DOI:
10.1039/C9RA08404E
(Paper)
RSC Adv., 2019,
9, 38355-38371
Drug-likeness of linear pentamidine analogues and their impact on the hERG K+ channel – correlation with structural features†
Received
14th October 2019
, Accepted 15th November 2019
First published on 2nd December 2019
Abstract
This work presents drug-likeness and the cardiotoxicity profiles of six potent pentamidine analogs 1–6 and three new compounds 7–9 as chemotherapeutics for therapy of Pneumocystis jiroveci pneumonia. A combination of experimental and computational approaches was used in the cardiotoxicity examination. The hERG trafficking and functionality of the hERG currents were tested by western blot analyses, immunofluorescent staining procedures, and patch-clamp electrophysiological assays. Cardiotoxicity combined with blocking the hERG K+ channel was predicted, and then simulated by docking to the CSM-TM model 732 protein. Location of pentamidines in the proximity of Leu622, Thr623, Ser649, Tyr652, Ala653, and Phe656, and the high energies of interactions were in accordance with probable blocking of the hERG channel. However, in the biochemical experiments, no significant changes in IhERG densities and a minor effect on hERG maturation were observed. Predicted metabolic transformation of pentamidines with S atoms in the aliphatic linker leads to oxidation of one S atom, but those with the phenyl sulfanilide moiety can be oxidized to chinones. The tested pentamidines characterized by the presence of sulfur atoms or sulfanilide groups, have favorable drug-likeness parameters and are promising lead structures in the development of new potent chemotherapeutics against PJP.
1. Introduction
A major problem after identifying lead compounds is to evaluate their drug-likeness parameters and cardiotoxicity. A lot of lead compounds and drugs developed for different treatments have been found to interfere with normal cardiac physiology, and have been retracted from early- and late-stage trials as well as from the medical practice.1,2 Cardiotoxicity associated with the hERG (human ether-á-go-go related gene) K+ ion channel named KCNH2 or Kv11.1 has received considerable attention.3 This gene is expressed in a variety of tissues (i.e. heart and brain), but the expression and function of the hERG protein are best described in cardiac myocytes. The hERG K+ ion channel shapes the rapidly activating component of the cardiac delayed rectifier potassium current (IKr), which is crucial for the repolarization of the cardiac action potential. Dysfunction of the hERG channel could cause long QT syndrome (LQTS) characterized by delayed repolarization and prolonged QT interval. These facts increase the risk of ventricular arrhythmias in the form of torsades de pointes (TdP) and sudden cardiac death.4–6 Diverse substances can induce LQTS via inhibition of hERG using a wide range of mechanisms. The mechanism often proposed for drug-induced QT interval is a block of hERG channels or its native current IKr. The drugs interact with a structurally unique receptor domain (pore helix aliphatic residues Thr-623, Ser-624, and Val-625 and aromatic residues Tyr-652 and Phe-656) in the pore-S6 region of the channel to suppress K+ ion permeation.7 Another mechanism for drug-induced LQTS is the disorganization of hERG channel protein trafficking to the cell surface membrane. Fewer mature hERG channels reach the surface membrane, thus reducing hERG K+ current (IhERG) or IKr.8 Whereas some drugs induce adverse effects via only one of the mechanisms, others act via a combination of both.9,10 The importance of hERG-related toxicity is driven by the tendency of hERG channels to bind many compounds, including anti-biotics (e.g., erythromycin, clarithromycine),11 anti-viral (e.g., amantadine),12 anti-fungal (e.g., ketoconazole, itraconazole),13,14 anti-cancer (e.g., arsenic trioxide),15 anti-protozoal and anti-malarial (e.g., pentamidine, chloroquine),16,17 anti-psychotic (e.g., chlorpromazine),18 anti-histamine (e.g., astemizole),19 and anti-arrhythmic (e.g., quinidine, dofetilide)20 drugs, which strongly block or disorder the membrane trafficking of hERG channels.
In recent years, Pneumocystis jiroveci pneumonia (PJP) has become one of the potentially fatal illness encountered in immunocompromised patients.21–23 Although the development of highly active anti-retroviral therapy, and the extensive use of Pneumocystis chemoprophylaxis, PJP remains an important cause of morbidity and mortality in patients with acquired immunodeficiency syndrome (AIDS).24 At present, interest in PJP infection goes beyond the people which suffer from AIDS due to the increasing number of patients receiving immunosuppressive therapies because of massive organ transplantations, malignancies, and autoimmune diseases, for which PJP is increasingly diagnosed.23,25,26 The effective treatment for PJP is trimethoprim–sulfamethoxazole (TMP–SMX) drug combination, but also alternative drugs have been developed,27,28 including pentamidine, dapsone, and atovaquone. Among them, intravenous pentamidine is known as the most potent for the second-line treatment of PJP.29 Structurally, pentamidine belongs to the aromatic bis-amidines which are lead compounds in many drug discovery pathways, but no substance has passed over the phases of clinical trials because of toxicity and improper drug-likeness properties.30 Therefore, there is an urgent need for an evaluation of these properties for newly available compounds. Many efforts were made for pentamidine cardiotoxicity examinations. Therapy with pentamidine can cause the prolongation of the QT interval on the electrocardiogram and in some cases TdP tachycardia's which can be transformed into ventricular fibrillation and cause sudden cardiac death.31 Several studies have shown that pentamidine prolongs the cardiac action potential by the impact on hERG trafficking and decreasing of the number of functional hERG channels at the cell surface.32–34
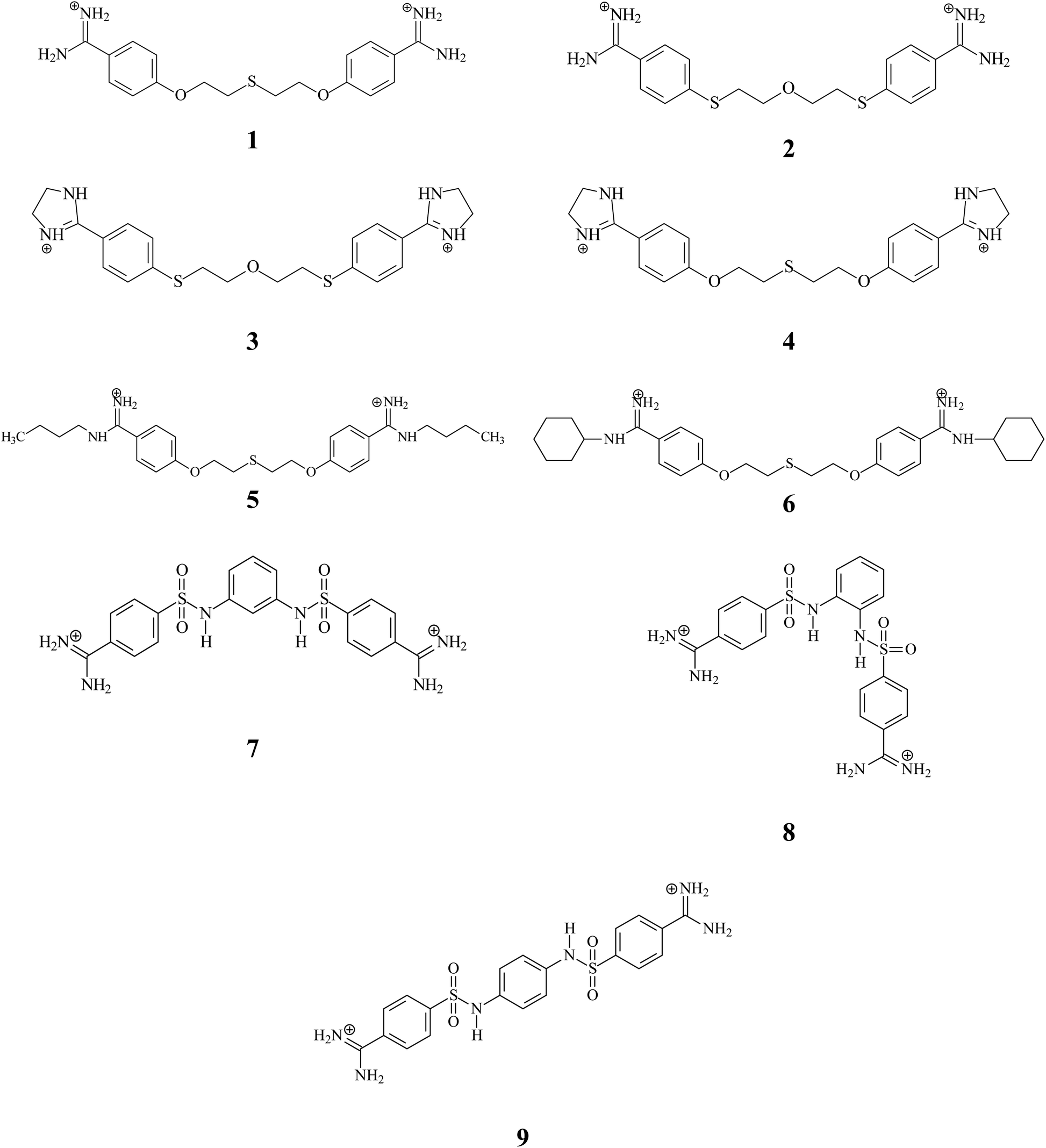
In an effort to find a more efficient therapeutic agent for the treatment of PJP, our laboratory has developed a number of linear pentamidine analogs which showed excellent anti-Pneumocystis activity.35–37 Among then, we have designed, synthesized and evaluated six bis-benzamidines 1–6 characterized by the presence of one or two sulfur atoms in the linker and introduction of alkyl substituents to the amidine groups (see Fig. 1). The tests for 1–3 revealed the high activities (IC50 with range 0.01–1.18 μM) and less or no cytotoxicity in mammalian cell lines compared to the parent compound pentamidine. In continuing the search for new drug candidates, we have taken into account the promising activity and very low cytotoxicity received for 3-phenylsulfonyl-1,5-bis(4-amidino-2,6-dimethylphenoxy)-3-azapentane dihydrochloride.37 We synthesized three new bis-benzamidines 7–9 in which two sulfanilide groups are in the ortho-, meta- and para-positions of the benzene ring. Moreover, these derivatives represent molecules with less mobile linker connecting bis-benzamidine moieties, which can have an impact on cardiotoxicity.
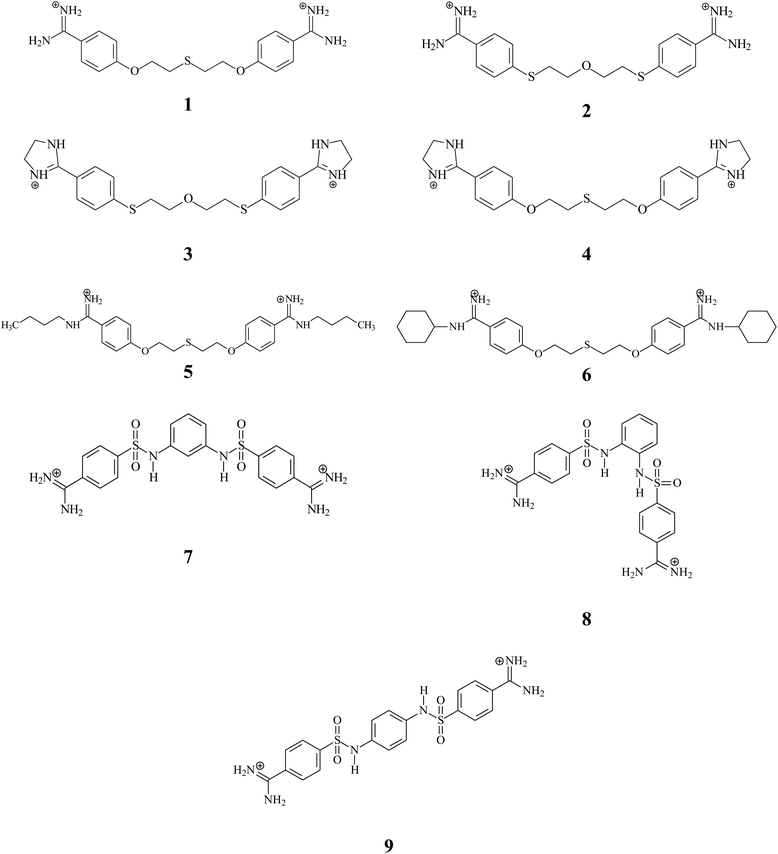 |
| | Fig. 1 Chemical formulas of pentamidine analogs 1–9 used in this study. | |
To get full insight into the drug-likeness of tested molecules, we calculated the theoretical values of the physio-chemical and biopharmaceutical parameters associated with adsorption, distribution, metabolism, elimination, and toxicity (ADMET) in the human body. Next, we elucidated the nature of the interaction between pentamidines and the hERG K+ channel for all nine compounds 1–9 using a combination of experimental and computational approaches. The impact of tested compounds on hERG trafficking and functionality of the hERG currents were examined by traditional western blot analyses, immunofluorescent staining procedures, and patch-clamp electrophysiological assays. The specific interactions governing the predicted blockade of the cardiac K+ channel was scrutinized by molecular docking and free enthalpy calculations.
2. Theoretical calculations
2.1. Ligands and hERG channel
Nine compounds synthesized in our laboratory (Fig. 1) were evaluated: 1,5-bis(4-amidinophenoxy)-3-thiapentane dihydrochloride (1), 1,5-bis[4-(amidino)phenylthio]-3-oxapentane dihydrochloride (2), 1,5-bis[4-(4,5-dihydro-2-imidazolyl)phenylthio]-3-oxapentane dihydrochloride (3), 1,5-bis[4-(4,5-dihydro-2-imidazolyl)phenoxy]-3-thiapentane dihydrochloride (4), 1,5-bis[4-(N-butylamidino)phenoxy)]-3-thiapentane dihydrochloride (5), 1,5-bis[4-(N-cyclohexylamidino)phenoxy)]-3-thiapentane dihydrochloride (6), 4,4′-[1,3-phenylenebis(aminosulfonyl)]dibenzamidine dihydrochloride (7), 4,4′-[1,2-phenylenebis(aminosulfonyl)]dibenzamidine dihydrochloride (8) and 4,4′-[1,4-phenylenebis(aminosulfonyl)]dibenzamidine dihydrochloride (9). Three-dimensional structures of ligand were prepared using Discovery Studio 2017R2 visual interface BIOVIA.38 Geometries of all compounds were optimized using the density functional theory (DFT) with the B3LYP/6-311G (d,p) hybrid functional, as implemented in Gaussian 16.39 ESP-atomic partial charges on all atoms were computed using the Breneman model reproducing the molecular electrostatic potential.40 Out of the generated structural models of the hERG K+ channel41–44 we used the open state model named CSM-TM-model 732 originated from the studies of S. Y. Noskov group.41 The experimentally validated model selected by us is a valuable tool in the theoretical studies of blockade hERG current also for cationic compounds.45–47
2.2. Prediction of drug-likeness descriptors and toxicity
To find out the drug-likeness properties for the synthesized compounds, ADMET calculations were performed by the mathematical models48 implemented into ADMET Predictor™ software version 8.5 Simulations Plus.49 We have predicted various drug-likeness parameters like molecular weight (MWt), distribution coefficient at pH = 7.4 (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) D), number of rotatable bonds (NRB), number of aromatic rings (NAR), the topological polar surface area (TPSA), the volume of distribution (Vd), the solubility (Sw), the effective permeability (Peff), Madin–Darby Canine Kidney cells apparent permeability (MDCK), qualitative likelihood of penetrating the blood–brain barrier (BBB) filter express as high/low, the logarithm of the blood–brain barrier partition coefficient logCbrain/Cblood (logBB), percentage of unbound drug to proteins within blood plasma (% Unbnd) and blood-to-plasma concentration ratio (RBP = Cwhole-blood/Cplasma) were estimated for 1–9 at pH 7.4. Prediction of metabolic indicators was based on phase I tests including cytochrome P450 (CYP 450) forms CYP 2C19, 1A2, 2D6 and 3A4, and the intrinsic hepatic clearance (Clint). All the CYP-metabolites generated for each compound were tested for risk potential whenever it was possible. Phase II metabolism of pentamidine derivatives was evaluated to determine the probability that human uridine 5′-diphosphate-glucuronosyltransferases (UGT) were involved. Whereas, virtual screenings were also performed to evaluate toxicological properties including Maximum Recommended Therapeutic Dose (MRTD), cardiac toxicity by affinity towards hERG-encoded potassium channels (described by the qualitative estimation of likelihood of the hERG potassium channel inhibition (hERG_Filter) and the pIC50 as measure of affinity for hERG K+ channel (hERG_pIC50)), and the hepatotoxicity (described by the level of alkaline phosphatase (AlkPhos), γ-glutamyl transferase (GGT), serum glutamate oxaloacetate transaminase (SGOT), serum glutamate pyruvate transaminase (SGPT) and lactate dehydrogenase (LDH)).
D), number of rotatable bonds (NRB), number of aromatic rings (NAR), the topological polar surface area (TPSA), the volume of distribution (Vd), the solubility (Sw), the effective permeability (Peff), Madin–Darby Canine Kidney cells apparent permeability (MDCK), qualitative likelihood of penetrating the blood–brain barrier (BBB) filter express as high/low, the logarithm of the blood–brain barrier partition coefficient logCbrain/Cblood (logBB), percentage of unbound drug to proteins within blood plasma (% Unbnd) and blood-to-plasma concentration ratio (RBP = Cwhole-blood/Cplasma) were estimated for 1–9 at pH 7.4. Prediction of metabolic indicators was based on phase I tests including cytochrome P450 (CYP 450) forms CYP 2C19, 1A2, 2D6 and 3A4, and the intrinsic hepatic clearance (Clint). All the CYP-metabolites generated for each compound were tested for risk potential whenever it was possible. Phase II metabolism of pentamidine derivatives was evaluated to determine the probability that human uridine 5′-diphosphate-glucuronosyltransferases (UGT) were involved. Whereas, virtual screenings were also performed to evaluate toxicological properties including Maximum Recommended Therapeutic Dose (MRTD), cardiac toxicity by affinity towards hERG-encoded potassium channels (described by the qualitative estimation of likelihood of the hERG potassium channel inhibition (hERG_Filter) and the pIC50 as measure of affinity for hERG K+ channel (hERG_pIC50)), and the hepatotoxicity (described by the level of alkaline phosphatase (AlkPhos), γ-glutamyl transferase (GGT), serum glutamate oxaloacetate transaminase (SGOT), serum glutamate pyruvate transaminase (SGPT) and lactate dehydrogenase (LDH)).
2.3. Molecular docking and dynamic simulations
Molecular docking was performed using CDOCKER module of Discovery Studio 2017R2. The binding site sphere of hERG K+ channel was defined as the regions that come within radius 15 Å from the geometric centroid of the ligands. The poses with the lowest CDOCKER interaction energies were selected as the best conformations for the binding with the active site of the hERG K+ channel through hydrogen bonds and hydrophobic interactions. The best poses predicted by CDOCKER were used as the starting points in the molecular dynamics (MD) simulation.
All MD simulations were run using the CHARMm force field50 implemented in the module of Discovery Studio 2017R2. The hERG channel complexes were surrounded by a cubic box of water molecules (TIP3P models)51 extending up to a distance of 10 Å from any solute atom. Additional K and Cl ions were added randomly to each complex at a concentration of ∼0.15 M, close to physiological conditions52 using the Solvation module of Discovery Studio 2017R2. All energy minimization and molecular dynamics simulations were performed using the Particle Mesh Ewald (PME) algorithm53 for the correct treatment of electrostatic interactions54 and periodic boundary conditions. Before simulations, 2500 steps of steepest descent algorithm followed by 2500 conjugate gradient energy-minimization steps (until the RMS gradient of the structure was below 0.01 kcal mol−1 Å−1) were performed. Subsequently, each simulation started with gradually heating from 50 to 300 K for 100 ps followed by equilibration of the systems for up to 500 ps, after which potential energies were sufficient. The equilibrated system was taken as the starting structure for 10 ns production runs in NPT ensemble, at a temperature of 300 K and 1 bar maintained using a Berendsen thermostat algorithm.55 During minimized, heating and equilibration, the protein and ligand were restrained with a force constant of started with 10 kcal mol−1 Å−2 and gradually decreased to 2 kcal mol−1 Å−2 and then subjected to a 5.5 ns production run with removing all constraint. The SHAKE method was used to constrain hydrogen atoms and the time step was set to 2 fs.56 The coordinates were saved every 10 ps for subsequent analysis.
2.4. Prediction the free enthalpy of binding
Even though that docking combined with MD simulations can provide a clear image of the shape complementarity between the ligand and the receptor, it is required to have the additional and essential information of the free enthalpy of binding, which will assess to quantify the affinity of a ligand to its target. The obtained stable MD trajectory of each complex was used to calculated the binding free enthalpy by the MM-PBSA approach57 using explicit solvent molecules. The binding free enthalpy (ΔGbind) of pentamidine derivatives to hERG channel was calculated by means of eqn (1):| | |
ΔGbind = GhERG–ligand − GhERG − Gligand
| (1) |
where GhERG–ligand is free enthalpy of complex, GhERG is free enthalpy of hERG channel and Gligand is free enthalpy tested pentamidines. Binding free enthalpy was calculated based on the average structures obtained from the last 6 ns of MD trajectories. The components of each complex were minimized using the conjugate gradient method for 10000 steps after 100 steps of the steepest descent algorithm and a dielectric constant of 4 for the electrostatic interactions until the RMS gradient of the structure was <0.001 kcal mol−1 Å−1.
3. Experimental section
3.1. Chemical compounds
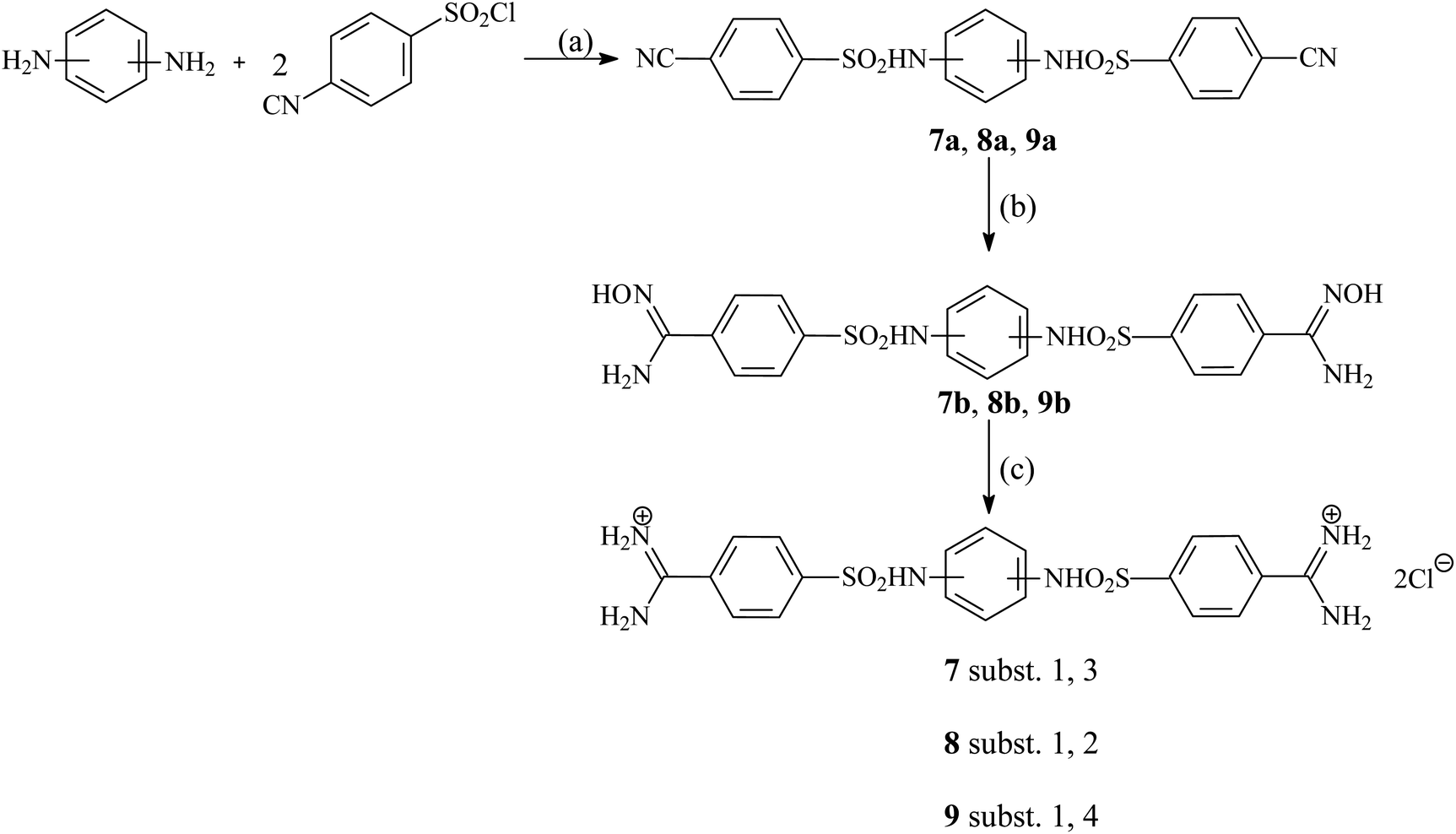
Six pentamidine analogues 1–6 have been described previously,35–37 three 7–9 are newly synthesized. The synthesis of the three isomeric compounds 7–9 was carried out in three stages according to the general procedure shown in Fig. 2. In the first step, 4-cyanobenzenesulfonyl chloride reacted with an appropriate isomer of phenylenediamine in the presence of pyridine to give the initial dinitriles in a high yield. Only 1,3-phenylenediamine was replaced with 1,3-phenylenediamine dihydrochloride, which is much more stable than a free amine and gives the expected product in a significantly higher yield. There was also used as an appropriate excess of pyridine. In the second step, the previously obtained dinitriles were treated with an excess of 50% aqueous solution of hydroxylamine furnishing desired intermediate diamidoximes in high yields. In the last stage formerly received diamidoximes were catalytically (Pd/C) reduced with formic acid in boiling acetic acid to give finally expected diamidines. Treating with an aqueous hydrochloric acid solution led to dihydrochlorides.
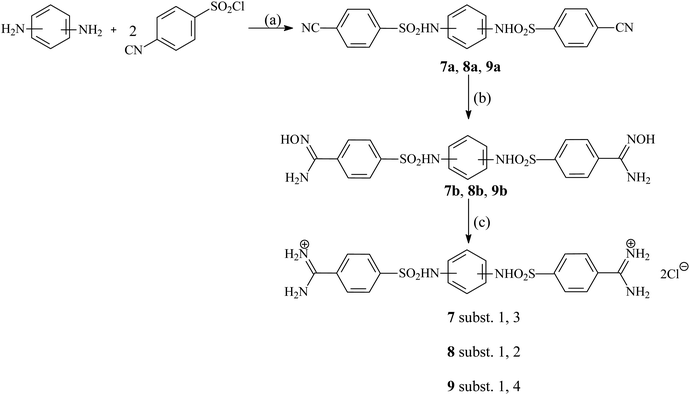 |
| | Fig. 2 Synthetic route of new pentamidine analogs 7–9. Reagents and conditions: (a) NMP, 60 °C, 1 h; (b) 50% NH2OH/ EtOH, r.t., 24 h; (c) HCOOH/10%PdC/AcOH, reflux, 3 h, then aq. HCl. | |
3.2. General procedure for the synthesis of phenylenebis(aminosulfonyl)dibenzonitriles
An appropriate phenylenediamine dihydrochloride (1.08 g, 10 mmol) and pyridine (1.92 g, 24 mmol or 3.80 g, 48 mmol) were dissolved in N-methyl-2-pyrrolidone (20 mL). To the stirred solution 4-cyanobenzenesulfonyl chloride (4.23 g, 21 mmol) was added portion-wise keeping the temperature below 40 °C. Next, the mixture was heated at 60 °C for 1 h and finally poured into ice water (80 mL). The formed solid was filtered, washed with plenty of water and dried giving the almost pure product. They may be purified by refluxing with ethanol (10–20 mL) for several minutes.
3.2.1 4,4′-[1,3-Phenylenebis(aminosulfonyl)]dibenzonitrile (7a). Yield 3.29 g (75%), mp 245–246 °C, 1H NMR (300 MHz, DMSO-d6, δ ppm): 10.59 (s, 2H), 8.05 (d, J = 9 Hz, 4H), 7.82 (d, J = 9 Hz, 4H), 7.10 (t, J = 6 Hz, 1H), 7.00 (s, 1H), 6.74 (d, J = 6 Hz, 2H). 13C NMR (75.4 MHz, δ ppm): 143.2, 137.8, 133.4, 130.2, 127.3, 117.4, 116.2, 115.4, 111.6.
3.2.2 4,4′-[1,2-Phenylenebis(aminosulfonyl)]dibenzonitrile (8a). Yield 3.42 g (78%), mp 238–239 °C, 1H NMR (300 MHz, DMSO-d6, δ ppm): 9.63 (s, 2H), 8.05 (d, J = 8.1 Hz, 4H), 7.87 (d, J = 8.1 Hz, 4H), 7.09–7.06 (m, 2H), 6.95–6.92 (m, 2H). 13C NMR (75.4 MHz, δ ppm): 143.4, 133.5, 129.9, 127.5, 126.7, 124.4, 117.6, 115.5.
3.2.3 4,4′-[1,4-Phenylenebis(aminosulfonyl)]dibenzonitrile (9a). Yield 3.94 g (90%), mp 270–271 °C, 1H NMR (300 MHz, DMSO-d6, δ ppm): 10.41 (s, 2H), 8.02 (d, J = 9 Hz, 4H), 7.82 (d, J = 9 Hz, 4H), 6.95 (s, 4H). 13C NMR (75.4 MHz, δ ppm): 143.2, 133.7, 133.4, 127.4, 122.1, 117.5, 115.1.
3.3. General procedure for the synthesis of phenylenebis(aminosulfonyl)dibenzamide dioximes
An appropriate phenylenebis(aminosulfonyl)dibenzonitrile (1.10 g, 2.5 mmol) was suspended in THF (15 mL). The 50% aqueous solution of hydroxylamine (1.12 g, 16.8 mmol) was subsequently added and the whole was stirred for 24 h at room temperature. Next, the mixture was poured into water (75 mL). The solid was filtered, washed with water and dried giving pure products. They may be used in the next step without additional purification.
3.3.1 4,4′-[1,3-Phenylenebis(aminosulfonyl)]dibenzamide dioxime (7b). Yield: 1.14 g (90%), mp 130–140 °C (decomp.), 1H NMR (300 MHz, DMSO-d6, δ ppm): 10.34 (s, 2H), 9.94 (s, 2H), 7.80 (d, J = 8.4 Hz, 4H), 7.69 (d, J = 8.4 Hz, 4H), 7.11 (s, 1H), 7.03 (t, J = 8.1 Hz, 1H), 6.68 (d, J = 8.1 Hz, 2H), 5.92 (s, 4H). 13C NMR (75.4 MHz, δ ppm): 149.6, 139.3, 138.4, 137.4, 129.8, 126.6, 125.9, 115.1, 110.8.
3.3.2 4,4′-[1,2-Phenylenebis(aminosulfonyl)]dibenzamide dioxime (8b). Yield: 1.06 g (86%), mp 214–215 °C, 1H NMR (300 MHz, DMSO-d6, δ ppm): 9.96 (s, 2H), 9.38 (s, 2H), 7.83 (d, J = 8.4 Hz, 4H), 7.71 (d, J = 8.4, 4H), 7.03–6.95 (m, 4H), 5.95 (s, 4H). 13C NMR (75.4 MHz, δ ppm): 149.5, 138.9, 137.6, 129.9, 126.8, 126.1, 126.0.
3.3.3 4,4′-[1,4-Phenylenebis(aminosulfonyl)]dibenzamide dioxime (9b). Yield: 1.16 g (92%), mp 253–254 °C, 1H NMR (300 MHz, DMSO-d6, δ ppm): 10.17 (s, 2H), 9.93 (s, 2H), 7.78 (d, J = 9 Hz, 4H), 7.66 (d, J = 9 Hz, 4H), 6.93 (s, 4H), 5.92 (s, 4H). 13C NMR (75.4 MHz, δ ppm): 149.7, 139.4, 137.4, 133.9, 126.5, 126.0, 121.7.
3.4. General procedure for the synthesis of phenylenebis(aminosulfonyl)dibenzamidine dihydrochlorides
An appropriate phenylenebis(aminosulfonyl)dibenzamide oxime (1.00 g, 2 mmol) was suspended in glacial acetic acid (8 mL). Then palladium on carbon (Pd/C 10% w/w) (80 mg) and subsequently formic acid (0.8 mL) were added. The mixture was refluxed for 3 h, after which cooled to room temperature. The catalyst and other solid impurities were filtered off. To the filtrate, a 35% aqueous solution of HCl (0.5 mL) was added dropwise and all the solvents evaporated to dryness. The solid residue was purified by refluxing with ethanol (5–10 mL).
3.4.1 4,4′-[1,3-Phenylenebis(aminosulfonyl)]dibenzamidine dihydrochloride (7). Yield: 1.00 g (92%), mp 290 °C (decomp.), 1H NMR (300 MHz, DMSO-d6, δ ppm): 10.80 (s, 2H), 9.60 (s, 4H), 9.43 (s, 4H), 8.01–7.88 (m, 8H), 7.20 (s, 1H), 7.08 (t, J = 7.8 Hz, 1H), 6.78 (d, J = 7.8 Hz, 2H). 13C NMR (75.4 MHz, δ ppm): 164.8, 143.6, 138.2, 132.2, 130.0, 129.4, 127.0, 115.2, 110.7. TOF MS ES+ [M − 2Cl − H]+ calcd for C28H21N6O4S2 (473.1066) found 473.1082.
3.4.2 4,4′-[1,2-Phenylenebis(aminosulfonyl)]dibenzamidine dihydrochloride (8). Yield: 0.30 g (28%), mp 290 °C (decomp.), 1H NMR (300 MHz, DMSO-d6, δ ppm): 10.30 (s, 2H), 9.57 (s, 4H), 9.37 (s, 4H), 8.03–7.96 (m, 8H), 7.01 (s, 4H). 13C NMR (75.4 MHz, δ ppm): 164.7, 144.2, 132.1, 129.8, 129.3, 127.1, 126.0, 123.8. TOF MS ES+ [M − 2Cl − H]+ calcd for C28H21N6O4S2 (473.1066) found 473.1053.
3.4.3 4,4′-[1,4-Phenylenebis(aminosulfonyl)]dibenzamidine dihydrochloride (9). Yield: 0.86 g (79%), mp 270 °C (decomp.), 1H NMR (300 MHz, DMSO-d6, δ ppm): 10.64 (s, 2H), 9.95 (s, 4H), 9.37 (s, 4H), 7.97–7.91 (m, 8H), 7.02 (s, 4H). 13C NMR (75.4 MHz, δ ppm): 164.8, 143.7, 133.7, 132.2, 129.4, 126.9, 121.5. TOF MS ES+ [M − 2Cl − H]+ calcd for C28H21N6O4S2 (473.1066) found 473.1052.
3.5. Biological data
All analogues were dissolved in DMSO as a stock solution at 50 mM and further diluted in ddH2O. The final working solution for all compounds was 3 mM. Pentamidine-isethionate (Pentacrit® 300, Sanofi Aventis, Gouda, The Netherlands) was dissolved in ddH2O and final working solution was 10 mM. Dofetilide (Procter and Gamble, Cincinnati, USA) was dissolved in DMSO and further diluted in ddH2O, final working solution was 1 mM. Concentrations of these controls were used as in previous research.34 All compounds were sterilized by filtration (0.2 μm), aliquoted and stored at −20 °C until use.
3.5.1 Cell culture. HEK-293T cells stably expressing hERG (HEK-hERG) were obtained from C. January.58 The cells were maintained in DMEM (Gibco-Fisher Scientific, Landsmeer, The Netherlands) with 10% fetal calf serum (Sigma-Aldrich, Zwijndrecht, The Netherlands), 2 mM L-glutamine, 50 μg mL−1 streptomycin and 1 U mL−1 penicillin (all three Lonza, Breda, The Netherlands). Cells were split twice a week and cultured at 37 °C and 5% CO2. The amount of DMSO added to the cells was kept below 0.1%.
3.6. Western blotting
Cells were cultured in 60 mm dishes for 20–24 h till confluency of 60–70%. Cells were treated with pentamidine (hERG trafficking blocker, 10 μM), dofetilide (hERG trafficking activator, 1 μM) or pentamidine analogues 1–9 (1 and 10 μM) for 24 h. Full dose–response experiments were performed with 1, 2 and 5 (0.5, 1, 3, 5, 10 and 20 μM). Following incubations, cells were washed with PBS (Lonza, Basel, Switzerland) containing 1 mM MgCl2 and CaCl2 (PBS++) twice, and lysed in 130–180 μL of lysis buffer (buffer D 20 mM HEPES, 125 mM NaCl, 10% (v/v) glycerol, 1 mM EDTA, 1 mM EGTA, 1 mM dithiothreitol and 1% (v/v) Triton X-100), 6.8 μg mL−1 aprotinin (Sigma-Aldrich, Zwijndrecht, The Netherlands and 1 mM PMSF) per dish was added depending on cell confluency. Lysates were centrifuged at 14000 × g for 10 min and the supernatant was collected. Protein concentrations were determined by bicinchoninic acid (Pierce™ BCA protein assay kit, Thermo Scientific, Waltham, USA) in combination with the Nanodrop 2000 (Thermo Scientific, Waltham, USA). Ten μg samples were run by 7% SDS-PAGE and proteins were blotted on a nitrocellulose membrane (Bio-Rad Laboratories, Veenendaal, The Netherlands). Reversible Ponceau red staining was used to determine equal loading and subsequent quantification. Membranes were blocked with 5% (w/v) non-fat milk powder (in TBST) and subsequently incubated overnight at 4 °C with the primary antibody, anti-Kv11.1 (1:2500; Alomone Labs, Jerusalem, Israel). Secondary antibody GαRb HRP (Bio-Rad Laboratories, Veenendaal, The Netherlands) was applied for 2 h at room temperature at 1:10.000. Membranes were developed using Amersham ECL™ prime western blotting detection reagent (GE Lifesciences, Chicago, USA) with Chemidoc XRS+ (Bio-Rad Laboratories, Veenendaal, The Netherlands). Quantification was done by Image Lab™ software version 5.2.1 (Bio-Rad Laboratories, Veenendaal, The Netherlands).
3.7. Immunofluorescence microscopy
HEK-hERG cells were cultured on ∅ 15 mm glass coverslips coated with 0.1% gelatin. After 24 h treatment with pentamidine (10 μM), dofetilide (1 μM) or 1, 2 or 5 (10 μM) coverslips were washed with PBS++, fixated with 3%-paraformaldehyde and permeabilized with 0.5% Triton X-100 (in PBS). After permeabilization cells were quenched with PBS/glycine (50 mM) and incubated twice with NET-gel (0.25% gelatin, 50 mM Tris–Cl, 150 mM NaCl, 4 mM EDTA, 0.05% Igepal, ±0.01% NaN3, pH 7.4). Cells were incubated with the primary antibody in NET-gel overnight (anti-Kv11.1, Alomone Labs, Jerusalem, Israel). After washing the cells, samples were incubated with the secondary antibody (Jackson). Coverslips were mounted with Vectashield (Vector Laboratories Inc., Burlingame, USA) and imaged using a Nikon Eclipse 80i (Nikon, Amsterdam, The Netherlands) and NIS elements Basic Research (Nikon, Amsterdam, The Netherlands) software.
3.8. Electrophysiological recordings
HEK-hERG cells were cultured on ∅ 12 mm glass coverslips coated with poly-L-lysine (Sigma-Aldrich, Germany). IhERG was recorded in whole-cell voltage mode. Pipettes (Harvard apparatus, Holliston, USA) were pulled and polished (Model P-2000, Sutter instruments Co., Novato, USA) and had a resistance of 2–4 MΩ once filled with solution. Potassium currents were measured with Axopatch 200B (Axon Instruments, USA) and Clampex 10.0 software.59 The following protocol was used: holding potential was −80 mV, a hyperpolarizing step was applied for 50 ms at −120 mV to correct for leak currents. Thereafter 4 s test pulses between −80 mV and +60 mV in 10 mV increments were applied. And it was followed by a 5 s deactivation pulse at −50 mV. After 3.5 min when the current had stabilized a control measurement was done. Subsequently, acute treatment with 4, 5, 6, 7, 8 or 9 (1 μM) was applied with a flow of ∼1 mL min−1. Currents were measured at 3.5, 7 and 15 min. All measurements were obtained at room temperature (22 °C). Bath solution consisted of 140 mM NaCl, 4 mM KCl, 10 mM HEPES, 2 mM CaCl2, 1 mM MgCl2 and adjusted to pH 7.4 (NaOH). Pipette solution consisted of 10 mM EGTA, 110 mM KCl, 10 mM HEPES, 4 mM K2-ATP, 5.17 mM CaCl2, 1.42 mM MgCl2, 15 mM sucrose and adjusted to pH 7.2 (KOH). The data were quantified by Clampfit software.60
3.9. Data analysis
All results are presented as mean ± standard deviation. Differences were tested with a one-way ANOVA and a Bonferroni post hoc test. Only for the difference between 1 and 10 μM treated cells a two-way ANOVA has been performed. All western blot data were normalized for control before statistical analysis. For the patch-clamp data values at +10 mV were used for statistical analysis. Differences were considered statistically significant with a p-value <0.05. All analyses were performed using GraphPad Prism version 7.04.61
4. Results and discussion
4.1. Assessing of drug-likeness parameters
All studied pentamidines 1–9 are analyzed in the dicationic form. The predicted drug-likeness parameters (the topological polar surface area (TPSA), volume of distribution (Vd), water solubility (Sw), effective permeability (Peff), apparent permeability (MDCK), percentage of unbound drug to blood plasma proteins (% Unbnd), blood-to-plasma concentration ratio (RBP), BBB_filter, blood–brain barrier partition coefficient (logBB)) for tested pentamidines are shown in Table 1. TPSA serves as an important parameter for the evaluation of molecular transport properties, especially in intestinal absorption and blood–brain barrier penetration. Its values for 1–6 were found within range 58.01–118.2 Å2 and these compounds are predicted to have a high capacity for penetrating cell membranes.62 New compounds 7–9 with sulfanilide groups show values close to 192 Å2 and it may accounts for their easy penetration in hydrophilic environments, such as the core of transporter proteins.63 The volume Vd is the pharmacokinetic parameter which can show the dose required to give a certain plasma concentration. The volume Vd of all compounds under study is in the range of 0.454–10.621 L kg−1 and probably they are not distributed to all tissues of the body. Compounds 7–9 which have Vd below 0.7 L kg−1 are confined to the blood plasma, but compounds 1–6 with one or two S atoms in the linker have values higher than 4.5 L kg−1 and are predicted to be distributed in whole blood. The aqueous solubility (Sw) of all tested pentamidine derivatives (we should remember that they are dihydrochlorides) are good and are within the range of 0.043–1.192 mg mL−1. The best solubility's have two dihydrochlorides: 1,5-bis(4-amidinophenoxy)-3-thiopentane (1) and 1,5-bis[4-(amidino)phenylthio]-3-oxapentane (2) (1.192 and 0.697 mg mL−1, respectively). Human jejunal permeability (Peff), which reflects the passive transport velocity in cm s−1 across the epithelial barrier in the human jejunum, was predicted high for 1–6 (0.701–2.063 × 10−4 cm s−1). The values of Madin–Darby canine kidney (MDCK), a parameter for assessing the apparent membrane permeability properties, were also high (47.23–804.27 × 10−7 cm s−1). New dihydrochlorides with phenylenebis(aminosulfonyl) linker (7, 8 and 9) have these two parameters decreased (Peff below 0.5 × 10−4 cm s−1) and MDCK (below 7.04 × 10−7 cm s−1), showing lower jejunal permeation and apparent membrane permeability. An ability to bind to plasma proteins, which is in most cases considered undesirable, is characterized by two parameters: the percentage of drug unbound to protein within blood plasma (% Unbnd) and the concentration of the drug in whole blood compared to plasma (RBP). For the majority of tested pentamidines the values of % Unbnd are in the region 10–41%, and the values of RBP in the region 1.01–1.17, which indicates low or moderate binding to plasma protein. Only values of % Unbnd and RBP for compounds 5 and 6 were below 10% and 1, respectively, which suggests that the alkyl substituents at amidine groups increase the plasma protein binding. The next molecular properties BBB_filter and logBB determine the likelihood of crossing the blood–brain barrier (BBB). Analysis of both parameters revealed that 3–6 derivatives remain in the qualitative likelihood of crossing the BBB because logBB was more than 0.3 values, suggesting that alkyl substituent at amidine groups increases the potential for BBB penetration. Whilst, bis-benzamidines 1, 2 and new 7–9 have the lower logBB values (below 0 and even below −1) suggesting the favorable impact of phenylenebis(aminosulfonyl) linker for toxicity joined with BBB penetration.
Table 1 Theoretical values of topological polar surface area (TPSA), distribution (Vd), water solubility (Sw), effective permeability (Peff), apparent permeability MDCK, percentage of unbound drug to blood plasma proteins (% Unbnd), blood-to-plasma concentration ratio (RBP), BBB filter and blood–brain barrier partition coefficient (logBB) for 1–9
| Compound |
TPSA |
Vd |
Sw |
Peff |
MDCK |
% Unbnd |
RBP |
BBB filter |
logBB |
| Expected values |
| (≤140 Å2) |
(≤3.7 L kg−1) |
(≥0.010 mg mL−1) |
(≥0.5 cm s−1 × 104) |
(≥30 cm s−1 × 107) |
(>10%) |
(<1) |
(High/low) |
|
| 1 |
118.20 |
4.606 |
1.192 |
0.701 |
59.926 |
16.085 |
1.122 |
High |
−0.058 |
| 2 |
108.97 |
6.410 |
0.697 |
0.728 |
47.227 |
15.652 |
1.116 |
High |
−0.231 |
| 3 |
58.01 |
7.178 |
0.183 |
2.063 |
638.620 |
10.100 |
1.163 |
High |
0.592 |
| 4 |
67.24 |
4.570 |
0.509 |
2.003 |
804.274 |
9.687 |
1.173 |
High |
0.783 |
| 5 |
90.22 |
10.621 |
0.096 |
0.810 |
407.340 |
2.841 |
0.912 |
High |
0.576 |
| 6 |
90.22 |
5.462 |
0.043 |
0.807 |
374.797 |
1.776 |
0.993 |
High |
0.653 |
| 7 |
192.08 |
0.454 |
0.160 |
0.141 |
4.343 |
34.676 |
1.076 |
Low |
−0.677 |
| 8 |
192.08 |
0.462 |
0.102 |
0.180 |
7.039 |
22.170 |
1.104 |
Low |
−1.251 |
| 9 |
192.08 |
0.617 |
0.287 |
0.131 |
3.953 |
41.859 |
1.012 |
Low |
−1.083 |
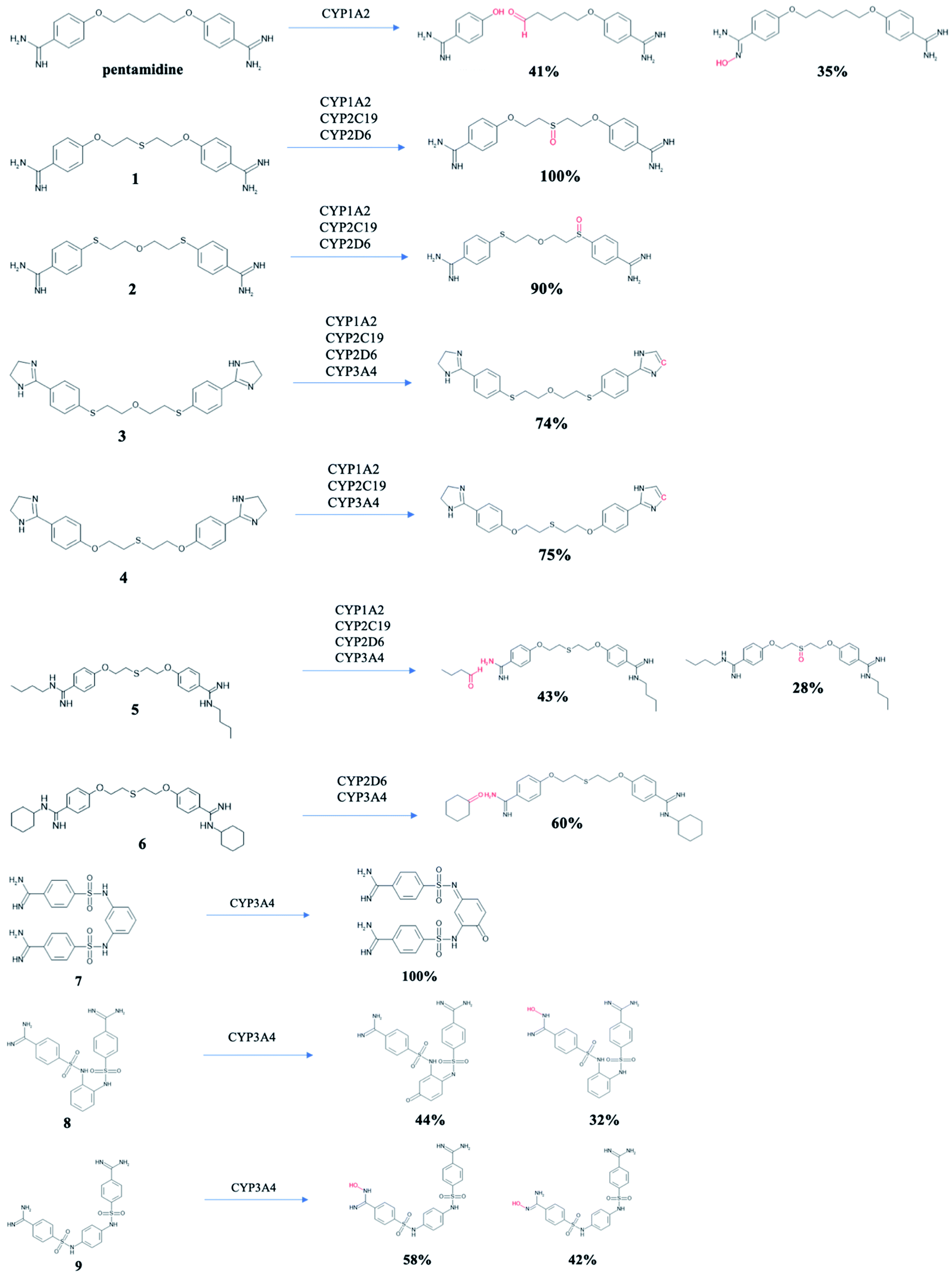
Pentamidines were targeted for the evaluation of the metabolic biotransformations. The predictions were made for phase I (cytochrome P450s hepatic enzyme system) and phase II (the enzymatic conjugation of substrates with the UGT enzymes).64 Modes of action (substrate/non-substrate/inhibition) and intrinsic hepatic clearance (Clint) are predicted for four isoforms cytochrome P450 (1A2, 2C19, 2D6 and 3A4),65 which are considered as the most important in xenobiotic metabolism (Table 2). Compounds 1 and 2 cannot be metabolized by CYP3A4, while the other tested pentamidines can be its substrates. Pentamidine derivatives 3 and 4 with amidine groups closed in imidazoline rings can be substrates or inhibitors of all isoforms, but 6 cannot be metabolized by CYP2C19, 7 and 9 by CYP2D6, and 8 by three isoforms CYP1A2, CYP2C19, and CYP2D6. Since inhibition of cytochrome P450 is the main source of adverse drug interactions, we examined intrinsic hepatic clearance (Clint) which describes the intrinsic ability of the liver to remove the drug in absence of restrictions imposed on drug delivery to the liver cell by blood flow or protein binding. The recommended range for predicted Clint values is >30 (μL min−1 mg−1). All studied pentamidines demonstrated microsomal metabolic liabilities with wide range of Clint values ranging from 15.3 to 80.3 (μL min−1 mg−1) for CYP1A1, from 64.0 to 437.7 (μL min−1 mg−1) for CYP2C19, from 104.5 to 812.0 (μL min−1 mg−1) for CYP2D6, and from 5.0 to 1790.0 (μL min−1 mg−1) for CYP3A4. The highest value (1790.0 (μL min−1 mg−1) for CYP3A4) was observed for compound 6 with cyclohexyl substituent at amidine groups, while the lowest value Clint <30 (μL min−1 mg−1) also for CYP3A4 was seen for new compounds 7, 8 and 9 with benzenesulfanilide linker (11.24, 7.95 and 5.03 (μL min−1 mg−1), respectively). It can mean that hepatic flow will have minimal influence on its metabolism. Further, nine main microsomal hepatic UGTs were checked to identify which isoforms may be responsible for pentamidines glucuronidation reaction leading to their easier elimination. Compounds 1–6 are evaluated as a substrate for three isoenzymes 1A1, 1A3 and 1A4, but new bis-benzamidines 7–9 are predicted rather not conjugate in phase II. Only isoforms 1A8 or 1A9 have some potential to metabolize these compounds (see Table S1 in ESI†). Potential structures of metabolites for tested pentamidines are shown in Fig. 3. Experimental data are approachable for pentamidine itself in rats.66 Two major pentamidine metabolites have the hydroxyl groups at the pentyl linker. Proposed metabolic transformations in humans lead to carboxylic acid derivative and 4-hydroxybenzamidine. For pentamidines 1–6 in which S atoms are present in the aliphatic linker, oxidation of one S atom was predicted for compounds 1–3 and 5, while for compound 4 dehydrogenation of imidazoline ring was predicted. Moreover, in 5 and 6 the aliphatic substituents may also be removed. New pentamidines 7–9 with phenyl sulfanilide moiety can be oxidized to chinones or one amidine group can be substituted by the hydroxyl group.
Table 2 Mode of action (inhibition/induction) and intrinsic hepatic clearance (Clint) for selected cytochrome P450 enzymes of 1–9a
| Compound |
CYP |
| Mode of action |
Clint (μL min−1 mg−1) |
| 1A2 |
2C19 |
2D6 |
3A4 |
1A2 |
2C19 |
2D6 |
3A4 |
| I – denotes inhibition of CYP isoforms; S – denotes substrate of CYP isoform; NS – denotes non-substrate for isoform. |
| 1 |
S/I |
S |
S/I |
NS |
16.878 |
63.994 |
104.452 |
— |
| 2 |
S/I |
S |
S/I |
NS |
15.311 |
85.408 |
229.701 |
— |
| 3 |
S/I |
S |
S/I |
S |
28.593 |
157.004 |
811.987 |
43.544 |
| 4 |
S/I |
S |
I |
S |
80.280 |
153.788 |
— |
46.525 |
| 5 |
S |
S |
S/I |
S |
70.659 |
437.686 |
367.709 |
377.690 |
| 6 |
I |
NS |
S/I |
S |
— |
— |
727.161 |
1789.969 |
| 7 |
I |
I |
NS |
S |
— |
— |
— |
11.238 |
| 8 |
NS |
NS |
NS |
S |
— |
— |
— |
7.949 |
| 9 |
I |
I |
NS |
S |
— |
— |
— |
5.025 |
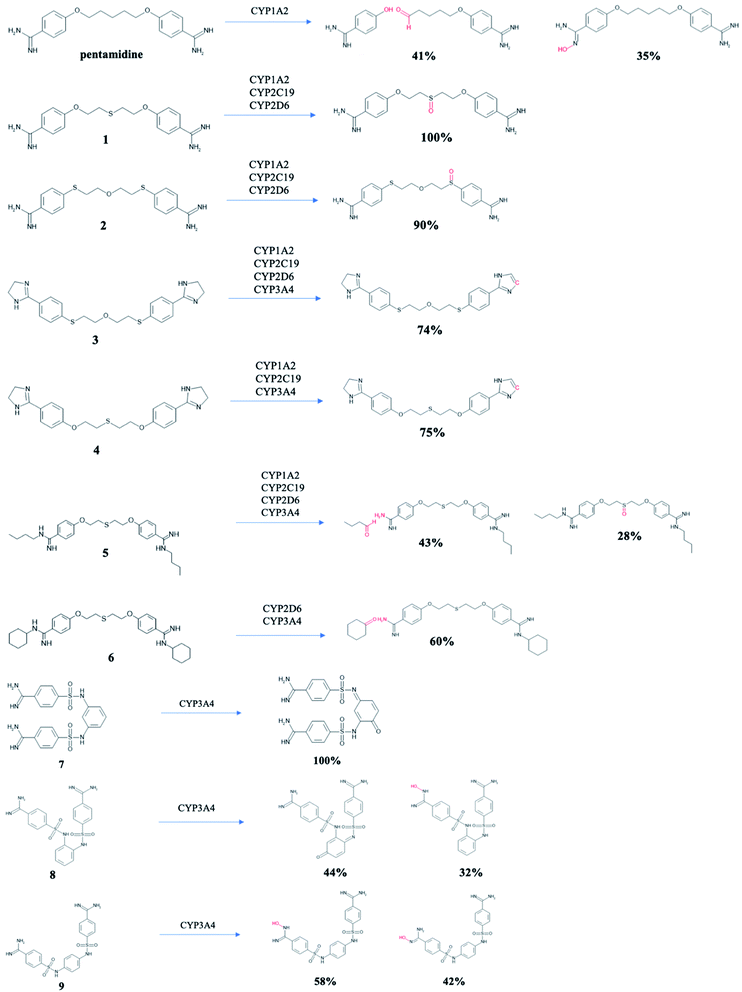 |
| | Fig. 3 Products of metabolism of pentamidine and 1–9 derivatives. | |
In the evaluation of the toxicity profile of pentamidines 1–9 several predictors were used including human maximum tolerated dose and hepatotoxicity (see Table 3). A maximum recommended therapeutic dose (MRTD) in a unit of mg per kg of body weight per day should be greater than 3.16 mg per kg bw per day, which can show that the tested compound has fewer side effects. All by one pentamidines have MRTD value >3.16, indicating that the appearance of side effects is less likely. Only 1,5-bis[4-(N-cyclohexylamidino)phenoxy]-3-thiapentane dihydrochloride (6) has MRTD value below 3.16. In the test of the hepatic injury, the majority of studied pentamidines are predicted as “non-toxic”. Level of LDH, SGOT, and SGPT are elevated only for 1 and 4 indicating some hepatic problems. The cytotoxicity of 1–6 were tested experimentally in our former works35–37 with other groups of pentamidines, which can explain observed data. In these assays, the majority of compounds were not cytotoxic (also those with the sulfobenzene group), only compounds 1 and 4 exhibited less or moderate cytotoxicity. The predicted hepatic problems for 1 and 4 have thrown more light on the factors affecting experimental data.
Table 3 Predicted hepatoxicity parameters for 1–9: maximum recommended therapeutic dose MRTD, level of alkaline phosphatase (AlkPhos), level of γ-glutamyl transferase (GGT), level of serum glutamate oxaloacetate transaminase (SGOT), level of serum glutamate pyruvate transaminase (SGPT), level of lactate dehydrogenase (LDH)
| Compound |
MRDT |
AlkPhos |
GGT |
LDH |
SGOT |
SGPT |
| Expected values |
| (>3.16 mg per kg per day) |
| 1 |
Above 3.16 |
NT |
NT |
T |
T |
T |
| 2 |
Above 3.16 |
NT |
NT |
NT |
T |
T |
| 3 |
Above 3.16 |
NT |
NT |
NT |
T |
T |
| 4 |
Above 3.16 |
NT |
NT |
T |
T |
T |
| 5 |
Above 3.16 |
NT |
NT |
T |
NT |
T |
| 6 |
Below 3.16 |
NT |
NT |
T |
NT |
T |
| 7 |
Above 3.16 |
NT |
T |
T |
NT |
NT |
| 8 |
Above 3.16 |
T |
T |
T |
T |
NT |
| 9 |
Above 3.16 |
T |
T |
T |
NT |
NT |
4.2. Impact of molecular descriptors on hERG binding
Cardiotoxicity is a severe problem in pharmacology; therefore, the theoretical parameters can be very useful in the drug discovery pipeline. Selected theoretical physio-chemical properties are presented in Table 4. The molecular weights (MWt) are in the range defined for orally available compounds and only compound 6 has the MWt of >500 g mol−1, since N,N′-cyclohexyl substituent attached to the amidine group increase its MWt to 522.76 g mol−1. Many studies suggest that the relationship between hERG blockage and molecular size is complex – the probability of inhibition is very low for small molecules with MWt <250 g mol−1, but larger molecules can be also non-blockers of the hERG channel.67–69 The logP values (for pentamidines acting as neutral molecules) were within the range 1.21–3.88 for the majority of compounds which is an agreement with a probability of lack inhibition of hERG channel (logP ≤ 4). For two pentamidine derivatives, 5 and 6, the logP values were predicted to be above 4 indicating a higher risk for hERG K+ channel inhibition.68 We also examined logarithm of the distribution coefficient values (logD) at pH 7.4, which give an estimate of the lipophilicity of a drug at the pH of blood plasma. The average values of logD are within the range −0.03 to 2.75 and not exceeding the traditionally cutoff value of 3.5. Molecular flexibility is an important contributor to hERG affinity, as rigid molecules with little conformational freedom are less likely to occupy an optimal pose within the binding region. We have chosen to describe this property by the ratio of rotatable bonds to all bonds in the molecule (FRB = NRB/Ntotal bonds) rather than the overall count of rotatable bonds. According to the suggestion of Didziapetris et al.,69 FBR below 0.4 becomes a major indicator of poor hERG binding for compounds with highly constrained ring systems. The obtained results indicate that among the analyzed pentamidine derivatives, only compound 5 with 4-(N,N′-butylamidino)phenoxy substituent has FRB above 0.4 i.e. should show some binding affinity to the hERG K+ channel. The impact of π-stacking interactions is another molecular property essential for hERG affinity,70 even though the currently accepted view is that aromaticity is not essential for efficient hERG binding, as long as the molecule is sufficiently lipophilic and has a suitable size and shape.71 The number of aromatic rings (NAR) is 2 or 3 for all tested pentamidine analogs, which is consistent with several published pharmacophore models that predict the ability to induce the LQTS by blocking the hERG K+ channel. The minimal structural requirements in these models are three phenyl rings suitably spaced and one proton at nitrogen.72,73
Table 4 Physio-chemical and cardiotoxicity (hERG_filter and affinity for hERG K+ (hERG_ pIC50)) parameters for 1–9
| Compound |
MWt |
logP |
logD |
NRB |
NAR |
FRB |
hERG filter |
hERG pIC50 |
| Expected values |
| (≤500 g mol−1) |
(≤4) |
(≤3.5) |
(≤10) |
(≤3) |
(<0.4) |
(Yes/No) |
(>5.5) |
| 1 |
358.46 |
2.969 |
0.334 |
8 |
2 |
0.3 |
No |
4.874 |
| 2 |
374.53 |
2.934 |
0.627 |
8 |
2 |
0.3 |
No |
4.492 |
| 3 |
426.61 |
3.746 |
−0.453 |
8 |
2 |
0.25 |
No |
5.684 |
| 4 |
410.54 |
3.877 |
−1.180 |
8 |
2 |
0.25 |
Yes |
6.154 |
| 5 |
470.68 |
6.042 |
2.541 |
16 |
2 |
0.47 |
Yes |
6.366 |
| 6 |
522.76 |
6.828 |
2.748 |
12 |
2 |
0.3 |
Yes |
6.676 |
| 7 |
472.55 |
1.211 |
0.550 |
6 |
3 |
0.2 |
No |
6.414 |
| 8 |
472.55 |
1.355 |
0.985 |
6 |
3 |
0.2 |
No |
5.802 |
| 9 |
472.55 |
1.407 |
−0.025 |
6 |
3 |
0.2 |
No |
6.078 |
The calculated qualitative parameters of the likelihood of the hERG potassium channel inhibition as hERG_filter and the pIC50 as a measure of affinity for hERG K+ channel (hERG_pIC50) are promising for compounds 1–3 and new 7–9 i.e. they can be classified as non-cardiotoxic. For the other three pentamidines (4, 5 and 6 with one S atom in the linker and the alkyl substituent in the amidine moiety) hERG_filter is marked as Yes and hERG_pIC50 values are in the range 6.154–6.676, exceeding the cutoff value 5.5. It means that only these compounds can potentially block hERG channel in the heart cells leading to cardiac problems. To explain the molecular mechanism of cardiotoxicity for those pentamidine analogues, compounds 4, 5 and 6 have been chosen to perform hERG protein affinity examination by molecular modeling technique and the experimental measurements.
4.3. Theoretical evaluation of interactions with hERG channel
First, the molecular docking calculations have been employed in order to find the optimal position of compounds in the binding pocket of hERG K+ channel. CDOCKER program successfully docked compounds 4, 5 and 6 into the binding cavity of hERG K+ channel producing ten poses for every compound with scores shown in Table S2 in ESI.† The poses which demonstrated the strongest interaction energies were selected as the ligand starting structures to the extensive molecular dynamics' simulations and the binding free enthalpy calculations. The predicted binding mode by CDOCKER is presented in ESI, Fig. S1† for 4, 5 and 6. The results demonstrate that analyzed compounds could interact with hERG K+ channel using hydrogen bonds, hydrophobic and van der Waals interactions. The amino acids Leu622, Thr623, Ser624 and Ser649 were consistently involved in the hydrogen bonds and the amino acids Tyr652, Ala653, and Phe656 in the hydrophobic and the π–π interactions. Compounds 5 and 6 interacted with the amino acid Phe656, while 4 with Tyr652 amino acid what is characteristic for the hERG K+ channel blockers.74
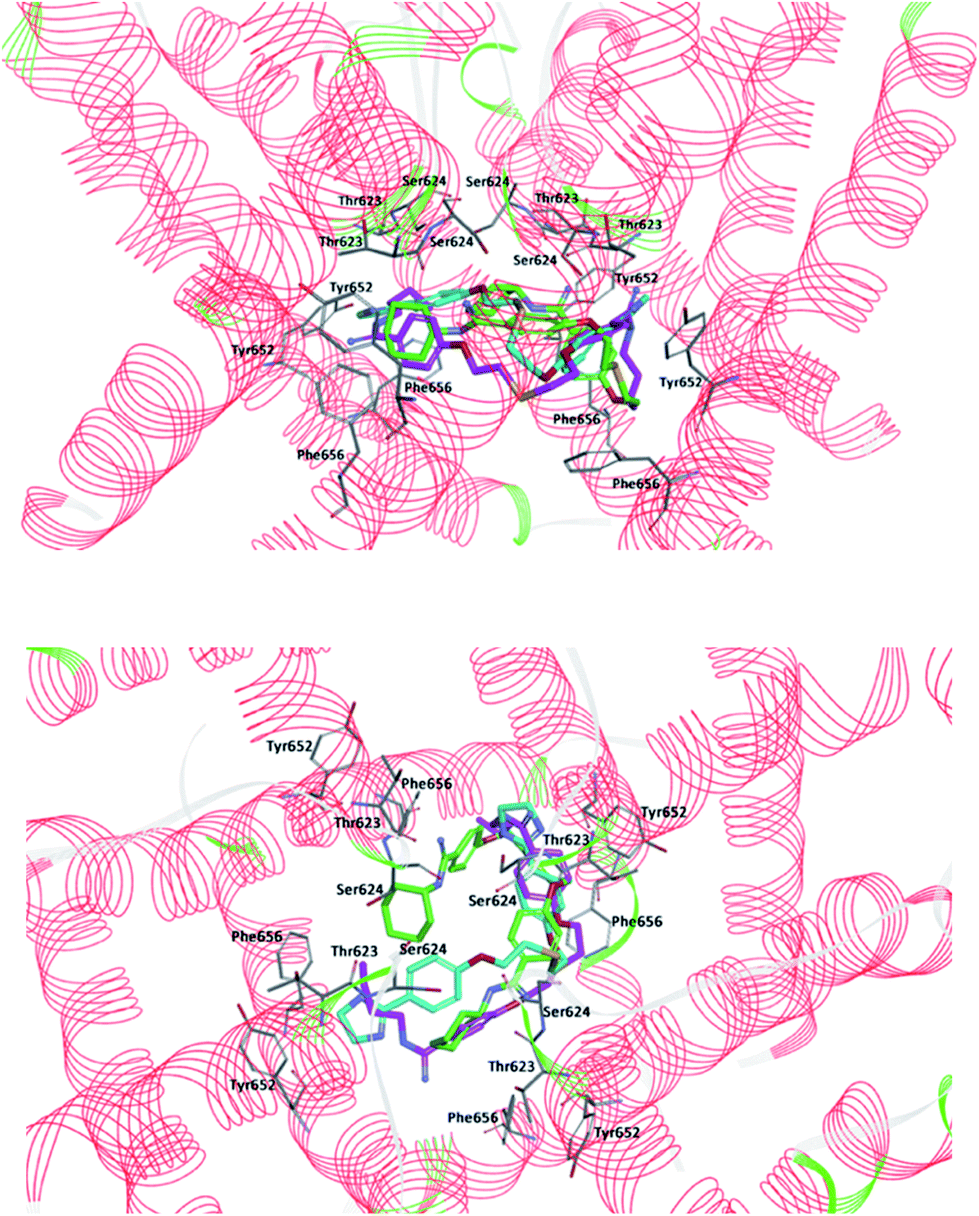
Next, the molecular dynamics simulation and binding free enthalpy calculations were performed for better understanding the various interactions between the ligand and the active site of hERG and to rationalize the obtained biological results. The structures of hERG complexes with 4–6 were optimized with water molecules and ions. The RMSD values between the average structures of replicate MD simulations as well as between the ligand starting and average structures were low in all cases. The binding free enthalpy values calculated using the MM-PBSA method were as follows −23.34 kcal mol−1 (4), −31.55 kcal mol−1 (5) and −13.70 kcal mol−1 (6), indicating the strongest interactions with compound 5 where butyl substituents at amidine groups are present. All compounds are in the same region of the hERG K+ cavity and are located perpendicularly to the cavity walls (Fig. 4). Our modelling suggests that tested pentamidines are accommodated in the proximity of amino acids Leu622, Thr623, Ser649, Tyr652, Ala653, and Phe656. The binding cavity is highly hydrophobic, and this feature is following the biochemical function of the hERG fragment which is embedded in the membrane bilayer.
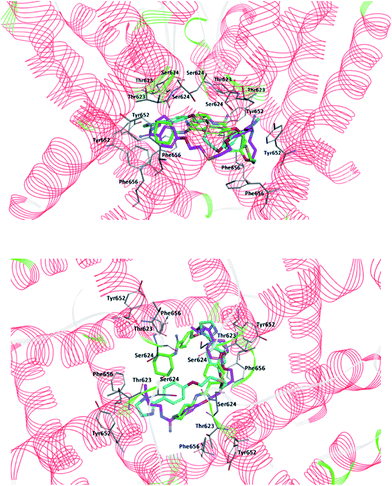 |
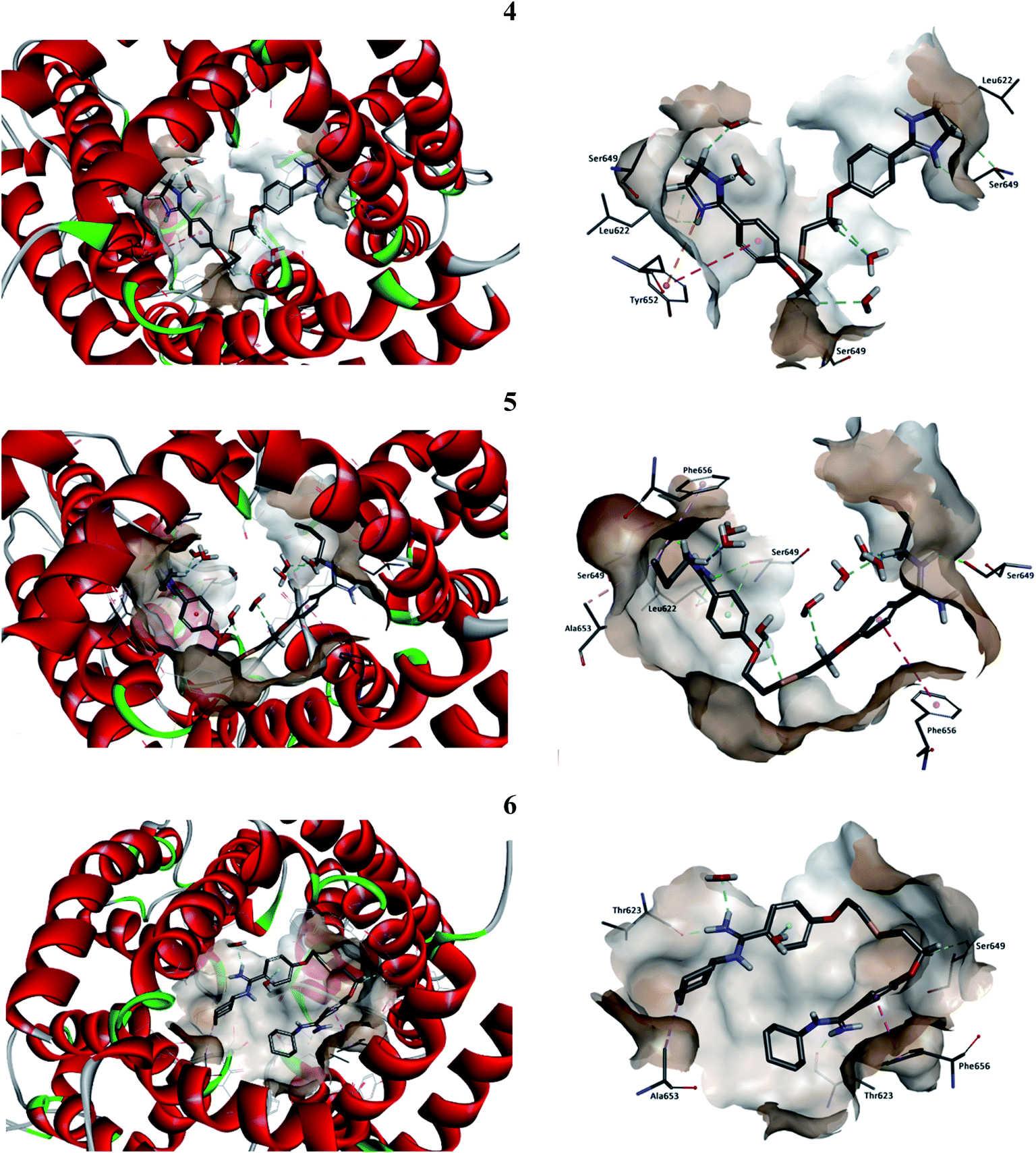
| | Fig. 4 Two views of predicted docking pose in the cavity of hERG K+ channel for compounds: 4 (cyan), 5 (fuchsia) and 6 (green). For clarity, subunits of the hERG K+ channel were removed and key residues (Thr623, Ser624, Tyr652, and Phe656) interacting with the ligands are visible. Upper part: projection of the compounds from the wall side seen perpendicular. Lower part: projection of the compounds from the upper side of the channel. | |
The interactions inside complexes 4–6 are depicted in Fig. 5. These compounds are highly similar structurally and differ only by the kind of N,N′-substituent at amidine groups. Compound 4 with amidine groups closed in 4-(4,5-dihydro-2-imidazolyl) substituents is predicted to be accommodated in the hERG channel in a bent conformation. Such orientation in the binding cavity leads to the π–π stacking (a distance of 5.61 Å) and electrostatic interaction (a distance of 4.72 Å) with the phenyl ring of amino acid Tyr652. Additionally, the position of 4 allows for creating strong hydrogen bonds with two copies of amino acids Leu622 (2.07 and 2.85 Å) and Ser649 (2.11 and 2.25 Å). Compound 5 with the N,N′-butylamidino substituents adopts a similar orientation like that of 4 and interacts with two copies of Phe656 involving T-shaped π–π stacking (4.93 and 5.70 Å). Aliphatic substituent interacts with side chains of two Ser649 (2.26 and 2.45 Å), one Leu622 (2.28 Å) and forms the hydrophobic interactions with Ala653 (4.26 Å). Compound 5 takes part in the hydrogen bond network, which can affect a higher affinity to the hERG channel than 4. Binding of compound 6 with N,N′-cyclohexylamidino substituent is predicted in the hERG cavity in U-shaped conformation. In this mode, the phenyl ring of compound 6 forms a strong π–π stacking interaction (a distance of 3.73 Å) with the aromatic ring of Phe656, whereas the N,N′-cyclohexyl substituents interact with both Thr623 (1.71 and 2.34 Å) and Ala653 (4.83 Å). However, we have to make clear that drug–channel interactions do not necessarily result in the full inhibition of the hERG channel.
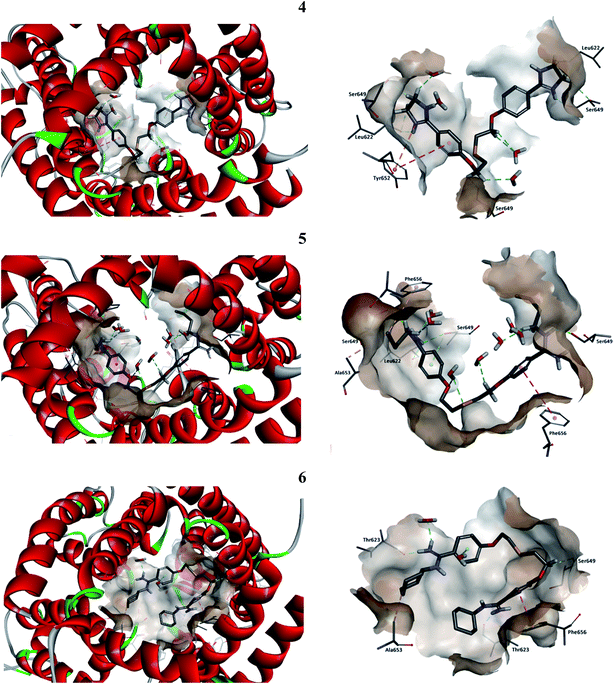 |
| | Fig. 5 Binding modes of compounds 4, 5 and 6 in the hERG K+ channel optimized by MD. | |
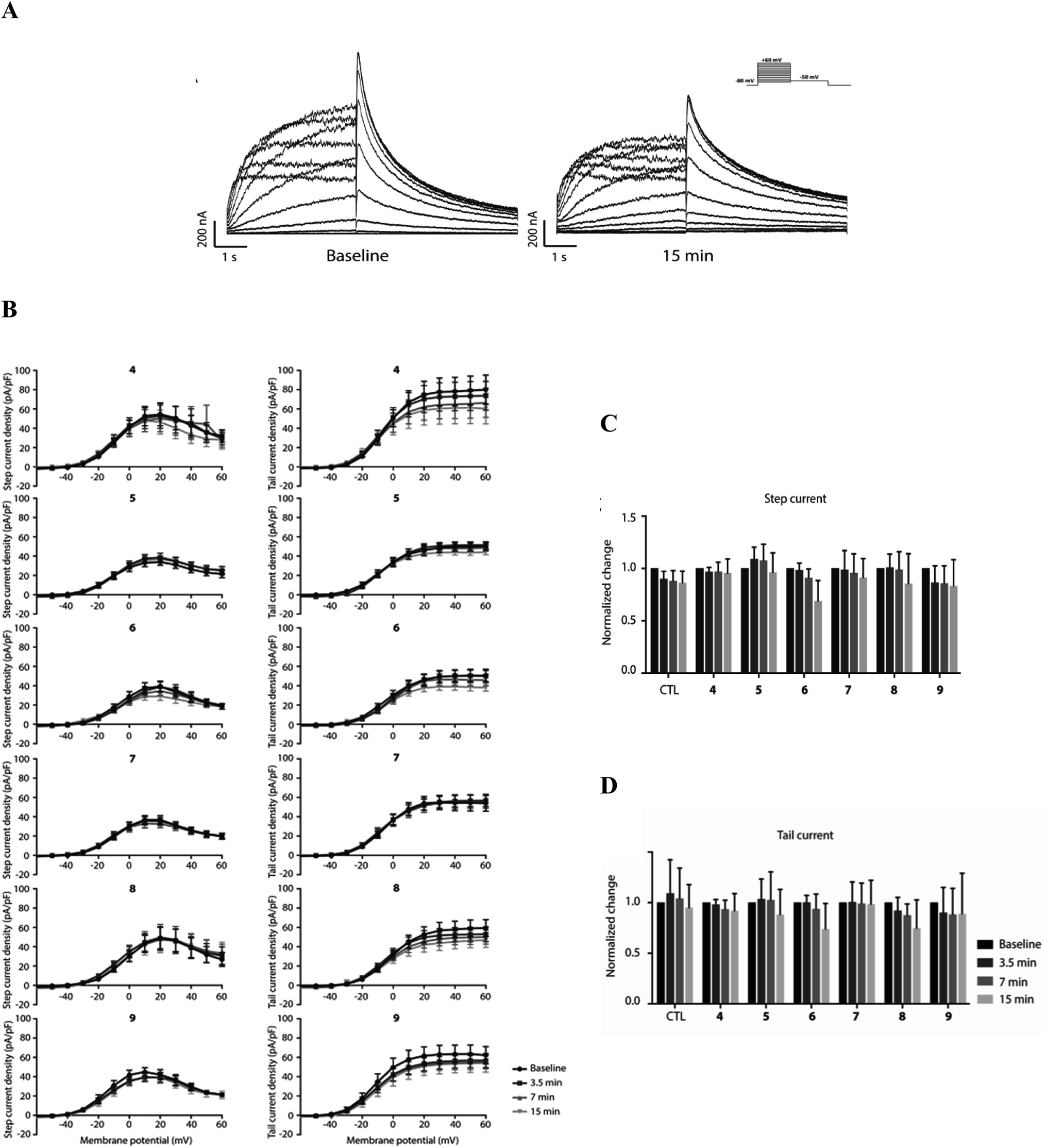
4.4. Electrophysiological effects of pentamidine analogues on hERG channel activity
To explore potential functional hERG channel blockade of 4–6 as indicated above, and the newly developed compounds 7–9, an electrophysiological patch-clamp assay on HEK-293T cells stably expressing hERG channels were used. hERG channel gating is complex and displays slow activation upon depolarization, followed by fast inactivation especially upon further depolarization, restricting outward K+ flow during early phases of the cardiac action potential. Upon initial the repolarization, fast recovery from inactivation then drives repolarization of the action potential. In both the step-current pulse, evaluating slow activation and rapid inactivation, and tail-current, evaluating fast recovery, measurements, no significant changes in IhERG densities were observed in the 15 min timeframe at a concentration of 1 μM (Fig. 6). Furthermore, no apparent effects on the voltage dependence of the hERG channel were observed with any of the compounds. Finally, we did not observe any obvious changes in channel dynamics when performing or analyzing the patch-clamp experiments and data, respectively. Compared to pentamidine, which shows no acute block at 1 μM using similar pulse protocols,32 modifications resulting in compounds 4–9 do not induce acute hERG channel blockade. It can mean that theoretically predicted interactions with hERG channel for 4–6 does not block the conduction path, however these findings do not exclude potential long-term effects on IhERG densities.
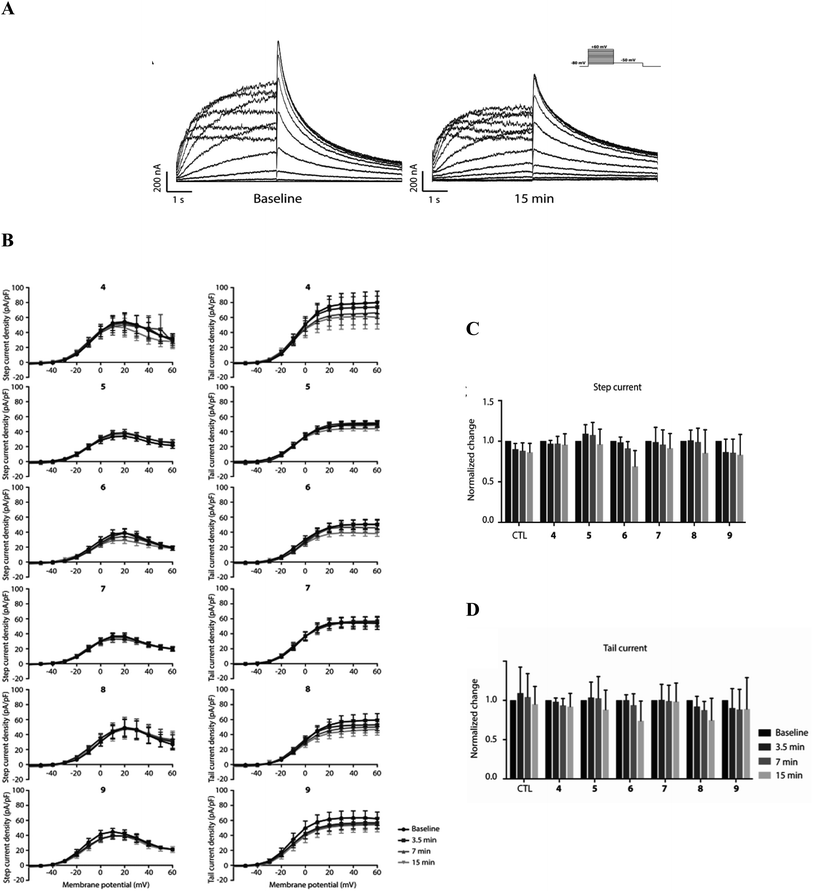 |
| | Fig. 6 Absence of IhERG inhibition in response to treatment with 4–9 (1 μM). (A) Representative traces of IhERG treated with 6 (1 μM) under baseline and 15 min perfusion. Stimulation protocol is shown at the right panel. (B) I–V relationship curves of step (left panels) and tail (right panels) current densities following treatment with 4–9 at baseline, 3.5 min, 7 min and 15 min. (C, D) Summary of step current (C) and tail current (D) densities, determined at +10 mV, under control (untreated) condition and following treatment with 4–9. Summarized data are normalized by their baseline. All data are presented as the mean ± SD. N-values for baseline, 3.5, 7 and 15 min. Measurements are as follows: 4: 17, 10, 9, 6; 5: 8, 7, 7, 6; 6: 17, 8, 8, 6; 7: 10, 10, 9, 8; 8: 10, 9, 8, 8; 9: 9, 9, 9, 5. | |
4.5. Impact of pentamidine analogues on hERG trafficking
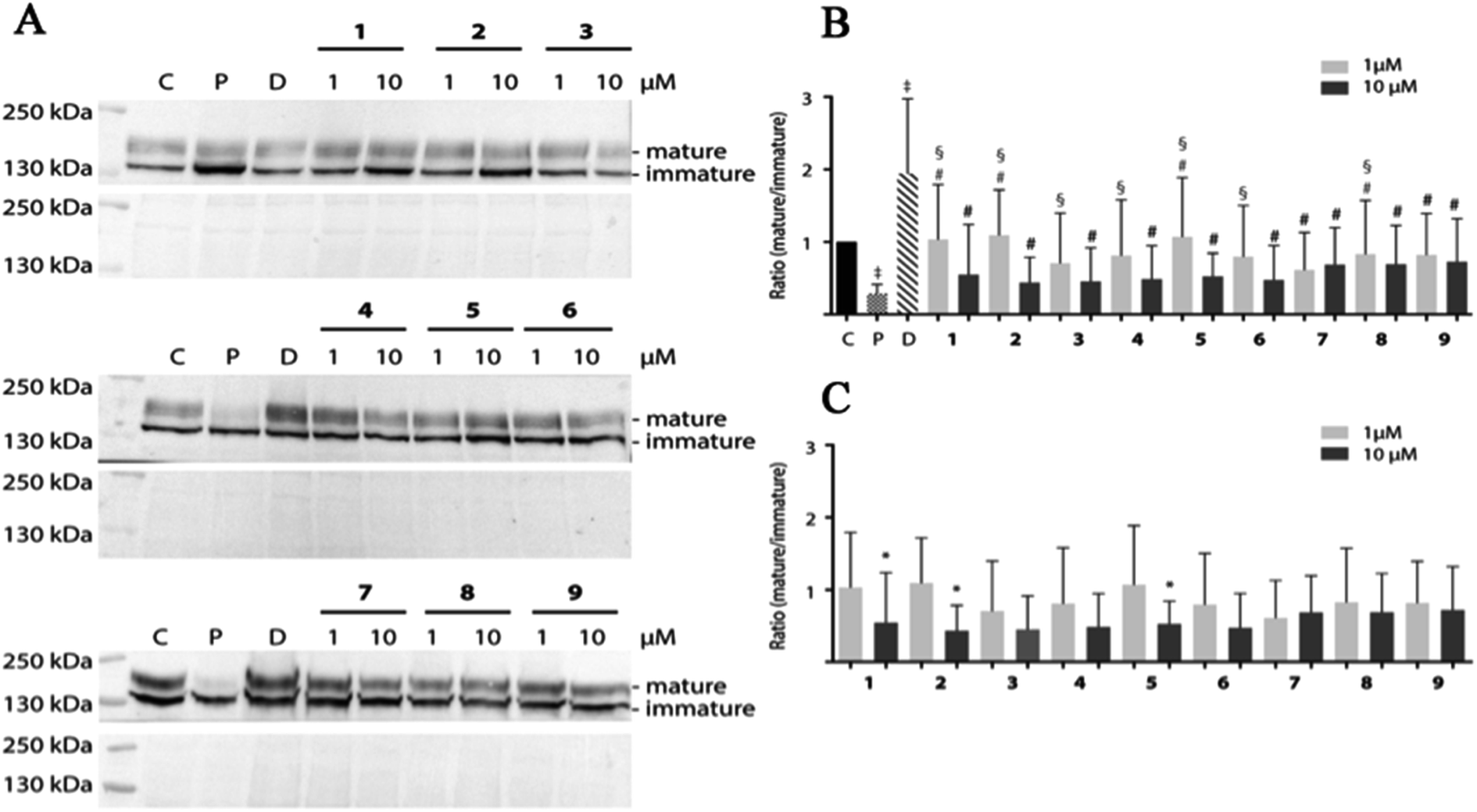
Pentamidine affects forward the trafficking of the hERG channel resulting in decreased mature/immature protein ratios.32 Pentamidine likely interferes with the process of proper hERG protein folding, in which the compound interacts with one or more intermediate folding states and thus inhibits subsequent folding steps required for endoplasmic reticulum export.33 One could imagine that pentamidine-bound folding intermediates cannot interact with chaperones in the endoplasmic reticulum that normally steer – and/or assist in – the process of proper folding.75 The high-affinity drug binding site Phe656 of the hERG channel is essential for intracellular pentamidine interaction, which may also explain the capacity of high-affinity hERG blockers to correct pentamidine-induced hERG trafficking defects.34 Decreased levels of mature hERG expression are one of the life-threatening side effects of pentamidine and the major trigger of Torsade de Pointes arrhythmias. To identify potentially trafficking inhibition structures, pentamidine and 1–9 were tested for effects on hERG mature/immature ratios.
HEK-hERG cells were treated with 1 and 10 μM pentamidine and its analogues for 24 h (Fig. 7). We did not observe induction of cell toxicity by routine visual inspection using phase-contrast microscopy and lysate protein concentration. As also observed in earlier studies,33,34 pentamidine treatment significantly decreased maturation (mature/immature ratio) of hERG. Dofetilide, which promotes forward trafficking, increases mature/immature ratios. In contrast to pentamidine, its analogues displayed minor effect on hERG maturation resulting in slightly lower mature/immature ratios, especially at 10 μM (pentamidine vs. 1 1 μM p < 0.0001, 2 1 μM p < 0.0001; 3 1 μM p < 0.05; 4 1 μM p < 0.01; 5 1 μM p < 0.0001; 6 1 μM p < 0.05; 8 1 μM p < 0.05) (Fig. 7B). No significant changes between 1 and 10 μM of each compound except for 1, 2 and 5 (Fig. 7C) were seen.
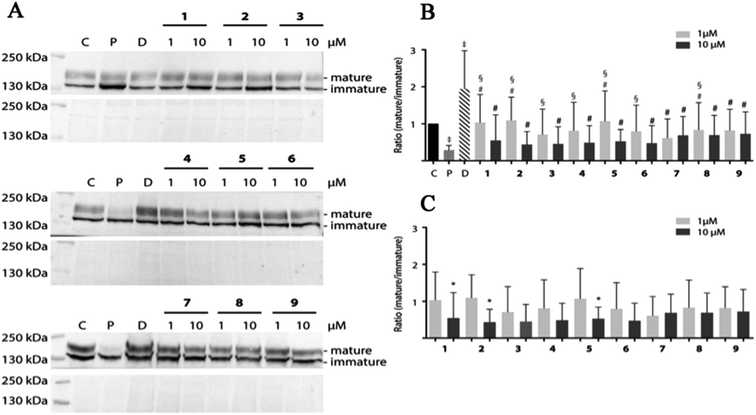 |
| | Fig. 7 The maturation of hERG is affected differently by pentamidine and its analogues 1–9 in HEK-hERG cells. (A) Western blot results of HEK-hERG cells exposed to pentamidine (10 μM), dofetilide (1 μM) and 1–9 (1 and 10 μM) for 24 h. Total protein ponceau staining was used as a loading control. (B) Summarized data of the ratios of mature and immature hERG expression in HEK-hERG cells after 24 h treatment with pentamidine (10 μM), dofetilide (1 μM) or analogues. ‡ indicated P < 0.05 vs. control, § P < 0.05 vs. pentamidine, # P < 0.05 vs. dofetilide. Control protein ratios were designated as 1. (C) Two concentration comparisons in the maturation ratio of hERG. * P < 0.05 between 1 and 10 μM treatment. Data are presented in mean ± SD. N-values for are as follows: control: 26; pentamidine: 27; dofetilide: 24; 1 (1, 10 μM): 10, 10; 2: 13, 10; 3: 4, 4; 4: 4, 4; 5: 7, 10; 6: 4, 3; 7: 4, 4; 8: 4, 4; 9: 4, 3. | |
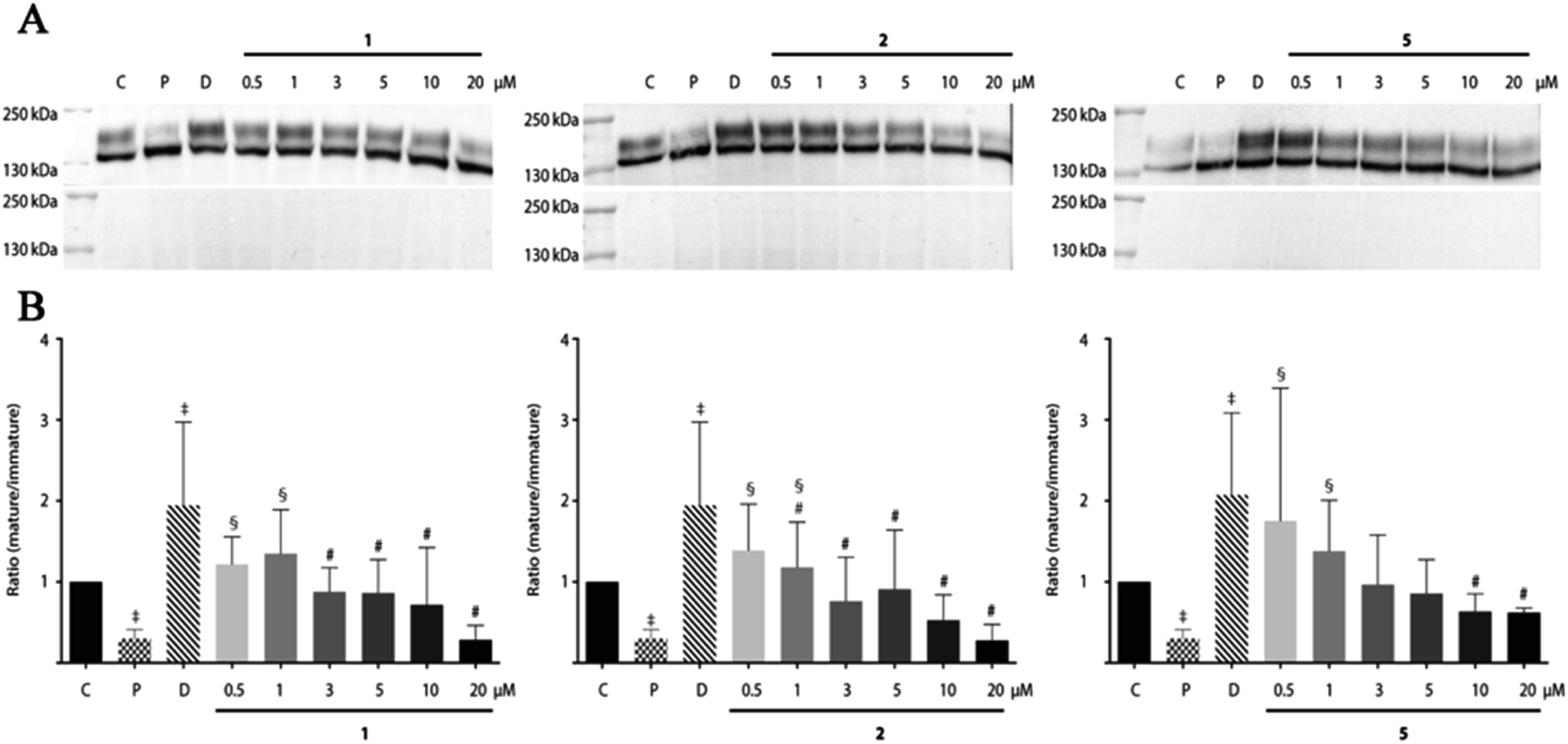
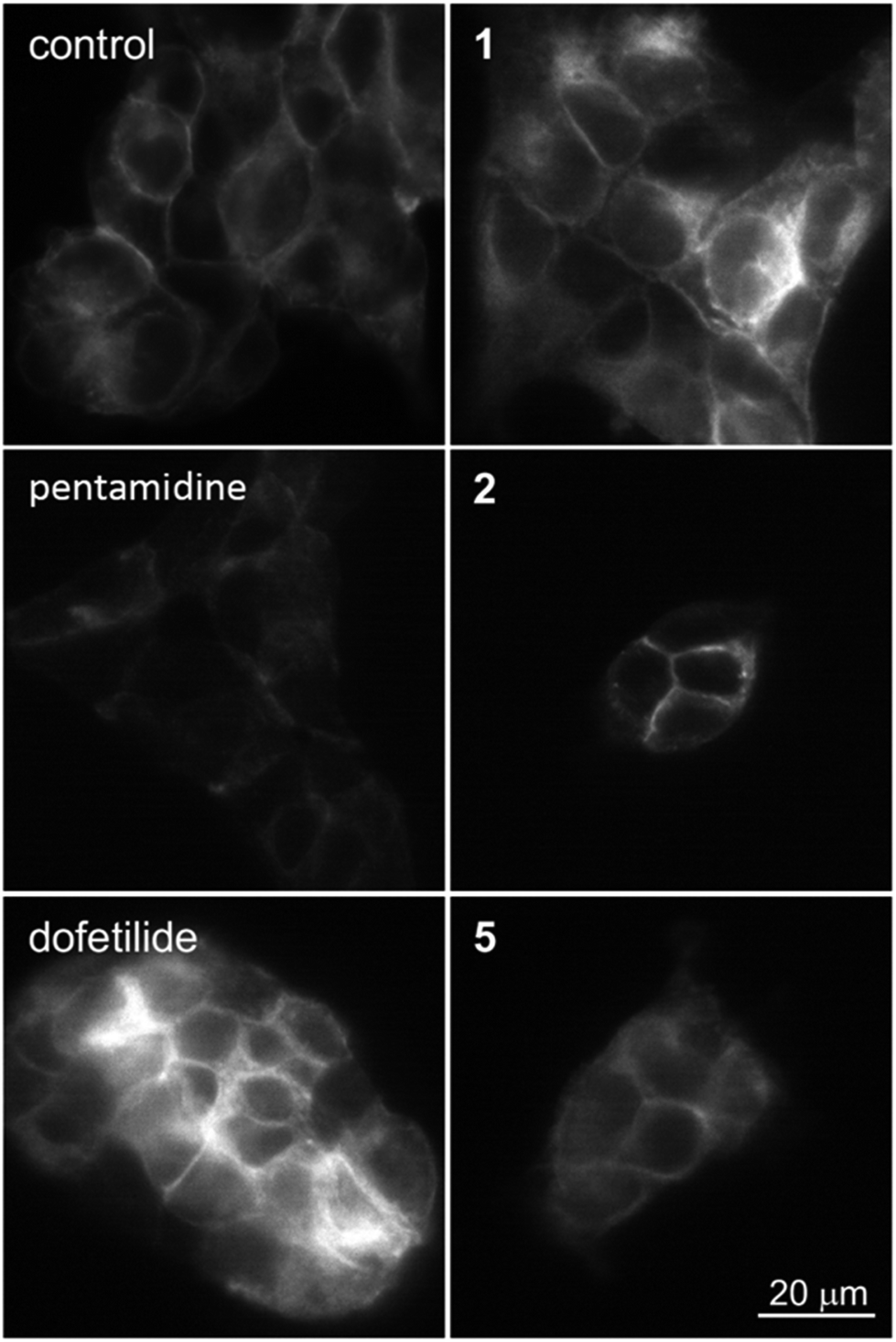
Since the compounds 1, 2 and 5 indicated dose-dependent effects in hERG maturation (Fig. 7C) we performed a full dose–response analysis. The three analogues were applied to HEK-hERG cells in different concentrations ranging from 0.5 to 20 μM (Fig. 8). A 20–25% decrease in total lysate protein concentration was observed with the highest concentration. However, this will not affect the experimental outcomes since identical amounts of protein were loaded and mature/immature hERG ratios were determined to assess forward trafficking. As expected, hERG maturation was dose-dependently decreased following 1, 2 and 5 application (pentamidine n = 27 vs. 1 0.5 μM p < 0.01; 1 1 μM p < 0.001; 2 0.5 μM p < 0.05; 2 1 μM p < 0.0001; 5 0.5 μM p < 0.05; 5 1 μM p < 0.05). Interestingly, the lowest concentration (0.5 μM) showed a trend for increasing maturation. The subcellular localization of the hERG protein after exposure to pentamidine and its analogues 1, 2 and 5 (10 μM) was studied by immunofluorescence microscopy on HEK-hERG cells (Fig. 9). In untreated control cells, hERG was distributed throughout the cell with clear membrane staining. The membrane hERG protein expression was remarkably decreased after pentamidine treatment and the intracellular expression level was relatively lower than an untreated cell, in contrast to dofetilide (1 μM). The intracellular localization of hERG after 1, 2 and 5 treatment revealed a pattern comparable with control cells and clear membrane staining was detected. These results are in accordance with observations displayed in Fig. 7 and 8. These qualitative results indicate that the modifications resulting in 1, 2 and 5 reduce, but not completely abolish, the effects seen on hERG trafficking by pentamidine, whereas the newly developed compounds 7–9 show the least effects on hERG trafficking.
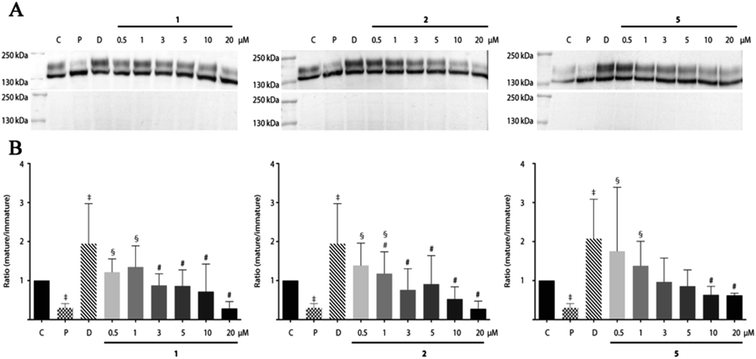 |
| | Fig. 8 Dose–response effects of 1, 2 and 5 on hERG maturation. (A) Dose-dependent effect on hERG expression after 24 h treatment with 1, 2 and 5 (0.5–20 μM), pentamidine (10 μM) or dofetilide (1 μM). In the lower panels, ponceau staining reveals equal loading. (B) Summarized results of (A). Data are presented as the ratios of mature and immature hERG. ‡ indicates P < 0.05 vs. control, § P < 0.05 vs. pentamidine, # P < 0.05 vs. dofetilide. N-values for are as follows: control: 29; pentamidine: 27; dofetilide: 27; 1 (all concentrations): 6; 2 (all concentrations): 6; 5: (all concentrations) 3. | |
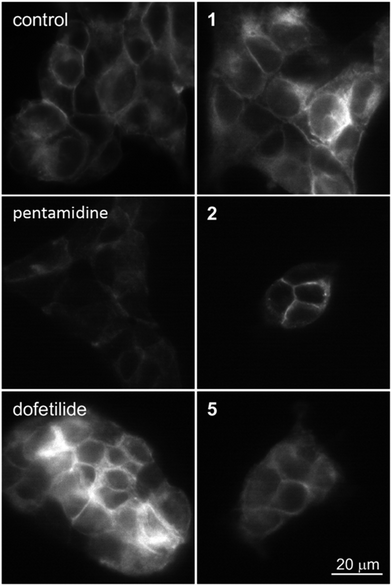 |
| | Fig. 9 Expression of hERG at the membrane in control (untreated) and HEK-hERG cells treated for 24 h with pentamidine (10 μM), dofetilide (1 μM), 1, 2 or 5 (10 μM). The scale bar represents 20 μm. | |
5. Conclusions
The tested linear pentamidine analogues 1–9 have favorable drug-likeness parameters (the topological polar surface area (TPSA), volume of distribution (Vd), water solubility (Sw), effective permeability (Peff), apparent permeability (MDCK), percentage of unbound drug to blood plasma proteins (% Unbnd), blood-to-plasma concentration ratio (RBP), BBB_filter, blood–brain barrier partition coefficient (logBB)) as the potential oral drugs. Nevertheless, some cardiotoxicity joined with hERG channel was suggested for tested analogs with one S atom in the linker and alkyl part in the amidine groups. The theoretical analysis of complexes between the hERG K+ channel and the selected compounds showed the binding free enthalpy values as follows −23.34 kcal mol−1 (4 with amidine groups closed in imidazolyl ring), −31.55 kcal mol−1 (5 with butyl substituent) and −13.70 kcal mol−1 (6 with cyclohexyl substituent), predicting the moderate strength of interactions. All compounds are located in the same region of the hERG K+ cavity in the proximity of amino acids Leu622, Thr623, Ser649, Tyr652, Ala653 and Phe656, what is in accordance with the possibility blocking hERG K+ channel. However, in the electrophysiological patch-clamp experiments for 1–9 on HEK-293T cells, no significant changes in IhERG densities were observed in the 15 min timeframe at a concentration of 1 μM. In the next biochemical experiments, in contrast to pentamidine, its analogues displayed minor effect on hERG maturation resulting in slightly lower mature/immature ratios, especially at 10 μM. The intracellular localization of hERG after 1, 2 and 5 treatment revealed a pattern comparable with control cells and clear membrane staining was detected. After a combined theoretical and experimental approach, we can suggest that the pentamidines characterized by the presence of sulfur atoms or sulfanilide groups are promising, not-toxic lead structures in the developments of new potent chemotherapeutics against PJP.
Authorship contributions
The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The cell line was made and provided by Professor C. T. January's team. M. Q. is the recipient of a scholarship from the Chinese Scholarship Council.
References
- S. Kongsamut, J. Kang, X. L. Chen, J. Roehr and D. Rampe, Eur. J. Pharmacol., 2002, 450, 37–41 CrossRef CAS PubMed.
- R. Netzer, A. Ebneth, U. Bischoff and O. Pongs, Drug Discovery Today, 2001, 6, 78–84 CrossRef CAS PubMed.
- M. C. Sanguinetti and M. Tristani-Firouzi, Nature, 2006, 440, 463–469 CrossRef CAS PubMed.
- J. C. Hancox, M. J. McPate, A. El Harchi and Y. H. Zhang, Pharmacol. Ther., 2008, 119, 118–132 CrossRef CAS PubMed.
- D. M. Roden, Circulation, 2008, 118, 981–982 CrossRef PubMed.
- H. Morita, J. Wu and D. P. Zipes, Lancet, 2008, 372, 750–763 CrossRef CAS.
- M. C. Sanguinetti and J. S. Mitcheson, Trends Pharmacol. Sci., 2005, 26, 119–124 CrossRef CAS PubMed.
- L. L. Eckhardt, S. Rajamani and C. T. January, Br. J. Pharmacol., 2005, 145, 3–4 CrossRef CAS PubMed.
- M. A. van der Heyden, M. E. Smits and M. A. Vos, Br. J. Pharmacol., 2008, 153, 406–409 CrossRef CAS PubMed.
- K. C. de Git, T. P. de Boer, M. A. Vos and M. A. van der Heyden, Pharmacol. Ther., 2013, 139, 24–31 CrossRef CAS PubMed.
- D. Justo and D. Zeltser, Mycoses, 2006, 49, 463–470 CrossRef PubMed.
- M. Schwartz, M. Patel, Z. Kazzi and B. Morgan, J. Med. Toxicol., 2008, 4, 173–179 CrossRef PubMed.
- P. K. Honig, D. C. Wortham, K. Zamani, D. P. Conner, J. C. Mullin and L. R. Cantilena, JAMA, J. Am. Med. Assoc., 1993, 269, 1513–1518 CrossRef CAS PubMed.
- P. K. Honig, D. C. Wortham, R. Hull, K. Zamani, J. E. Smith and L. R. Cantilena, J. Clin. Pharmacol., 1993, 33, 1201–1206 CrossRef CAS PubMed.
- E. Ficker, Y. A. Kuryshev, A. T. Dennis, C. Obejero-Paz, L. Wang, P. Hawryluk, B. A. Wible and A. M. Brown, Mol. Pharmacol., 2004, 66, 33–44 CrossRef CAS PubMed.
- A. J. Taylor, R. W. Hull, P. E. Coyne, R. L. Woosley and A. H. Eliasson, Clin. Pharmacol. Ther., 1991, 49, 698–700 CrossRef CAS PubMed.
- J. E. Touze, P. Heno, L. Fourcade, J. C. Deharo, G. Thomas, S. Bohan, P. Paule, P. Riviere, E. Kouassi and A. Buguet, Am. J. Trop. Med. Hyg., 2002, 67, 54–60 CrossRef CAS PubMed.
- K. S. Kim and E. J. Kim, Drug Chem. Toxicol., 2005, 28, 303–313 CrossRef CAS PubMed.
- Z. Zhou, V. R. Vorperian, Q. Gong, S. Zhang and C. T. January, J. Cardiovasc. Electrophysiol., 1999, 10, 836–843 CrossRef CAS PubMed.
- L. C. Macdonald, R. Y. Kim, H. T. Kurata and D. Fedida, Sci. Rep., 2018, 8, 289 CrossRef PubMed.
- N. G. Mansharamani, R. Garland, D. Delaney and H. Koziel, Chest, 2000, 118, 704–711 CrossRef CAS PubMed.
- C. F. Kelley, W. Checkley, D. M. Mannino, C. Franco-Paredes, C. Del Rio and F. Holguin, Chest, 2009, 136, 190–197 CrossRef PubMed.
- H. Chang, M. C. Kuo, T. L. Lin, J. H. Wu and P. N. Wang, J. Intern. Med., 2018, 48, 81–83 CrossRef PubMed.
- E. J. Calderón, S. Gutiérrez-Rivero, I. Durand-Joly and E. Dei-Cas, Expert Rev. Anti Infect. Ther., 2010, 8, 683–701 CrossRef PubMed.
- E. M. Carmona and A. H. Limper, Ther. Adv. Respir. Dis., 2011, 5, 41–59 CrossRef CAS PubMed.
- Y. Liu, L. Su, S. J. Jiang and H. Qu, Oncotarget, 2017, 8, 59729–59739 Search PubMed.
- F. R. Sattler, R. Cowan, D. M. Nielsen and J. Ruskin, Ann. Intern. Med., 1988, 109, 280–287 CrossRef CAS PubMed.
- S. Safrin, D. M. Finkelstein, J. Feinberg, P. Frame, G. Simpson, A. Wu, T. Cheung, R. Soeiro, P. Hojczyk and J. R. Black, Ann. Intern. Med., 1996, 124, 792–802 CrossRef CAS PubMed.
- B. Wispelwey and R. Pearson, Drug Saf., 1990, 5, 212–219 CrossRef CAS PubMed.
- A. Porollo, J. Meller, Y. Joshi, V. Jaiswal, A. G. Smulian and M. T. Cushion, Curr. Drug Targets, 2012, 13, 1575–1585 CrossRef CAS PubMed.
- C. R. Kroll and L. S. Gettes, J. Cardiovasc. Electrophysiol., 2002, 13, 936–938 CrossRef PubMed.
- Y. A. Kuryshev, E. Ficker, L. Wang, P. Hawryluk, A. T. Dennis, B. A. Wible, A. M. Brown, J. Kang, X. L. Chen, K. Sawamura, W. Reynolds and D. Rampe, J. Pharmacol. Exp. Ther., 2005, 312, 316–323 CrossRef CAS PubMed.
- A. T. Dennis, L. Wang, H. Wan, D. Nassal, I. Deschenes and E. Ficker, Mol. Pharmacol., 2012, 81, 198–209 CrossRef CAS PubMed.
- R. Varkevisser, M. J. Houtman, T. Linder, K. C. de Git, H. D. Beekman, R. R. Tidwell, A. P. Ijzerman, A. Stary-Weinzinger, M. A. Vos and M. A. van der Heyden, Br. J. Pharmacol., 2013, 169, 1322–1334 CrossRef CAS PubMed.
- D. Maciejewska, J. Zabinski, P. Kazmierczak, M. Rezler, B. Krassowska-Swiebocka, M. S. Collins and M. T. Cushion, Eur. J. Med. Chem., 2012, 48, 164–173 CrossRef CAS PubMed.
- D. Maciejewska, J. Zabinski, P. Kazmierczak, K. Wojciuk, M. Kruszewski and H. Kruszewska, Bioorg. Med. Chem. Lett., 2014, 24, 2918–2923 CrossRef CAS PubMed.
- D. Maciejewska, J. Zabinski, M. Rezler, P. Kazmierczak, M. S. Collins, L. Ficker and M. T. Cushion, MedChemComm, 2017, 8, 2164 RSC.
- Dassault Systèmes BIOVIA, Discovery Studio 2017R2, Dassault Systèmes, San Diego, 2017 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian, Inc., Wallingford, CT, 2016.
- C. M. Breneman and K. B. Wiberg, J. Comput. Chem., 1990, 11, 361–373 CrossRef CAS.
- S. Durdagi, S. Deshpande, H. J. Duff and S. Y. Noskov, J. Chem. Inf. Model., 2012, 52, 2760–2774 CrossRef CAS PubMed.
- J. Louvel, J. F. Carvalho, Z. Yu, M. Soethoudt, E. B. Lenselink, E. Klaasse, J. Brussee and A. P. Ijzerman, J. Med. Chem., 2013, 56, 9427–9440 CrossRef CAS PubMed.
- P. Schmidtke, M. Ciantar, I. Theret and P. Ducrot, J. Chem. Inf. Model., 2014, 54, 2320–2333 CrossRef CAS PubMed.
- W. Wang and R. MacKinnon, Cell, 2017, 169, 422–430 CrossRef CAS PubMed.
- S. Durdagi, H. J. Duff and S. Y. Noskov, J. Chem. Inf. Model., 2011, 51, 463–474 CrossRef CAS PubMed.
- J. P. Lees-Miller, J. O. Subbotina, J. Guo, V. Yarov-Yarovoy, S. Y. Noskov and H. J. Duff, Biophys. J., 2009, 96, 3600–3610 CrossRef CAS PubMed.
- Y. Wang, J. Guo, L. L. Perissinotti, J. Lees-Miller, G. Teng, S. Durdagi, H. J. Duff and S. Y. Noskov, Sci. Rep., 2016, 6, 32536 CrossRef CAS PubMed.
- J. Ghosh, M. S. Lawless, M. Waldman, V. Gombar and R. Fraczkiewicz, Methods Mol. Biol., 2016, 1425, 63–83 CrossRef CAS PubMed.
- ADMET Predictor™ software provided by Simulation Plus, Inc., Lancaster, California, USA.
- B. R. Brooks, R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan and M. Karplus, J. Comput. Chem., 1983, 4, 187–217 CrossRef CAS.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- J. Aqvist, J. Phys. Chem., 1990, 94, 8021–8024 CrossRef.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS.
- C. Sagui and T. A. Darden, Annu. Rev. Biophys. Biomol. Struct., 1999, 28, 155–179 CrossRef CAS PubMed.
- H. J. C. Berendsen, J. P. M. Postma, W. F. Vangunsteren, A. Dinola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- J. P. Ryckaert, G. Ciccotti and H. J. C. Berendsen, J. Comput. Phys., 1977, 23, 327–341 CrossRef CAS.
- P. A. Kollman, I. Massova, C. Reyes, B. Kuhn, S. Huo, L. Chong, M. Lee, T. Lee, Y. Duan, W. Wang, O. Donini, P. Cieplak, J. Srinivasan, D. A. Case and T. E. Cheatham III, Acc. Chem. Res., 2000, 33, 889–897 CrossRef CAS PubMed.
- Z. Zhou, Q. Gong, B. Ye, Z. Fan, J. C. Makielski, G. A. Robertson and C. T. January, Biophys. J., 1998, 74, 230–241 CrossRef CAS PubMed.
- Clampex software version 10.7.0.3, 2016, Molecular Devices, San Jose, USA Search PubMed.
- Clampfit software version 10.7.0.3, 2016, Molecular Devices, San Jose, USA Search PubMed.
- One-way ANOVA followed by Bonferroni multiple comparisons test was performed using GraphPad Prism version 7.04 for Windows, GraphPad Software, La Jolla California, USA, http://www.graphpad.com Search PubMed.
- K. Palm, P. Stenberg, K. Luthman and P. Artursson, Pharm. Res., 1997, 14, 568–571 CrossRef CAS PubMed.
- J. Fernandes and C. R. Gattass, J. Med. Chem., 2009, 52, 1214–1218 CrossRef CAS PubMed.
- P. J. van Bladeren, Chem.-Biol. Interact., 2000, 129, 61–76 CrossRef CAS PubMed.
- S. Rendic and F. P. Guengerich, Chem. Res. Toxicol., 2015, 28, 38–42 Search PubMed.
- B. J. Berger, N. A. Naiman, J. E. Hall, J. Peggins, T. G. Brewer and R. R. Tidwell, Antimicrob. Agents Chemother., 1992, 36, 1825–1831 CrossRef CAS PubMed.
- M. J. Twiner, G. J. Doucette, A. Rasky, X. P. Huang, B. L. Roth and M. C. Sanguinetti, Chem. Res. Toxicol., 2012, 25, 1975–1984 Search PubMed.
- C. Buyck, An in silico model for detecting potential hERG blocking, Blackwell Publishing, 2003 Search PubMed.
- R. Didziapetris and K. Lanevskij, J. Comput.-Aided Mol. Des., 2016, 30, 1175–1188 CrossRef CAS PubMed.
- A. M. Aronov, Drug Discovery Today, 2005, 10, 149–155 CrossRef CAS PubMed.
- D. Fernandez, A. Ghanta, G. W. Kauffman and M. C. Sanguinetti, J. Biol. Chem., 2004, 279, 10120–10127 CrossRef CAS PubMed.
- A. Cavalli, R. Buonfiglio, C. Ianni, M. Masetti, L. Ceccarini, R. Caves, M. W. Chang, J. S. Mitcheson, M. Roberti and M. Recanatini, J. Med. Chem., 2012, 55, 4010–4014 CrossRef CAS PubMed.
- A. Cavalli, E. Poluzzi, F. De Ponti and M. Recanatini, J. Med. Chem., 2002, 45, 3844–3853 CrossRef CAS PubMed.
- B. O. Villoutreix and O. Taboureau, Adv. Drug Delivery Rev., 2015, 86, 72–82 CrossRef CAS PubMed.
- J. L. Smith, C. L. Anderson, D. E. Burgess, C. S. Elayi, C. T. January and B. P. Delisle, Journal of Arrhythmia, 2016, 32, 373–380 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra08404e |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Muge Qileb,
Paweł Kaźmierczaka,
Meye Bloothooftb,
Marcel A. G. van der Heydenb and
Dorota Maciejewska
*a,
Muge Qileb,
Paweł Kaźmierczaka,
Meye Bloothooftb,
Marcel A. G. van der Heydenb and
Dorota Maciejewska