
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign, synthesis and evaluation of novel, branched trident small interfering RNA nanostructures for sequence-specific RNAi activity†
Akash Chandelaa and
Yoshihito Ueno*ab
aThe United Graduate School of Agricultural Science, Gifu University, Gifu, Japan
bCourse of Applied Life Science, Faculty of Applied Biological Sciences, Gifu University, 1-1 Yanagido, Gifu 501-1193, Japan. E-mail: uenoy@gifu-u.ac.jp
First published on 23rd October 2019
Abstract
Small interfering RNAs (siRNAs) are potential candidates for gene regulation with efficient activity, but off-target effects and limited systemic delivery. Herein, we report the design and synthesis of the branched siRNA nanostructures with highly improved resistance against exonucleases. Also, these branched siRNAs showed suppression of off-target gene silencing through selection of the passenger strand as the branching unit. The physical characterization of branched siRNAs showed that they form a compact assembly with a hydrodynamic diameter of 6.9 nm against 2.8 nm of the duplex. We demonstrated that a branched siRNA synthesized with a trebling solid-support selectively exhibits RNAi activity and suppresses the off-target effect.
Introduction
Nucleic acid therapeutics have emerged as a potential agent for gene therapy with the advances in a clinical tool, namely, RNA interference (RNAi).1–4 Specifically, small interfering RNAs with 19 to 21 nucleotides (nt) in length regulate the expression of a gene through catalytic degradation of the target mRNA.5–7 But, several limitations such as nuclease degradation, off-target effect, and renal clearance impede the systemic delivery of siRNAs to the target site.8,9 Therefore, several investigations with the carrier assisted delivery of siRNAs to elude nucleases and kidney filtration have been performed.10–14 Our review on the systemic delivery of siRNAs has also enlightened several delivery carriers such as lipid nanoparticles, cell penetrating peptides, aptamers, lipid bioconjugates and dendrimers for the targeted delivery.15 However, a self-assembled delivery system with branched RNA structure has not yet been exploited to facilitate the efficient delivery and silencing activity. Large molecular size of such self-assemblies will also assist in evading the filtration through the glomerulus.16Developments in this direction have led to the exploration of RNA architectonics, the scientific study of the principles of RNA architecture with the aim of constructing RNA nanostructures of arbitrary size and shape.17,18 Accordingly, branched RNAs can be formulated through chemical modifications to evade the barriers in systemic administration and display prominent regulation of gene expression. But, the complexity and difficulty in the synthesis of these branching units, have restricted investigations with this class of pharmaceutics. Although, commercially available doubling or tripling synthons improve the complexity and yields of such structures but they are only available as phosphoramidites and, the downstream processing is still troublesome.
Previously, the construction of branched RNA architectures have been reported through hybridization of three or more strands coding for different genes or, incorporating the commercially available doubling and tripling phosphoramidite synthons for yielding the branched structures.19–24 And such branched RNA nanostructures have evidently showed reduced mRNA levels and marked gene silencing against the linear duplex counterparts. Also, branched oligonucleotides have been reported to exhibit high affinity for single-strand oligonucleotides to form alternated strand triplexes.25–27 However, the simultaneous synthesis of three identical arms of the RNA using a modified solid-support, to yield branched RNA structure forming a nano-assembly, remains unexplored.
Therefore, we envisioned that a branched RNA structure could be afforded with a modified solid-support that will yield trident RNAs (td RNAs) that will impart high serum stability and on-target RNAi activity with sequence selectivity. Also, the physical characterization of these structures with dynamic light scattering (DLS) could reveal their size and nature of assembly. To this end, we designed and synthesized, a novel branched RNA structure originating from a modified single solid-support to accommodate the branched structure with a molecular weight of approximately 22 kDa which results into gross molecular weight of 44 kDa of the td siRNA.9,28–31 These novel branched siRNAs were terminally modified with alkyl-halide nucleobases (previously reported) at 3′-end, and showed consistent thermal stability but branching RNA sequence-dependent gene silencing (Fig. 1).32 The hydrodynamic diameter of these nanostructures was found to be increased, showing that they forms a compact structure. Moreover, these branched RNAs were also found to be considerably resistant against the 3′-exonuclease, revealing a 10-fold increase in the half-life. Thereby, the moiety described here may provide a starting point for further endeavours with nucleic acid pharmaceutics.
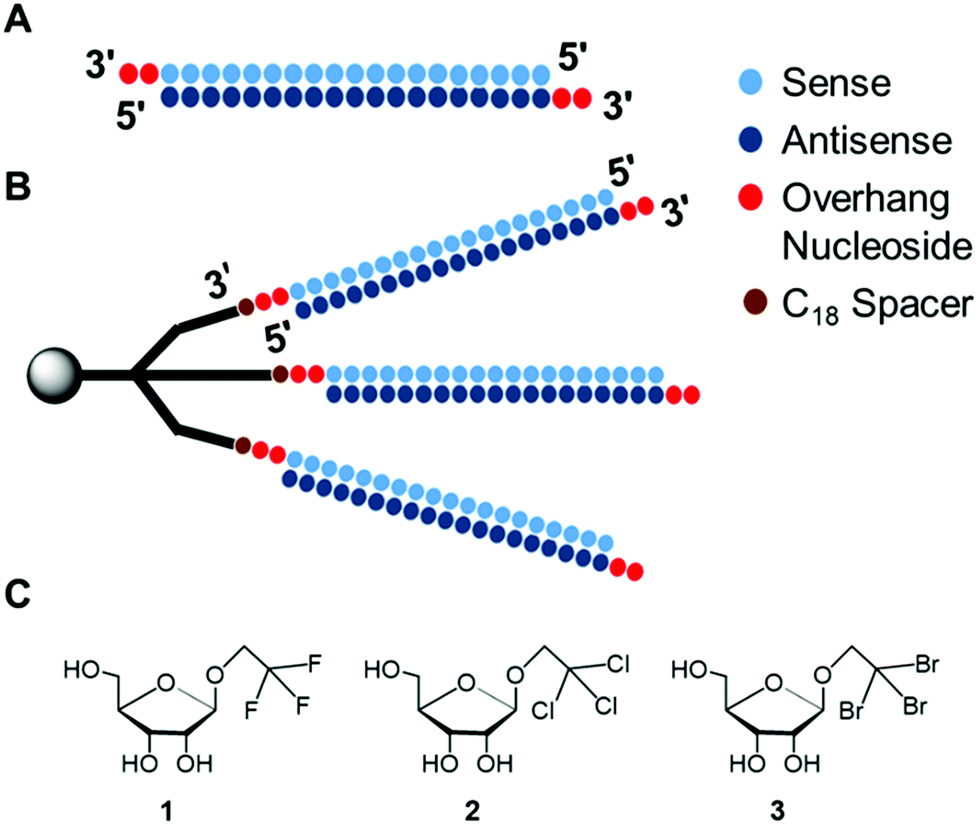 | ||
| Fig. 1 Graphical representation of siRNA structures and modified analogs, (A). Duplex siRNA (conventional) having 2-nucleotide overhang at 3′-ends, for RNAi. (B). Trident siRNA (this study) for RNAi. (C). Haloalkyl nucleobase analogs introduced at 3′-overhangs of non-branching RNA for analysing thermal stability and RNAi activity. | ||
Results and discussion
Synthesis of trebler solid-support
The modified solid-support, D9 for the synthesis of branching oligonucleotides was obtained through the divergent synthesis approach. The synthetic route has been shown in Scheme 1. Commercially available pentaerythritol (D1) was mono-protected with the silyl-ether, tert-butyldimethylsilyl chloride (TBDMSCl) to afford D2 in 65% yield. Thereupon, the elongation of the three arms was achieved through coupling reaction following Williamson ether synthesis. The primary hydroxyl groups were reacted with allyl bromide in the presence of NaH to give D3 in 81% yield. Subsequently, D4 was obtained in 83% yield through anti-Markovnikov hydroboration oxidation of D3 with 9-BBN. D4 was again reacted with allyl bromide in the presence of NaH to give D5 in 64% yield. Next, D6 was obtained in 93% yield through hydroboration oxidation of D5 with 9-BBN. Subsequently, the hydroxyl groups of D6 were protected by DMTr group to give the corresponding DMTr derivative D7 in 90% yield. The deprotection of the silyl group of D7 was performed with TBAF to afford D8 in 91% yield. Lastly, the solid-support for oligonucleotide synthesis was attained by converting compound D8 to the corresponding succinate, which was then reacted with LCAA-CPG to produce the solid supports D9 with 27.4 μmol g−1 of loading activity.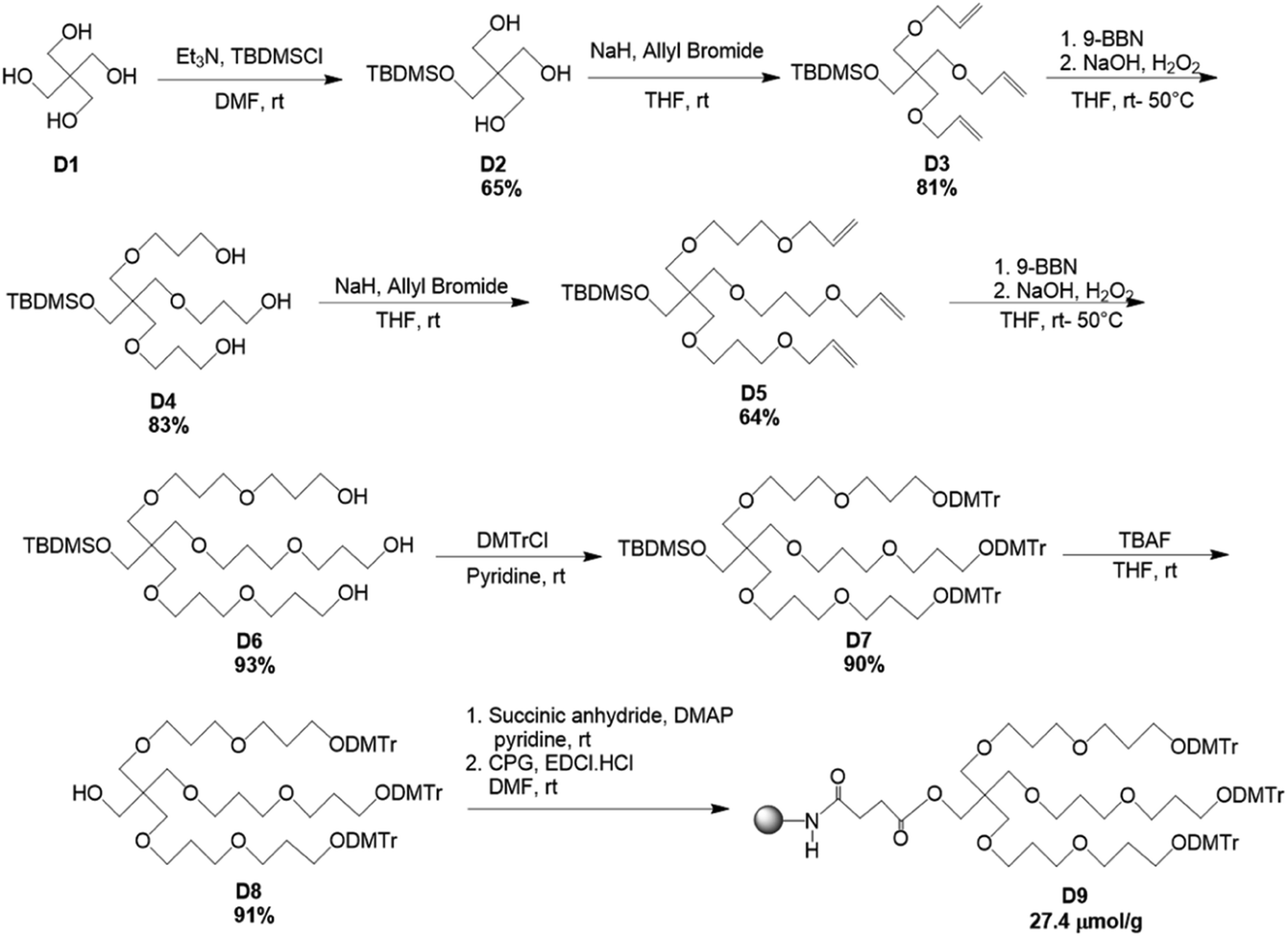 | ||
| Scheme 1 Synthetic route for the production of trebling solid-support, D9. | ||
Synthesis of trident RNA
The modified trebling solid-support D9, natural RNA phosphoramidites and hexaethylene glycol (C18) spacer phosphoramidite, were used to synthesize the td siRNAs by a DNA/RNA synthesizer. The RNAs were synthesized with a modified cycle having increased coupling time of the phosphoramidite on a DMT-ON mode for obtaining the target full length sequences. The C18 spacer have been used for obtaining efficient oligonucleotide synthesis as initially with small arms of trebling support, the synthesis yield was very low. However, the target sequence was also obtained without the use of spacer but in low yield. The mass analysis of these trident RNAs revealed unconsolidated peaks. And, further screening with reverse-phase high-performance liquid chromatography, RP-HPLC showed the presence of bident and trident mixture with merged peaks. Similar observations about the co-existence of the double and triple branched RNAs were also made with ultra-performance liquid chromatography-mass spectrometry, UPLC-MS (Fig. S1†). Thus, purification via polyacrylamide gel electrophoresis (PAGE) was executed to produce two distinctive bands (Fig. 2), which were analysed again by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF/MS), to confirm the branched RNAs (Table S1†). The two bands corresponded to the td RNA and bd RNA, respectively.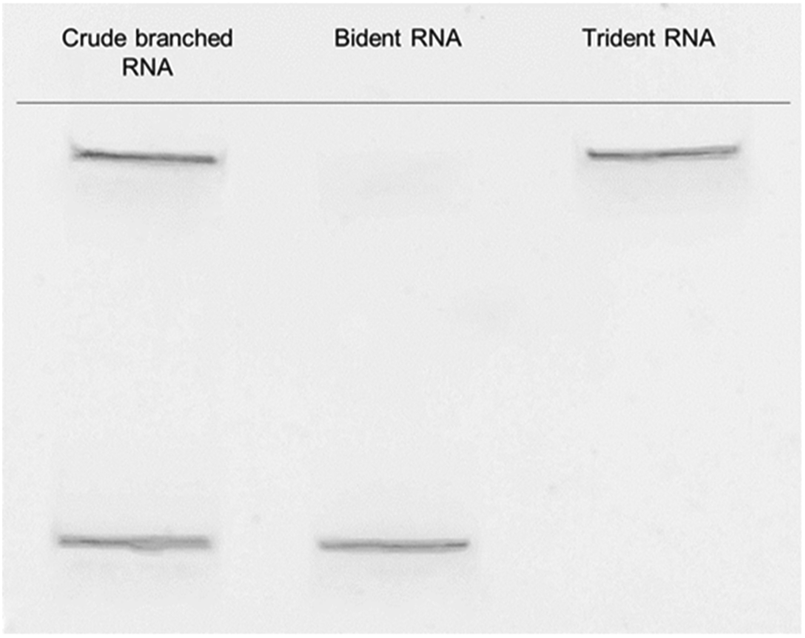 | ||
| Fig. 2 Purification of branched RNA with PAGE to yield bident and trident RNAs. | ||
Thermodynamic stability of td siRNAs
Next, we examined the thermodynamic stability of the branched siRNAs and compared with the duplex siRNAs. Herein, the passenger strand formed the branching RNA whereas the guide strand was the single strand RNA (ssRNA) with alkyl-halide nucleobase modified 3′-overhang. These modified branched siRNAs were subjected to evaluation by UV melting experiments in a buffer composed of 10 mM sodium phosphate (pH 7.0) and 100 mM NaCl (Fig. 3). The Tm value of the unmodified siRNA 1 was found to be 77.6 °C, whereas those of the modified siRNAs 2–4 were 77.0, 77.1, and 77.2 °C, respectively. And, with the modified td siRNAs 2–4, the Tm values were found to be 77.0, 77.5, 77.5 °C, respectively (Table S3†). Thereby, revealing that these branched structures have similar thermodynamic stability like the duplex siRNAs and the structural difference didn't alter it any significantly. Hence, they can be utilized for the same function of RNAi activity due to these conserved thermal stability.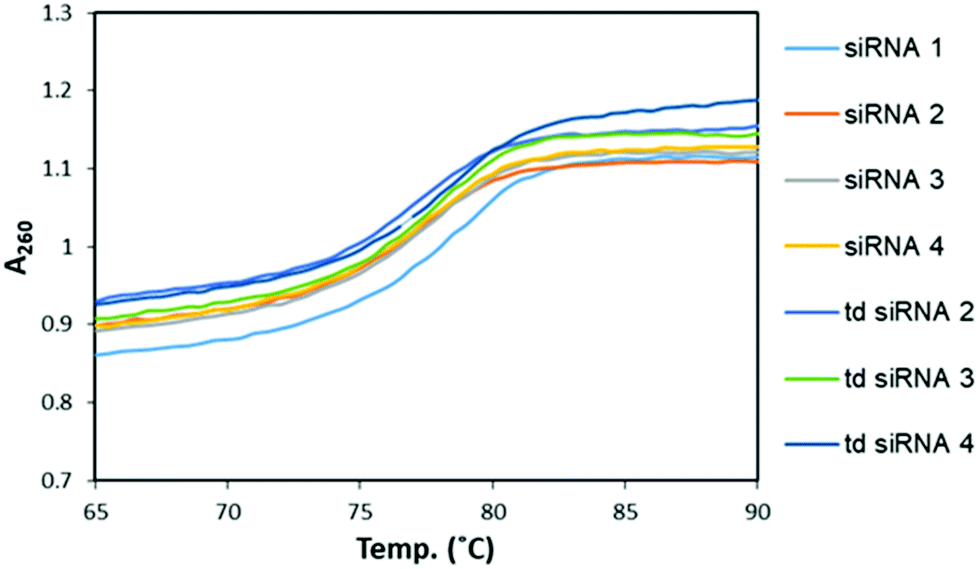 | ||
| Fig. 3 UV melting profiles for the comparative study of melting temperatures of modified, siRNAs and td siRNAs. | ||
Size measurement with dynamic light scattering (DLS)
Physical characterization of these branched siRNAs using Dynamic Light Scattering (DLS) showed the resulting hydrodynamic diameters of 6.9 nm of td siRNA against the 2.8 nm of the duplex siRNA, which revealed the compactness of these nanostructures (Fig. 4). Therefore, such compact nanostructures with a very high molecular weight, could easily escape the barrier of kidney filtration unit during systemic administration and also the circulation time could be increased as the threshold for glomerular filtration have been reported around 6 nm.33–35 The sequences have been shown in Table S2.†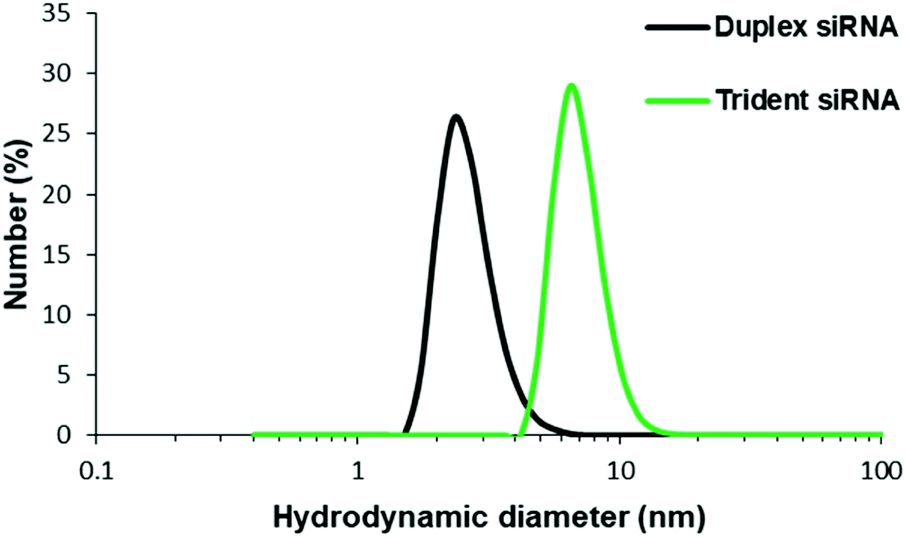 | ||
| Fig. 4 Hydrodynamic diameter determination using DLS method for siRNA and td siRNA. | ||
Gene silencing determined by dual luciferase assay
The RNAi activities of the unmodified and modified td siRNAs were investigated with a dual luciferase reporter assay using HeLa cells, in which the target luciferase genes were constitutively expressed. All modified td siRNAs targeted the Renilla luciferase genes, while firefly luciferase genes were used as controls. All td siRNAs were transfected using RNAimax. The expression levels of both luciferase genes were analyzed after 24 h of treatment with the modified td siRNAs. The silencing activity was expressed as the ratios of Renilla:firefly luciferase activities with respect to the no siRNA control.We hypothesized that these structures exhibit sequence selectivity and the td siRNAs with sense strand as the branching unit will display profound silencing activity whereas those with guide strands will have reduced activity due to the caging of the 3′-end. To this end, we observed that all of the alkyl-halide base modified td siRNAs 2–4 showed comparable activity by inhibiting 78% gene expression in each case whereas 81% inhibition was observed with the unmodified branched td siRNA 1, at 10 nM concentration. The duplex siRNA 1 also showed 78% of inhibition (Fig. 5, Table S4†). Hence, the RNAi activity was found to be conserved with these structurally different moieties.
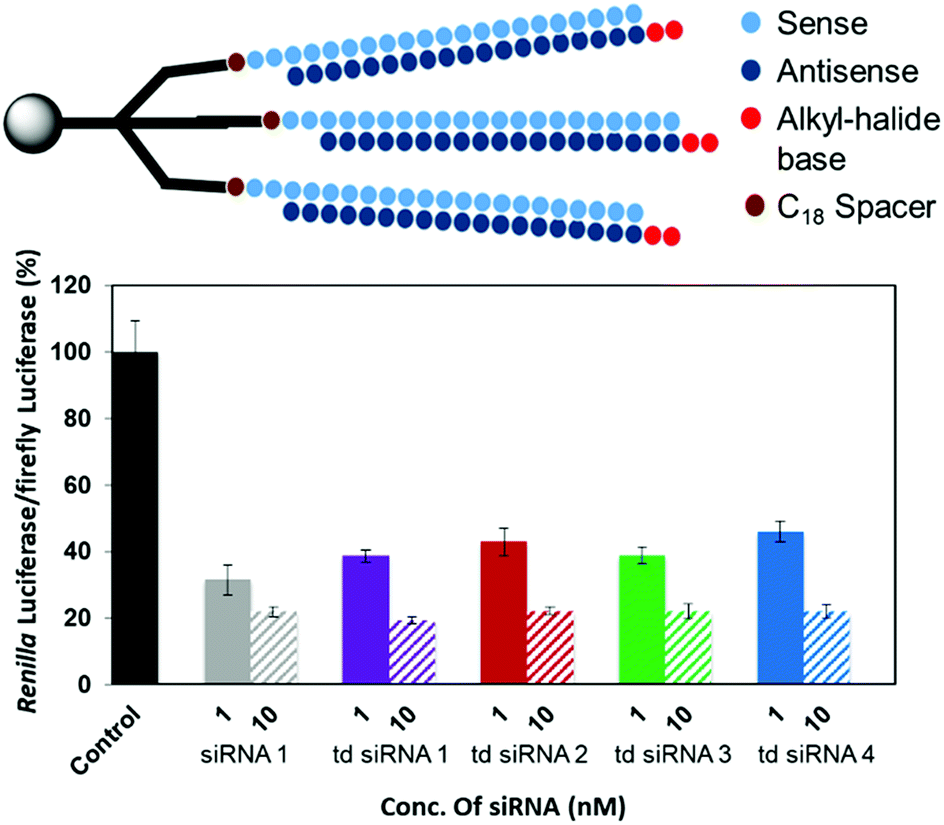 | ||
| Fig. 5 RNAi activity of the modified td siRNAs with sense strand as branching unit. | ||
However, an anomalous behaviour as hypothesized for the td siRNAs constituting antisense strand as the branching unit was observed. The gene silencing was considerably reduced for each of the td siRNAs 5–8. The inhibition for td siRNAs 5–8 at 10 nM were found to be 58%, 66%, 69% and 72%, respectively (Fig. 6, Table S5†).
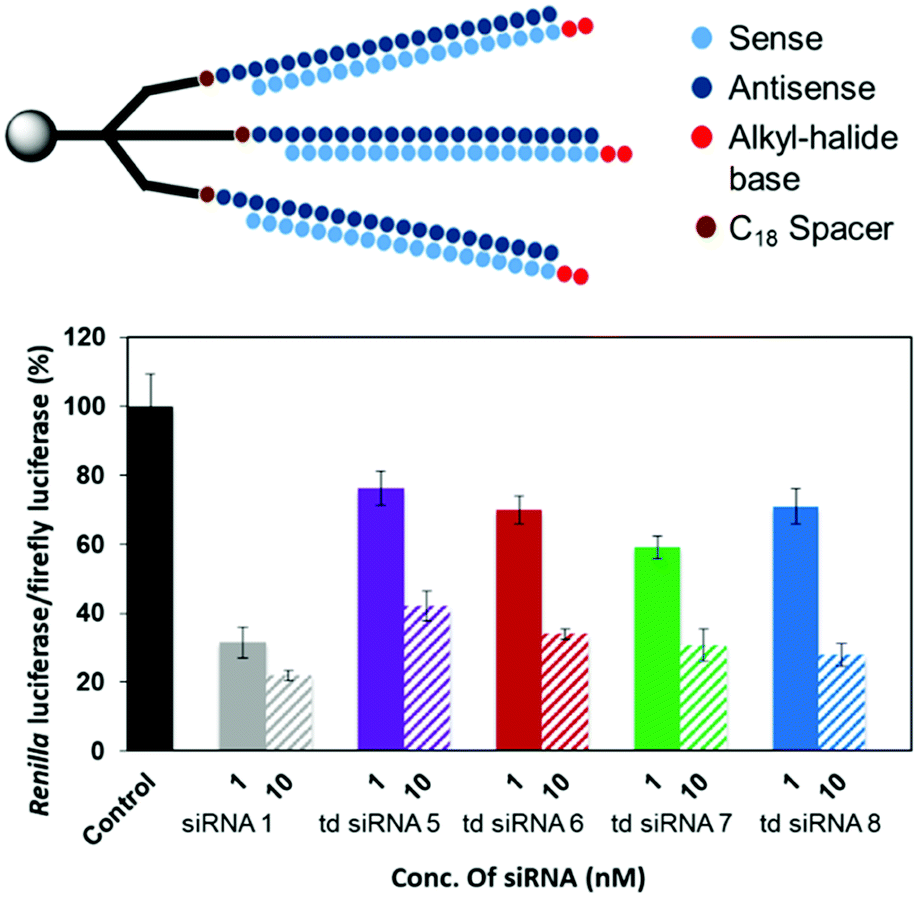 | ||
| Fig. 6 RNAi activity of the modified td siRNA with antisense strand as the branching unit. | ||
Hence, it can be inferred that the branching structures are tolerant for siRNA mediated RNAi, if the branching RNA backbone constitutes passenger strand, which lies in agreement with our hypothesis of reduced recognition of the termini of guide strand by the Ago protein due to caging effect. Hence, these structures can be successfully utilized for the suppression of off-target effects and regulate gene silencing. In addition, we also examined the cellular uptake of these structures in the presence of transfecting agent and found a similar response for cellular internalization alike the duplex siRNA (Fig. S2†).
Stability against exonucleases
Instability of unmodified siRNAs in serum is another critical limitation in the systemic administration of nucleic acid therapeutics. Since, we earlier stated the presence of caged-structure at the 3′-end of these RNAs, we further hypothesized that the existence of such an assembly will improve stability in the serum. Thus, we analysed the branched RNAs for resistance against snake venom phosphodiesterase (SVPD), a 3′-exonuclease. The unmodified RNA and the branched td RNA were labelled with fluorescein at their 5′-ends, incubated with SVPD, and then examined by denaturing PAGE. As shown in Fig. 7, the unmodified RNA underwent instantaneous degradation with fairly no full length RNA after 5 min of incubation; whereas, the branched RNA exhibited enhanced stability to the exonuclease enzyme and showed sustained full length structures even after 30 min of treatment with the enzyme. The half-lives (t1/2) of unmodified RNA was 1.2 min, while that of the branched td RNA displayed over 10 folds increase and valued at 14 min. Therefore, we can be firmly conclude upon these observations that the branched RNAs provide better stability against the exonucleases in the serum.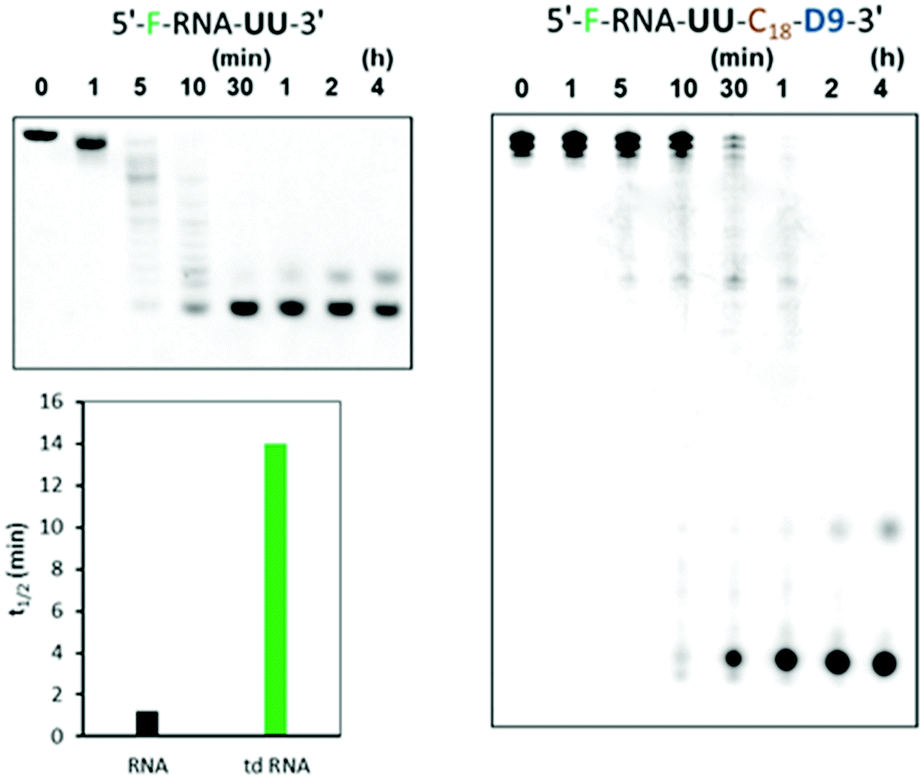 | ||
| Fig. 7 Serum stability of unmodified ssRNA and unmodified td RNA against SVPD with 20% and 6% denaturing PAGE respectively. F denotes fluorescein, C18 denotes spacer and D9 denotes the trebling solid-support. The sequence of the RNA part is 5′-GGCCUUUCACUACUCCUAC-3’. | ||
Experimental
Synthesis of the solid support
N,N-Dimethyl-4-aminopyridine (DMAP) (0.23 g, 1.84 mmol), and succinic anhydride (0.37 g, 3.68 mmol) were added to a solution of D8 (1.28 g, 0.92 mmol) in pyridine (10 mL) under argon atmosphere and the mixture was stirred for 20 h at room temperature. The mixture was extracted with H2O and EtOAc. The organic layer was washed with sat. NaHCO3 solution, brine, dried over Na2SO4 and concentrated in vacuo. The residue was purified by column chromatography on silica gel (hexane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethyl acetate, 2:1) to give succinate mixture as a colorless, viscous liquid in 82% yield. Aminopropyl controlled pore glass (0.49 g, 60 μmol) and 1-ethyl-3-[3-(dimethylamino)propyl] carbodiimide hydrochloride (46 mg, 0.24 mmol) were added to a solution of succinate (0.36 g, 0.24 mmol) in DMF and the mixture was kept at room temperature for 4 days. This resin was washed with pyridine, 15 mL of capping solution (0.1 M DMAP in pyridine:Ac2O, 9:1) were added to the resin and the mixture was kept at room temperature for 1 day. The resin was washed with pyridine, ethanol, acetonitrile and dried under vacuum to give solid support D9. The amount of nucleoside loaded to the solid support was calculated by release of dimethoxytrityl cation using solution of 70% HClO4:EtOH (3:2, v/v).
:ethyl acetate, 2:1) to give succinate mixture as a colorless, viscous liquid in 82% yield. Aminopropyl controlled pore glass (0.49 g, 60 μmol) and 1-ethyl-3-[3-(dimethylamino)propyl] carbodiimide hydrochloride (46 mg, 0.24 mmol) were added to a solution of succinate (0.36 g, 0.24 mmol) in DMF and the mixture was kept at room temperature for 4 days. This resin was washed with pyridine, 15 mL of capping solution (0.1 M DMAP in pyridine:Ac2O, 9:1) were added to the resin and the mixture was kept at room temperature for 1 day. The resin was washed with pyridine, ethanol, acetonitrile and dried under vacuum to give solid support D9. The amount of nucleoside loaded to the solid support was calculated by release of dimethoxytrityl cation using solution of 70% HClO4:EtOH (3:2, v/v).
Oligonucleotide synthesis
Synthesis was carried out with a DNA/RNA synthesizer by phosphoramidite method according to the normal protocol. Post synthesis, the oligomers were cleaved from CPG beads and deprotected with concentrated NH3 solution/40% methylamine (1:1, v/v) for 10 min at 65 °C. 2′-O-TBDMS groups were removed by Et3N·3HF in a solution of trimethylamine and DMSO at 65 °C for 2.5 h. The reaction was quenched with quenching buffer (Tris–HCl) and loaded onto Glen-pak cartridge. The sample was washed with 2% TFA and eluted with 1 M ammonium bicarbonate in 30% MeCN. The oligonucleotides were later purified by 6% denaturing PAGE to give highly purified trident RNAs.
MALDI-TOF/MS analysis of RNAs
The spectra were obtained with a time-of-flight mass spectrometer equipped with nitrogen laser (337 nm, 3 ns pulse). A solution of 3-hydroxypicolinic acid (3-HPA) and diammonium hydrogen citrate in 0.1 M MeCN/H2O (1:1, v/v) was used as matrix. An improvised, sandwich method was used to measure very high masses. Initially, matrix was applied and upon drying, the mixture of matrix with sample was applied. When the crystallization appeared, one more coating with matrix and was performed and let dry.
Thermal denaturation study
The solution containing 3.0 μM duplex in a buffer of 10 mM sodium phosphate (pH 7.0) containing 100 mM NaCl was heated at 100 °C, then gradually cooled to room temperature and used for this study. Thermally induced transitions of each mixture were monitored at 260 nm with a UV/vis spectrometer fitted with temperature controller in quartz cuvettes with a path length of 1.0 cm. The sample temperature was increased by 0.5 °C min−1.Dynamic light scattering
Hydrodynamic diameters of siRNA were determined by dynamic light scattering (DLS) using a Zetasizer nano ZS (Malvern Instruments,UK). siRNAs solutions (9 nmol in 1 mL PBS) were analyzed at 25 °C in triplicate. Subsequently, the trident siRNAs solution were also formed by annealing of equimolar amounts. All the scattered photons were collected at a 173°-scattering angle. The scattering intensity data was processed using instrumental software to obtain the hydrodynamic diameter and the size distribution of each sample.Dual-luciferase reporter assay
HeLa cells were transfected with the psiCHECK-2 (Promega) reporter and the pcDNA3.1 containing a hygromycin resistance gene (Thermo Fisher Scientific). Cells were cultured in the presence of 0.5 mg mL−1 hygromycin for one week. Stable HeLa-psiCHECK-2 cells expressing both Renilla and firefly luciferases were grown in Dulbecco's Modified Eagle Medium (D-MEM) supplemented with 10% bovine serum (BS) and 0.25 mg mL−1 hygromycin at 37 °C. HeLa-psiCHECK-2 cells (8.0 × 104/mL) were cultured on a 96-well microplate (100 μL per well) for 24 h and transfected with siRNA targeting the Renilla luciferase gene using lipofectamine RNAi max in Opti-MEM I reduced serum medium. Transfection without siRNA was used as a control. After 1 h, each well was seeded with 50 μL of D-MEM containing 10% BS and cells were further incubated for another 24 h. The activities of Renilla and firefly luciferases in the cells were determined with Dual-Luciferase Reporter Assay System (Promega) according to a manufacture's protocol. The activity of Renilla luciferase was normalized by the firefly luciferase activity. The results were confirmed by at least three independent transfections and expressed as the average from four experiments as mean ± SD.Partial digestion of RNAs by 3′-exonuclease SVPD
Each ON (600 pmol) labeled with fluorescein at the 5′-end was incubated with SVPD (0.075 unit) in a buffer (150 μL) comprising 0.1 M Tris–HCl (pH 8.0) and 20 mM MgCl2 at 37 °C. After 0, 1, 5, 10, 30, 60, 120, or 240 min, an aliquot (5 μL) of the reaction mixture was mixed with the loading buffer (15 μL), comprising Tris–borate–EDTA (TBE) buffer and 20% glycerin, on ice. Each sample was analyzed by 20% denaturing PAGE for ssRNA and 6% denaturing PAGE for td RNA at room temperature for 2 h at 20 mA. The gel was visualized by use of a Luminescent Image analyser, LAS-4000 (Fujifilm).Conclusion
In summary, we have architected a novel branched trident siRNA nanostructure originating from a trebling solid-support, with highly enhanced serum stability and suppression of off-target silencing. Our findings suggest that these branching td siRNAs form a stable, compact nanostructure, which is thermodynamically stable alike the duplex siRNA. The gene silencing have also been showed to be conserved with these branched structures and comparable to the unmodified siRNAs.The RNAi activity have been sequence-specific dependent on the sequence of the branching unit. The promising results of these td siRNAs with in vitro experiments provide us the basis for their incorporation in the in vivo studies.
Furthermore, since branched siRNAs have been sparingly investigated, we suggest that the formulation of such branched siRNAs and their compatibility with siRNA mediated RNAi signify them as potential building blocks for further developments with branched siRNAs. Therefore, we anticipate that these branched td siRNAs could further be examined for in vivo experiments and be useful in the systemic administration to evade the barrier of renal clearance and efficiently deliver the drug to the target site as based on the previous reports, they overcome the threshold of hydrodynamic diameter for glomerular filtration.16
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Japan Agency for Medical Research and Development (AMED) through its Funding Program for Basic Science and Platform Technology Program for Innovative Biological Medicine, development of siRNA conjugates with tissue-specific delivery functions. The authors thank Professor Yoko Hirata (Gifu University) for supplying cells and giving technical advice.References
- A. Fire, S. Xu, M. K. Montgomery, S. A. Kostas, S. E. Driver and C. C. Mello, Nature, 1998, 391, 806–811 CrossRef CAS
.
- A. Fire, Trends Genet., 1999, 15, 358–363 CrossRef CAS
- P. A. Sharp, Genes Dev., 2001, 15, 485–490 CrossRef CAS
- C. C. Mello and D. Conte, Nature, 2004, 431, 338–342 CrossRef CAS
- T. Tuschl, ChemBioChem, 2001, 2, 239–245 CrossRef CAS
- S. M. Elbashir, J. Harborth, W. Lendeckel, A. Yalcin, K. Weber and T. Tuschl, Nature, 2001, 411, 494–498 CrossRef CAS PubMed
- S. M. Elbashir, W. Lendeckel and T. Tuschl, Genes Dev., 2001, 15, 188–200 CrossRef CAS PubMed
- Z. Paroo and D. R. Corey, Trends Biotechnol., 2004, 22, 390–394 CrossRef CAS
- C. V. Pecot, G. A. Calin, R. L. Coleman, G. Lopez-Berestein and A. K. Sood, Nat. Rev. Cancer, 2011, 11, 59–67 CrossRef CAS
- Y. Ueno, T. Inoue, M. Yoshida, K. Yoshikawa, A. Shibata, Y. Kitamura and Y. Kitade, Bioorg. Med. Chem. Lett., 2008, 18, 5194–5196 CrossRef CAS PubMed
- I. V. Chernikov, D. V. Gladkikh, M. I. Meschaninova, A. G. Ven'yaminova, M. A. Zenkova, V. V. Vlassov and E. L. Chernolovskaya, Mol. Ther.–Nucleic Acids, 2017, 6, 209–220 CrossRef CAS
- S. R. Suter, J. Sheu-Gruttadauria, N. T. Schirle, R. Valenzuela, A. A. Ball-Jones, K. Onizuka, I. J. Macrae and P. A. Beal, J. Am. Chem. Soc., 2016, 138, 8667–8669 CrossRef CAS
- M. M. Janas, M. K. Schlegel, C. E. Harbison, V. O. Yilmaz, Y. Jiang, R. Parmar, I. Zlatev, A. Castoreno, H. Xu, S. Shulga-Morskaya, K. G. Rajeev, M. Manoharan, N. D. Keirstead, M. A. Maier and V. Jadhav, Nat. Commun., 2018, 9, 723 CrossRef
- B. Ozpolat, A. K. Sood and G. Lopez-Berestein, J. Intern. Med., 2010, 267, 44–53 CrossRef CAS
- A. Chandela and Y. Ueno, Reviews in Agricultural Science, 2019, 7, 10–28 CrossRef
- M. Longmire, P. L. Choyke and H. Kobayashi, Nanomedicine, 2008, 3, 703–717 CrossRef CAS
- L. Jaeger and A. Chworos, Curr. Opin. Struct. Biol., 2006, 16, 531–543 CrossRef CAS
- Y. Nakashima, H. Abe, N. Abe, K. Aikawa and Y. Ito, Chem. Commun., 2011, 47, 8367–8369 RSC
- C. Il Chang, T. Y. Lee, S. Kim, X. Sun, S. W. Hong, J. W. Yoo, P. Dua, H. S. Kang, S. Kim, C. J. Li and D. K. Lee, J. Gene Med., 2012, 14, 138–146 CrossRef
- S. Sajeesh, T. Y. Lee, J. K. Kim, D. S. Son, S. W. Hong, S. Kim, W. S. Yun, S. Kim, C. Chang, C. Li and D. K. Lee, J. Controlled Release, 2014, 196, 28–36 CrossRef CAS PubMed
- S. Sajeesh, T. Y. Lee, J. Y. Choe and D. Lee, J. Controlled Release, 2015, 213, e95 CrossRef CAS
- E. Utagawa, A. Ohkubo, M. Sekine and K. Seio, J. Org. Chem., 2007, 72, 8259–8266 CrossRef CAS
- B. G. Nair, Y. Zhou, K. Hagiwara, M. Ueki, T. Isoshima, H. Abe and Y. Ito, J. Mater. Chem. B, 2017, 5, 4044–4051 RSC
- A. Aviñó, S. M. Ocampo, J. C. Perales and R. Eritja, J. Nucleic Acids, 2011, 2011, 1–7 CrossRef
- Y. Ueno, M. Takeba, M. Mikawa and A. Matsuda, J. Org. Chem., 1999, 64, 1211–1217 CrossRef CAS
- M. D. Sorensen, M. Meldgaard, V. K. Rajwanshi and J. Wengel, Bioorg. Med. Chem. Lett., 2000, 10, 1853–1856 CrossRef CAS
- A. Aviño, M. G. Grimau, M. Frieden and R. Eritja, Helv. Chim. Acta, 2004, 87, 303–316 CrossRef
- K. A. Whitehead, R. Langer and D. G. Anderson, Nat. Rev. Drug Discovery, 2009, 8, 129–138 CrossRef CAS
- A. Wittrup and J. Lieberman, Nat. Rev. Genet., 2015, 16, 543–552 CrossRef CAS
- H. Borna, S. Imani, M. Iman and S. Azimzadeh Jamalkandi, Expert Opin. Biol. Ther., 2015, 15, 269–285 CrossRef CAS
- M. Gujrati and Z. R. Lu, in Gene Therapy of Cancer: Translational Approaches from Preclinical Studies to Clinical Implementation, 3rd edn, 2013, pp. 47–65 Search PubMed
- A. Chandela, T. Watanabe, K. Yamagishi and Y. Ueno, Bioorg. Med. Chem., 2019, 27(7), 1341–1349 CrossRef CAS
- A. P. Chapman, P. Antoniw, M. Spitali, S. West, S. Stephens and D. J. King, Nat. Biotechnol., 1999, 17, 780–783 CrossRef CAS
- H. Kobayashi, T. M. Yoo, I. S. Kim, M. K. Kim, N. Le, K. O. Webber, I. Pastan, C. H. Paik, W. C. Eckelman and J. A. Carrasquiillo, Cancer Res., 1996, 56, 3788–3795 CAS
- S. S. Olmsted, J. L. Padgett, A. I. Yudin, K. J. Whaley, T. R. Moench and R. A. Cone, Biophys. J., 2001, 81, 1930–1937 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra08071f |
| This journal is © The Royal Society of Chemistry 2019 |