
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLanthanide-doped mesoporous MCM-41 nanoparticles as a novel optical–magnetic multifunctional nanobioprobe†
Jun Liu *ab,
Siqian Liua,
Yaling Lia,
Jiayan Xuea,
Youyi Hea,
Fuchi Liua,
Li Yanga,
Junhui Hua,
Zhengye Xiong*c and
Lizhen Long*a
*ab,
Siqian Liua,
Yaling Lia,
Jiayan Xuea,
Youyi Hea,
Fuchi Liua,
Li Yanga,
Junhui Hua,
Zhengye Xiong*c and
Lizhen Long*a
aCollege of Physics Science and Technology & Guangxi Key Laboratory of Nuclear Physics and Technology, Guangxi Normal University, Guilin 541004, P. R. China. E-mail: liujun719@163.com; longlzh@foxmail.com; Fax: +86-773-5846479; Tel: +86-773-5846479
bState Key Laboratory for Chemistry and Molecular Engineering of Medicinal Resources, Guilin 541004, P. R. China
cSchool of Electronics and Information Engineering, Guangdong Ocean University, Zhanjiang 524088, China. E-mail: xiongzhengye@139.com
First published on 10th December 2019
Abstract
To research and develop potential multifunctional nanoprobes for biological application, lanthanide-doped MCM-41 (Ln-MCM-41, Ln = Gd/Eu) silica nanoparticles with excellent pore structure and optical–magnetic properties were synthesized via a facile and economical sol–gel method. The microstructure and pore distribution of Ln-MCM-41 nanoparticles were obviously affected by the Ln-doping. As the Ln/Si mole ratio increased, the specific surface area and total pore volume of Ln-MCM-41 nanoparticles rapidly decreased. However, the Ln-MCM-41 nanoparticles still retained the typical well-ordered mesoporous structure, and exhibited excellent drug release behavior. Moreover, the drug release rate of Ln-MCM-41 was remarkably pH-dependent and increased gradually upon decreasing pH. Additionally, these nanoparticles also exhibit considerable photoluminescence properties, living cells photoluminescence imaging in vitro, and paramagnetism behavior at room temperature due to the Ln3+-ions doping. Our research shows the possibility of our Ln-MCM-41 nanoparticles as multifunctional nanoprobes for application in bioseparation, bioimaging, and drug delivery.
1. Introduction
With fascinating optical properties like a large Stokes shift, long excited-state lifetimes, photo-chemical stability, and narrow emission bands, trivalent lanthanide ion (Ln3+) doped nanoparticles have attracted significant attention in biological imaging,1–4 biomolecule detection,5–7 security encoding,8–10 and cancer therapeutics.11–13 Many efforts have been made to improve the luminescence performance by optimizing the size, shape, and doping concentration of the Ln3+-doped luminescence nanoparticles. Among the investigated materials, Ln3+-doped NaYF4 nanoparticles are of particular interest due to their lowest phonon cutoff energy and highly controllable synthesis process.14–16 In recent years, the partial or complete substitution of Y3+ by Gd3+ in NaYF4 was demonstrated, and Gd3+ in the resulted NaGdF4 matrix can mediate energy migration from Tm3+ to other emitting centers (e.g., Tb3+, Eu3+, Sm3+, and Dy3+) to achieve enriched luminescence.17,18 Meanwhile, the Gd also exhibits magnetic properties to fabricate optically and magnetically active bifunctional materials.19,20 However, the preparation of the Ln3+-doped NaYF4 nanoparticles is not satisfactory, owing to their complex process, low production and source reagent limitation. Compared to fluorides, the oxide counterparts (e.g., Y2O3, Gd2O3, and ZrO2) also has been widely used as luminescence-magnetic host due its high chemical stability, low bio-toxicity, and more stable in aqueous circumstances.21–23 Unfortunately, the development of mass-scale synthesis methods for homogeneous, ultrafine, and robust Ln3+-doped oxide nanoparticles is one of the key materials challenges for commercialization and advanced applications.24,25In addition, much effort has been expended to develop the biocompatible and multifunctional Ln3+-doped luminescent nanoparticles in recent years, in order to meet the clinical needs. Silica coating is particularly promising for this purpose because these particles reveal a low cytotoxicity, they are readily available in various thicknesses, and their surface can be functionalized almost at will through facile surface modification. Therefore, silica-coated Ln3+-doped nanoparticles have successfully used in biomedical applications, such as biomarkers, biosensors, and in anti-cancer therapy.26–28 Moreover, the silica coating may be also useful as drug delivery vehicle, e.g. for mesoporous-silica (m-SiO2), active biological compounds, like anticancer drug or photosensitizers can be adsorbed, due to its unique porous ordered structure.29–31 What is more, the readily chemically modifiable surface enable the use of such materials in controlled drug delivery.32 However, the preparation of these core–shell materials is generally related to a multistep process, including the synthesis of uniform luminescence nanoparticles, coating of silica, and tedious wash process, which is not only complicated, but also poor reproducibility.
In recent years, tremendous efforts have been devoted to synthesis the Ln3+-doped silica materials, e.g., silica nanoparticles,33–35 silica nanorods,36 silica film.37 In addition to the excellent luminescence properties, the Ln3+-doped silica materials possess many advantages, such as high uniformity, easy surface modification and easy control of reaction conditions. Despite the substantial merits, the application of Ln3+-doped silica materials as nanoprobes in biomedical application is still insufficient. Inspired by these researches, we dedicated to synthesis the porous structured luminescence silica nanoparticles for multifunctional bioapplication in this work. With ordered cylindrical channels, the mesoporous MCM-41 silica nanoparticles can be used as the nanocontainer for excellent drug storage and sustained drug release properties.31,38 Therefore, the MCM-41 nanoparticles were selected as host, and Ln3+ ions as activators to synthesis Ln3+-doped MCM-41 (Ln-MCM-41) nanoparticles via the sample CTAB assisted sol–gel method. The pore distribution and luminescence properties can be easily tailored by the adjusting of Ln3+-doping concentration and selection of activators. This system seems to be ideal for bioseparation, targeting and tracking drug delivery based on its pore-structure, magnetic and luminescent properties.
2. Experimental
2.1 Materials and preparation of samples
Gd2O3 (99.99%), Eu2O3 (99.99%), tetraethylorthosilicate (TEOS, 99.99%), cetyltrimethylammonium bromide (C16TAB), and doxorubicin (DOX, 98%) were purchased from Aladdin Reagent Co., Ltd. (China). Polyethylene glycol-4000 (PEG-4000) and ammonia (NH3·H2O, 25%) was purchased from Xilong Scientific (China). Deionized (DI) water was produced using Direct-Q3 water purification system.The Ln2O3 (Ln = Gd/Eu) powders with definite ratio Gd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Eu = 95:5 were firstly dissolved in HNO3 while heating with the water-bath method and stirring till excess nitric acid evaporated, and a certain amount of deionized water was subsequently added with stirring for 10 min to form 2.4 M Ln(NO3)3 solution. The Ln-MCM-41 nanoparticles was synthesized by the one-pot sol–gel method. The typical synthesis process is described as follows: 0.2 g of C16TAB and 0.505 g PEG-4000 was dissolved in 100 mL of deionized water. 2 mL of 25% NH3·H2O was then added to the vigorously stirred solution at room temperature, followed by the addition of 1 mL TEOS and a certain amount of Ln(NO3)3 solution. After one hour of stirring, the fine particle precipitate was centrifuged and dried in a freezer dryer. Finally, the samples were calcined at 500 °C for 5 h to remove the templates.
:Eu = 95:5 were firstly dissolved in HNO3 while heating with the water-bath method and stirring till excess nitric acid evaporated, and a certain amount of deionized water was subsequently added with stirring for 10 min to form 2.4 M Ln(NO3)3 solution. The Ln-MCM-41 nanoparticles was synthesized by the one-pot sol–gel method. The typical synthesis process is described as follows: 0.2 g of C16TAB and 0.505 g PEG-4000 was dissolved in 100 mL of deionized water. 2 mL of 25% NH3·H2O was then added to the vigorously stirred solution at room temperature, followed by the addition of 1 mL TEOS and a certain amount of Ln(NO3)3 solution. After one hour of stirring, the fine particle precipitate was centrifuged and dried in a freezer dryer. Finally, the samples were calcined at 500 °C for 5 h to remove the templates.
2.2 Characterization
The X-ray diffraction (XRD) patterns were obtained on the X-ray diffractometer (Bruker D8 Advance, Germany) instrument operated using the Cu Kα radiation. Transmission electron microscopy (TEM) images and energy dispersive X-ray spectroscopy (EDS) analysis were acquired using a JEM-3200FS microscopy (JEOL, Japan). The nitrogen adsorption/desorption measurements were performed on a specific surface and pore size analysis instrument (3H-2000PS, Beishide Instrument Technology (Beijing) Co., Ltd., China) at 77 K, and all samples were degassed at 200 °C for 3 h prior to the measurement. Fourier transform infrared spectroscopy (FTIR) spectra were collected in Agilent Cary 630 FTIR spectrometer in the range of 500 to 4000 cm−1. The photoluminescence (PL) spectra were measured at ambient conditions by a fluorescence spectrofluorophotometer (Shimadzu RF-5301PC, Japan) using a Xe lamp as the light source. Magnetic hysteresis curve were measured at room temperature using vibrating sample magnetometer (VSM, LakeShore 7404, American).2.3 Optical imaging of live cells
Raw264.7 cell lines were inoculated on a 96-well culture plate and then cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, penicillin (100 units per mL) and streptomycin (100 mg mL−1) at 37 °C under 5% CO2 atmosphere. After growing to logarithmic growth phase, the cells were rinsed with phosphate buffered saline (PBS) and then were incubated with 50 μg mL−1 of the Ln-MCM-41 in the same environment for another 2 h. After incubation, the cells were washed with PBS to remove the remaining particles and dead cells and then observed under a confocal fluorescence microscope (Leica TCS SP8, Germany).2.4 Preparation of drug storage and delivery system
For the loading of DOX on the Ln-MCM-41 nanoparticles, 480 mg of the Ln-MCM-41 nanoparticles were dispersed in 80 mL phosphate buffer solution (PBS, pH = 7.4) with 40 mg DOX. After stirring for 2 h at room temperature, 3 mL of PA solution (5.0 mg mL−1) was added and stirred for another 30 min. Subsequently, the pH value of the mixture was adjusted to alkaline. After then, 0.5 mL of 0.24 M (NH4)3PO4·3H2O solution and 0.5 mL of 0.29 M Ca(NO3)2·4H2O solution were injected 8 times in turn at 10 min intervals. The as-fabricated product was collected by centrifugation at 10000 rpm for 8 minutes and then freeze-dried under vacuum for 12 h dried at 60 °C, and the solid was named as DOX-Ln-MCM-41. UV-vis was used to determine the concentration of the separated supernatant solution. To evaluate the DOX-release behavior, 30 mg DOX-Ln-MCM-41 samples were immersed in 10 mL PBS solution (pH = 7.4, 6.5, or 5.5) at 37 °C under continuous stirring. At predetermined time intervals, 3 mL of the released medium was taken and centrifuged. The amount of released DOX was measured by a UV-vis spectrophotometer (Shimadzu UV-2700, Japan) at a wavelength of 480 nm. Finally, the medium taken out for measurement was returned to the original released medium.
3. Results and discussion
To investigate the influence of Ln3+-ions doping on the pore properties of the Ln-MCM-41 nanoparticles, three samples with zero, low (0.07), high (0.13) mole ratio of Ln/Si (Ln = Gd/Eu, Gd:Eu = 95:5) were synthesized and denotes as pristine MCM-41, LLn-MCM-41, and HLn-MCM-41, respectively (see the details in Table S1†). The wide-angle XRD pattern (Fig. 1a) of pristine MCM-41 exhibit a broad diffraction peak at 2θ values between 15.0° and 35.0°, which is typical for amorphous silica. By doping with Ln3+-ions, the amorphous diffraction peak remarkably reduced for both the LLn-MCM-41 and HLn-MCM-41. Small-angle XRD patterns (Fig. 1b) recorded at the range of 0.5° to 10° show a very intense (100) Bragg diffraction and two less intense (110) and (200) diffraction peaks reflections associated with a p6 mm hexagonal symmetry of the ordered MCM-41-type materials.38 Obviously, the incorporation of heteroatoms into the silica not only decreased the peak intensity but also caused the shift of d100-spacing relative to undoped MCM-41. However, the shift of d100-spacing for LLn-MCM-41 toward to the large value, while the d100-spacing for HLn-MCM-41 shifted to a small value. The incorporation of heteroatoms into the silica pore framework or the formation of Gd and Eu oxide clusters may cause the shift in d100-spacing.
 | ||
| Fig. 1 (a) Wide-angle XRD and (b) small-angle XRD patterns of the mesoporous MCM-41, LLn-MCM-41, and HLn-MCM-41 nanoparticles. | ||
The morphology of the Ln-MCM-41 nanoparticles were further observed by TEM measurements. As presented in Fig. 2a and b, the TEM image of the MCM-41 and HLn-MCM-41 nanoparticles revealed their well-defined ordered pore structure. The pore structure of MCM-41 nanoparticles is clearer than that of HLn-MCM-41. The mean diameters based on the corresponding size distribution histogram (Fig. 2c and d) were determined to ∼86.28 for MCM-41 and ∼75.12 nm for HLn-MCM-41, respectively. Additionally, the HLn-MCM-41 nanoparticles possess a broader size distribution compared to MCM-41. These results revealed that the Ln3+-doping promotes the formation of small-sized particles.
 | ||
| Fig. 2 Typical TEM image of MCM-41 (a) and HLn-MCM-41 (b) nanoparticles, and their corresponding size distribution histogram (c and d). | ||
In addition, we also noticed that some regions of the Ln-doped MCM-41 nanoparticles are darker than other regions or pristine MCM-41 nanoparticles, or some particles seem have coating layer on the surface, according to the TEM images in Fig. 2b. These zones may be rich in the incorporation of heteroatoms Eu and Gd. Thus, the TEM-EDS mapping (Fig. 3a) were employed to check these nanoparticles. The elemental mappings clearly reveal that the elements of Gd and Eu are unevenly distributed, or there is aggregation in Gd and Eu. From the TEM-EDS (Fig. 3b) of HLn-MCM-41 nanoparticles, signals of O, Si, Gd, and Eu can be surveyed, demonstrating the successful doping of Gd and Eu into MCM-41. The carbon (C) and copper (Cu) peaks in EDS spectrum probably comes from the TEM-grid.
 | ||
| Fig. 3 (a) Elemental mapping and (b) EDS spectrum of the HLn-MCM-41 nanoparticles. | ||
To confirm the reason of the aggregation in Gd and Eu, the SAED pattern and high-resolution TEM (HR-TEM) analysis of the HLn-MCM-41 nanoparticles was further studied. In the TEM image (Fig. 4a) taken with the electron beam parallel to the pore direction of the HLn-MCM-41 nanoparticles, the pores present an ordered hexagonal structure. The SAED pattern (the insert in Fig. 4a) of the HLn-MCM-41 nanoparticles is consistent with the amorphous structure diffraction pattern, which is in accordance with the wide-angel XRD results. However, some tiny lattice streaks can be observed in HR-TEM image (Fig. 4b). These tiny lattice fringes indicate the presence of fine crystalline grains in the HLn-MCM-41 nanoparticles, and these grains may be the Gd or Eu oxide clusters.
 | ||
| Fig. 4 (a) TEM image and SAED pattern taken with the electron beam parallel to the pore direction of the HLn-MCM-41 nanoparticles; (b) HR-TEM image of the HLn-MCM-41 nanoparticles. | ||
Fig. 5 shows the nitrogen adsorption/desorption isotherms and pore-size distribution curves of all samples. The isotherm of MCM-41 exhibit a typical type IV behavior with an H1-type hysteresis loop (Fig. 5a), which is typical for mesoporous materials with two-dimensional hexagonal structures.39 The N2 uptake at low relative pressure (P/P0 < 0.1) for all samples indicates the existence of micropores. No obvious hysteresis loop is observed above a relative pressure of 0.9, but a sharp step increase at P/P0 = 0.25–0.35 in the isotherm of the MCM-41 both the well-ordered mesoporous structure and the uniformity of the pore size distribution of the sample. The decrease of the absorption amount of LLn-MCM-41 and HLn-MCM-41 can be attributed to the reduced surface area, whereas the shift of inflection point of the step to lower relative pressure P/P0 is caused by the smaller mesopores size, which can be associated with the pore-filling effect. The pore-size distribution curves (Fig. 5b), derived from the desorption branch based on the density functional theory (DFT), reveals the presence of relatively uniform micropores and mesopores in all samples. The calculated structural parameters of all samples are listed in Table 1. The specific surface area (SBET) was calculated using the Brunauer–Emmett–Teller (BET) based on adsorption data in the partial pressure (P/P0) range of 0.04–0.32. The specific surface area rapidly drops from 910 m2 g−1 of MCM-41 to 281 m2 g−1 of HLn-MCM-41, while the total pore volume decreases from 1.30 mL g−1 to 0.77 mL g−1, respectively. Mesopores are the major pores in Ln-MCM-41 nanoparticles, while micropores are minor with a decreased proportion by increasing of Ln3+ doping amount.
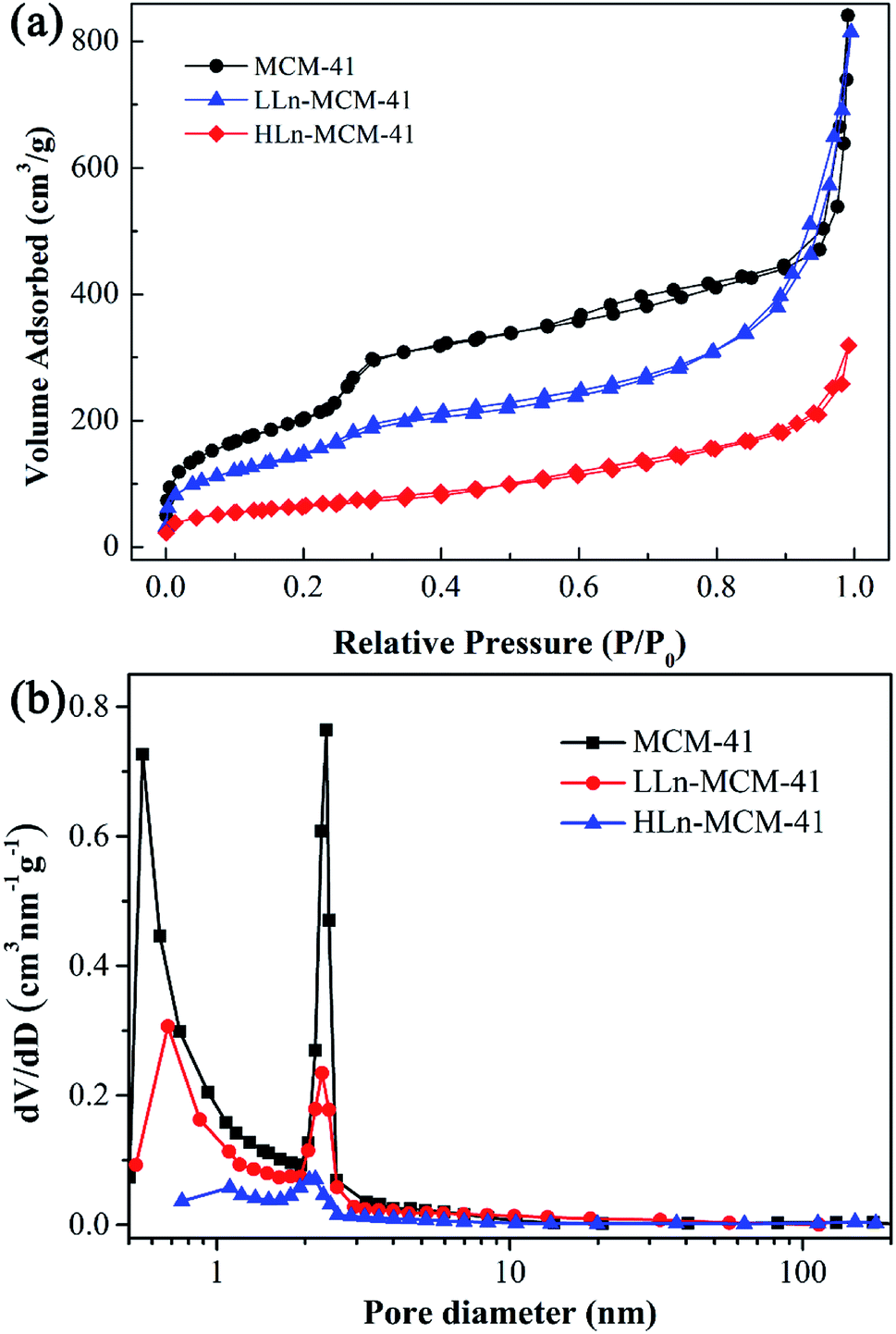 | ||
| Fig. 5 Porous textures of the samples. (a) Nitrogen adsorption–desorption isotherms, and (b) pore size distribution curves calculated from the desorption branch of the isotherm. | ||
| Samples | SBET (m2 g−1) | Total pore volume (mL g−1) | Micropore volume (mL g−1) | Proportion of micropore | ||
|---|---|---|---|---|---|---|
| T-plot | H–K | DR | ||||
| MCM-41 | 910 | 1.30 | 0.31 | 0.27 | 0.30 | <24% |
| 0.07 Gd/Eu-MCM-41 | 612 | 1.26 | 0.16 | 0.20 | 0.21 | <17% |
| 0.13 Gd/Eu-MCM-41 | 281 | 0.77 | 0.06 | 0.07 | 0.08 | <10% |
Lanthanide-based nanoparticles have excellent and diverse photoluminescence properties, thus we further studied the photoluminescence properties of the Ln-MCM-41 nanoparticles. Fig. 6a gives the luminescence photograph of the MCM-41, LLn-MCM-41, and HLn-MCM-41 powders, which was recorded by a digital camera without using any optical filters under excitation with ultraviolet light (365 nm). Red emission can be clearly observed from Ln-MCM-41 nanoparticles at room temperature, and no emission for undoped MCM-41. Obviously, the luminescence intensity of Ln-MCM-41 nanoparticles increased with the increase of Ln3+ doping amount. Fig. 6b demonstrates the room temperature photoluminescence excitation (PLE) and photoluminescence emission (PL) spectra of HLn-MCM-41 nanoparticles. The PLE spectrum monitored by 613 nm (5D4 → 7F2 energy levels transition of Eu3+ ions) exhibit a strong broad excitation peak centering at 244 nm corresponding to the O2+–Eu3+ charge transfer band (CTB) adsorption, and a weak sharp excitation peak at 274 nm may assigned to Gd3+ (8S7/2 → 6I7/2), suggesting the energy transfer occurred from the Gd3+ ions to the Eu3+ ions.40 Upon excitation at 244 and 274 nm, the Gd3+/Eu3+ co-doped MCM-41 nanoparticles exhibit characteristic emission spectrum. The emission peaks originate from 5Di → 7FJ energy levels transitions of Eu3+, i.e., 5D4 → 7F0 (362 nm), 5D2 → 7F0 (464 nm), 5D0 → 7F1 (594 nm), 5D0 → 7F2 (613 nm), 5D0 → 7F3 (653 nm), and 5D0 → 7F4 (701 nm).41 To obtain multicolored Ln-MCM-41 fluorescence nanoparticles, the different ion-combinations including Gd3+/Tm3+ and Gd3+/Tb3+, were also introduced to synthesis the blue and green Ln-MCM-41 luminescence nanoparticles (Fig. S1†).
 | ||
| Fig. 6 (a) Photoluminescence photograph of MCM-41, LLn-MCM-41, and HLn-MCM-41 nanoparticles under excitation at 256 nm; (b) room temperature photoluminescence excitation (PLE) and photoluminescence emission (PL) properties of the HLn-MCM-41 nanoparticles. | ||
In addition to the excellent fluorescent properties, lanthanide-based nanoparticles also possess commendable magnetic properties and thus could be used as important materials for magnetic resonance imaging (MRI) and bioseparation. Magnetization as a function of magnetic fields of the HLn-MCM-41 nanoparticles is shown in Fig. 7. Keeping the total doping concentration and Gd content as the constants, the magnetization curves obtained by using Gd/Eu, Gd/Tb and Gd/Tm co-doped nanoparticles are very similar (as shown in Fig. S2†). The HLn-MCM-41 nanoparticles show paramagnetism at RT, unlike the magnetic response behavior of Gd atoms which exhibit ferromagnetism below 289 K. In general, the magnetic properties of Gd3+ arise from seven unpaired inner 4f electrons which are closely bound to the nucleus and effectively shielded by the outer closed shell electrons 5s2 5p6. According to the M–H relationship, the magnetic mass susceptibilities of HLn-MCM-41 nanoparticles are 5.26 × 10−5 emu g−1 Oe−1, respectively. The magnetization of HLn-MCM-41 nanoparticles at 20 kOe is about 1.04 emu g−1, which is close to the reported magnetite in bioseparation and bioimaging (as shown in Table S2†).
 | ||
| Fig. 7 RT magnetization as a linear function of magnetic field for the HLn-MCM-41 nanoparticles. | ||
To verify the feasibility of using Ln-MCM-41 nanoparticles as bioimaging probes, the Raw264.7 living cells incubated with the HLn-MCM-41 nanoparticles were imaged under the laser confocal fluorescence microscope. The preprocessed cells were imaged by laser scanning microscope excited at 488 nm. As shown in Fig. 8, the HLn-MCM-41 nanoparticles maintain intrinsically significant photoluminescence characteristics after being swallowed by the cells. The overlay images reveal that the red fluorescence are unevenly and mainly distributed in the cytoplasm, which is regarded to the heterogeneous distribution of the sample inside the cell. The result validate that the HLn-MCM-41 nanoparticles, verified to possess both good cell compatibility and cell internalization, are qualified for the function of live cell imaging.
 | ||
| Fig. 8 Confocal laser microscope images of Raw 264.7 cells incubated with HLn-MCM-41 nanoparticles: (a) fluorescence, (b) bright-field, and (c) overlay images (scale bar: 10 μm). | ||
The mesoporous nanoparticles can be used as the nanocontainer for excellent drug storage and sustained drug release properties. Although the pore properties of the Ln-MCM-41 nanoparticles are affects by the Ln3+-doping, we found that they still possess commendable properties of nanocarriers. As shown in Fig. 9a, we selected the HLn-MCM-41 nanoparticles as nanocarriers and doxorubicin (DOX) as a model drug to evaluate the drug storage and release behavior in our experiment. It can be calculated that the loading mount of DOX-Ln-MCM-41 nanoparticles were ∼0.06 mg of the DOX per mg of nanoparticles, based on the UV-vis absorbance analysis of the PBS medium measured at different loading time (as shown in Fig. S3†). FT-IR (Fig. 9b) was used to investigate the drug loading. A broad band in the region of 3437 cm−1 was attributed to the symmetric stretching of OH groups from Si–OH in HLn-MCM-41 and DOX-HLn-MCM-41. They also show the main peaks characteristic of silica in the region around 1088 and 789 cm−1 corresponding to symmetrical and a symmetrical stretching vibration of Si–O–Si. Two characteristic absorption peaks at 1629 and 1576 cm−1 are ascribed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration from the anthraquinone ring of DOX in DOX-HLn-MCM-41 sample. The supernatant solution was determined to obtain the residual DOX content by UV-vis measurement. As seen in Fig. 9c, the DOX release amount could be determined by the characteristic DOX absorbance intensity peaked at 480 nm. The in vitro real time monitoring release profiles and absorption spectra of DOX from DOX-HLn-MCM-41 nanoparticles in PBS buffer solution at different pH value are shown in Fig. 9d and S4.† The drug release rate of DOX-HLn-MCM-41 was remarkably pH-dependent and increased gradually upon decreasing pH. The pH-sensitive DOX released from nanoparticles might be beneficial for controlled drug delivery and release into cancer cells since the microenvironments in intracellular lysosomes, endosomes and certain cancerous tissues is acidic.
O stretching vibration from the anthraquinone ring of DOX in DOX-HLn-MCM-41 sample. The supernatant solution was determined to obtain the residual DOX content by UV-vis measurement. As seen in Fig. 9c, the DOX release amount could be determined by the characteristic DOX absorbance intensity peaked at 480 nm. The in vitro real time monitoring release profiles and absorption spectra of DOX from DOX-HLn-MCM-41 nanoparticles in PBS buffer solution at different pH value are shown in Fig. 9d and S4.† The drug release rate of DOX-HLn-MCM-41 was remarkably pH-dependent and increased gradually upon decreasing pH. The pH-sensitive DOX released from nanoparticles might be beneficial for controlled drug delivery and release into cancer cells since the microenvironments in intracellular lysosomes, endosomes and certain cancerous tissues is acidic.
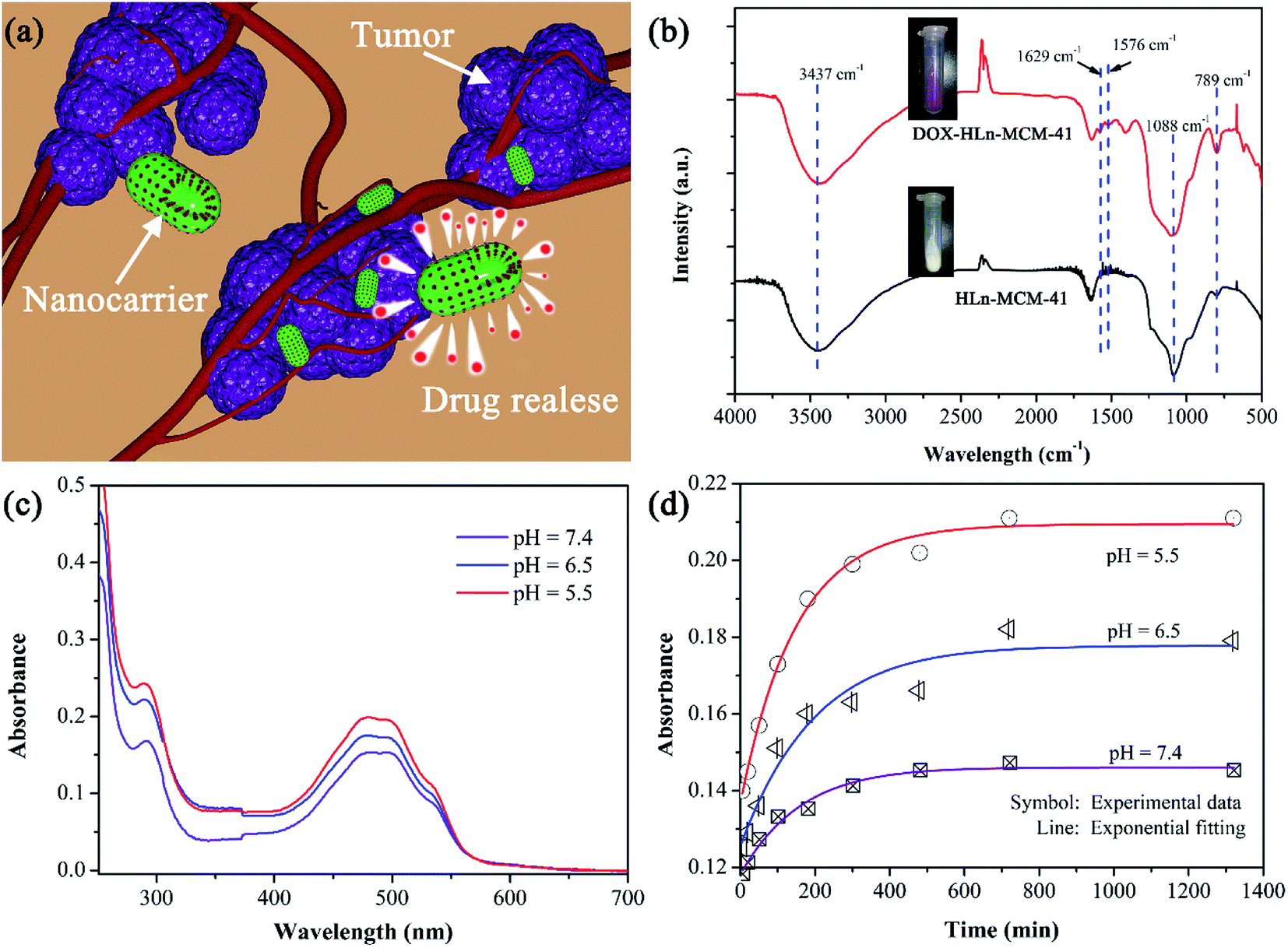 | ||
| Fig. 9 (a) Schematic illustrations of DOX release behavior for HLn-MCM-41 nanoparticles; (b) FTIR spectra of HLn-MCM-41 and DOX-HLn-MCM-41 samples; (c) UV-vis absorbance spectra of the PBS medium after 5 h release at three different pH values; (d) real time monitoring absorbance in 480 nm of the released medium at three different pH values and the corresponding exponential curve fitting. | ||
4. Conclusion
In summary, the Ln3+-doped MCM-41 (Ln-MCM-41) nanoparticles with excellent pore structure and optical–magnetic properties have been successfully prepared via a simple one-pot route. The prepared Ln-MCM-41 nanoparticles exhibit typical well-ordered mesoporous structure. As the Ln/Si mole ratio increased from 0 to 0.13, the specific surface area of Ln-MCM-41 nanoparticles drops from 910 to 281 m2 g−1, and the total pore volume decrease from 1.30 to 0.77 mL g−1. Further work demonstrated that although the pore volume is reduced, the capability of Ln-MCM-41 nanoparticles as a drug carrier is still satisfactory. Moreover, the drug release behavior of Ln-MCM-41 nanoparticles was pH-sensitive, and the drug release rate was increased gradually upon decreasing pH. The Ln-MCM-41 nanoparticles also exhibit excellent photoluminescence properties and considerable magnetic mass susceptibility. In fact, this work is expected to develop an optical–magnetic MCM-41 nanoparticles, which promises potential applications in a wide range of fields such as bioseparation, optical–magnetic bioimaging, and drug delivery.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Science Foundation of Guangxi (No. 2018GXNSFBA138007, 2018GXNSFBA281073, 2018GXNSFAA294021), the National Natural Science Foundation of China (No. 11665007, 11664003), the Guangxi Key Research and Development Program (No. AB18221033), the State Key Laboratory for Chemistry and Molecular Engineering of Medicinal Resources (Guangxi Normal University, No. CMEMR2018-B11), the PhD Start-up Fund of Guangxi Normal University (No. 2017BQ023, 2017BQ006), and the Innovation Project of Guangxi Graduate Education (No. XYCSZ2019058).Notes and references
- T. Maldiney, A. Bessière, J. Seguin, E. Teston, S. K. Sharma, B. Viana, A. J. J. Bos, P. Dorenbos, M. Bessodes, D. Gourier, D. Scherman and C. Richard, Nat. Mater., 2014, 13, 418–426 CrossRef CAS PubMed.
- Q. Liu, W. Feng, T. Yang, T. Yi and F. Li, Nat. Protoc., 2013, 8, 2033 CrossRef CAS PubMed.
- D. H. Ortgies, M. Tan, E. C. Ximendes, B. del Rosal, J. Hu, L. Xu, X. Wang, E. Martín Rodríguez, C. Jacinto, N. Fernandez, G. Chen and D. Jaque, ACS Nano, 2018, 12, 4362–4368 CrossRef CAS PubMed.
- A. Bednarkiewicz, E. M. Chan, A. Kotulska, L. Marciniak and K. Prorok, Nanoscale Horiz., 2019, 4, 881–889 RSC.
- Z. Zhang, S. Shikha, J. Liu, J. Zhang, Q. Mei and Y. Zhang, Anal. Chem., 2019, 91, 548–568 CrossRef CAS PubMed.
- X. Lei, R. Li, D. Tu, X. Shang, Y. Liu, W. You, C. Sun, F. Zhang and X. Chen, Chem. Sci., 2018, 9, 4682–4688 RSC.
- Z. Chen, W. Zheng, P. Huang, D. Tu, S. Zhou, M. Huang and X. Chen, Nanoscale, 2015, 7, 4274–4290 RSC.
- Y. Lu, J. Zhao, R. Zhang, Y. Liu, D. Liu, E. M. Goldys, X. Yang, P. Xi, A. Sunna and J. Lu, Nat. Photonics, 2014, 8, 32–36 CrossRef CAS.
- Y. Lu, J. Zhao, R. Zhang, Y. Liu, D. Liu, E. M. Goldys, X. Yang, P. Xi, A. Sunna, J. Lu, Y. Shi, R. C. Leif, Y. Huo, J. Shen, J. A. Piper, J. P. Robinson and D. Jin, Nat. Photonics, 2013, 8, 32 CrossRef.
- X. Wen, T. K. Lee, B. E. Moon, H. Song, C. Xu, B. Chun, Y. I. Kim, K. K. Sang, C. Peng and D. Kim, Adv. Opt. Mater., 2018, 6, 1701119 CrossRef.
- L. Cheng, K. Yang, Y. Li, X. Zeng, M. Shao, S.-T. Lee and Z. Liu, Biomaterials, 2012, 33, 2215–2222 CrossRef CAS PubMed.
- N. M. Idris, M. K. Gnanasammandhan, J. Zhang, P. C. Ho, R. Mahendran and Y. Zhang, Nat. Med., 2012, 18, 1580–1585 CrossRef CAS PubMed.
- J. Zuo, L. Tu, Q. Li, Y. Feng, I. Que, Y. Zhang, X. Liu, B. Xue, L. J. Cruz, Y. Chang, H. Zhang and X. Kong, ACS Nano, 2018, 12, 3217–3225 CrossRef CAS PubMed.
- F. Wang, Y. Han, C. S. Lim, Y. Lu, J. Wang, J. Xu, H. Chen, C. Zhang, M. Hong and X. Liu, Nature, 2010, 463, 1061–1065 CrossRef CAS PubMed.
- R. Deng, F. Qin, R. Chen, W. Huang, M. Hong and X. Liu, Nat. Nanotechnol., 2015, 10, 237–242 CrossRef CAS PubMed.
- J. Zhao, D. Jin, E. P. Schartner, Y. Lu, Y. Liu, A. V. Zvyagin, L. Zhang, J. M. Dawes, P. Xi, J. A. Piper, E. M. Goldys and T. M. Monro, Nat. Nanotechnol., 2013, 8, 729–734 CrossRef CAS PubMed.
- S. Han, X. Qin, Z. An, Y. Zhu, L. Liang, Y. Han, W. Huang and X. Liu, Nat. Commun., 2016, 7, 13059 CrossRef CAS PubMed.
- C. Dong, A. Korinek, B. Blasiak, B. Tomanek and F. C. J. M. van Veggel, Chem. Mater., 2012, 24, 1297–1305 CrossRef CAS.
- X. Zhang, Z. Zhao, X. Zhang, D. B. Cordes, B. Weeks, B. Qiu, K. Madanan, D. Sardar and J. Chaudhuri, Nano Res., 2015, 8, 636–648 CrossRef CAS.
- G. Chen, T. Y. Ohulchanskyy, W. C. Law, H. Ågren and P. N. Prasad, Nanoscale, 2011, 3, 2003–2008 RSC.
- Z. Liu, F. Pu, S. Huang, Q. Yuan, J. Ren and X. Qu, Biomaterials, 2013, 34, 1712–1721 CrossRef CAS PubMed.
- J. Liu, L. Huang, X. Tian, X. Chen, Y. Shao, F. Xie, D. Chen and L. Li, Int. J. Nanomed., 2017, 12, 1–14 CrossRef PubMed.
- M. Wu, G. Guan, B. Yao, C.-P. Teng, S. Liu, S. Y. Tee, B. C. Ong, Z. Dong and M.-Y. Han, ACS Appl. Nano Mater., 2019, 2, 1421–1430 CrossRef CAS.
- J. Liu, X. Tian, N. Luo, C. Yang, J. Xiao, Y. Shao, X. Chen, G. Yang, D. Chen and L. Li, Langmuir, 2014, 30, 13005–13013 CrossRef CAS PubMed.
- J. Leng, J. Chen, D. Wang, J.-X. Wang, Y. Pu and J.-F. Chen, Ind. Eng. Chem. Res., 2017, 56, 7977–7983 CrossRef CAS.
- M. Back, E. Trave, G. Zaccariello, D. Cristofori, P. Canton, A. Benedetti and P. Riello, Nanoscale, 2019, 11, 675–687 RSC.
- M. Runowski, N. Stopikowska, D. Szeremeta, S. Goderski, M. Skwierczyńska and S. Lis, ACS Appl. Mater. Interfaces, 2019, 11, 13389–13396 CrossRef CAS PubMed.
- R. Tian, S. Zhao, G. Liu, H. Chen, L. Ma, H. You, C. Liu and Z. Wang, Biomaterials, 2019, 212, 64–72 CrossRef CAS PubMed.
- J. Xu, P. Yang, M. Sun, H. Bi, B. Liu, D. Yang, S. Gai, F. He and J. Lin, ACS Nano, 2017, 11, 4133–4144 CrossRef CAS PubMed.
- C.-C. Hsu, S.-L. Lin and C. A. Chang, ACS Appl. Mater. Interfaces, 2018, 10, 7859–7870 CrossRef CAS PubMed.
- P. K. Pandey, A. K. Sharma, S. Rani, G. Mishra, G. Kandasamy, A. K. Patra, M. Rana, A. K. Sharma, A. K. Yadav and U. Gupta, ACS Biomater. Sci. Eng., 2018, 4, 2860–2869 CrossRef CAS.
- Y. Zhang, K. Ren, X. Zhang, Z. Chao, Y. Yang, D. Ye, Z. Dai, Y. Liu and H. Ju, Biomaterials, 2018, 163, 55–66 CrossRef CAS PubMed.
- Y. Mou, M. Kang, F. Wang, M. Liu, K. Chen and R. Sun, J. Sol-Gel Sci. Technol., 2017, 83, 447–456 CrossRef CAS.
- J. Chen, X. Zhou, Y. Sheng, X. Xie, M. M. A. Abualrejal, M. Chang, Z. Shi and H. Zou, Ceram. Int., 2017, 43, 4440–4449 CrossRef CAS.
- J. Chen, Y. Sheng, Y. Song, M. Chang, X. Zhang, L. Cui, D. Meng, H. Zhu, Z. Shi and H. Zou, ACS Sustainable Chem. Eng., 2018, 6, 3533–3545 CrossRef CAS.
- C. Lin, Y. Song, F. Gao, H. Zhang, Y. Sheng, K. Zheng, Z. Shi, X. Xu and H. Zou, J. Sol-Gel Sci. Technol., 2014, 69, 536–543 CrossRef CAS.
- J. Weimmerskirch-Aubatin, M. Stoffel, A. Bouché, P. Boulet, M. Vergnat and H. Rinnert, J. Alloys Compd., 2015, 622, 358–361 CrossRef CAS.
- A.-M. Brezoiu, M. Deaconu, I. Nicu, E. Vasile, R.-A. Mitran, C. Matei and D. Berger, Microporous Mesoporous Mater., 2019, 275, 214–222 CrossRef CAS.
- C. Huo, J. Ouyang and H. Yang, Sci. Rep., 2014, 4, 3682 CrossRef PubMed.
- A. G. Macedo, R. A. S. Ferreira, D. Ananias, M. S. Reis, V. S. Amaral, L. D. Carlos and J. Rocha, Adv. Funct. Mater., 2010, 20, 624–634 CrossRef CAS.
- H. Deng, F. Chen, C. Yang, M. Chen, L. Li and D. Chen, Nanotechnology, 2018, 29, 415601 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra08116j |
| This journal is © The Royal Society of Chemistry 2019 |