
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConcise synthesis of 3-alkylthieno[3,2-b]thiophenes; building blocks for organic electronic and optoelectronic materials†
Koray T. Ilhana,
Sebahat Topala,
Mehmet S. Eroglubc and
Turan Ozturk *ac
*ac
aChemistry Department, Istanbul Technical University, Istanbul, Turkey. E-mail: ozturktur@itu.edu.tr
bChemical Engineering, Marmara University, Istanbul, Turkey
cChemistry Group Laboratories, TUBITAK UME, Gebze, Kocaeli, Turkey
First published on 25th November 2019
Abstract
Four step synthesis of 3-alkylthieno[3,2-b]thiophenes in the literature was reduced to two steps in good yields, through the preparation of the mono ketone, i.e. 1-(thiophene-3-ylthio)alkan-2-one, from 3-bromothiophene and ring formation reaction. This convenient method provides an easy access with good yields to the preparation of 3-alkylthieno[3,2-b]thiophenes, which are important materials for organic electronic and optoelectronic applications. SEM, AFM and contact angle (CA) analyses of their electropolymers on indium tin oxide (ITO) indicated that as the alkyl chains became longer, the polymers provide a more hydrophobic layer with CA up to 107°.
1. Introduction
Conjugated organic materials are the focus of intense research due to their use as organic semiconductors, which find applications as transistors, solar cells, organic light emitting diodes (OLED), etc.1 Compared with their silicon counterparts, as they can be deposited faster from solution during device fabrication, including over large areas, they have significant advantages in terms of time and cost.2 Stability of the organic material to oxidation is a necessary criterion as oxidation can diminish device performance.3Various important organic materials in this area are made of thiophene based compounds,4 which represent thermally and environmentally stable organic materials for electronic and optoelectronic applications.3,4c Fused thiophenes play important role in designing building blocks for polymers and small molecules.5 Thieno[3,2-b]thiophene (TT) is among the widely used fused thiophenes, possessing two fused thiophene units. Thienothiophenes, in general, have four isomers formed through the orientations of the sulfur atoms of the thiophene rings, among which thieno[3,2-b]thiophene belongs to the most widely used TTs as it provides continuous conjugation through two fused thiophenes and polymer backbone. Moreover, presence of two sulfur atoms makes them electron-rich, enabling to be used as electron donating moieties in construction of semiconductors.5
In spite of great achievements, preparation of organic materials, particularly polymers and the molecules having fused aromatic systems with desired electronic/optoelectronic properties and soluble in common organic solvents, is still a challenge. It appears that (i) involvement of flat and fused electron rich moieties into the designed molecules to tune their electronic/optoelectronic properties and (ii) providing solubility to the designed molecules are among the important topics. As TTs have flat structures with extended π-conjugation, they are among highly desirable compounds for tuning band gaps of organic materials and increasing their intermolecular interactions in solid state.5a,e Thus, involvement of TT in designing organic materials helps tuning organic electronic/optoelectronic materials. Regarding the solubility of the materials, it is mainly provided by pendant alkyl groups on the backbone. Besides, the alkyl groups through their positions on the compound can also provide control on regularity and packing, for which 3-hexylthiophene is widely applied.6 Although TTs having alkyl chains are highly desired molecules, providing both (i) flat and fused thiophenes and (ii) alkyl chains on the same molecule, their applications are not as wide as 3-hexylthiophene. One of the important reasons is their cumbersome four step synthesis.7–11 It involves Friedel–Crafts acylation of 3-bromothiophene 1, which can give a mixture of isomers 2 and 3 as minor and major products, respectively (Scheme 1).7g Treatment of the major product 3 with ethyl thioglycolate in the presence of NaOH produces 3-alkylthieno[3,2-b]thiophene-2-carboxylate 4, hydrolysis of which with NaOH yields the corresponding acid 5. In the last step, the acid is decarboxylated to give the desired 3-alkylthieno[3,2-b]thiophene 6 (3-alkyl-TT).
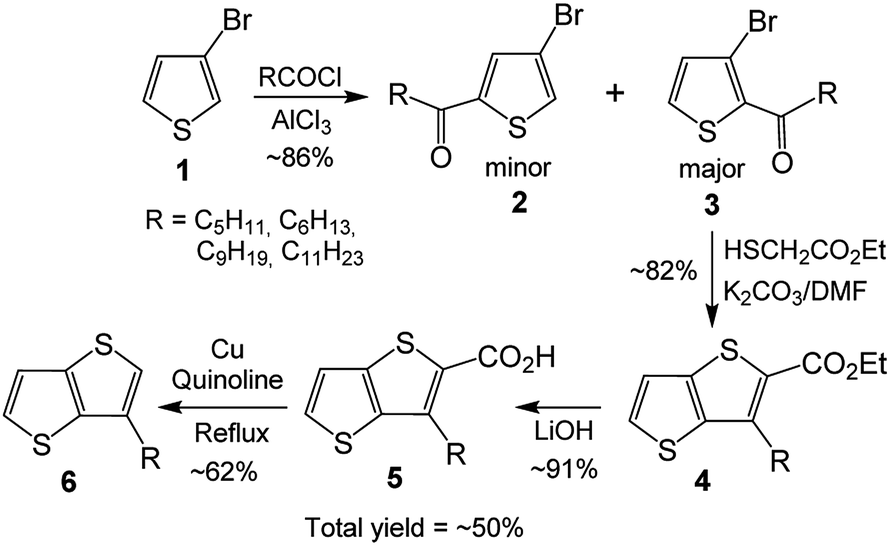 | ||
| Scheme 1 The literature synthesis of 3-alkyl-TTs. | ||
As its methodology could be applied to the synthesis of 3-alkyl-TT, synthesis of 3,6-dialkylthieno[3,2-b]thiophene (3,6-dialkyl-TT) was depicted in Scheme 2, which also involved four step synthesis.12 Thienothiophene 7 was tetrabrominated with Br2 to obtain tetrabromothieno[3,2-b]thiophene 8, which was converted to 3,6-dibromo-TT 9 by treating it with Zn in acetic acid. Coupling reaction of 9 with 1-alkyne produced 3,6-dialkyne-TT 10, hydrogenation reaction of which finally gave 3,6-dialkylthieno[3,2-b]thiophene 11.
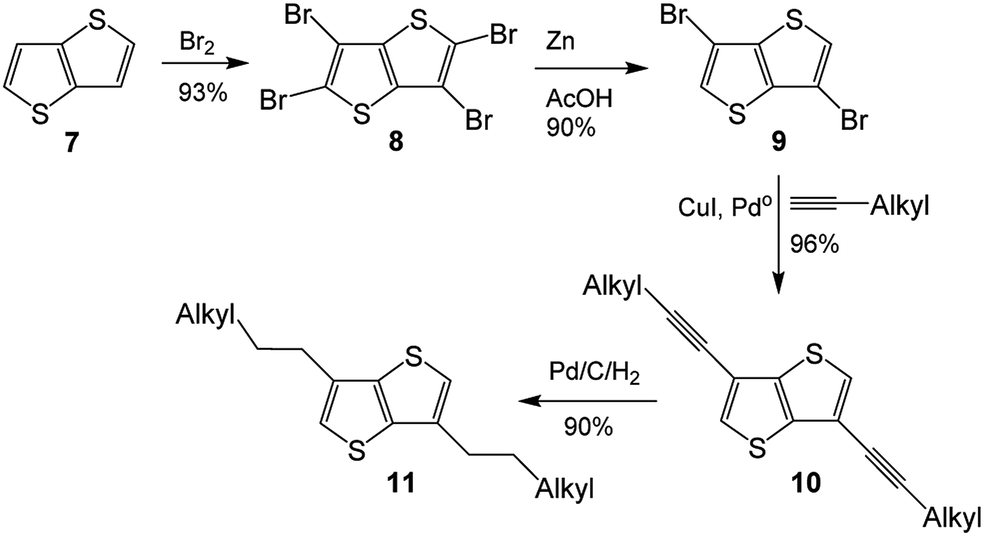 | ||
| Scheme 2 The literature synthesis of dialkyl-TT.8 | ||
In the light of our experience on the syntheses of substituted thienothiophenes (TTs) and dithienothiophenes (DTTs),5a we decided to employ our ring formation reaction13 to the syntheses of 3-alkylthieno[3,2-b]thiophenes, as thieno[3,2-b]thiophene is the most popular TT among its six isomers due to its better conjugation.
2. Result and discussion
2.1. Synthesis
The synthesis started with the preparation of the monoketones 14 through one-pot three-step reaction (Scheme 3), i.e. (i) lithiation of 3-bromothiophene 12 with BuLi, (ii) addition of elemental sulfur and, then, (iii) addition of α-haloketone 13. The yields of the ketones varied between 65–92%. The next step involved ring closure to obtain the alkyl-TTs 15. Refluxing the mono-ketones 14 in chlorobenzene in the presence of polyphosphoric acid (PPA) produced the desired alkyl-TTs between 75–90% yields. The overall yield for the two-step reaction varied between 49–80% (Table 3). Compare with the literature total yields, which are around 17–52%, these are very good yields (Table 1). Although the syntheses of TTs with 3-alkyl chains between 5 (15e)–9 (15j) are available in the literature, the TT with a longer alkyl chain of 12 is not available. Besides, the TTs with shorter alkyl chains (15a–15f) either not available or available with lower yields.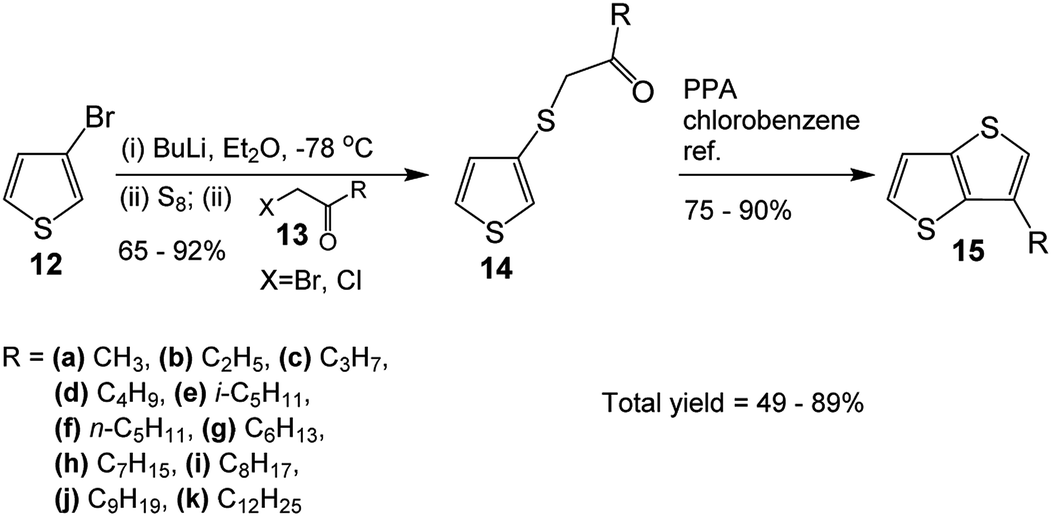 | ||
| Scheme 3 Synthesis of 3-alkyl-TTs. | ||
| R | Ketone (%) 14 | TT (%) 15 | Total yield (%) | Literature yield (%) | References |
|---|---|---|---|---|---|
| Methyl (a) | 65 | 75 | 49 | 17 | 14 |
| Yield is not given | 7g and 8 | ||||
| Ethyl (b) | 67 | 78 | 52 | — | — |
| Propyl (c) | 72 | 85 | 61 | Yield is not given | 9 |
| Butyl (d) | 70 | 82 | 60 | — | — |
| iso-pentyl (e) | 75 | 90 | 68 | — | — |
| Pentyl (f) | 73 | 85 | 62 | ∼35 | 7e |
| Hekzyl (g) | 77 | 83 | 64 | 50 | 10 |
| Heptyl (h) | 80 | 77 | 62 | ∼50 | 11 and 7g |
| Octyl (i) | 84 | 79 | 66 | 50 | 12 |
| Nonyl (j) | 92 | 87 | 80 | ∼52 | 7g |
| Undecyl (k) | 80 | 90 | 72 | — | — |
Interestingly, the literature synthesis of 15a (Scheme 4)14 has some close similarities with the methodology presented in this study. One may think that this study is an extension of the literature one. We honestly declare that this study is a logical extension of our previous studies.1h,5a,5b,13 As the literature reaction with a very low total yield was almost hidden in the published manuscript,14 we could only be aware of it after the start of this project. While the synthesis of the mono ketone had the same yield with almost the same reaction method, the second step, ring closure reaction, was totally different and with very different yields. The literature reaction was conducted in CS2 using AlCl3, which gave low yield of 26%. The total yield was only 17%. Obviously such reaction condition could not be extended to the synthesis of TTs with various alkyl chains. On the other hand, our ring closure reaction had reaction condition of PPA in refluxing chlorobenzene resulting in higher yield of 75%. As our reaction is simpler and more applicable, it was extended to the synthesis of the TTs with various alkyl chains, even the one having branched alkyl group (15e).
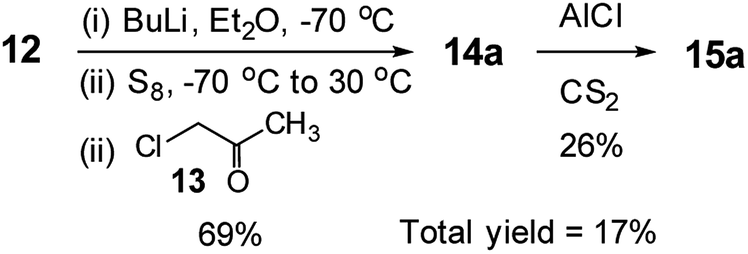 | ||
| Scheme 4 The literature synthesis of 3-methyl-TT.14 | ||
2.2. Electrochemical studies
As the 3-alkyl-TTs 15 are in hand and, to our best knowledge, their electropolymerization and UV-CV behaviors were not studied, they were electropolymerized on ITO in 0.1 M Bu4PF6/acetonitrile, using Ag and Pt electrodes as reference and counter electrodes, respectively, to obtain the polymers P15a–j. The monomer concentrations were kept at 1 × 10−3 M and the scan rate was 1.0 V s−1. Polymerizations of the 3-alkyl-TTs 15a–j were observed by means of deposition of the polymers on ITO electrode surface and through the increase of oxidation and reduction peaks (Fig. S1†). Their spectroelectrochemistry behaviors were investigated in a monomer-free CH3CN solution in the presence of Bu4NPF6 as a supporting electrolyte (Fig. S2†). The absorbances of the polymers were monitored in situ as a function of potential, ranging from 0.0 to 2.0 V. While the intensity of the π–π* transitions decreased, an increasing polaron (700–760 nm) and bipolaron (around 1050 nm) bands appeared with the increasing potential. They all demonstrated orange-green-blue color changes around 0.0, 1.2 and 1.5 V, respectively. As the polymers demonstrated similar color changes almost at the same potentials, as an example, cyclic voltammetry-ultraviolet-visible (CV-UV) measurement of Pf6 is depicted in Fig. 1. | ||
| Fig. 1 The color transition of the polymer P15f film coated on ITO at different potentials. | ||
The polymers had UV-Vis absorption maxima between 390–445 nm, while their monomers had absorptions between 260–270 nm in solution (CH2Cl2). The optical band gaps varied between 1.90–2.30 eV (Table 2.)
| λmax (nm) | λonset (nm) | EOpticgap (eV) | |
|---|---|---|---|
| P15a | 445 | 615 | 2.02 |
| P15b | 391 | 540 | 2.30 |
| P15c | 404 | 605 | 2.05 |
| P15d | 402 | 580 | 2.13 |
| P15e | 413 | 590 | 2.10 |
| P15f | 420 | 625 | 1.98 |
| P15g | 396 | 570 | 2.18 |
| P15h | 393 | 535 | 2.32 |
| P15i | 406 | 645 | 1.92 |
| P15j | 441 | 650 | 1.90 |
2.3. Surface analyses
Surface studies of the polymers by scanning electron microscopy (SEM) and atomic force microscopy (AFM), which were correlated by contact angle (CA) measurements, were then performed, except Pe5, which had a branched alkyl chain (Fig. 2). SEM images were recorded applying two magnifications at 1.000× and 10.000× with a scale bars of 50 microns (top row) and 100 microns (bottom row). As evidenced by the higher magnified SEM images, the surfaces had a tendency of becoming more homogeneous with the increasing length of the side chains. While the polymers P15a–P15d had relatively rough surfaces, quasi spherical islets (aggregations), spreading over the scanned area, clearly appeared with the polymers P15f–P15i. These small islets formed a smooth surface with the polymer P15j. AFM-phase (Fig. S3†) and topography images, i.e. the cross-sections along the red lines showing the topographies of the samples (Fig. 2d), indicated that the islets had a different morphology from the polymer matrix, and their root mean square (Rms) values are collected in Table 3. The polymers P15a–P15d had relatively non-homogeneous surface morphology, and, while no apparent trend on their Rms values was observed, a decreasing trend appeared with the polymers P15f–P15j. Moreover, water contact angle studies demonstrated that the surface hydrophobicity of the polymers increased as the length of the lipidic side chains increased. In agreement with the results obtained by SEM, AFM and CA measurements, lipidic side chains were apparently oriented at the surface. Thus, a more homogeneous and highly hydrophobic layer with CA angle up to 107° (P15j) was formed with the increasing length of the chains. These values are higher than or comparable with the most of the well-known hydrophobic polymers such as polyethylene (96°), polypropylene (102.1°), paraffin (108.9°), etc.15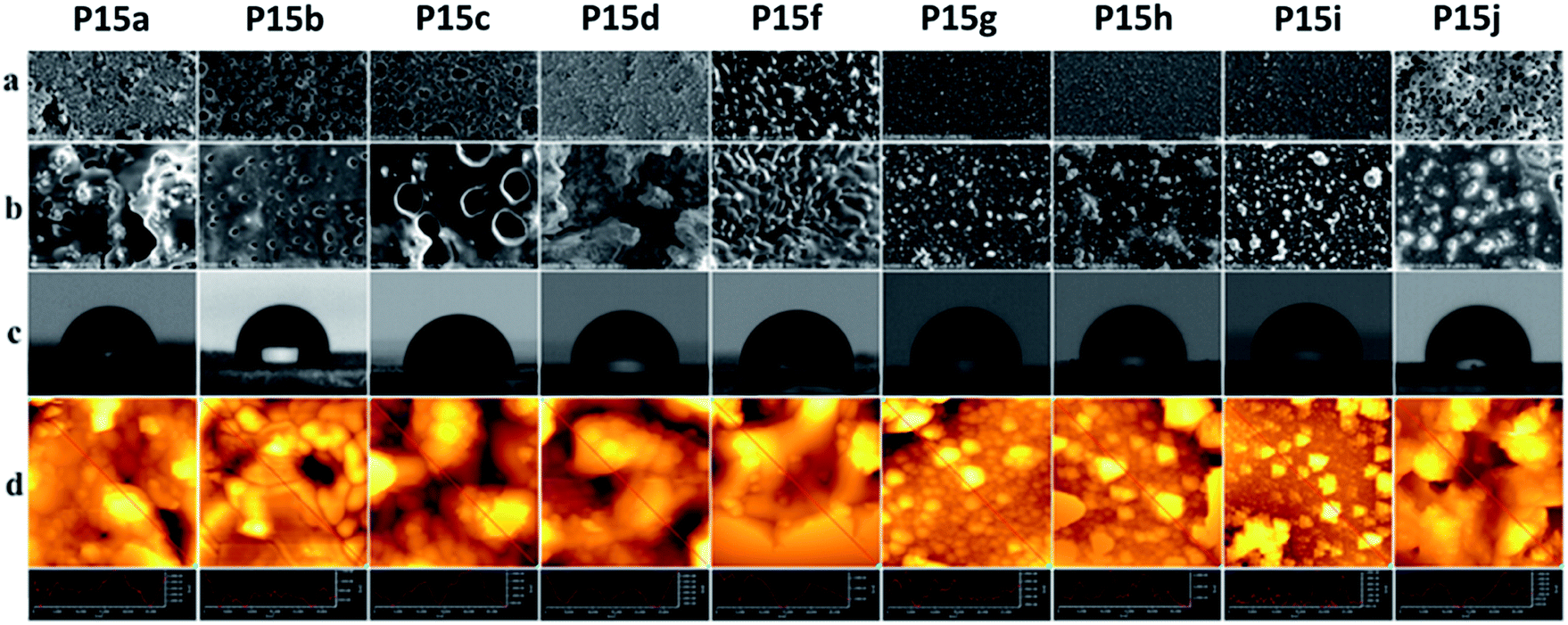 | ||
| Fig. 2 SEM (a) 1.000×, (b) 10.000×; (c) contact angle, CA; (d) AFM topography images and surface cross section analyses of P15a–P15j. | ||
| Polymers | Rms (nm) | Water contact angles (±2°) |
|---|---|---|
| P15a | 1.09 × 103 | 83 |
| P15b | 1.52 × 102 | 84 |
| P15c | 7.13 × 102 | 91 |
| P15d | 1.09 × 103 | 95 |
| P15f | 4.44 × 102 | 97 |
| P15g | 4.34 × 102 | 99 |
| P15h | 4.24 × 102 | 101 |
| P15i | 3.00 × 102 | 102 |
| P15j | 2.98 × 102 | 107 |
3. Conclusion
In conclusion, a convenient synthesis of 3-alkylthieno[3,2-b]thiophenes, important building blocks of various organic materials, with good yields has been developed. This new method reduces the four step cumbersome synthesis available in the literature to a simple two-step synthesis. The two steps, (i) synthesis of mono-ketone and (ii) ring-formation, had the yields between 65–92% and 75–90%, respectively, with overall yield between 77–90%. This convenient method led to the synthesis of a series of alkyl substituted thienothiophens, varying from 1 to 12 carbons. SEM, AFM and contact angle (CA) analyses of their electropolymers on indium tin oxide (ITO) indicated that, as the carbon chain of the alkyl group increases, lipidic side chains were apparently oriented at the surface, and more homogeneous and highly hydrophobic layer with CA angle up to 107° was obtained.4. Experimental
4.1. Materials method
Cyclic voltammetry (CV) studies were performed using CH-Instruments Model 400A as a potentiostat. UV-Vis measurements were studied on Hitachi U-0080D. 1H and 13C NMR spectra were recorded on Varian model NMR (500 MHz). Proton and carbon chemical shifts were reported in ppm downfield from tetramethylsilane (TMS). Mass spectra were recorded on Bruker MICROTOFQ and Thermo LCQ-Deca ion trap mass instruments.Scanning Electron Microscopy (SEM) images were recorded to examine the surface morphology of the samples using Hitachi SU 500 FEG-SEM instrument. Images were obtained at 10k and 20k magnifications in the range of 2.0–3.0 kV acceleration voltages, in high vacuum. Surface coating was not applied to samples to observe the morphology in their original form.
Atomic Force Microscopy (AFM) was used to determine the surface topology and morphology of the samples using Hitachi AFM5100N type instrument, which was operated under dynamic mode (DFM). In DFM mode, the tip implements intermittent contact to the sample surface to minimize the destructive lateral forces. Images were obtained by scanning 20 × 20 micron area, using SI-DF-3P2 (Hitachi) cantilever with a spring constant of 2.4 N m−1 at ambient temperature and humidity.
Contact angle (CA) measurments were conducted to determine the water wettability of the sample surfaces using a Kürüss GmbH DSA-100 model instrument controlled with DS3210 software equipped with direct dosing system and high speed camera. CA measurements were performed using sessile drop technique. At each measurement, 5 mL of deionized water was placed on the surface. The CA values are average of 5 measurements with a standard deviation of ±3°.
Spectroelectrochemistry of the polymers was investigated on ITO surface in monomer-free solution using a UV spectroscopy. The polymer coated ITO was placed into a quartz cuvette, filled with monomer-free electrolyte solution for UV-CV measurement. Pt and Ag wires were used as counter and reference electrodes, respectively. The changes in their absorbance of the polymers were measured in situ as a function of potential change, starting from 0 V, which was gradually increased up to 2.0 V.
4.2. Synthesis of mono ketones
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) solvent system to give the title compound 14a (1.36 g, 65%) as a colorless viscose liquid. IR: 1700 cm−1 (C
:1) solvent system to give the title compound 14a (1.36 g, 65%) as a colorless viscose liquid. IR: 1700 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O); MS m/z: 173 (M+ + 1). As it was found to be eye irritant, it was not further characterized and directly used for the next step.
O); MS m/z: 173 (M+ + 1). As it was found to be eye irritant, it was not further characterized and directly used for the next step.The followings were similarly prepared.
:1) solvent system to give the title compound 14b (1.52 g, 67%) as a colorless viscose liquid. IR: 1700 cm−1; MS m/z: 185 (M+ + 1). As it was found to be eye irritant, it was used for the next step without characterization.:1) solvent system to give the title compound 14c (1.76 g, 72%) as a colorless viscose liquid. IR: 1700 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.33 (dd, J = 5.0 Hz, 3.0 Hz, 1H), 7.22 (dd, J = 3.0 Hz, 1.3 Hz, 1H), 7.03 (dd, J = 5.0 Hz, 1.3 Hz, 1H), 3.58 (s, 2H), 2.57 (t, J = 7.3 Hz, 2H), 1.60 (m, 2H), 0.91 (t, J = 7.3 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 205.6, 130.0, 129.6, 126.5, 125.0, 45.0, 42.5, 17.3, 13.7. As it was found to be eye irritant, it was used for the next step without further characterization.:1) solvent system to give the title compound 14d (1.83 g, 70%) as a colorless viscose liquid. IR: 1700 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.33 (dd, J = 5.0 Hz, 3.0 Hz, 1H), 7.22 (dd, J = 3.0 Hz, 1.3 Hz, 1H), 7.03 (dd, J = 5.0 Hz, 1.3 Hz, 1H), 3.58 (s, 2H), 2.58 (t, J = 7.4 Hz, 2H), 1.56 (m, 2H), 1.29 (m, 2H), 0.90 (t, J = 7.3 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 205.7, 129.6, 128.7, 126.5, 125.0, 45.0, 40.4, 25.9, 22.2, 13.8. As it was found to be eye irritant, it was used for the next step without further characterization.:1) solvent system to give the title compound 14e (2.10 g, 75%) as a brown viscose liquid. IR: 1700 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.33 (dd, J = 4.9 Hz, 3.1 Hz, 1H), 7.23 (d, J = 3.1 Hz, 1H), 7.03 (d, J = 4.9 Hz, 1H), 3.59 (s, 2H), 2.58 (t, J = 7.3 Hz, 2H), 1.46 (m, J = 1.51–1.43, 3H), 0.88 (d, J = 6.4 Hz, 6H). As it was found to be eye irritant, it was used for the next step without further characterization.4.3. Synthesis of TTs
The following were similarly prepared.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We thank TUBITAK for supporting this work (216Z041), Istanbul Technical University for grant to KTI (MSc), YOK 100/2000 (ST) and Unsped Global Lojistik, Turkey, for financial support.References
- (a) J. Wang, K. Liu, L. Ma and X. Zhan, Chem. Rev., 2016, 116, 14675–14725 CrossRef CAS PubMed; (b) X. Gao and Z. Zhao, Sci. China: Chem., 2015, 58, 947–968 CrossRef CAS; (c) H. Dong, W. Hu and D. Zhu, Chem. Rev., 2012, 112, 2208 CrossRef PubMed; (d) A. Facchetti, Chem. Mater., 2011, 23, 733–758 CrossRef CAS; (e) G. Turkoglu, M. E. Cinar and T. Ozturk, Top. Curr. Chem., 2017, 375, 84–129 CrossRef PubMed; (f) A. Mishra and P. Bauerle, Angew. Chem., Int. Ed., 2012, 51, 2020–2067 CrossRef CAS PubMed; (g) S. Gunes, H. Neugebauer and N. S. Sariciftci, Chem. Rev., 2007, 107, 1324–1338 CrossRef PubMed; (h) T. Ozturk, E. Ertas and O. Mert, Tetrahedron, 2005, 61, 11055–11077 CrossRef CAS.
- A. C. Arias, J. D. MacKenzie, I. McCulloch, J. Rivnay and A. Salleo, Chem. Rev., 2010, 110, 3–24 CrossRef CAS PubMed.
- (a) E. Bundgaard, M. Helgesen, J. E. Carlé, F. C. Krebs and M. Jørgensen, Chem. Phys., 2013, 214, 1546–1558 CAS; (b) M. Manceau, E. Bundgaard, J. E. Carlé, O. Hagemann, M. Helgesen, R. Søndergaard, M. Jørgensen and F. C. Krebs, J. Mater. Chem., 2011, 21, 4132–4141 RSC.
- (a) M. Kertesz, C. H. Choi and S. Yang, Chem. Rev., 2005, 105, 3448–3481 CrossRef CAS PubMed; (b) I. F. Perepichka and D. F. Perepichka, Handbook of Thiophene-based Materials: Applications in Organic Electronics and Photonics, John Wiley & Sons, Chichester, UK, 2009 CrossRef; (c) R. McCullough, Adv. Mater., 1998, 10, 93–116 CrossRef CAS; (d) J. Roncali, Chem. Rev., 1997, 97, 173–206 CrossRef CAS.
- (a) M. E. Cinar and T. Ozturk, Chem. Rev., 2015, 115, 3036–3140 CrossRef CAS PubMed; (b) M. E. Cinar and T. Ozturk, Top. Heterocycl. Chem., 2015, 39, 161–202 CAS; (c) P. Kumaresan, S. Vegiraju, Y. Ezhumalai, S. L. Yau, C. Kim, W. Wen-Hsi Lee and M. Chen, Polymers, 2014, 6, 2645–2669 CrossRef; (d) A. C. Arias, J. D. MacKenzie, I. McCulloch, J. Rivnay and A. Salleo, Chem. Rev., 2010, 110, 3–24 CrossRef CAS PubMed; (e) P. J. Skabara, in Handbook of thiophene-based materials, ed. I. F. Perepichka and D. F. Perepichka, Wiley, Chichester, 2009, Ch. 3 Search PubMed.
- A. Marrocchi, D. Lanari, A. Facchetti and L. Vaccaro, Energy Environ. Sci., 2012, 5, 8457–8474 RSC.
- (a) Z. Xiao, J. Subbiah, K. Sun, D. J. Jones, A. B. Holmes and W. W. Wong, Polym. Chem., 2014, 5, 6710–6717 RSC; (b) X. Wang, Z. G. Zhang, H. Luo, S. Chen, S. Yu, H. Wang, X. Li, G. Yu and Y. Li, Polym. Chem., 2014, 5, 502–511 RSC; (c) Y. Lia, L. Chen, Y. Chen, C. Li, P. Zhang, L. Gao, X. Yang, Y. Tu and X. Zhu, Sol. Energy Mater. Sol. Cells, 2013, 108, 136–145 CrossRef; (d) X. Guo, M. Zhang, L. Huo, F. Xu and Y. Wu, J. Mater. Chem., 2012, 22, 21024–21031 RSC; (e) W. Tang, L. Ke, L. Tan, T. Lin, T. Kietzke and Z. Chen, Macromolecules, 2007, 40, 6164–6171 CrossRef CAS; (f) M. He and F. Zhang, J. Org. Chem., 2007, 72, 442–451 CrossRef CAS PubMed; (g) X. Zhang, M. Köhler and A. Matzger, Macromolecules, 2004, 37, 6306–6315 CrossRef CAS.
- H. Liu, F. Wu, B. Zhao, L. Meng, G. Wang, J. Zhang, P. Shen and B. Tan, Dyes Pigm., 2015, 120, 44–51 CrossRef CAS.
- X. Zong, M. Liang, T. Chen, J. Jia, L. Wang, Z. Sun and S. Xue, Chem. Commun., 2012, 48, 66–6647 RSC.
- J. H. Kim, S. Wood, J. B. Park, J. Wade, M. Song, S. C. Yoon, I. H. J. Jung, J. S. Kim and D. H. Hwang, Adv. Funct. Mater., 2016, 26, 1517–1525 CrossRef CAS.
- D. Gao, K. Tian, W. Zhang, J. Huang, Z. Chen, Z. Mao and G. Yu, Polym. Chem., 2016, 7, 4046–4052 RSC.
- (a) J. C. Bijleveld, R. A. M. Verstrijden, M. M. Wienk and R. A. J. Janssen, J. Mater. Chem., 2011, 21, 9224 RSC; (b) W. Li, Y. Guo, J. Shi, H. Yu, H. Meng, Macromolecules, 49, 7211–7219 Search PubMed.
- (a) M. E. Cinar, S. T. Cankaya, A. Capan, M. S. Eroglu and T. Ozturk, Eur. Polym. J., 2018, 104, 72–80 CrossRef CAS; (b) I. Osken, O. Sahin, A. S. Gundogan, H. Bildirir, A. Capan, E. Ertas, M. S. Eroglu, J. D. Wallis and T. Ozturk, Tetrahedron, 2012, 68, 1216–1222 CrossRef CAS; (c) A. Capan, H. Veisi, A. C. Goren and T. Ozturk, Macromolecules, 2012, 45, 8228–8236 CrossRef CAS; (d) I. Osken, A. S. Gundogan, E. Tekin, M. S. Eroglu and T. Ozturk, Macromolecules, 2013, 46, 9202–9210 CrossRef CAS; (e) C. Kutahya, A. Allushi, R. Isci, J. Kreutzer, T. Ozturk, G. Yilmaz and Y. Yagci, Macromolecules, 2017, 50, 6903–6910 CrossRef CAS; (f) T. Ozturk, Tetrahedron Lett., 1996, 37, 2821–2824 CrossRef CAS; (g) E. Ertas and T. Ozturk, J. Chem. Soc., Chem. Commun., 2000, 2039–2040 RSC; (h) T. Ozturk, N. Saygili, S. Ozkara, M. Pilkington, C. R. Rice, D. A. Tranter, F. Turksoy and J. D. Wallis, J. Chem. Soc., Perkin Trans. 1, 2001, 1, 407–414 RSC; (i) F. Turksoy, J. D. Wallis, U. Tunca and T. Ozturk, Tetrahedron, 2003, 59, 8012–8116 CrossRef; (j) O. Mert, E. Sahin, E. Ertas, T. Ozturk, E. A. Aydin and L. Toppare, J. Electroanal. Chem., 2006, 591, 53–58 CrossRef CAS; (k) G. Turkoglu, M. E. Cinar, A. Buyruk, E. Tekin, S. P. Mucur, K. Kaya and T. Ozturk, J. Mater. Chem. C, 2016, 4, 6045–6053 RSC.
- D. R. Rutherford, J. K. Stille, C. M. Elliott and V. R. Reichert, Macromolecules, 1992, 25, 2294–2306 CrossRef CAS.
- https://www.accudynetest.com/polytable_03.html?sortby=contact_angle.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra08023f |
| This journal is © The Royal Society of Chemistry 2019 |