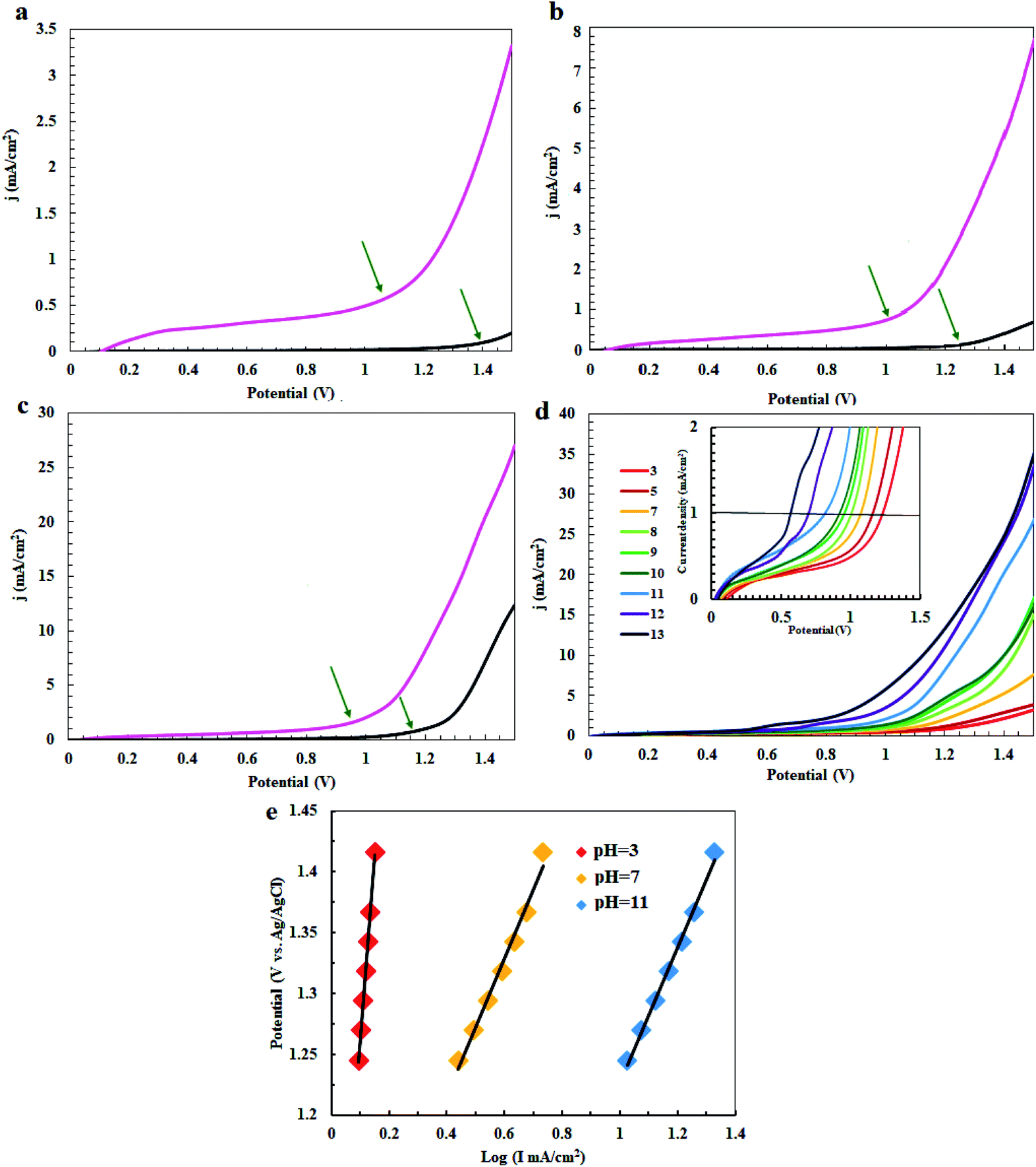
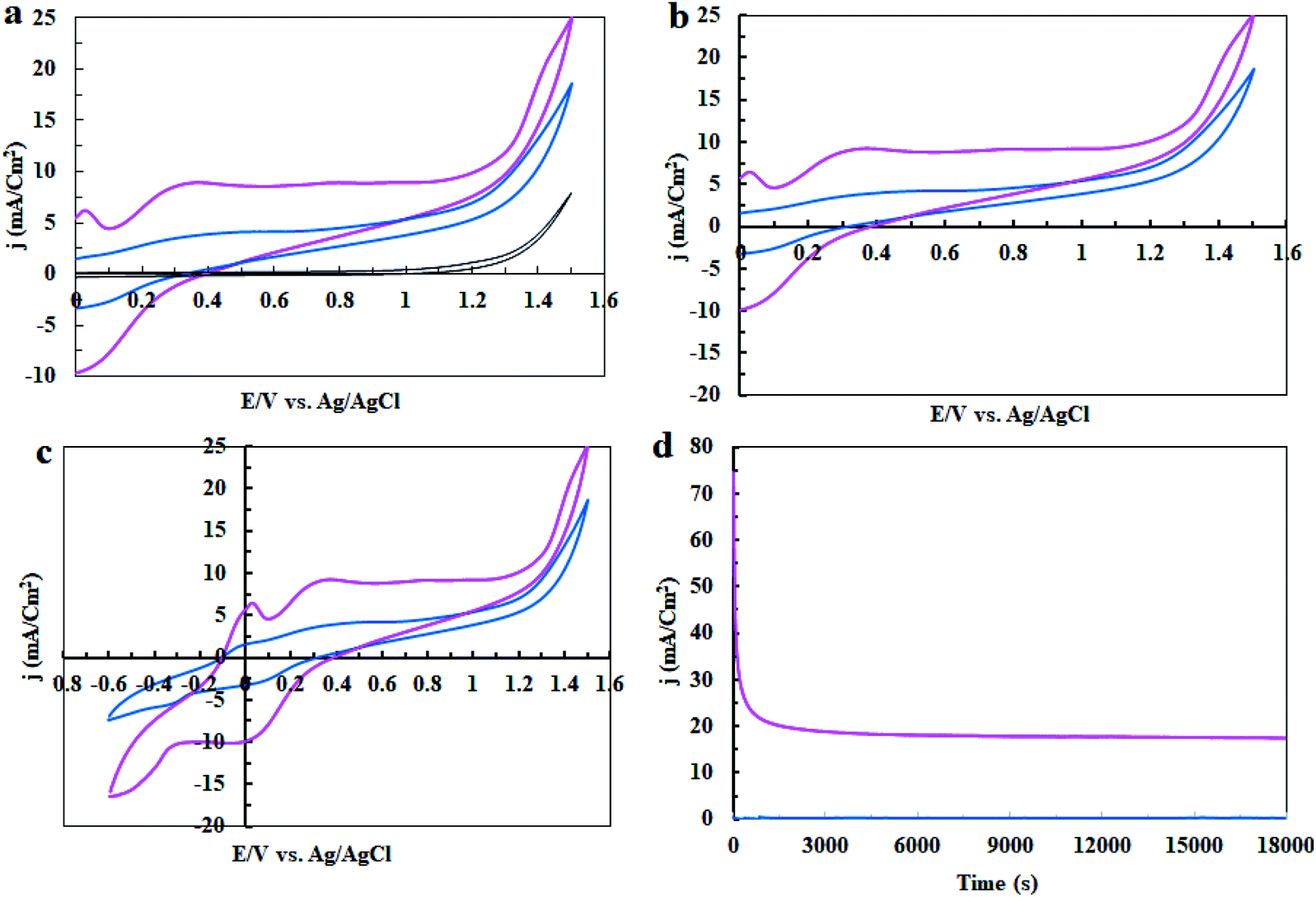
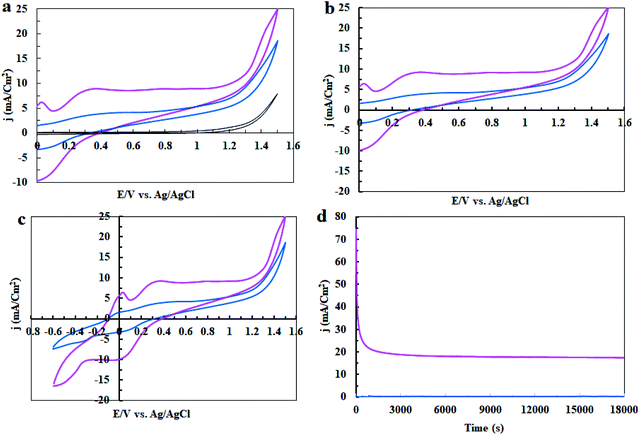
In the next step, the electrocatalytic activity of complex 1 for water oxidation was tested in 0.5 M of borate buffer at pH = 3, 7 and 11 in a typical three-electrode setup with a scan rate of 50 mV s−1. Fig. 5 shows the linear sweep voltammetry (LSV) curves of bare carbon paste electrode (CPE) and carbon paste electrode modified with complex 1 (CPE-complex 1) in a borate buffer solution and at pH = 3, 7 and 11. The bare CPE shows very poor water oxidation activity with the need of large overpotential 500, 470 and 290 mV vs. Ag/AgCl to drive 0.7, 0.8 and 2.0 mA cm−2 as the selected current density in pH = 3, 7 and 11, respectively, while complex 1 shows a good activity and only demands overvoltage of 140, 35 and 21 mV vs. Ag/AgCl for approaching 0.7, 0.8 and 2.0 mA cm−2 as the selected current density under the same conditions (Fig. 5a–c). Therefore, complex 1 exhibits a much superior activity and only needs 21 mV overvoltage to reach a geometrical catalytic current density of 2.0 mA cm−2 at pH = 11. Also, as expected, the onset potential decreases from 1.15 V to 0.67 V with an increase of pH from 3 to 13 under constant current density of 1.0 mA cm−2 (Fig. 5d). Tafel plots of CPE-complex 1 electrocatalyst at pH = 3, 7 and 11 are shown in Fig. 5e. The Tafel slopes increase for complex 1 in the following order: pH = 3 > pH = 7 > pH = 11. LSV results indicate a higher current density for CPE-complex 1 in pH = 11 than the others at a certain applied potential. The current density at pH = 3 rises more rapidly with the increase in voltage. The small Tafel slope for CPE modified with complex 1 at pH = 7 and 11 shows that the electron and mass transfers on the surface of the electrode are easier than pH = 3.
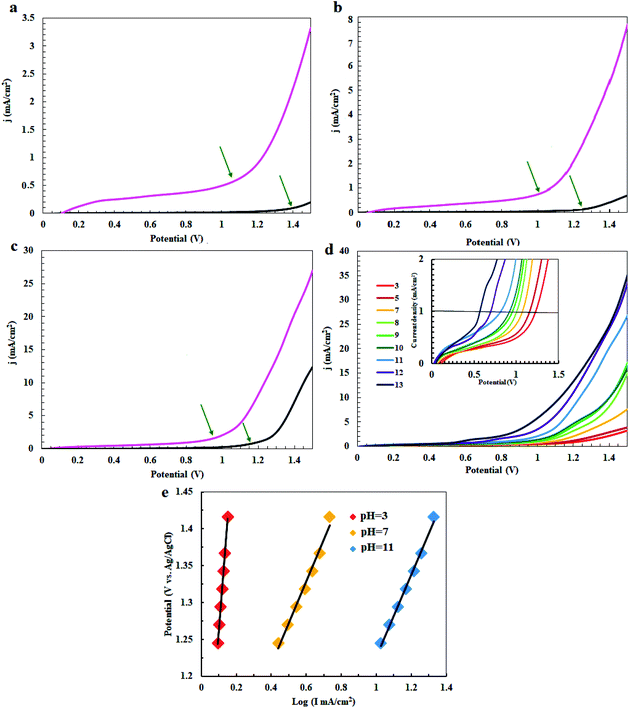 |
| | Fig. 5 Linear sweep voltammetry (LSV) curves of a fresh CPE (black) and CPE modified with complex 1 (pink) in buffer solution at pH = 3 (a), pH = 5 (b), pH = 11 (c), LSV curves for CPE-complex 1 at pH = 3–13 (d) and Tafel plots of CPE-complex 1 at pH = 3, 7 and 11 (e). | |
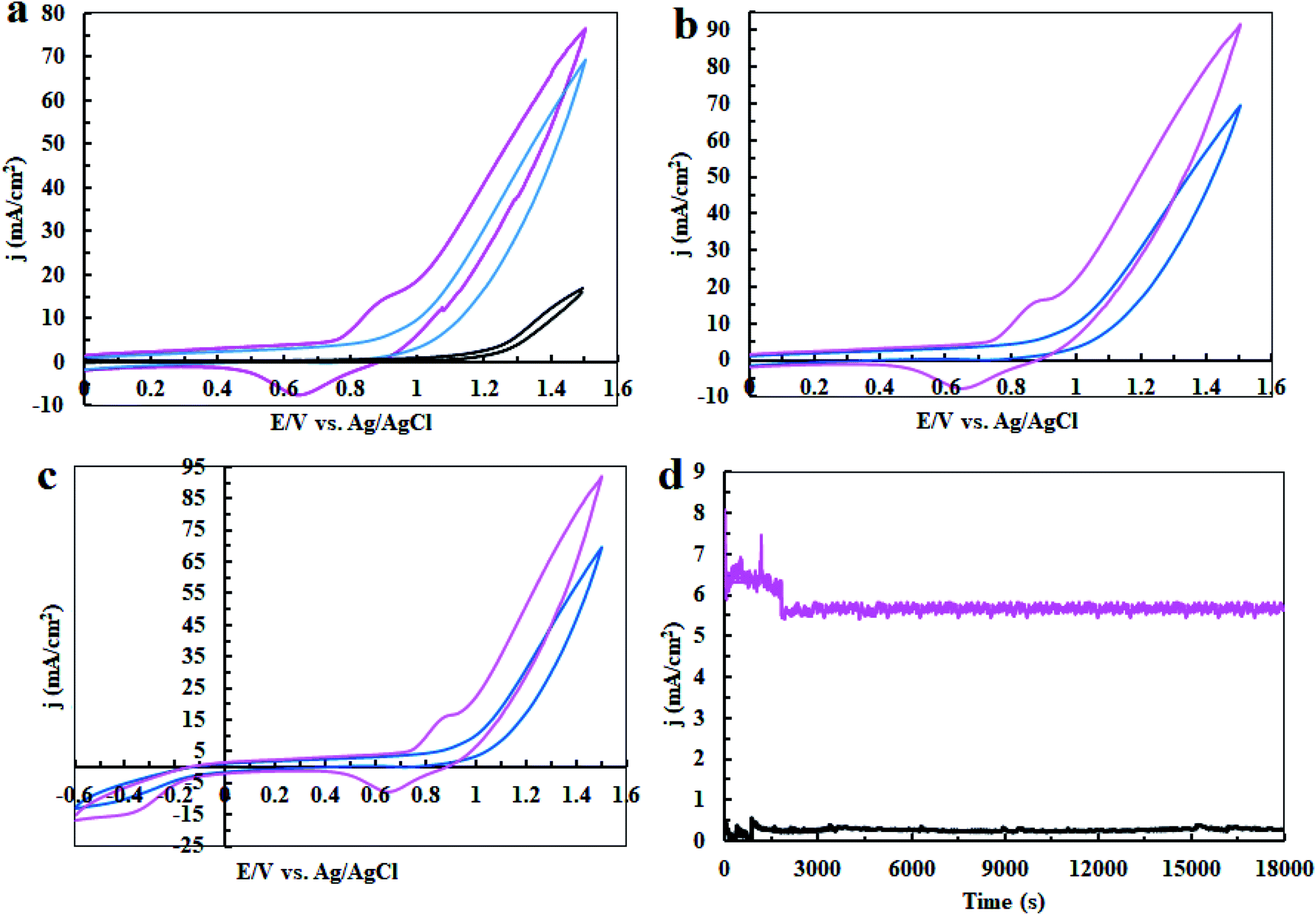
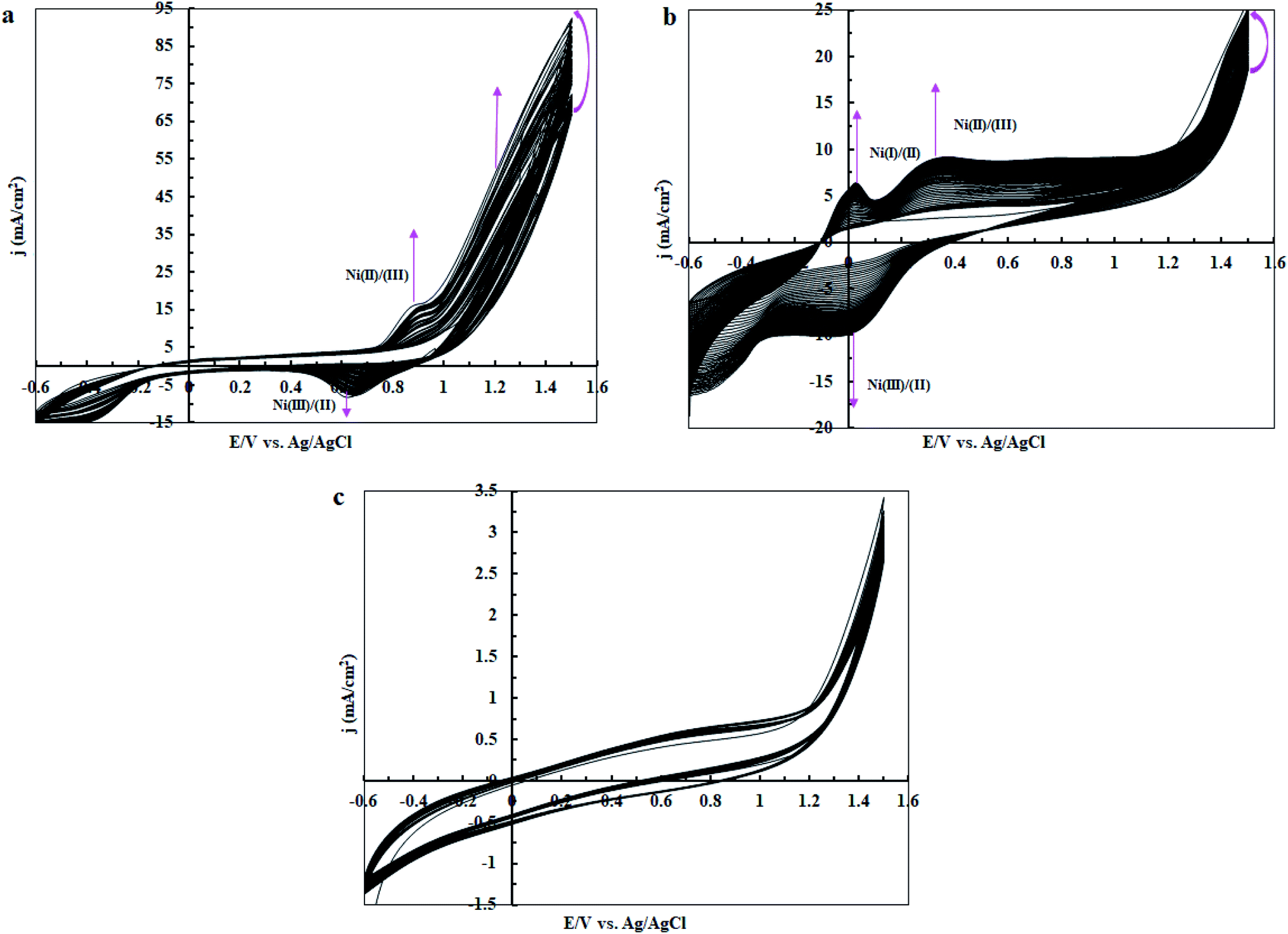
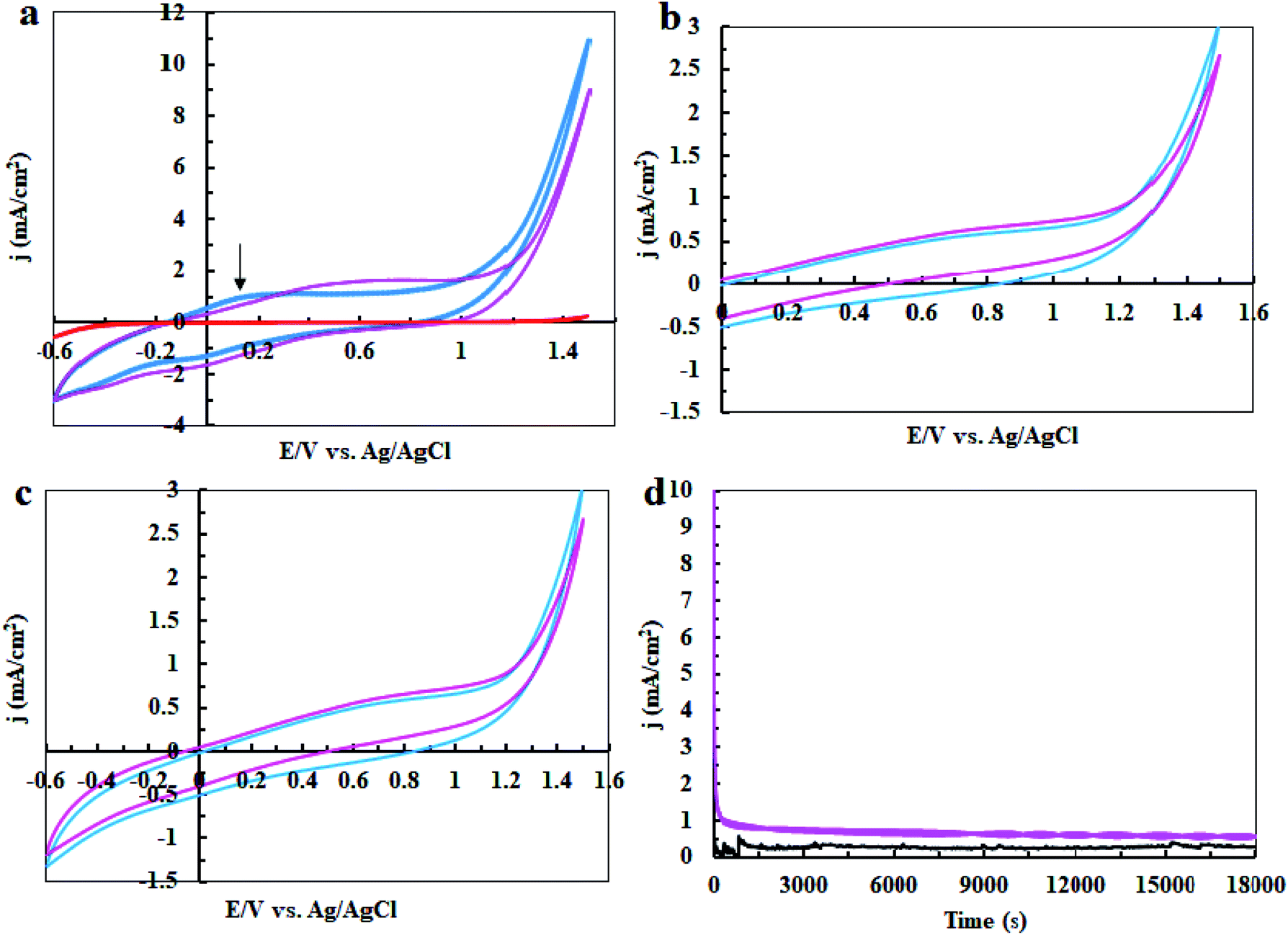
In the next step, cyclic voltammetry studies were conducted at pH = 3, 7 and 11 in order to determine the true catalyst for water oxidation reaction in the presence of complex 1. The results are presented in Fig. 6, 7, 10 and 13. As shown in Fig. 6a, the modified CPE with complex 1 is a water oxidizing catalyst in comparison to a fresh CPE at pH = 11. After carrying out the amperometry for a period of 5 hours at 1.12 V vs. Ag/AgCl, CPE-complex 1 becomes an efficient electrode for water oxidation. According to the literature,16 the increase in the efficiency of the CPE-complex 1 for oxidizing of water can be attributed to the formation of a solid on the surface of the electrode. With continuous CVs for complex 1, two new peaks at 0.887 V and 0.633 V (vs. Ag/AgCl) were formed (Fig. 6b and c), which can be attributed to redox couple of Ni(II)/Ni(III).16 The continuous CV also causes an increase in the water oxidation reaction for CPE-complex 1. It is likely that under continues CV in the presence of complex 1, NiO is produced which rises the oxidation of water. The amperometry (1.12 V vs. Ag/AgCl) for a fresh carbon paste electrode reveals low activity for water oxidation (Fig. 6d), but a CPE-complex 1 shows an increase due to Ni(II)/Ni(III) oxide formation. Then, a decrease and constant current density is observed (Fig. 6d). Finally, the continuous CVs (50 cycles) at pH = 11 for CPE-complex 1 are shown in Fig. 7a. Two peaks attributed to the oxidation of Ni(II)/Ni(III) and reduction of Ni(III)/Ni(II) are observed at 0.887 V and 0.633 V, respectively (E1/2 = 0.76 V vs. Ag/AgCl and ΔE = 0.254 V) for CPE-complex 1. Generally, the continuous CV displays a growth for Ni(II)/Ni(III) and water oxidation (Fig. 7a). According to the literature,15,16 it seems that a significant change slowly occurs for complex 1 during the continuous CV which can be related to the deposition of material on the surface of CPE.
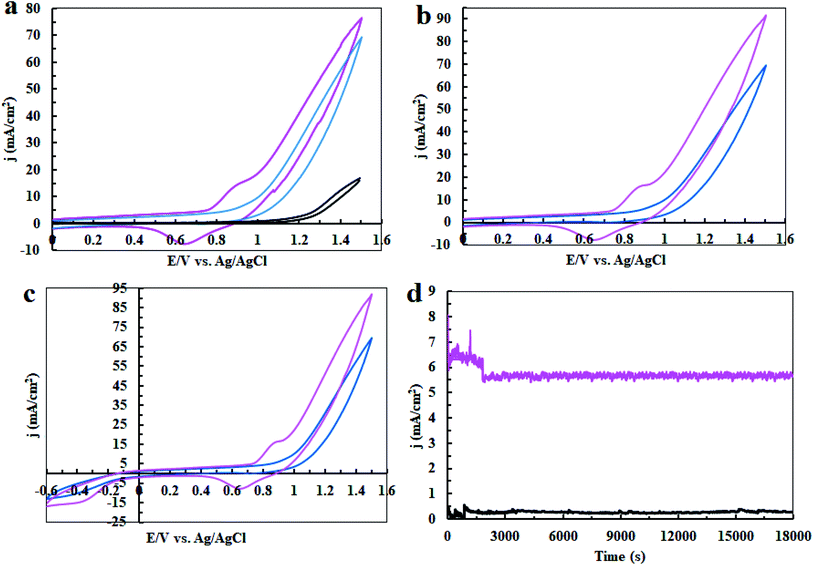 |
| | Fig. 6 Cyclic Voltammetry (CV) for a fresh CPE (black), CPE-complex 1 (blue) and CPE-complex 1 after performing amperometry of 5 hours (pink) in the buffer solution (0.5 M) in the range of 0–1.6 V (a); CV conditions: buffer solution at pH = 11 and scan rate of 50 mV s−1; amperometry conditions: 1.12 V vs. Ag/AgCl; CPE-complex 1 in a buffer solution (25 ml) at pH = 11. The 5th and 50th CV for CPE-complex 1 (blue and pink, respectively) in 25 ml of buffer solution (0.5 M) at pH = 11 and scan rate of 50 mV s−1 in the range of 0 to +1.6 V vs. Ag/AgCl (b); and in the range of −0.6 to +1.6 V (c). Amperometry for a fresh CPE (black) and CPE-complex 1 (pink) in buffer solution; amperometry conditions: 1.12 V vs. Ag/AgCl: in buffer solution (25 ml) at pH = 11 (d). | |
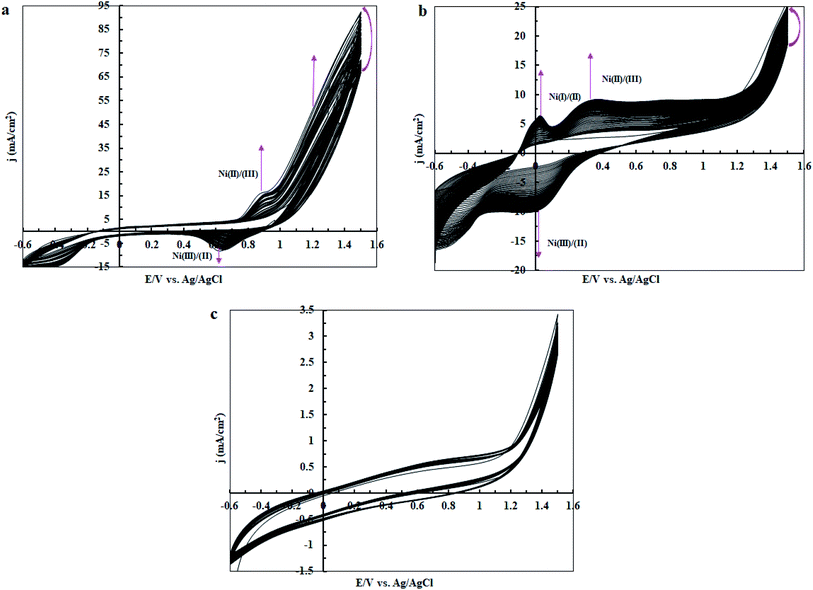 |
| | Fig. 7 Continuous CVs for CPE-complex 1 in 25 ml of borate buffer solution (0.5 M) at pH = 11 (a); pH = 7 (b); and pH = 3 (c). The scan rate in all cases is 50 mV s−1. | |
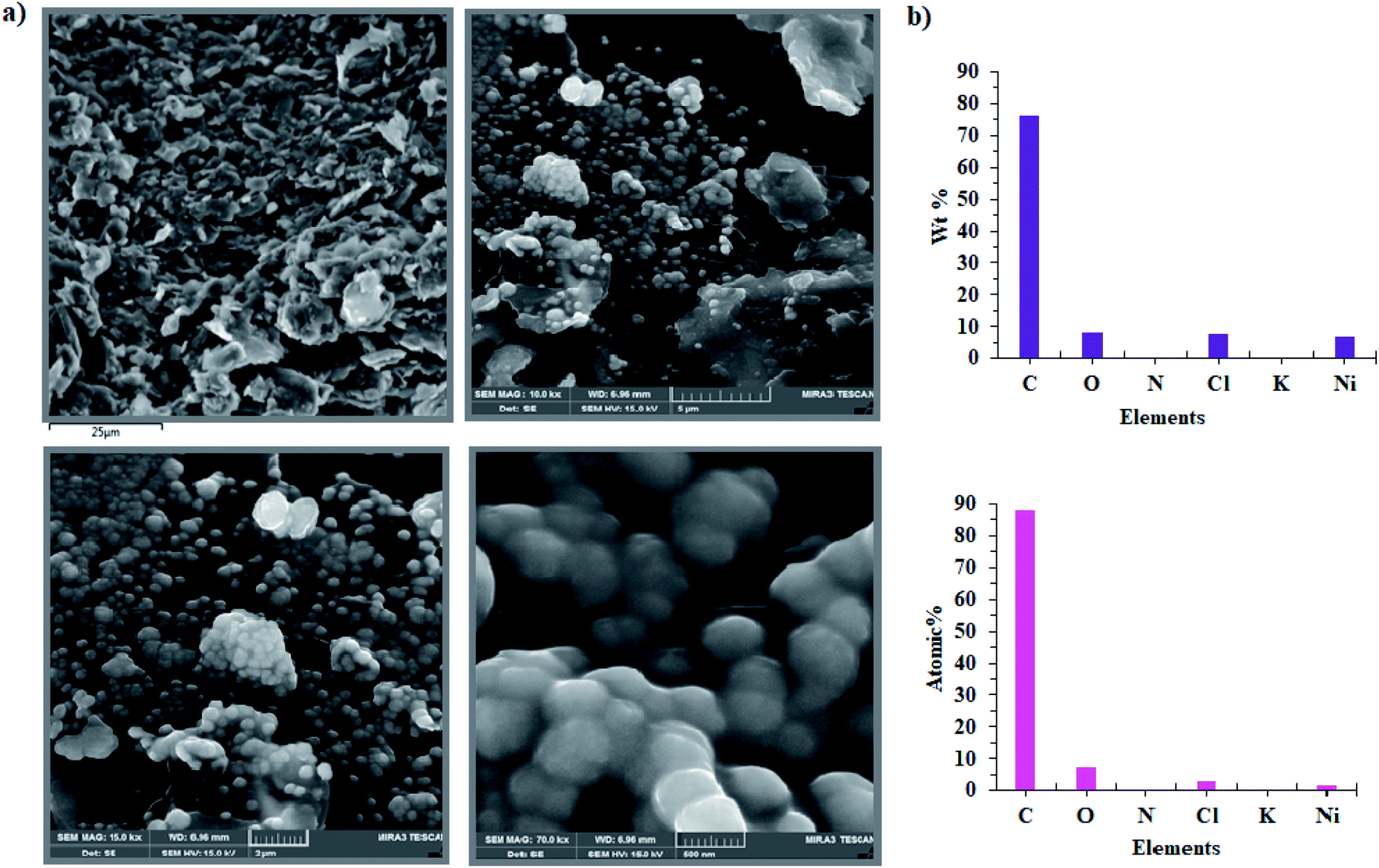
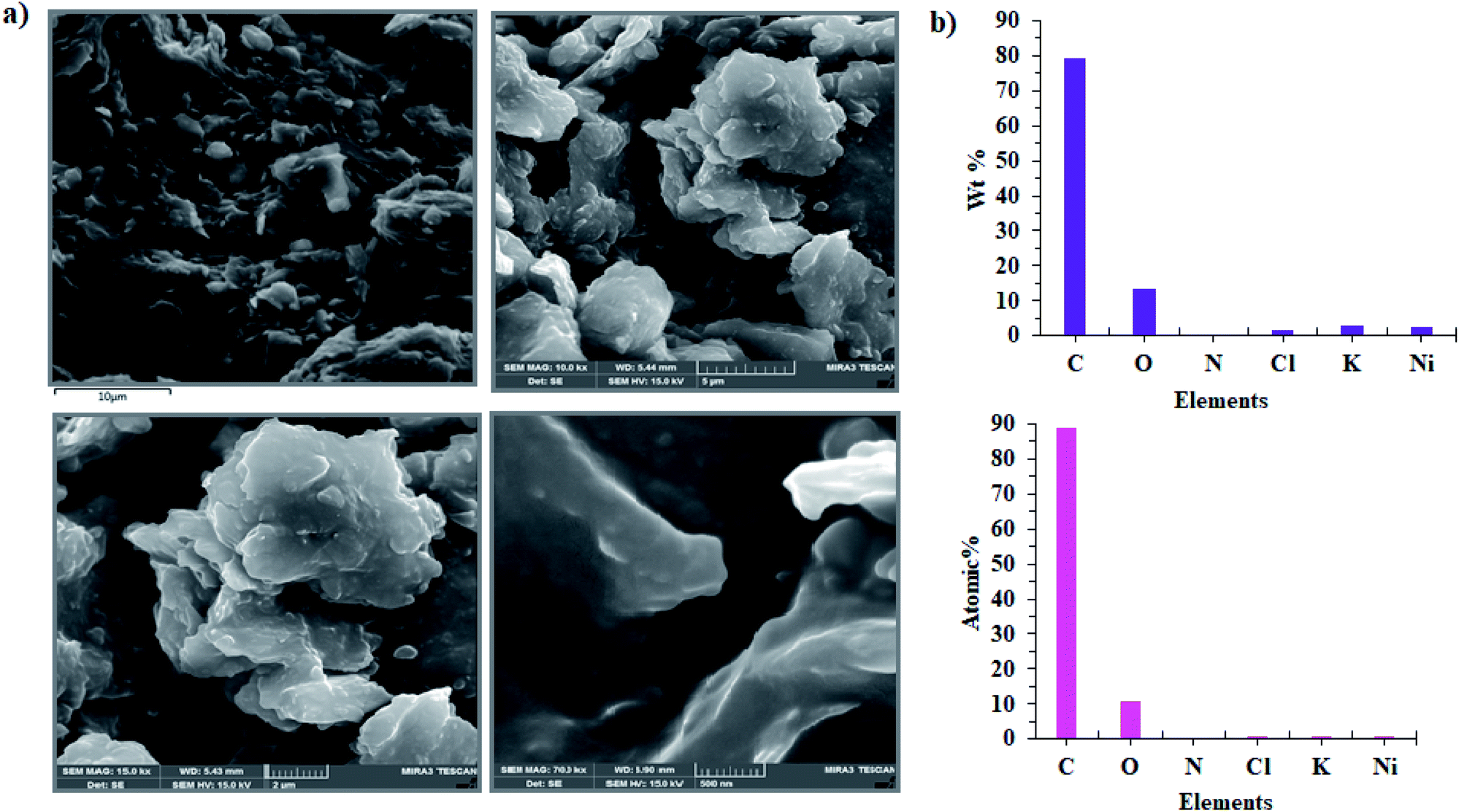
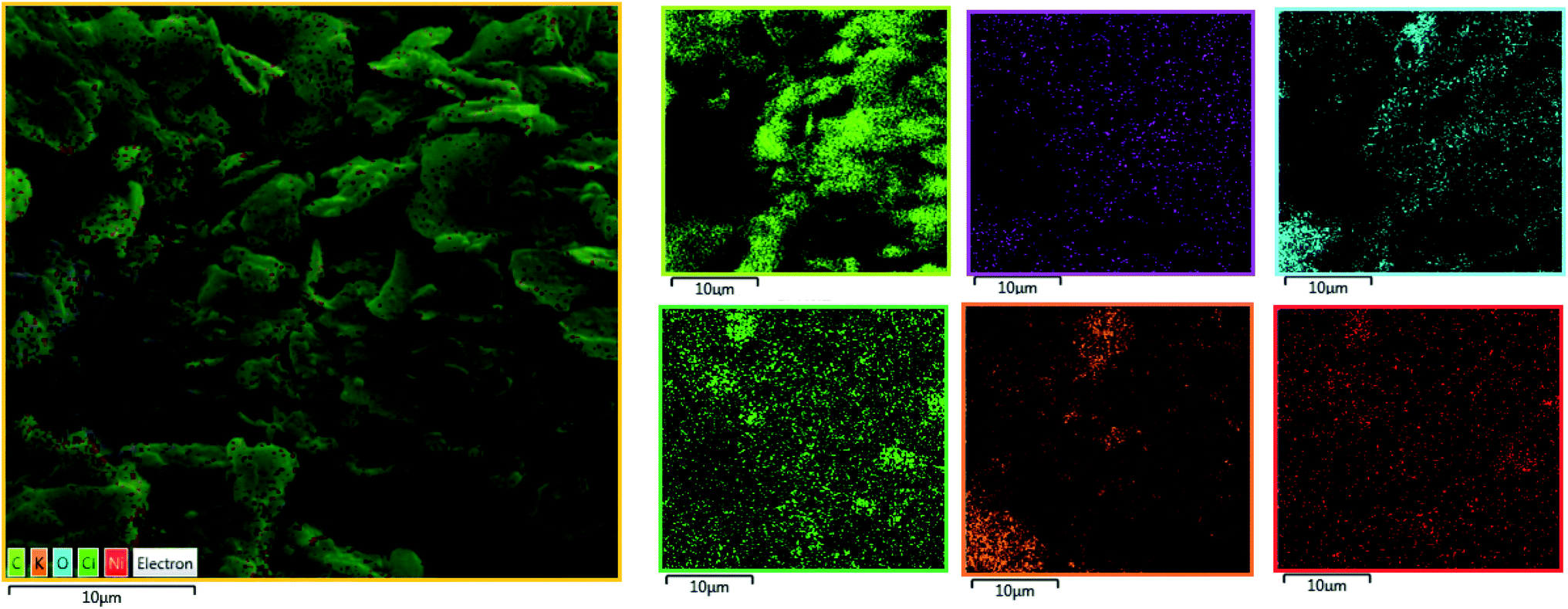
In the next step, the film on the surface of CPE-complex 1 was investigated by using field-emission scanning electron microscopy (FE-SEM). The results are shown in Fig. 8 and 9. The FE-SEM images clearly show the formation of NiO nanoparticles (∼325–475 nm) on the surface of the electrode. The images indicate the nickel oxide nanospheres are distributed uniformly in the film on the surface of electrode (Fig. 8a). The energy dispersive X-ray analysis (EDX) reveals that the surface of CPE-complex 1 at pH = 11 consists of C, N, O, Cl, Ni and low amount of K (Fig. 8b). Also, the EDX-SEM images of the CPE-complex 1 after amperometry for 5 h reveal approximately homogenous distribution of Ni and O atoms on the electrode surface (Fig. 9). From these observations, it seems that under the present conditions a heterogeneous (NiOx) catalyst acts as a true catalyst for the water oxidation reaction. Finally, the powder X-ray diffraction study (XRD) indicates the film is considerably amorphous which is consistent with previous studies.16 This means that NiO on the surface of electrode does not have pure, simple and crystalline structure (Fig. S6†), but, it can be effective electrocatalyst for oxidation of water.
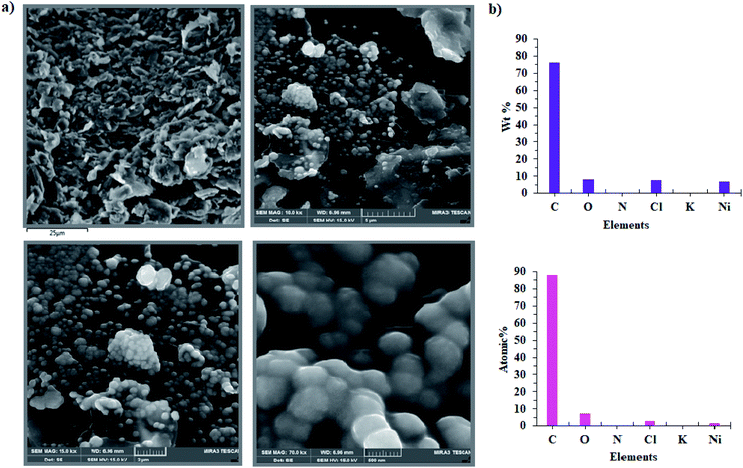 |
| | Fig. 8 FE-SEM images of CPE-complex 1 after the aerometry at 1.12 V vs. Ag/AgCl for 5 h under the water oxidation condition in buffer solution (pH = 11) in the bulk electrolysis of complex 1 (a) and EDX analysis for CPE-complex 1 (b). | |
 |
| | Fig. 9 EDX-SEM images of the CPE-complex 1 after the reaction (C (light green); N (blue); O (pink); Cl (dark green); K (orange); Ni (red)). | |
At pH = 7, as shown in Fig. 10a and b, the designed electrode converts to an efficient catalyst for water oxidation after performing amperometry for 5 hours at 1.12 V vs. Ag/AgCl. This behaviour is quite similar to the observation at pH = 11. With continuous CV for complex 1, two new anodic peaks at 0.344 and 0.0397 V (vs. Ag/AgCl) are formed (Fig. 10b and c), which can be attributed to the decomposition of complex 1 or Ni(II)/Ni(III) and Ni(I)/Ni(II) oxidation.16 Surprisingly, the continuous CV also causes an increase in the water oxidation reaction for complex 1. Therefore, we guess under CV conditions in the presence of complex 1, NiO or other compounds of Ni are formed on the surface of the electrode which can increase the water-oxidizing activity of complex 1. Amperometry (1.12 V) reveals sensible improvement in water oxidation in the presence of CPE-complex 1 (Fig. 10a and d). The amperometry for a fresh CPE shows a low activity for water oxidation, but the CPE-complex 1 shows a high and constant current density (Fig. 10d). The continuous CVs (50 cycles) at pH = 7 for CPE-complex 1 are shown in Fig. 7b. For complex 1, the continuous CV displays an increase in the attributed peaks to the redox couple of Ni(II)/Ni(III) and oxidation of Ni(I)/(II) at 0.344, 0.0299 and 0.0397 V vs. Ag/AgCl, respectively, which indicates that the complex is stable under these conditions. Anyway, in this condition the current density for water oxidation increases slightly in comparison with pH = 11 (Fig. 7a and b). This is not surprising because, in contrast to that of pH = 11, the nanosized NiO is relatively soluble at pH = 7.
 |
| | Fig. 10 CV for a fresh CPE (black), CPE-complex 1 (blue) and CPE-complex 1 after performing amperometry for 5 hours (pink) in the buffer solution (0.5 M) in the range of 0–1.6 V vs. Ag/AgCl (a); CV conditions: buffer solution at pH = 7 and scan rate of 50 mV s−1; amperometry conditions: 1.12 V vs. Ag/AgCl; CPE-complex 1 in buffer solution (25 ml). The 5th and 50th CV for CPE-complex 1 (blue and pink, respectively) in 25 ml of buffer solution (0.5 M) in the range of 0 to +1.6 V vs. Ag/AgCl (b); and in the range of −0.6 to +1.6 V (c); CV conditions: buffer solution at pH = 7 and scan rate of 50 mV s−1. Amperometry for a fresh CPE (black) and CPE-complex 1 (pink) in buffer solution (d); amperometry conditions: 1.12 V vs. Ag/AgCl: in buffer solution (25 ml) at pH = 7. | |
Our findings are surprising since the designed Ni(II) salophen complex can show high efficiency for water oxidation under applied electrochemical conditions at neutral solution as well as alkaline condition, while the Ni-salen complex (with the non-aromatic ligand) that was previously investigated by Najafpour et al.16, only revealed good electrocatalytic activity for water oxidation at pH = 11 and was not stable in neutral solution and showed low activity for water oxidation.16 This may be due to the effect of the aromatic ligand compared to its non-aromatic counterpart. Finally, the surface of the CPE-complex 1 is studied by FE-SEM images after the amperometry (Fig. 11a). The SEM images show no a special new morphology, but the EDX data from the surface of the electrode show low amounts of Ni which Ni is dispersed on the surface of the electrode (Fig. 11b).15 Although, compared to pH = 11, the formation of nickel oxide nanoparticles with the spherical morphology on the electrode surface is not clearly observed and in addition the amount of nickel on the surface of the electrode is low and also the nature of the Ni compound on CPE is not exactly known. But, it shows a high catalytic performance for water oxidation reaction. More interestingly, unlike that observed at pH = 11, Ni and O atoms are uniformly distributed on the electrode surface (Fig. 12). Consequently, we guess that a heterogeneous Ni-based compound catalyzes water oxidation reaction under the electrochemical conditions.
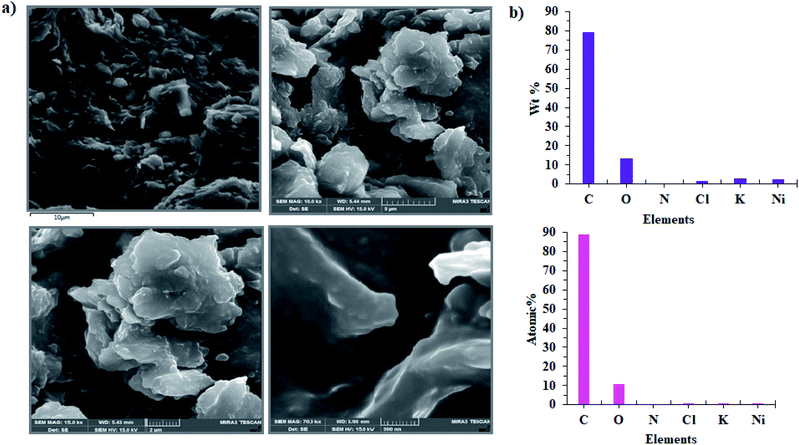 |
| | Fig. 11 FE-SEM images of CPE-complex 1 after the aerometry for 5 h under the water oxidation condition in buffer solution (pH = 7) in the bulk electrolysis of complex 1 at 1.12 V vs. Ag/AgCl (a) and EDX analysis for CPE-complex 1 (b). | |
 |
| | Fig. 12 EDX-SEM images of the CPE-complex 1 after the reaction (C (light green); N (violet); O (blue); Cl (dark green); K (orange); Ni (red)). | |
Finally, we studied the water oxidation activity of complex 1 in a buffer solution at pH = 3. As shown in Fig. 13a–d, CV and amperometry display no efficient water-oxidizing activity for complex 1 under these conditions. In the CV of fresh CPE-complex 1, one peak in the 0.20 V vs. Ag/AgCl is observed which can be attributed to the decomposition of complex 1 and Ni(II)/Ni(III) oxidation (Fig. 13a). This peak is not observed during continuous CV (Fig. 13b and c). This indicates that complex 1 is not stable under these conditions. During amperometry, complex 1 does not show water oxidation activity and only a low current density is observed (Fig. 13d). This can be related to the high solubility of NiO at pH = 3. Also, the continuous CVs (50 cycles) for complex 1 at pH = 3 are given in Fig. 7c which indicates the attributed peak for the complex disappears in this condition. This observation confirms that the complex is not stable under these conditions and no significant water oxidation was observed (Fig. 14).
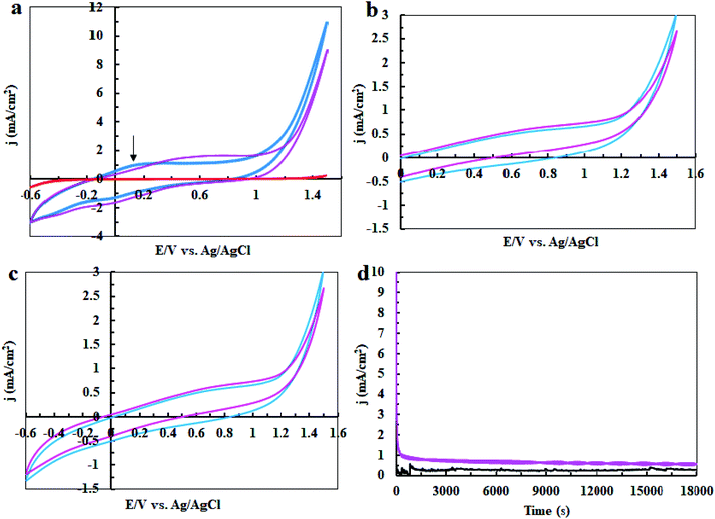 |
| | Fig. 13 CV for a fresh CPE (black), CPE-complex 1 (blue) and CPE-complex 1 after performing amperometry for 5 hours (pink) in the buffer solution (0.5 M) in the range of −0.6 to +1.6 V vs. Ag/AgCl (a); CV conditions: buffer solution at pH = 3 and scan rate of 50 mV s−1; amperometry conditions: 1.12 V vs. Ag/AgCl. The 5th and 50th CV for CPE-complex 1 (blue and pink, respectively) in 25 ml of buffer solution (0.3 M) at pH = 3 and scan rate of 50 mV s−1 in the range of 0 to +1.6 V (b); and in the range of −0.6 to +1.6 V (c). Amperometry for a fresh CPE (black) and CPE-complex 1 (pink) in buffer solution; amperometry conditions: 1.12 V vs. Ag/AgCl: in buffer solution (25 ml) at pH = 3 (d). | |
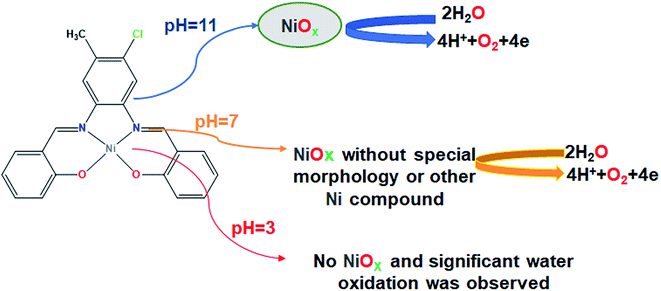 |
| | Fig. 14 A simple scheme of suggested mechanism for water oxidation reaction in the presence of complex 1 in different pH. | |
Conclusions
The design of efficient electrocatalysts for water splitting as a clean and renewable energy resource in recent years has been of a great interest. For this purpose, a new mononuclear Ni(II) salophen-type complex was synthesized by the reaction of a salophen-type N2O2-donor ligand and Ni(OAc)2·4H2O and was characterized by spectroscopic methods and single crystal X-ray analysis. The water-oxidizing activity of the obtained complex was studied at three different pH (pH = 3, 7 and 11). LSV curves showed that the CPE modified with complex 1 can exhibit a high electrocatalytic activity for the oxidizing of water in basic and neutral solutions in terms of the onset potential and Tafel slope. Also, cyclic voltammetry experiments and FE-SEM images showed that at pH = 11, NiOx is the heterogeneous catalyst for water oxidation under electrochemical water oxidation conditions and in the presence of complex 1. Surprisingly, our experiments showed that complex 1 can also act as an efficient electrocatalyst for water oxidation at pH = 7. FE-SEM images did not clearly show the formation of NiO on the surface of the electrode (and also a special new morphology), but the EDX data from the surface of CPE showed a low amount of Ni. Although the nature of the Ni compound on the electrode surface was not clear, but it could show a high catalytic performance for water oxidation reaction. Finally, at pH = 3, no significant water oxidation was observed.
Experimental section
Materials and instrumentation
4-Chloro-5-metyl-1,2-diaminobenzene, 2-hydroxybenzaldehyde and Ni(II) acetate tetrahydrate were purchased from Sigma-Aldrich company. Solvents with highest purity were provided from Merck and used without further purification. Graphite powder and paraffin oil from Merck were used for the preparation of carbon paste electrode. To adjust the pH of the electrolyte solution at pH = 3–11, 0.5 M of boric acid, 1.0 M of HCl and 1.0 M of NaOH solutions were used. NMR spectra were recorded on a Bruker Avance DPX-400 MHz spectrometer. The elemental analyses (carbon, hydrogen and nitrogen) of the compounds were obtained from a Carlo ERBA Model EA 1108 analyzer. FT-IR spectra were prepared with a FT-IR Spectrometer Bruker Tensor 27 after mixing the samples with KBr. Electronic absorption spectra were obtained with T 60 UV/Vis Spectrometer PG Instruments Ltd. X-ray diffraction patterns of electrodes were obtained using Bruker AXS model D8-Advance diffractometer equipped with Cu-Kα radiation at λ = 1.5418 Å from 2θ = 30° to 70°. Surface morphology of electrodes was studied using TESCAN mira 3 field-emission scanning electron microscope (FE-SEM) equipped with energy dispersive X-ray analysis (EDX).
Synthesis of 2,2′-((1E,1′E)-((4-chloro-5-methyl-1,2-phenylene)bis(azanylylidene))bis(methanylylidene))diphenol (H2L)
4-Chloro-5-metyl-1,2-diaminobenzene (1.00 g, 6.38 mmol) in 30 ml ethanol was slowly added to ethanol solution of salicylaldehyde (1.36 ml, 12.76 mmol) under stirring in room temperature. Immediately, the color of the solution turned to orange-brown and precipitate was appeared after a few minutes. The reaction was continued for 4 h at room temperature (the end of the reaction was controlled by thin layer chromatography (TLC)). The precipitate was filtered, washed with ethanol and dried at air. Yield: 84.2% (1.96 g). Anal. calc. for C21H17N2O2Cl (M = 364.82): C, 69.14; H, 4.70; N, 7.68%. Found: C, 69.19; H, 4.67; N, 7.61%. 1H NMR (400 MHz, DMSO-d6): δ 12.77 (s, 1H, –OH), 12.69 (s, 1H, –OH), 8.99 (s, 1H, –HC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–), 8.95 (s, 1H, –HCN–), 7.70 (s, 1H, Ar–H), 7.60 (s, 1H, Ar–H), 7.49 (d, 2H, J = 8.8 Hz, ArH), 7.45–7.42 (m, 2H, ArH), 7.02–6.97 (m, 4H, ArH), 2.34 (s, 3H, –CH3); FT-IR (KBr, cm−1): 3447 (b),1614 (vs.), 1578 (m), 1478 (s), 1558 (m), 1405 (m), 1276 (s), 1224 (w), 1190 (m), 1151 (m), 1115 (m), 1085 (s), 969 (m), 920 (w), 907 (w), 893 (m), 866 (w), 811 (m), 752 (s), 697 (w), 652 (w), 591 (w), 503 (w), 437 (w).
N–), 8.95 (s, 1H, –HCN–), 7.70 (s, 1H, Ar–H), 7.60 (s, 1H, Ar–H), 7.49 (d, 2H, J = 8.8 Hz, ArH), 7.45–7.42 (m, 2H, ArH), 7.02–6.97 (m, 4H, ArH), 2.34 (s, 3H, –CH3); FT-IR (KBr, cm−1): 3447 (b),1614 (vs.), 1578 (m), 1478 (s), 1558 (m), 1405 (m), 1276 (s), 1224 (w), 1190 (m), 1151 (m), 1115 (m), 1085 (s), 969 (m), 920 (w), 907 (w), 893 (m), 866 (w), 811 (m), 752 (s), 697 (w), 652 (w), 591 (w), 503 (w), 437 (w).
Synthesis of complex 1
Complex 1 was synthesized by the reaction of H2L (0.100 g, 0.274 mmol) and Ni(OAc)2·4H2O (0.068 g, 0.274 mmol) in ethanol by using a branched tube. Mentioned amounts of materials were placed in the main arm of a branched tube. Ethanol was carefully added to fill the arms. The tube was tight closed and the arm containing the reagents was immersed in an oil bath at 70 °C, while the other arm was kept at ambient temperature. After four days, red crystals were formed in the cooler arm. The crystals were filtered off, washed with ethanol and dried at air. Yield: 74% (0.085 g). Anal. calc. for C21H15N2NiO2Cl (Mw = 421.51 g mol−1): C, 59.84; H, 3.59; N, 6.65; Ni, 13.92%. Found: C, 59.89; H, 3.62; N, 6.61; Ni, 13.85%. 1H NMR (400 MHz, DMSO-d6): δ 9.02 (s, 1H, –HCN–), 8.97 (s, 1H, –HCN–), 8.32 (s, 1H, ArH), 8.19 (d, 1H, J = 8.8 Hz, ArH), 7.60 (d, 2H, J = 7.62, Ar–H), 7.34–7.36 (m, 2H, Ar–H), 6.92 (d, 2H, J = 8.4 Hz, ArH), 6.68–6.73 (m, 2H, ArH), 2.15 (s, 3H, –CH3). FT-IR (KBr, cm−1): 3420 (m, br), 1608 (s), 1574 (s), 1522 (m), 1488 (m), 1460 (m), 1441 (m), 1368 (m), 1333 (m), 1262 (s), 1231 (m), 1196 (m), 1149 (m), 1132 (m), 1090 (m), 1022 (m), 1057 (m), 946 (w), 900 (w), 846 (m), 799 (w), 752 (s), 709 (w), 595 (w), 556 (w), 449 (w).
Single crystal X-ray analysis
The molecular structure of complex 1 was determined by single crystal X-ray analysis at 293(2) K. The data collection was performed on an Oxford Sapphire CCD diffractometer using Mo Kα radiation (λ = 0.71073 Å) and ω–2θ method. The structure was solved by direct methods and refined with the full-matrix least-squares method on F2 with the use of SHELX-2014 program packages.32 The analytical absorption corrections were applied.33 Positions of hydrogen atoms have been found from the electron density maps and hydrogen atoms were constrained during refinement with the appropriate riding model as implemented in SHELX during refinement. A summary of the crystal data and refinement details for complex 1 is given in Table 2. The structural data have been deposited at the Cambridge Crystallographic Data Centre: (CCDC no. 1954803).†
Table 2 Crystallographic data and refinement details for complex 1
| Compound |
Complex 1 |
| Formula |
C21H15ClN2NiO2 |
| Mr/g mol−1 |
421.51 |
| Crystal shape, color |
Needle, red |
| Crystal size/mm3 |
0.19 × 0.08 × 0.02 |
| Crystal system |
Monoclinic |
| Space group |
P21/c |
| a/Å |
11.0230(14) |
| b/Å |
8.0024(8) |
| c/Å |
19.532(2) |
| β/° |
106.302(12) |
| V Å−3 |
1653.6(3) |
| Z |
4 |
| Dcalc/g cm−3 |
1.693 |
| μ/mm−1 |
1.36 |
| F(000) |
864 |
| θ range/° |
1.9–28.4 |
| h, k, l |
−14 → 14 |
| −10 → 8 |
| −18 → 26 |
| Measured reflections |
12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 101 101 |
| Independent reflections |
3825 |
| Reflections with I > 2σ(I) |
1214 |
| Rint |
0.215 |
| R[F2 > 2σ(F2)] |
0.094 |
| wR(F2) |
0.346 |
| S |
0.99 |
| Abs. correction |
Analytical |
| Parameters |
246 |
| Restraints |
0 |
| Δρmax/Δρmin |
0.53/−0.59 |
Electrochemical tests
Electrochemical analyses of complex 1 were done by using an Autolab PGSTAT204. The carbon paste electrode (CPE) modified with complex 1 was used as working electrode. The Ag/AgCl and Pt wire electrodes were used as reference and counter electrodes, respectively. CPE modified with complex 1 (CPE-complex 1) was fabricated as follows: graphite powder, complex 1 and paraffin oil with a mass ratio of 80:15:5 were mixed using a mortar until a homogenous paste was obtained. Then, the obtained homogenous paste was packed in a copper wire with a 1.10 mm radius. The unmodified CPE electrode was fabricated using the same method. The surface of CPE electrodes was smoothed and the electrodes were washed with double distilled water.34
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors are grateful to Azarbaijan Shahid Madani University, Imam Khomeini International University and Nicolaus Copernicus University in Torun for financial support of this study.
References
- R. Esinberg and D. G. Nocera, Inorg. Chem., 2005, 44, 6799–6801 CrossRef PubMed.
-
(a) Y. Gao, H. Chen, L. Ye, Z. Lu, Y. Yao, Y. Wei and X. Chen, Chin. J. Catal., 2018, 39, 479–486 CrossRef CAS;
(b) A. Han, H. Jia, H. Ma, S. Ye, H. Wu, H. Lei, Y. Han, R. Cao and P. Du, Phys. Chem. Chem. Phys., 2014, 16, 11209–11217 Search PubMed;
(c) M. Yagi and M. Kaneko, Chem. Rev., 2001, 101, 21–36 CrossRef CAS PubMed;
(d) W. Rüttinger and G. C. Dismukes, Chem. Rev., 1997, 97, 1–24 CrossRef PubMed;
(e) G. Liao, Y. Gong, L. Zhang, H. Gao, G.-J. Yang and B. Fang, Energy Environ. Sci., 2019, 12, 2080–2147 RSC;
(f) W. Zhong, Z. Lin, S. Feng, D. Wang, S. Shen, Q. Zhang, L. Gu, Z. Wang and B. Fang, Nanoscale, 2019, 11, 4407–4413 RSC;
(g) D. Wang, D. Zhang, C. Tang, P. Zhou, Z. Wu and B. Fang, Catal. Sci. Technol., 2016, 6, 1952–1956 RSC;
(h) W. Zhong, S. Shen, M. He, D. Wang, Z. Wang, Z. Lin, W. Tu and J. Yu, Appl. Catal. B Environ., 2019, 258, 117967 CrossRef CAS;
(i) Y. Zhang, X. Wang, F. Luo, Y. Tan, L. Zeng, B. Fang and A. Liu, Appl. Catal. B Environ., 2019, 256, 117852 CrossRef CAS;
(j) Z. Wu, B. Fang, A. Bonakdarpour, A. Sun, D. P. Wilkinson and D. Wang, Appl. Catal. B Environ., 2012, 125, 59–66 CrossRef CAS;
(k) W. Zhong, W. Tu, S. Feng and A. Xu, J. Alloys Compd., 2019, 772, 669–674 CrossRef CAS.
- M. A. Asraf, H. A. Younus, M. Yusubov and F. Verpoort, Catal. Sci. Technol., 2015, 5, 4901–4925 RSC.
- J. Lin, B. Ma, M. Chen and Y. Ding, Chin. J. Catal., 2018, 39, 463–471 CrossRef CAS.
-
(a) M. M. Najafpour, S. Mehrabani, Y. Mousazade and M. Holynska, Dalton Trans., 2018, 47, 9021–9029 RSC;
(b) J. P. McEvoy and G. W. Brudvig, Chem. Rev., 2006, 106, 4455–4483 CrossRef CAS PubMed;
(c) M. D. Karkas, O. Verho, E. V. Johnston and B. Ekermark, Chem. Rev., 2014, 114, 11863–12001 CrossRef PubMed;
(d) J. Limburg, J. S. Vrettos, L. M. Liable-Sands, A. L. Rheingold, R. H. Crabtree and G. W. Brudvig, Science, 1999, 283, 1524–1527 CrossRef CAS PubMed;
(e) W. J. Youngblood, S.-H. A. Lee, Y. Kobayashi, E. A. Hernandez-Pagan, P. G. Hoertz, T. A. Moore, A. L. Moore, D. Gust and T. E. Mallouk, J. Am. Chem. Soc., 2009, 131, 926–927 CrossRef CAS PubMed;
(f) T. W. Kim and K.-S. Choi, Science, 2014, 343, 990–994 CrossRef CAS PubMed.
- P. Du and R. Eisenberg, Energy Environ. Sci., 2012, 5, 6012–6021 RSC.
-
(a) M. Wiechen, M. M. Najafpour, S. Allakhverdiev and L. Spiccia, Energy Environ. Sci., 2014, 7, 2203–2212 RSC;
(b) M. M. Najafpour, G. Renger, M. Hołyńska, A. N. Moghaddam, E.-M. Aro, R. Carpentier, H. Nishihara, J. J. Eaton-Rye, J.-R. Shen and S. I. Allakhverdiev, Chem. Rev., 2016, 116, 2886–2936 CrossRef CAS PubMed;
(c) W. M. C. Sameera, C. J. McKenzie and J. E. McGrady, Dalton Trans., 2011, 40, 3859–3870 RSC;
(d) R. Brimblecombe, A. Koo, G. C. Dismukes, G. F. Swiegers and L. Spiccia, J. Am. Chem. Soc., 2010, 132, 2892–2894 CrossRef CAS PubMed.
-
(a) J. L. Fillol, Z. Codola, I. Garcia-Bosch, L. Gomez, J. J. Pla and M. Costas, Nat. Chem., 2011, 3, 807–813 CrossRef CAS PubMed;
(b) D. Hong, S. Mandal, Y. Yamada, Y.-M. Lee, W. Nam, A. Llobet and S. Fukuzumi, Inorg. Chem., 2013, 52, 9522–9531 CrossRef CAS PubMed;
(c) W.-P. To, T. W.-S. Chow, C.-W. Tse, X. Guan, J.-S. Huang and C.-M. Che, Chem. Sci., 2015, 6, 5891–5903 RSC;
(d) V. K. K. Praneeth, M. Kondo, M. Okamura, T. Akai, H. Izu and S. Masaoka, Chem. Sci., 2019, 10, 4628–4639 RSC.
-
(a) A. Singh, S. L. Y. Chang, R. K. Hocking, U. Bach and L. Spiccia, Catal. Sci. Technol., 2013, 3, 1725–1732 RSC;
(b) G. Chen, L. J. Chen, S. W. Ng and T. C. Lau, ChemSusChem, 2014, 7, 127–134 CrossRef CAS PubMed.
-
(a) M. M. Najafpour and H. Feizi, Catal. Sci. Technol., 2018, 8, 1840–1848 RSC;
(b) T. T. Li, J. Qian, Q. Zhou, J. L. Lin and Y. Q. Zheng, Dalton Trans., 2017, 46, 13020–13026 RSC;
(c) B. Huang, Y. Wang, S. Zhan and J. Ye, Appl. Surf. Sci., 2018, 396, 121–128 CrossRef;
(d) H. Chen, Z. Sun, X. Liu, A. Han and P. Du, J. Phys. Chem. C, 2015, 119, 8998–9004 CrossRef CAS;
(e) H. T. Shi, X. X. Li, F. H. Wu and W. B. Yu, Dalton Trans., 2017, 46, 16321–16326 RSC.
-
(a) M.-T. Zhang, Z. Chen, P. Kang and T. J. Meyer, J. Am. Chem. Soc., 2013, 135, 2048–2051 CrossRef CAS PubMed;
(b) S. M. Barnett, K. I. Goldberg and J. M. Mayer, Nat. Chem., 2012, 4, 498–502 CrossRef CAS PubMed;
(c) F. Yu, F. Li, J. Hu, L. Bai, Y. Zhu and L. Sun, Chem. Commun., 2016, 52, 10377–10380 RSC;
(d) P. Garrido-Barros, I. Funes-Ardoiz, S. Drouet, J. Benet-Buchholz, F. Maseras and A. Llobet, J. Am. Chem. Soc., 2015, 137, 6758–6761 CrossRef CAS PubMed.
- K. Kinoshita, Electrochemical Oxygen Technology, Wiley-Interscience, New York, 1992 Search PubMed.
-
(a) M. Gao, W. Sheng, Z. Zhuang, Q. Fang, S. Gu, J. Jiang and Y. Yan, J. Am. Chem. Soc., 2014, 136, 7077–7084 CrossRef CAS PubMed;
(b) M. Gong, W. Zhou, M. C. Tsai, J. Zhou, M. Guan, M. C. Lin, B. Zhang, Y. Hu, D. Y. Wang, J. Yang, S. J. Pennycook, B. J. Hwang and H. Dai, Nat. Commun., 2014, 5, 7077–7084 Search PubMed;
(c) K. Sun, N. Park, Z. Sun, J. Zhou, J. Wang, X. Pang, S. Shen, S. Y. Noh, Y. Jing, S. Jin, P. K. L. Yu and D. Wang, Energy Environ. Sci., 2012, 5, 7872–7877 RSC;
(d) X. Yu, P. Xu, T. Hua, A. Han, X. Liu, H. Wu and P. Du, Int. J. Hydrogen Energy, 2014, 39, 10467–10475 CrossRef CAS.
- J. Lin, P. Kang, X. Liang, B. Ma and Y. Ding, Electrochim. Acta, 2017, 258, 353–359 CrossRef CAS.
- G. Azadi, Z. Zand, Y. Mousazade, R. Bagheri, J. Cui, Z. Song, R. Bikas, K. Wozniak, S. I. Allakhverdiev and M. M. Najafpour, Int. J. Hydrogen Energy, 2019, 44, 2857–2867 CrossRef CAS.
- H. Feizi, F. Shiri, R. Bagheri, J. P. Singh, K. H. Chae, Z. Song and M. M. Najafpour, Catal. Sci. Technol., 2018, 8, 3954–3968 RSC.
- M. Zhang, M.-T. Zhang, C. Hou, Z.-F. Ke and T.-B. Lu, Angew. Chem. Int. Ed., 2014, 53, 13042–13048 CrossRef CAS PubMed.
- P. Garrido-Barros, S. Grau, S. Drouet, J. Benet-Buchholz, C. Gimbert-Surinach and A. Llobet, ACS Catal., 2019, 9, 3936–3945 CrossRef CAS.
- Y. Han, Y. Wu, W. Lai and R. Cao, Inorg. Chem., 2015, 54, 5604–5613 CrossRef CAS PubMed.
- L. Wang, L. Duan, R. B. Ambre, Q. Daniel, H. Chen, J. Sun, B. Das, A. Thapper, J. Uhlig, P. Dinér and L. Sun, J. Catal., 2016, 335, 72–78 CrossRef CAS.
- G. Zhu, E. N. Glass, C. Zhao, H. Lv, J. W. Vickers, Y. V. Geletii, D. G. Musaev, J. Songa and C. L. Hill, Dalton Trans., 2012, 41, 13043–13049 RSC.
-
(a) J. Ping, Y. Wang, Q. Lu, B. Chen, J. Chen, Y. Huang, Q. Ma, C. Tan, J. Yang, X. Cao, Z. Wang, J. Wu, Y. Ying and H. Zhang, Adv. Mater., 2016, 28, 7640–7645 CrossRef CAS PubMed;
(b) S. Nayak, L. Mohapatra and K. Parida, J. Mater. Chem. A, 2015, 3, 18622–18635 RSC;
(c) G. Y. Luo, H. H. Huang, J. W. Wang and T. B. Lu, ChemSusChem, 2016, 9, 485–491 CrossRef CAS PubMed;
(d) M. M. Najafpour and H. Feizi, Dalton Trans., 2012, 41, 10292–10297 RSC.
-
(a) A. Singh, S. L. Y. Chang, R. K. Hocking, U. Bachcde and L. Spiccia, Energy Environ. Sci., 2013, 6, 579–586 RSC;
(b) I. M. Sadiek, A. M. Mohammad, M. E. Shakre and M. S. Deab, Int. J. Hydrogen Energy, 2012, 37, 68–77 CrossRef CAS;
(c) X. Wang, H. Luo, H. Yang, P. J. Sebastian and S. A. Gamboa, Int. J. Hydrogen Energy, 2004, 29, 967–972 CrossRef CAS;
(d) M. Dinca, Y. Surendranath and D. G. Nocera, Proc. Natl. Acad. Sci. U.S.A., 2010, 107, 10337–10341 CrossRef CAS PubMed.
- L.-H. Zhang, F. Yu, Y. Shi, F. Li and H. Li, Chem. Commun., 2019, 55, 6122–6125 RSC.
-
(a) P. Mahapatra, M. G. B. Drew and A. Ghosh, Dalton Trans., 2018, 47, 13957–13971 RSC;
(b) R. Bikas, E. Shahmoradi, S. Reinoso, M. Emami, L. Lezama, J. Sanchiz and N. Noshiranzadeh, Dalton Trans., 2019, 48, 13799–13812 RSC;
(c) R. Bikas, M. Emami, K. Ślepokura and N. Noshiranzadeh, New J. Chem., 2017, 41, 9710–9717 RSC.
-
(a) S. Roy, A. Bhattacharyya, S. Purkait, A. Bauzá, A. Frontera and S. Chattopadhyay, Dalton Trans., 2016, 45, 15048–15059 RSC;
(b) R. Bikas, V. Kuncser, J. Sanchiz, G. Schinteie, M. Siczek, H. Hosseini-Monfared and T. Lis, Polyhedron, 2018, 147, 142–151 CrossRef CAS.
- S. Demir, H. Yılmaz, M. Dilimulati and M. Andac, Spectrochim. Acta, Part A, 2015, 150, 523–532 CrossRef CAS PubMed.
-
(a) K.-L. Kuo, C.-C. Huang and Y.-C. Lin, Dalton Trans., 2008, 3889–3898 RSC;
(b) R. Bikas, V. Lippolis, N. Noshiranzadeh, H. Farzaneh-Bonab, A. J. Blake, M. Siczek, H. Hosseini-Monfared and T. Lis, Eur. J. Inorg. Chem., 2017, 999–1006 CrossRef CAS.
- L. Rigamonti, A. Forni, S. Righetto and A. Pasinic, Dalton Trans., 2019, 48, 11217–11234 RSC.
- R. Atkins, G. Brewer, E. Kokot, G. M. Mockler and E. Sinn, Inorg. Chem., 1985, 24, 127–134 CrossRef CAS.
-
(a) C. Gosden, K. P. Healy and D. Pletcher, J. Chem. Soc., Dalton Trans., 1978, 972–976 RSC;
(b) P. Zanello, Inorganic electrochemistry: theory, practice and application, Royal Society of Chemistry, 2007 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., 2015, 71, 3–8 CrossRef PubMed.
- CrysAlis 171.38.43 package of programs, Rigaku Oxford Diffraction, 2015 Search PubMed.
- L. J. Foruzin, B. Habibi and Z. Rezvani, New J. Chem., 2018, 42, 13963–13970 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1954803. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra08585h |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Rahman Bikas
*a,
Rahman Bikas