
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of polyvinyl acetals on non-isothermal crystallization behaviour and mechanical properties of poly(ε-caprolactone)
Biao Yang *,
Xin Zhang,
Chun Wang,
Ran Liu,
Baomin Fan*,
Huijuan Zhang and
Hui Sun
*,
Xin Zhang,
Chun Wang,
Ran Liu,
Baomin Fan*,
Huijuan Zhang and
Hui Sun
School of Materials Science and Mechanical Engineering, Beijing Technology and Business University, Beijing 100048, P. R. China. E-mail: ybiao@btbu.edu.cn; fanbaomin@btbu.edu.cn
First published on 12th November 2019
Abstract
Polyvinyl acetaldehyde (PVAC) and polyvinyl butyral (PVB), as nucleating agents, were melt mixed with poly(ε-caprolactone) (PCL), and the non-isothermal crystallization behavior and crystalline morphology of PCL was characterized using differential scanning calorimetry (DSC) and polarizing microscopy (POM). It was found that melt shearing made the crystallinity of control PCL increase from 37.94% (as-received) to 41.15% and the crystallization temperature raised by 7.7 °C. As the mass fraction of polyvinyl acetals increased from 0.1 wt% to 10 wt%, both crystallization temperature and crystallinity of PCL were decreased; the spherulite size of PCL was increased from a few microns to 200 μm and a fast cooling procedure facilitated the homogeneity of spherulite size. When the content of nucleating agent was less than 1 wt%, no obvious microphase separation was observed under scanning electron microscopy; the tensile strength, yield strength and elongation at break of PCL increased slightly; and the Young's modulus of PCL was improved by 67% at a 0.1 wt% mass fraction of PVB which is beneficial to PCL in high load bearing applications. When the contents of nucleating agents were higher than 1 wt%, the microphase separation occurred obviously and a large amount of fine spherulites appeared with the deterioration of mechanical properties. The modified Avrami and Tobin models could well describe the non-isothermal crystallization kinetics.
Introduction
As an aliphatic polyester with excellent toughness and biocompatibility, poly(ε-caprolactone) (PCL) has been widely used in biomedical fields,1–3 such as a carrier for drug-controlled release,4–8 cell culture substrate,3,9 scaffold.10–12 However, the nature of fast crystallization behaviour made PCL disadvantageous in the field of rapid degradation and high load bearing applications. Therefore, how to improve the final properties of PCL by regulating the crystallization behaviour has attracted more attention.Based on molecular design, the crystallization behaviour of PCL can be tailored by copolymerization with flexible or rigid segments. The reported copolymer components include poly(lactic acid),13–18 polyethylene glycol,19–21 polystyrene,22 polyvinylpyrrolidone,23 poly(hydroxybutyrate-co-hydroxyvalerate),24,25 and so on. In most cases, the crystallization nature of PCL was retarded after copolymerization resulting a decreased crystallinity of PCL segment. For example, the crystallinity of PCL segment decreased from 54.7% to 2.2% in the six-armed star-shaped poly(ε-caprolactone)-b-poly(D,L-lactide-co-glycolide).26
Small molecules, macromolecules and nanoparticles were also used as nucleating agents to influence the crystallization behaviour of PCL and the effects were quite different. Generally, the addition of nucleating agent can increase nucleation density resulting in smaller grain size and higher crystallization temperature. For instance, after incorporating with cyanuric acid (0.2 wt%)27 and α-cyclodextrin inclusion complex (α-CD-IC)28,29 by solution casting, the non-isothermal crystallization temperature of PCL, overall isothermal melt crystallization rate, and nucleation density of PCL spherulites were improved. Carbon nanotubes,30,31 attapulgite (0.5–7 wt%)32 and pristine nanocrystalline cellulose33 act as heterogeneous nucleating agent, promoted the crystallization rate, and increased the non-isothermal crystallization temperature by 10–16 °C. The spherulite size of isothermally crystallized PCL decreased from nearly 200 μm to a few microns. Meanwhile, the crystallinity increased slightly.31 Although the particle size is much larger than nano-scale, basalt fibers with a diameter of 7–15 μm and average length of 5 mm still show excellent nucleation effect. The non-isothermal crystallization temperature of PCL was raised by 10 °C, and the crystallinity was increased from 38% to 45%.34
Contrary to what was mentioned above, although benzoxazine (10–50 wt%) was miscible with PCL but the non-isothermal crystallization temperature and isothermal crystallization rate of PCL were decreased.35 Moreover, when macromolecules were blended with PCL at high mass fraction, the nucleation effect was weakened due to the mobility constraints. Cai et al. found that thermoplastic starch (TPS) (ratios of TPS/PCL were higher than 2/10), had a restrain effect on the isothermal crystallization of PCL and resulted in a decreased crystallinity.36
The content of highly effective nucleating agent is preferably less than 1–2 wt%, and excessive dosage will affect the inherent properties of the substrate. In our previous works, we found that polyvinyl butyral (PVB) (less than 1 wt%) was an efficient macromolecular nucleating agent for biodegradable polyesters, such as poly(butylene succinate) (PBS)37 and poly(butylene succinate-co-terephthalate) (PBST).38 The spherulite size of PBS and PBST was reduced to several microns and the crystallization temperature was increased by 10 °C. Therefore, polyvinyl acetals are supposed to be effective nucleating agent of PCL. However, the few relevant reports of PCL/PVB based composite39,40 and blend41 with a PVB content higher than 2 wt%, which were based on solution casting process, are all focused on the isothermal crystallization behaviour and the banded spherulite. Non-isothermal crystallization is closely related to the forming process of PCL, which directly determines the crystalline morphology, biodegradation rate and mechanical properties of the final product. In this work, we prepared the blends of PCL with the content of polyvinyl acetals from 0.1 wt% to 10 wt%, and revealed the effect of polyvinyl acetaldehyde (PVAC) and PVB on the non-isothermal crystallization behaviour, crystalline morphology and mechanical properties of PCL.
Experimental
Materials
Poly(ε-caprolactone) with a molecular weight of 8 × 105 g mol−1 (CAPA 6800) was purchased from Solvay, USA. The self-made PVAC and PVB have the same polymerization degree of 1700, and the content of acetal group are 84% and 78%, respectively. The structures of PVAC and PVB are illustrated in Fig. 1.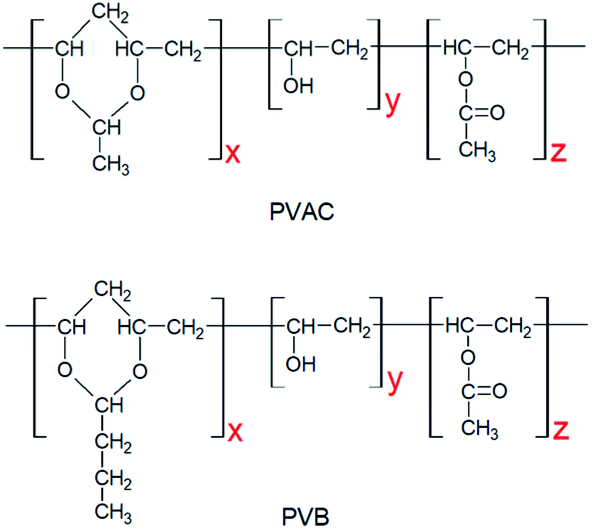 | ||
| Fig. 1 Molecular structures of PVAC and PVB. | ||
Sample preparation
PCL and polyvinyl acetals were dried in a vacuum oven at 50 °C for 8 h prior to melt mixing. PVAC and PVB were dissolved in alcohol to obtain a 10 wt% solution, then the solution was mixed with the PCL granules. After being dried at 50 °C for 8 h, the mixture was blended in a torque rheometer (XSS-300, Shanghai Kechuang Rubber & Plastic Machinery Co. Ltd., China) at 100 °C and 50 rpm for 8 min. As-received PCL was mixed under the same conditions as the control sample. The result materials were cut into small particles and oven-dried at 50 °C for 8 h. The standard type 5A dumbbell shaped specimens (GB/T 1040.2-2006/ISO 527-2-1993) were prepared by injection moulding (MiniJet Pro, Thermo Fisher Scientific, USA) at a cylinder temperature of 100 °C and a mould temperature of 25 °C; the injection pressure is 500 bar and the pressure holding time is 20 seconds. All the specimens were conditioned at room temperature (about 25 °C) for 48 h, followed by mechanical properties test and differential scanning calorimetry analysis.Characterization
All mechanical properties were examined using an electronic universal testing machine (CMT6104, Shenzhen Xinsansi Metrology Technology Co. Ltd., China) at a tensile rate of 70 mm min−1. The final values were averages of at least five specimens.The non-isothermal crystallization behaviour was analysed by differential scanning calorimeter (Q20, TA Instruments Inc.) under nitrogen atmosphere. Initially, the samples of 5–10 mg were firstly rapidly heated to 110 °C and maintained for 5 min to eliminate the heat history; subsequently, samples were cooled to −20 °C at a cooling rate of 10 °C min−1 and held for 5 min, then the temperature raised to 110 °C at a heating rate of 10 °C min−1. The relative crystallinity during the cooling process (Xc) can be calculated according the eqn (1).
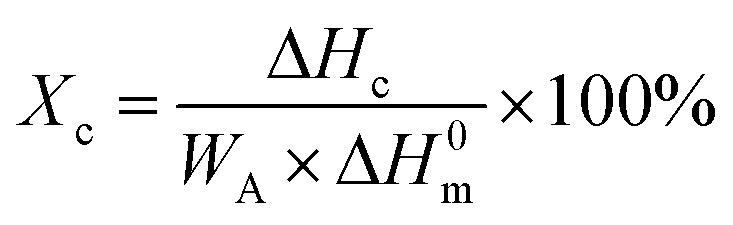 | (1) |
The crystallinity of PCL during the heating process 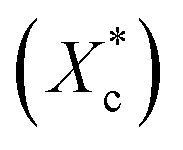 is calculated according to the eqn (2).
is calculated according to the eqn (2).
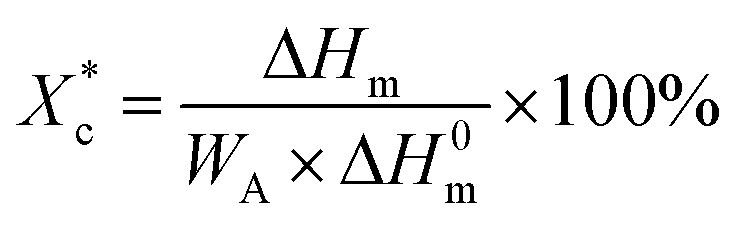 | (2) |
The crystalline morphology was observed under a polarizing optical microscope (Olympus BX51) equipped with a heating stage (THMS600, Linkam Scientific Instrument Ltd.). Samples sandwiched between two glass slides were heated to 110 °C and kept for 3 min to eliminate any thermal history, then the melt was cooled to 22 °C at a rate of 10 °C min−1 and POM micrographs were recorded. To obtain the crystalline morphology of injection moulded (rapidly cooled) PCL, samples used for POM observation undergone cooling conditions similar to those experienced during injection. After eliminating thermal history as mentioned above, the set of slides and clamped sample was quickly moved into a 25 °C water bath for 10 min before POM observation at room temperature.
The microphase separation of PCL blends was observed on scanning electron microscope (SEM, Quanta FEG 250, FEI Company). The samples for SEM analysis were brittle fractured after cooling in liquid nitrogen, and the images of fracture surface were obtained under an accelerating voltage of 10 kV.
Results and discussion
Compatibility of PCL/polyvinyl acetals
Two polyvinyl acetals with different side groups, PVAC and PVB, as shown in Fig. 1, were used as macromolecular nucleating agents to regulate the crystallization behaviour and crystalline morphology of PCL. Due to the melting temperature of PCL is about 60 °C, which overlaps with the glass transition temperature of PVB and PVAC, neither DSC nor dynamic mechanical analysis (DMA) can directly characterize the compatibility of the blending system. Therefore, SEM was employed to observe the microphase separation in the blends and the microphotographs are shown in Fig. 2. | ||
| Fig. 2 SEM photomicrographs for the fractured morphology of PCL blends. (a) Control PCL; (b)–(f) PCL with 0.1 wt%, 0.3 wt%, 0.7 wt%, 1.0 wt% and 10.0 wt% PVAC; (g)–(k) PCL with 0.1 wt%, 0.3 wt%, 0.7 wt%, 1.0 wt% and 10.0 wt% PVB. | ||
When the content of nucleating agent was less than 1%, nanoparticles of PVAC and PVB seldom appeared in the field of view, indicating that the system had good compatibility. While at nucleating agent dosage of 1% (Fig. 2d and i), obvious microphase separation occurred, nanoparticles of PVAC and PVB with a size of 150–200 nm were densely dispersed in PCL matrix. With the further increase of the dosage (10 wt%), the particle size of polyvinyl acetals increased obviously, indicating that the compatibility became poor.
Effect of polyvinyl acetal on crystallization behaviour of PCL
Although the molecular structures of PVAC and PVB are different, their effects on the crystallization behaviour of PCL are similar. From the DSC curves (Fig. 3) and the crystallization parameters (Table 1) of the PCL and PCL/acetal blends, we found a very unusual phenomenon, i.e., simply mixing made the crystallinity of control PCL increased from 37.94% (as-received) to 41.15% and the crystallization temperature (Tc) raised by 7.7 °C. The undercooling degree, Tonset − Tc, is only 2.52 °C, which is significantly lower than other samples, indicating that the crystallization rate is improved dramatically. This phenomenon can be explained by carding effect induced by melt shearing.43 The intertwisted chains of as-received PCL are partly disentangled during mixing, and thus the chain's spatial freedom was improved, which is favourable for crystallization. As a consequence, the crystallization rate and crystallinity are improved.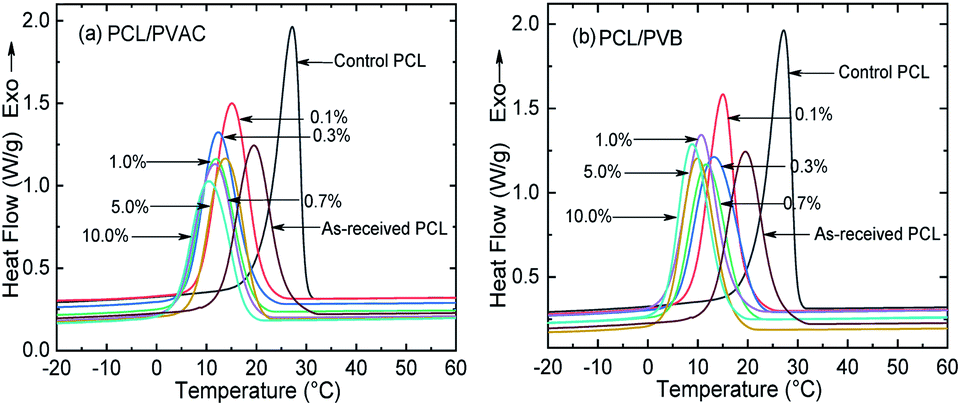 | ||
| Fig. 3 DSC curves of (a) PCL/PVAC and (b) PCL/PVB blends under a cooling rate of 10 °C min−1. | ||
| PCL sample | PCL/PVAC | PCL/PVB | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Tc (°C) | Xc (%) | Tonset − Tc (°C) | Tm (°C) |
|
Tc (°C) | Xc (%) | Tonset − Tc (°C) | Tm (°C) |
|
|
| As-received | 19.51 | 37.94 | 6.34 | 56.8 | 38.93 | 19.51 | 37.94 | 6.34 | 56.8 | 38.93 |
| Control | 27.21 | 41.15 | 2.52 | 54.8 | 40.95 | 27.21 | 41.15 | 2.52 | 54.8 | 40.95 |
| 0.1% acetal | 15.21 | 40.28 | 5.99 | 53.33 | 38.91 | 15.01 | 37.30 | 4.26 | 54.85 | 38.67 |
| 0.3% acetal | 12.39 | 37.32 | 7.13 | 53.51 | 36.84 | 13.20 | 37.94 | 8.27 | 53.92 | 36.49 |
| 0.7% acetal | 11.87 | 34.51 | 7.13 | 54.58 | 34.01 | 11.69 | 33.52 | 7.06 | 53.88 | 32.49 |
| 1.0% acetal | 11.71 | 35.40 | 6.77 | 55.27 | 34.74 | 10.68 | 36.57 | 6.07 | 55.06 | 35.33 |
| 5.0% acetal | 12.60 | 34.49 | 5.29 | 54.83 | 35.39 | 9.89 | 32.99 | 6.53 | 54.16 | 35.37 |
| 10.0% acetal | 10.47 | 32.00 | 7.25 | 55.21 | 36.46 | 8.86 | 33.03 | 6.48 | 53.53 | 34.24 |
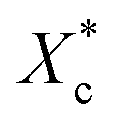
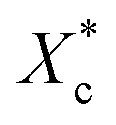
Furthermore, incorporating acetals decreased the crystallization temperature of PCL. Compared with the as-received PCL, Tc of samples with 10 wt% PVAC and 10 wt% PVB decreased by 9.04 °C and 10.65 °C, respectively. Meanwhile, the as-received and acetal-added samples have a similar undercooling degree. This suggests that the crystallization ability of PCL is weakened, which needs to crystallize at a lower temperature. In spite of this, the crystallinity has just a slight drop, and the minimum value of 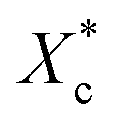 appeared at 0.7 wt% content of polyvinyl acetals. It can be understood from the difference of glass transition temperature (Tg) between polyvinyl acetals and PCL. PVAC and PVB act as macromolecular nucleating agents with a Tg of 50–60 °C, which is much higher than that of pristine PCL (−60 °C). During the cooling process, the molecular chains of polyvinyl acetals were frozen firstly and acted as heterogeneous nucleating centres to induce crystallization via hydrogen bond between hydroxyl group on acetal chains and ester groups of PCL as well as the steric effect of side chains of acetal group, meanwhile entanglement between polyvinyl acetals and PCL restricting the rearrangement of flexible PCL chains.
appeared at 0.7 wt% content of polyvinyl acetals. It can be understood from the difference of glass transition temperature (Tg) between polyvinyl acetals and PCL. PVAC and PVB act as macromolecular nucleating agents with a Tg of 50–60 °C, which is much higher than that of pristine PCL (−60 °C). During the cooling process, the molecular chains of polyvinyl acetals were frozen firstly and acted as heterogeneous nucleating centres to induce crystallization via hydrogen bond between hydroxyl group on acetal chains and ester groups of PCL as well as the steric effect of side chains of acetal group, meanwhile entanglement between polyvinyl acetals and PCL restricting the rearrangement of flexible PCL chains.
Effect of polyvinyl acetal on crystalline morphology of PCL
To gain insight into the nucleating effect of acetals, we observed the crystalline morphology through POM. Fig. 4 shows the polarized photomicrographs of the program cooling samples (they were kept at 110 °C for 3 min, then cooled to room temperature at 10 °C min−1), which presents the evolution of crystal morphology of PCL. | ||
| Fig. 4 POM photomicrograph of PCL undergone a program cooling (10°C min−1): (a) control PCL; (b)–(g) PCL with 0.1 wt%, 0.3 wt%, 0.7 wt%, 1.0 wt%, 5.0 wt% and 10.0 wt% PVAC; (h)–(m) PCL with 0.1 wt%, 0.3 wt%, 0.7 wt%, 1.0 wt%, 5.0 wt% and 10.0 wt% PVB. | ||
Nucleating agent usually increases the nucleation density and reduces spherulite size, and thus elevates the crystallization rate and/or crystallinity.27,34,44,45 POM observation indicates that control PCL has a uniform small grain size (5–10 μm) (Fig. 4a). However, polyvinyl acetals (less than 1 wt%) increase the spherulite size of PCL to 170–220 μm (PVAC) and 160–200 μm (PVB), as well as measly fine grains appear in the field of view. The sample of 1 wt% PVB (Fig. 4j) visualizes that the large spherulite dominated the crystallization and fine grains disappeared completely. When the content of polyvinyl acetals beyond 1 wt%, some fine grains with the similar size as control PCL appear again, and “large” spherulites with different size co-existed.
The increase of spherulite size mainly depends on the unique molecular nature of PCL, extremely low glass transition temperature and excellent flexibility of aliphatic polyester, as well as the competition between self-nucleation of PCL and heterogeneous nucleation of acetals. At lower acetal content (less than 1 wt%), PVB and PVAC are miscible with PCL and the heterogeneous nucleation of acetals suppresses the homogeneous self-nucleation of PCL. With the increases of acetal content (greater than 1 wt%), as a result of compatibility deterioration, chain clustering of acetals even phase separation occurred easily. Accordingly, the nucleation density decreased and the distribution of nucleation centers was non-uniform, which is helpful to self-nucleation of PCL.
Nucleation induced grain sizes growing-up during non-isothermal crystallization is similar with reports of isothermal crystallization,39,41,46 whereas no banded spherulites of PCL have been observed, which generally appeared at higher temperatures and longtime crystallization.
Mechanical properties of PCL
Generally, the mechanical properties of PCL depend to a large extent on the microstructure and morphology. Therefore, controlling the content of amorphous and crystalline phase in a semicrystalline polymer offers the final desired properties.There are three stages during the stretching of PCL (Fig. 5a and b) i.e. elastic zone, cold drawing, stress hardening. The molecular structure and dosage of acetals have different effects on the mechanical properties. It can be found from Fig. 5c, PVAC significantly reduced the Young's modulus more than 50%, from 1.03 MPa of control sample to a steady level (about 0.45 MPa) in the whole range of dosage. On the contrary, PVB enhanced Young's modulus significantly at low dosage (less than 1 wt%). The maximum of Young's modulus increased about 67% at 0.1 wt% content of PVB. At higher PVB contents (1 wt%, 5 wt%, 10 wt%), the Young's modulus decreased to the similar level as PCL/PVAC samples. For materials with very low glass transition temperature, the modulus of linear viscoelastic zone is very important to ensure the stability of the products. Therefore, the improvement in Young's modulus is of great significance for PCL in high load applications, such as stent or scaffold.
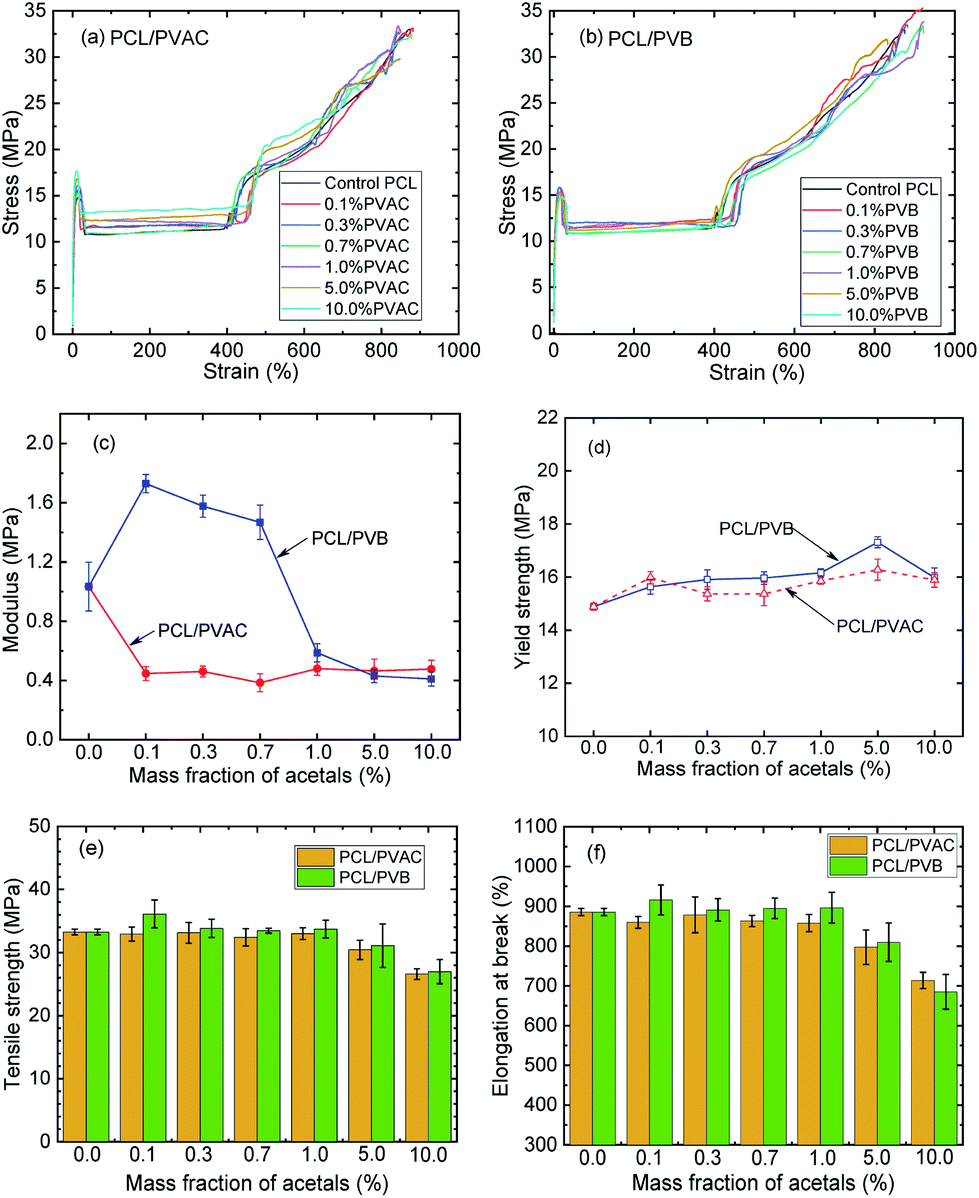 | ||
| Fig. 5 Effects of acetals on mechanical properties of PCL: (a) stress–strain curves of PCL/PVAC; (b) stress–strain curves of PCL/PVB; (c) modulus; (d) yield strength; (e) tensile strength; (f) elongation at break. | ||
As shown in Fig. 5d, crystallinity and crystal morphology have little effect on the yield strength of PCL. At lower contents of polyvinyl acetals (≤1 wt%), the tensile strength and elongation at break of PVB added samples were a little higher than that of PVAC added sample, but they were comparable with the control sample. When the mass fractions beyond 1%, both PVB and PVAC made the tensile strength and elongation at break decrease obviously (Fig. 5e and f).
It is generally accepted that the larger the crystalline size, the lower the mechanical properties. Quite to the contrary, herein, polyvinyl acetals enlarged the spherulite size of PCL to 160–200 μm and PVB enhanced its Young's modulus. Considering that the samples in the nucleation analysis were program cooled (10 °C min−1), while the samples used for mechanical property test were rapidly cooled (the melt at 100 °C was injected directly into a mould at 25 °C), the injection samples were also analysed by DSC, and the crystalline morphology of samples under simulated injection cooling conditions was observed. The details were discussed in the following section.
Effect of cooling rate on crystallization of PCL
Fig. 6 shows the crystallinities of PCL which undergone program cooling (10 °C min−1) or quenched (injection moulded) procedure, and the variation of crystallinity and melting temperature of stretched PCL. As seen from Fig. 6a, the crystallinities of injection samples are almost above 45%, which is much higher than that of programmed cooling samples. This indicated that fast cooling is beneficial to increase crystallinity. Fig. 6b and c show the different crystallization parameters of injection sample before and after tensile test. Stretching is beneficial to the orientation and regular arrangement of PCL chains. Thus, the crystallinity was promoted, e.g. the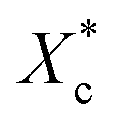 of control samples increased from 45.8% to 52% (Fig. 6b). When the mass fraction of polyvinyl acetals was less than 1%, the melting temperatures of stretched samples had an increase of more than 2 °C (Fig. 6c). The root cause for this result is the extremely low glass transition temperature of PCL, which makes the orientation and rearrangement of molecules possible at room temperature. During fragmentation of the lamellae, the chains escaped from the initial chain-folded lamellae into the amorphous region, became extended, and easily crystallized into new chain-extended lamellae.47
of control samples increased from 45.8% to 52% (Fig. 6b). When the mass fraction of polyvinyl acetals was less than 1%, the melting temperatures of stretched samples had an increase of more than 2 °C (Fig. 6c). The root cause for this result is the extremely low glass transition temperature of PCL, which makes the orientation and rearrangement of molecules possible at room temperature. During fragmentation of the lamellae, the chains escaped from the initial chain-folded lamellae into the amorphous region, became extended, and easily crystallized into new chain-extended lamellae.47
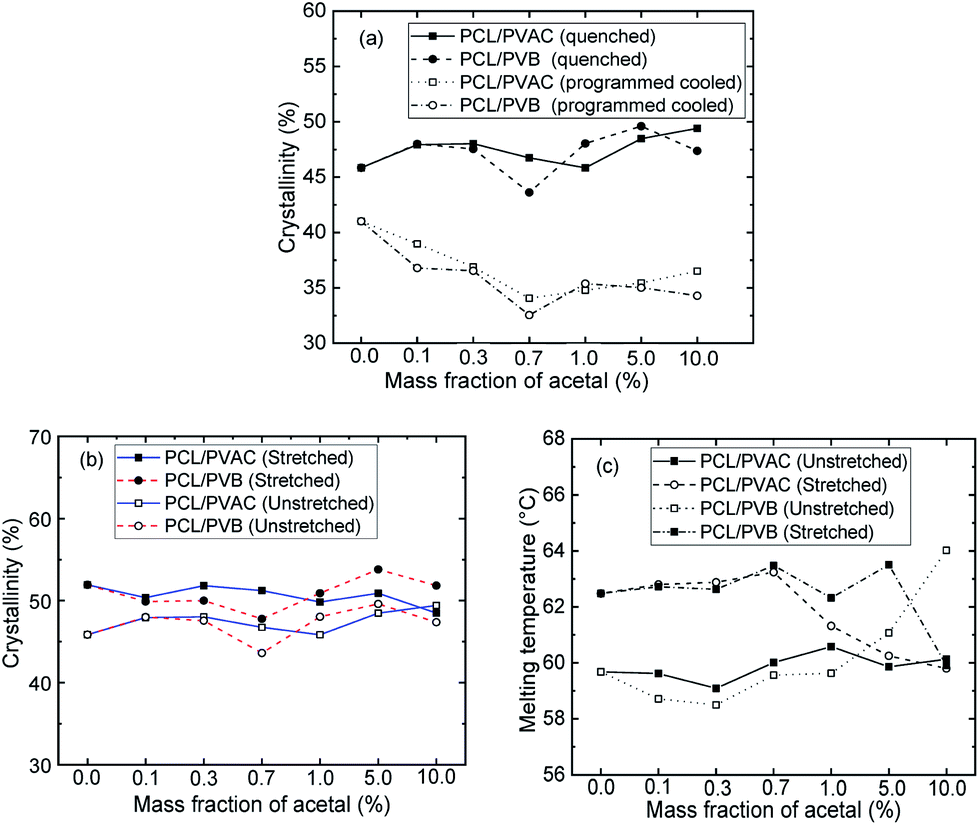 | ||
Fig. 6 DSC analysis of program cooling and injection moulded specimens. (a) Crystallinity calculated from the melting enthalpy 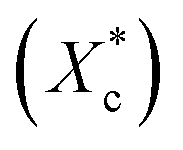 of program cooling (10 °C min−1) and injection moulded samples; (b) of program cooling (10 °C min−1) and injection moulded samples; (b) 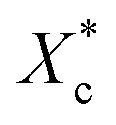 and (c) melting temperature of injection sample (quenched). and (c) melting temperature of injection sample (quenched). | ||
POM observation also confirmed the clear distinction between program cooling samples (10 °C min−1) and the fast cooling sample which undergone a simulated cooling conditions of the injection process (Fig. 7).
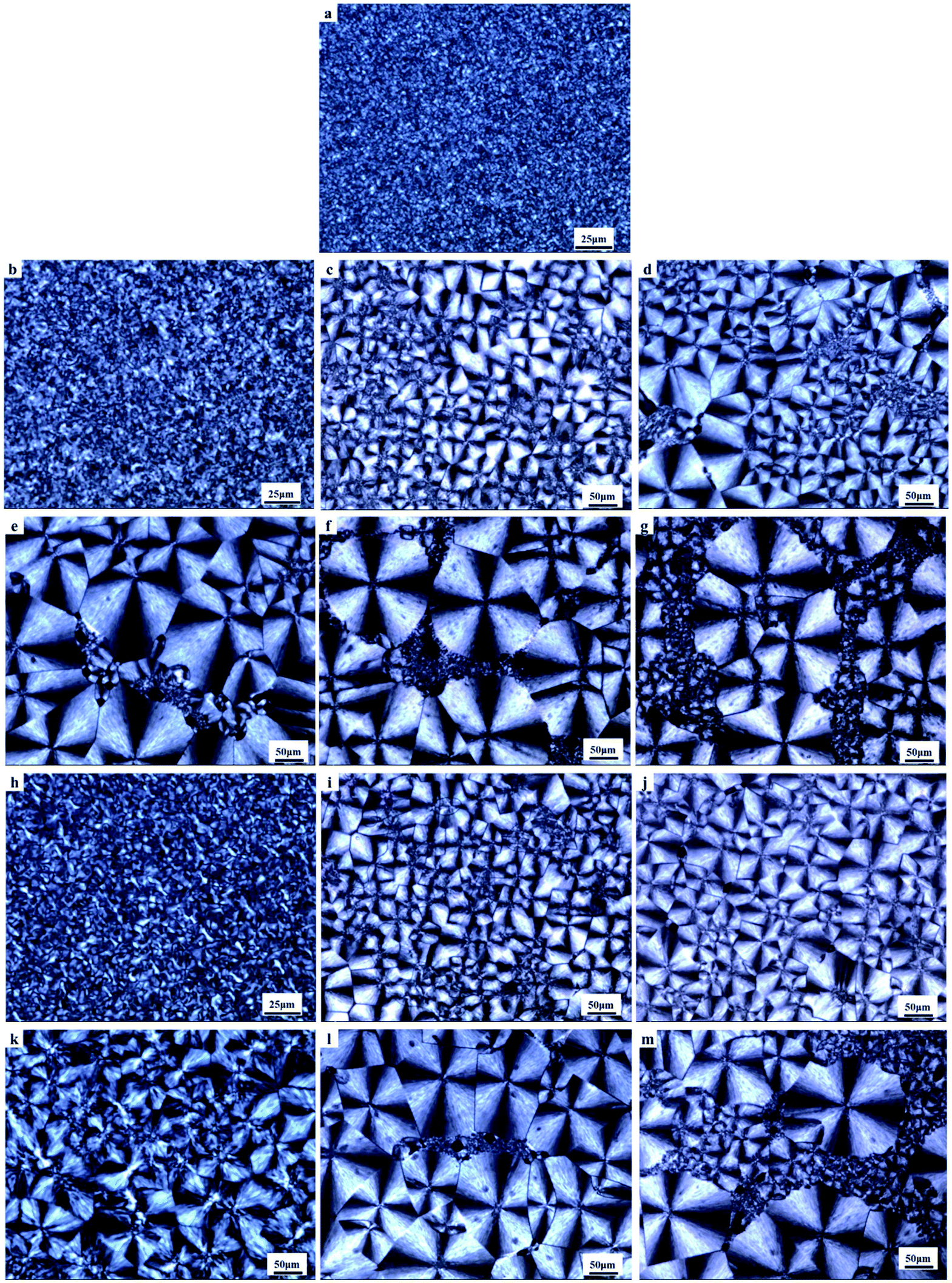 | ||
| Fig. 7 POM microphotographs of PCL undergo a fast cooling process: (a) control PCL; (b)–(g) PCL with 0.1 wt%, 0.3 wt%, 0.7 wt%, 1.0 wt%, 5.0 wt% and 10.0 wt% PVAC; (h)–(m) PCL with 0.1 wt%, 0.3 wt%, 0.7 wt%, 1.0 wt%, 5.0 wt% and 10.0 wt% PVB. | ||
Compared with the programmed cooling sample in Fig. 4, the spherulite size of the fast cooling samples decreased significantly at low content of polyvinyl acetals. For example, the grain size decreased from 100 μm to about 20 μm at 0.1 wt% polyvinyl acetals. When the content of polyvinyl acetals was less than 1 wt%, the grain size of PVB nucleated samples was more uniform and no fine grains appeared, which was consistent with POM results of program cooling samples.
Combining the crystallization behaviour and crystal morphology, we propose that the initial distortion during tensile test was originated from amorphous regions and small spherulite. All the samples containing PVAC and samples with high mass fraction of PVB (1%, 5%, 10%) are tend to deform firstly from the “weak region”, i.e. fine grains, resulting in the decrease of Young's modulus. However, the samples with lower content PVB (less than 1%) content have uniform grain size and no fine grain aggregation area, which ensured macroscopic uniformity and correspondingly increased modulus.
Crystallization kinetics of PCL
| X(t) = 1 − exp(−Ktn) | (3) |
log(−ln(1 − X(t))) = log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K + nlogt K + nlogt
| (4) |
By plotting log(−ln(1 − X(t))) versus logt, a series of lines with slope n and intercept logK can be obtained. To apply the Avrami model to depicting non-isothermal crystallization, Jeziorny51 corrected the logK by introducing the cooling rate, Φ:
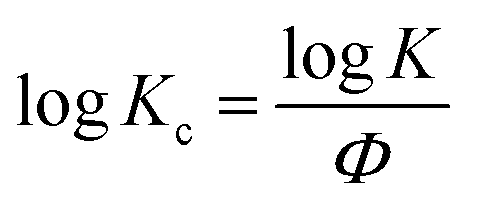 | (5) |
The half-crystallization time t1/2 can be obtained from eqn (6):
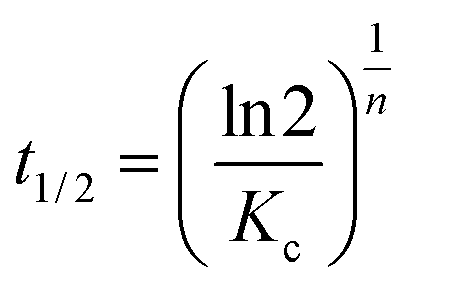 | (6) |
Fig. 8 gives the linear relationship of log(−ln(1−X(t))) versus logt of control and nucleated PCL at various cooling rates, where the data of intermediate crystallization were used so as to apply the modified Avrami model. There is no deviation for all samples, indicating that the modified Avrami model is suitable to describe the non-isothermal crystallization kinetics of PCL. The crystallization kinetics parameters derived from Fig. 8 are summarized in Table 2. With the increase of cooling rate, half-crystallization time (t1/2) is shortened, while the non-isothermal crystallization rate constant (Kc) is increased. This indicates that the crystallization rate increases with the cooling rate, which is consistent with the above discussion about the effect of cooling rate on PCL crystallization.
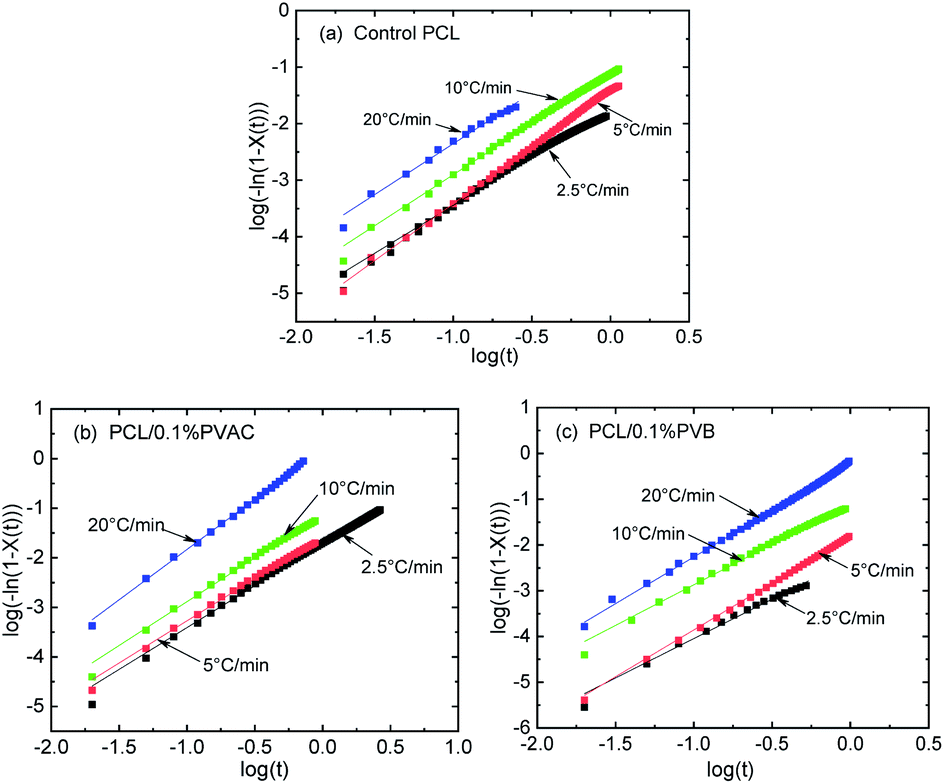 | ||
| Fig. 8 Avrami plots under various cooling rates for control and nucleated PCL. | ||
| Sample | Φ (°C min−1) | n | Kc | t1/2 (min) |
|---|---|---|---|---|
| Control PCL | 2.5 | 1.697 | 0.200 | 1.643 |
| 5.0 | 2.014 | 0.525 | 1.057 | |
| 10.0 | 1.804 | 0.777 | 0.989 | |
| 20.0 | 1.801 | 0.938 | 0.985 | |
| 0.1% PVAC | 2.5 | 1.688 | 0.205 | 1.628 |
| 5.0 | 1.703 | 0.485 | 1.074 | |
| 10.0 | 1.767 | 0.773 | 0.989 | |
| 20.0 | 2.031 | 1.023 | 0.981 | |
| 0.1% PVB | 2.5 | 1.722 | 0.118 | 2.032 |
| 5.0 | 2.048 | 0.435 | 1.098 | |
| 10.0 | 1.775 | 0.777 | 0.989 | |
| 20.0 | 2.042 | 0.975 | 0.983 |
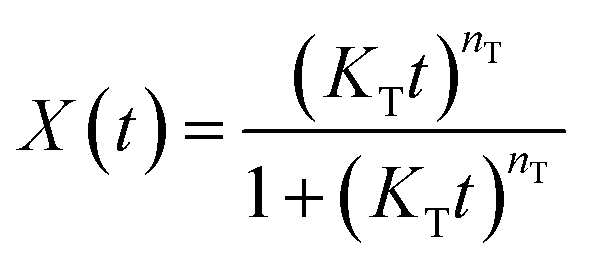 | (7) |
The curves of relative crystallinity versus time during non-isothermal crystallization of PCL at different cooling rate are shown in Fig. 9. The typical “S” curve shifts to the left with the increase of cooling rate. Tobin model well describes the relationship of relative crystallization and time at higher cooling rate (10 °C min−1 and 20 °C min−1). Under lower cooling rate (2.5 °C min−1 and 5 °C min−1), slight deviation appears at the beginning and end of crystallization.
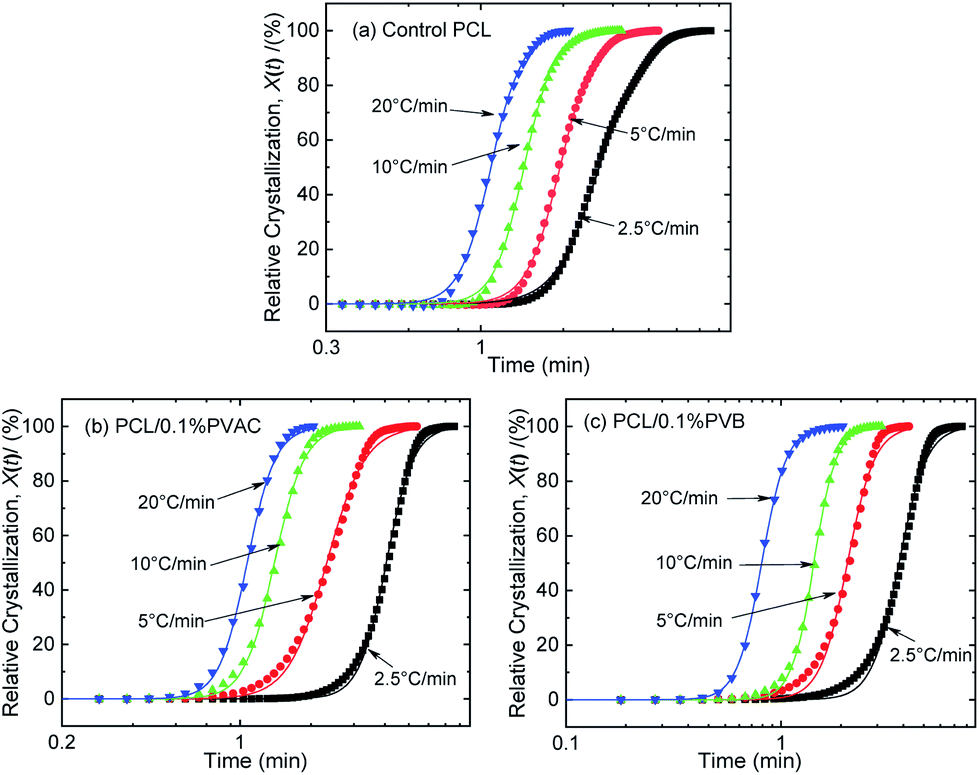 | ||
| Fig. 9 Relative crystallinity versus time for (a) control PCL, (b) PCL/0.1%PVAC, and (c) PCL/0.1%PVB. | ||
The Tobin crystallization rate constant and exponent are list in Table 3. The rate constant of crystallization increases with the cooling rate, and the values are similar at the same cooling rate except the rate of 2.5 °C min−1. The Tobin exponent of control and nucleated PCL falls in the range of 5.7–8.4, and is larger than the modified Avrami index.
| Sample | Φ/°C min−1 | ||||
|---|---|---|---|---|---|
| 2.5 | 5 | 10 | 20 | ||
| Control PCL | KT | 0.40 | 0.54 | 0.74 | 0.93 |
| nT | 5.89 | 7.70 | 8.53 | 8.27 | |
| 0.1% PVAC | KT | 0.27 | 0.46 | 0.75 | 0.94 |
| nT | 8.12 | 5.73 | 7.36 | 7.83 | |
| 0.1% PVB | KT | 0.28 | 0.49 | 0.70 | 1.23 |
| nT | 6.78 | 6.85 | 8.39 | 7.57 | |
Conclusions
As a lineal aliphatic polyester, PCL has a very low glass transition temperature, and the flexible polymer chains are easily entangled, which is unfavourable to crystallization. Interestingly, benefit from the disentangled effect of melt shearing (mixing), the crystallization temperature (increased by 7.7 °C) and crystallinity of control PCL were improved obviously. This discovery provides a new approach to improve the crystallization capacity of PCL.Using polyvinyl acetals as macromolecular nucleating agent, we investigated the non-isothermal crystallization behaviour, crystalline morphology and mechanical properties of PCL. The blends showed good compatibility in the acetal contents range from 0.1% to 0.7%. When the nucleating agent contents were beyond 0.7% (1–10 wt%), microphase separation occurred. PVAC and PVB significantly increased the spherulite size of PCL and reduce the crystallization temperature. Compared with PVAC, PVB has more powerful heterogeneous nucleation effect due to the longer side groups. When the contents were less than 1 wt%, PVB inhibited the formation of fine grains which was closely related with the initial distortion during the tensile test; the fine grains completely disappeared under fast cooling and the Young's modulus increased dramatically by 67%. Both the modified Avrami and Tobin models well described the non-isothermal crystallization of PCL, and the crystallization parameters had no significant change. This means that the addition of nucleating agents has no effect on the crystallization kinetics of PCL.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors specially acknowledge the support by National Natural Science Foundation of China (51473007, 21606005, 31570575), Fundamental Research Funds of Beijing Technology and Business University (19005902015) and Natural Science Foundation of Beijing Municipality (2192016).References
- D. Goldberg, J. Environ. Polym. Degrad., 1995, 3, 61–67 CrossRef CAS.
- O. Persenaire, M. Alexandre, P. Degee and P. Dubois, Biomacromolecules, 2001, 2, 288–294 CrossRef CAS.
- M. A. Woodruff and D. W. Hutmacher, Prog. Polym. Sci., 2010, 35, 1217–1256 CrossRef CAS.
- L. Qiu, B. Hu, H. Chen, S. Li, Y. Hu, Y. Zheng and X. Wu, Int. J. Nanomed., 2015, 10, 1415–1423 CAS.
- X. Tang, Y. Lyu, D. Xie, A. Li, Y. Liang and D. Zheng, J. Biomed. Nanotechnol., 2018, 14, 396–403 CrossRef CAS.
- N. Upho, B. Tangtrakulwanich, P. Pripatnanont, P. Thitiwongsawet and N. Ingviya, J. Pharm. Innov., 2015, 10, 211–221 CrossRef.
- D. de Cassan, S. Sydow, N. Schmidt, P. Behrens, Y. Roger, A. Hoffmann, A. L. Hoheisel, B. Glasmacher, R. Hansch and H. Menzel, Colloids Surf., B, 2018, 163, 309–320 CrossRef CAS PubMed.
- V. Tamboli, G. P. Mishra and A. K. Mitra, Colloid Polym. Sci., 2013, 291, 1235–1245 CrossRef CAS.
- M. Gumusderelioglu, F. B. Kaya and I. G. Beskardes, J. Colloid Interface Sci., 2011, 358, 444–453 CrossRef PubMed.
- V. Guarino, F. Veronesi, M. Marrese, G. Giavaresi, A. Ronca, M. Sandri, A. Tampieri, M. Fini and L. Ambrosio, Biomed. Mater., 2016, 11, 015018 CrossRef CAS.
- L. M. He, B. Liu, X. P. Guan, G. Y. Xie, S. S. Liao, D. P. Quan, D. Z. Cai, J. Lu and S. Ramakrishna, Eur. Cells Mater., 2009, 18, 63–74 CrossRef CAS.
- H. Y. Mi, X. Jing, E. Yu, X. Wang, Q. Li and L. S. Turng, J. Mech. Behav. Biomed. Mater., 2018, 78, 433–441 CrossRef CAS.
- M. J. Liu, S. C. Chen, K. K. Yang and Y. Z. Wang, RSC Adv., 2015, 5, 42162–42173 RSC.
- M.-H. Huang, S. Li and M. Vert, Polymer, 2004, 45, 8675–8681 CrossRef CAS.
- J. M. Ugartemendia, M. E. Munoz, A. Santamaria and J. R. Sarasua, J. Mech. Behav. Biomed. Mater., 2015, 48, 153–163 CrossRef CAS.
- W. Z. Yuan, X. Z. Tang, X. B. Huang and S. X. Zheng, Polymer, 2005, 46, 1701–1707 CrossRef CAS.
- I. Kelnar, J. Kratochvil, L. Kapralkova, A. Zhigunov and M. Nevoralova, J. Mech. Behav. Biomed. Mater., 2017, 71, 271–278 CrossRef CAS.
- W. Q. Han, X. Liao, Q. Yang, G. X. Li, B. He, W. L. Zhu and Z. H. Hao, RSC Adv., 2017, 7, 22515–22523 RSC.
- S. X. Li, C. N. Ye, G. D. Zhao, M. J. Zhang and Y. L. Zhao, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 3135–3148 CrossRef CAS.
- W. W. Wang, L. L. Chang, X. Li, Y. L. Wu, J. F. Xing, L. D. Deng and A. J. Dong, Soft Matter, 2012, 8, 1575–1583 RSC.
- C. He, J. Sun, J. Ma, X. Chen and X. Jing, Biomacromolecules, 2006, 7, 3482–3489 CrossRef CAS.
- P. Zhang, H. Y. Huang, Y. Chen, S. Yu, C. Krywka, S. K. Vayalil, S. V. Roth and T. B. He, Chin. J. Polym. Sci., 2014, 32, 1188–1198 CrossRef CAS.
- J. A. Pena, S. J. Gutierrez, J. C. Villamil, N. A. Agudelo and L. D. Perez, Mater. Sci. Eng., C, 2016, 58, 60–69 CrossRef CAS.
- S. N. Xia, Y. Shen, Y. Q. Zhou, P. F. Yao, Q. S. Liu and B. Y. Deng, Colloid Polym. Sci., 2018, 296, 1667–1677 CrossRef CAS.
- Z. B. Qiu, W. T. Yang, T. Ikehara and T. Nishi, Polymer, 2005, 46, 11814–11819 CrossRef CAS.
- W. Z. Yuan, J. Y. Yuan, X. B. Huang and X. Z. Tang, J. Appl. Polym. Sci., 2007, 104, 2310–2317 CrossRef CAS.
- K. J. Zhang and Z. B. Qiu, Chin. J. Polym. Sci., 2017, 35, 1517–1523 CrossRef CAS.
- T. Dong, Y. He, B. Zhu, K. M. Shin and Y. Inoue, Macromolecules, 2005, 38, 7736–7744 CrossRef CAS.
- T. Dong, W. H. Kai, P. J. Pan, A. Cao and Y. Inoue, Macromolecules, 2007, 40, 7244–7251 CrossRef CAS.
- J. F. Vega, J. Fernandez-Alcazar, J. V. Lopez, R. M. Michell, R. A. Perez-Camargo, B. Ruelle, J. Martinez-Salazar, M. L. Arnal, P. Dubois and A. J. Muller, J. Polym. Sci., Part B: Polym. Phys., 2017, 55, 1310–1325 CrossRef CAS.
- T. H. Zhao, K. L. Yang, R. T. Zeng, A. K. Du, M. Wang and J. B. Zeng, Ind. Eng. Chem. Res., 2016, 55, 1881–1889 CrossRef CAS.
- Q. Liu, Z. Q. Peng and D. J. Chen, Polym. Eng. Sci., 2007, 47, 460–466 CrossRef CAS.
- Q. L. Lv, C. J. Xu, D. F. Wu, Z. F. Wang, R. Y. Lan and L. S. Wu, Composites, Part A, 2017, 92, 17–26 CrossRef CAS.
- Q. L. Lv, Z. R. Ying, D. F. Wu, Z. F. Wang and M. Zhane, Ind. Eng. Chem. Res., 2017, 56, 2746–2753 CrossRef CAS.
- H. J. Chiu, R. S. Tsai and J. M. Huang, E-Polymers, 2010, 10, 75–86 Search PubMed.
- J. Cai, Z. Y. Xiong, M. Zhou, J. Tan, F. B. Zeng, M. H. Ma, S. Lin and H. G. Xiong, Carbohydr. Polym., 2014, 102, 746–754 CrossRef CAS.
- B. Yang, H. Ni, J. Huang and Y. Luo, Macromolecules, 2013, 47, 284–296 CrossRef.
- B. Yang, S. J. Zhu, Y. Luo and J. J. Huang, J. Polym. Sci., Part B: Polym. Phys., 2017, 55, 658–672 CrossRef CAS.
- K. N. Jeong-Chang Lee, T. Ikehara and T. Nishi, Public Prod. Manag. Rev., 1997, 21, 230–250 Search PubMed.
- T. M. Wu, J. C. Cheng and M. C. Yan, Polymer, 2003, 44, 2553–2562 CrossRef CAS.
- Y. Nozue, R. Kurita, S. Hirano, N. Kawasaki, S. Ueno, A. Iida, T. Nishi and Y. Amemiya, Polymer, 2003, 44, 6397–6405 CrossRef CAS.
- M. R. Lostoeeo, C. A. Murphy, J. A. Cameron and S. J. Huang, Polym. Degrad. Stab., 1998, 59, 303–307 CrossRef.
- J. X. Fu, Y. X. Wang, K. Z. Shen, Q. Fu and J. Zhang, J. Polym. Sci., Part B: Polym. Phys., 2019, 57, 598–606 CrossRef CAS.
- M. Trujillo, M. L. Arnal, A. J. Muller, M. A. Mujica, C. U. de Navarro, B. Ruelle and P. Dubois, Polymer, 2012, 53, 832–841 CrossRef CAS.
- G. M. Chen, T. M. Zou, L. Chen and Y. P. Huang, Asian J. Chem., 2010, 22, 5902–5912 CAS.
- A. Toda, K. Taguchi and H. Kajioka, Polymer, 2012, 53, 1765–1771 CrossRef CAS.
- T. Kamal, T. J. Shin and S.-Y. Park, Macromolecules, 2012, 45, 8752–8759 CrossRef CAS.
- M. Avrami, J. Chem. Phys., 1939, 7, 1103–1112 CrossRef CAS.
- M. Avrami, J. Chem. Phys., 1940, 8, 212–224 CrossRef CAS.
- M. Avrami, J. Chem. Phys., 1941, 9, 177–184 CrossRef CAS.
- A. Jeziorny, Polymer, 1978, 19, 1142–1144 CrossRef CAS.
- M. C. Tobin, J. Polym. Sci., Part B: Polym. Phys., 1976, 14, 2253–2257 CAS.
- M. C. Tobin, J. Polym. Sci., Part B: Polym. Phys., 1977, 15, 2269–2270 CAS.
- G. Eder, H. Janeschitz-Kriegl and S. Liedauer, Prog. Polym. Sci., 1990, 15, 629–714 CrossRef CAS.
- G. Z. Papageorgiou, D. S. Achilias and D. N. Bikiaris, Macromol. Chem. Phys., 2007, 208, 1250–1264 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |