
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Comparative DFT study of metal-free Lewis acid-catalyzed C–H and N–H silylation of (hetero)arenes: mechanistic studies and expansion of catalyst and substrate scope†
Pan Du‡
a and
Jiyang Zhao‡ *b
*b
aSchool of Life Science and Chemistry, Jiangsu Second Normal University, Nanjing 210013, China
bSchool of Environmental Science, Nanjing Xiaozhuang University, Nanjing 211171, China. E-mail: jyzhao1981@163.com
First published on 19th November 2019
Abstract
Direct selective dehydrogenative silylation of thiophenes, pyridines, indoles and anilines to synthesize silyl-substituted aromatic compounds catalyzed by metal-free Lewis acids was achieved recently. However, there is still insufficient mechanistic data for these transformations. Using density functional theory calculations, we conducted a detailed investigation of the mechanism of the B(C6F5)3-catalyzed dehydrogenative silylation of N-methylindole, N,N-dimethylaniline and N-methylaniline. We successfully located the most favourable reaction pathways that can explain the experimental observations notably well. The most favourable pathway for B(C6F5)3-catalyzed C–H silylation of N-methylindole includes nucleophilic attack, proton abstraction and hydride migration. The C–H silylation of N,N-dimethylaniline follows a similar pathway to N-methylindole rather than that proposed by Hou's group. Our mechanism successfully explains that the transformations of N-methylindoline to N-methylindole produce different products at different temperatures. For N-methylaniline bearing both N–H and para-phenyl C–H bonds, the N–H silylation reaction is more facile than the C–H silylation reaction. Our proposed mechanism of N–H silylation of N-methylaniline is different from that proposed by the groups of Paradies and Stephan. Lewis acids Al(C6F5)3, Ga(C6F5)3 and B(2,6-Cl2C6H3)(p-HC6F4)2 can also catalyze the C–H silylation of N-methylindole like B(C6F5)3, but the most favourable pathways are those promoted by N-methylindoline. Furthermore, we also found several other types of substrates that would undergo C–H or N–H silylation reactions under moderate conditions. These findings may facilitate the design of new catalysts for the dehydrogenative silylation of inactivated (hetero)arenes.
1 Introduction
(Hetero)arylsilanes are highly important species in molecular and materials synthesis,1 medicinal chemistry,2 and synthetic chemistry.3 The direct selective C–H silylation of (hetero)arenes with hydrosilanes is atom-economical, efficient and convenient and is one of the most attractive methods for synthesizing silyl-substituted aromatic compounds.4 To date, various metal-catalyzed C–H silylation reactions between (hetero)arenes and hydrosilanes have been reported.5 Generally, the use of metal catalysts has several shortcomings such as the high cost of the catalysts, difficulty of catalyst recycling, and addition of additives. Therefore, the development of a cheap and environmentally friendly synthetic method for the preparation of silylated (hetero)arenes is still a challenging task.Recently, boron-catalyzed hydrosilylation of (hetero)arenes has attracted considerable attention. The pioneering work in this area was performed by Kawashima6 and Ingleson groups.7 They accomplished intramolecular dehydrogenative silylation of 2-(SiR2H)-biphenyls using Lewis acid B(C6F5)3 or Ph3CB(C6F5)4 as the catalysts. Since then, C–H silylations of thiophenes, pyridines, indoles and anilines with hydrosilanes using Lewis acid B(C6F5)3, Al(C6F5)3 or Brønsted acid [H(OEt2)2]+[B(C6F5)4]− as the catalysts have been reported (Scheme 1(a and b)).8–13 Furthermore, N–H silylations of aniline with hydrosilanes catalyzed by B(C6F5)3 and [(C6F5)3PF][B(C6F5)4] were observed by the Paradies and Stephan groups (Scheme 1(c)).14
 | ||
| Scheme 1 Metal-free catalyzed C–H silylation of indoles (a) and anilines (b), and N-H silylation of anilines (c). | ||
Although many catalytic cycles for the silylation of aromatic C–H bonds using hydrosilanes have been proposed in previous experimental studies, few mechanistic investigations have been performed.15 Ingleson8 and Zhang11 groups proposed a possible reaction pathway for B(C6F5)3-catalyzed C–H silylation of indole, which includes nucleophilic attack, proton abstraction and hydride migration (exhibited in Scheme 2A). However, Hou's group9b proposed a different mechanism for the C–H silylation of aniline catalyzed by B(C6F5)3, which involves nucleophilic attack and H2 release (Scheme 2B). For the N–H silylation of indole, Paradies and co-workers14c proposed a catalytic cycle involving N-silylation, rearrangement and reduction (Scheme 2C). Stephan group investigated the mechanism of N–H silylation of anilines catalyzed by [(C6F5)3PF][B(C6F5)4] based on density functional theory (DFT) calculations. They found a dehydrocoupling mechanism that contains Si–N bond formation and H2 liberation (Scheme 2D). Moreover, the mechanism of B(C6F5)3-catalyzed silylation of carbonyl group have been established both experimentally and theoretically.16 Nevertheless, the mechanism for N–H silylation of aniline catalyzed by B(C6F5)3 has not been elucidated yet. And it is not clear which C–H silylation mechanism (Scheme 2A and B) is more reasonable. More importantly, if a substrate bears both C–H and N–H bonds, which type of silylation reaction is more likely to occur? Hence, in this work, we performed DFT calculations to investigate the molecular mechanisms of B(C6F5)3-catalyzed dehydrogenative silylation of indole and aniline with hydrosilanes and extend the scope of the silylation catalysts and substrates.
 | ||
| Scheme 2 Proposed mechanisms for the Lewis acid catalyzed C–H silylation of indole (A) and aniline (B), N–H silylation of indole (C) and aniline (D). | ||
2 Computational details
All calculations were performed with the Gaussian09 program.17 Geometry optimizations and energy calculations were performed using the M06-2X functional18 that was proven to be accurate for describing weak interactions. The 6-311G(d,p) basis set was used for all atoms in the substrates and catalyst.19 Geometry optimizations are conducted in chlorobenzene and benzene solutions (using the self-consistent reaction field (SCRF) method with the IEFPCM solvation model20 with default parameters). We have performed harmonic-frequency-analysis calculations at the same level of theory to obtain the relevant thermodynamic energy corrections and to determine whether the optimized stationary points are minima or saddle points. At the M06-2x/6-311G(d,p) geometries, the energies were further refined by M06-2x/6-311++G(d,p) single-point energy calculations with the effect of the solvent taken into account by the IEFPCM solvation model. To determine the reasonability of our computational method, the wb97xd functional was also used to investigate the rate-limiting step in our mechanism. The calculated Gibbs free energies are for T = 298.15 K and 1 atm. Furthermore, important transition states were determined by intrinsic reaction coordinate (IRC) analysis.21 The 3D-optimized structures in this paper were displayed using the CYLview visualization program.22Since our studied reactions involve multicomponent changes, the entropy contributions to the free energies for the reactions in the solvent will be overestimated. In addition, there are no standard quantum mechanics-based methods to accurately calculate the entropy in solution. In this study, based on “the theory of free volume”,23 corrections were added to calculate free energies; generally, for 2-to-1 (or 1-to-2) reactions, a correction of −2.6 (or 2.6) kcal mol−1 was added.
3 Results and discussion
In this section, we will discuss the B(C6F5)3-catalyzed dehydrosilylation of N-methylindole, N,N-dimethylaniline and N-methylaniline with PhSiH3 successively. Then, we explore other possible catalysts and select other substrates to undergo C–H or N–H silylation as in the case of indoles or anilines.3.1 C–H silylation of N-methylindole
Based on our calculations, the B(C6F5)3-catalyzed C–H silylation of N-methylindole with PhSiH3 including three elementary steps: (1) nucleophilic attack of the indole to the Si centre of PhSiH3, (2) proton abstraction by a second N-methylindole, and (3) hydride migration to form indoline and regenerate B(C6F5)3. Moreover, we also examined the conversion of indoline to indole and the second silylation step of N-methylindole. | ||
| Fig. 1 Free-energy profile for the B(C6F5)3-catalyzed C–H silylation of N-methylindole with PhSiH3, along with the optimized structures of the stationary points (path A in (A) and paths B and C in (B)). Key bond lengths are given in Å. | ||
First, a stable Lewis adduct add1 is formed from indole and B(C6F5)3. Because the add1 is 3.7 kcal mol−1 lower than that of free Lewis acid and base, it can be expected that there is an equilibrium between Lewis adduct add1 and the free B(C6F5)3 and indole. When PhSiH3 participates in the reaction, a B–Si complex 1 is generated. Natural population analysis shows that charge of the Si center of complex 1 increases to 1.14 from 0.87 in PhSiH3, suggesting that the Si center becomes more electrophilic. Then, the C3 atom of N-methylindole attacks complex 1 at the Si center via a barrierless transition state TS2/3 (6.6 kcal mol−1, relative to add1) through the concerted linear backside SN2 attack, giving intermediate 3. The C–Si bond formation and Si–H bond braking take place simultaneously in TS2/3. It is worth noting that the mechanism of the silylation reaction of acetone with Me3SiH proposed by the group of Sakata16c is analogous to the one in our proposed mechanism. The distance of C3 and Si is 2.04 Å in intermediate 3, indicating that the Si–C bond is nearly formed. Additionally, the C3–H bond distance in intermediate 3 is 1.09 Å, which is longer than that in free N-methylindole (1.08 Å), suggesting that the C3–H bond is weakened.
 | ||
| Scheme 3 Metal-free B(C6F5)3-catalyzed transformation of N-methylindoline to N-methylindole ((a) producing indole and H2, (b) producing indole and ion pair). | ||
 | ||
| Fig. 2 Free-energy profile for B(C6F5)3-catalyzed transformation of indoline to indole, along with the optimized structures of the stationary points. Key bond lengths are given in Å. | ||
First, B(C6F5)3 selectively abstracts a hydride from the C2 position of N-methylindoline through transition state TS7/8 to give an ion pair 7 over a low barrier of 12.5 kcal mol−1 (as compared to 8). Starting with ion pair 7, two pathways are possible. In path A, a second N-methylindoline abstracts a proton from ion pair 7 through transition state TS15/16, leading to the formation of intermediate 16. Subsequently, intermediate 16 is dissociated to form N-methylindole and a stable ion pair 13 (ΔG = −10.2 kcal mol−1). This proton abstraction is very facile because a barrier of only 11.3 kcal mol−1 must be overcome. Finally, H2 is released from the ion pair 13 to regenerate N-methylindoline and B(C6F5)3. This step requires the overcoming of a free energy barrier of 26.8 kcal mol−1 (relative to ion pair 13). The three elementary steps in path A are analogous with that proposed by the group of Paradies.24a Nevertheless, we found a new pathway (path B in Fig. 2) in which N-methylindole can also facilitate the reaction through proton abstraction (TS17/18, 24.5 kcal mol−1) and H2 generation (TS19/20, 26.0 kcal mol−1). Cleanly, the reaction barrier of path B is close to that of path A, indicating that both paths A and B are possible.
It is worth mentioning that the proton abstraction in path A is more facile than that in path B (11.3 kcal mol−1 vs. 24.5 kcal mol−1). Therefore, when the reaction occurs at room temperature, it will follow pathway A to give indole and stable ion pair 13, as was found in Zhang's experiments (Scheme 2(B)).25 However, when the reaction temperature is increased to 120 °C (Scheme 2(A)), pathways A and B are both possible because their reaction barriers are very close (path A, 26.8 kcal mol; path B, 26.0 kcal mol−1). Therefore, our mechanism is in good consistent with the experimental observations. To further confirm the reaction barriers of paths A and B, we used the wB97XD functional to study the same reaction. The barriers obtained using the wB97XD functional are 22.6 (TS13/14) and 21.2 (TS19/20) kcal mol−1, respectively, in agreement with the values obtained using the M062X functional.
 | ||
| Scheme 4 Silylation of N-methylindole with the silylated-indole as hydrosilane to yield bis(indol-3-yl)-substituted product. | ||
3.2 C–H silylation of N,N-dimethylaniline
The second reaction we studied was the B(C6F5)3-catalyzed C–H silylation of N,N-dimethylaniline. The reaction is started with nucleophilic attack on the B–Si complex by N,N-dimethylaniline, followed by three different reaction pathways (paths A, B, and C) to generate the silylated product. The free energy profiles and related optimized geometric structures of these pathways are shown in Fig. 3 (path B) and S6–S7 (paths A and C) in the ESI.† In path A, species N1-3 undergoes an proton-hydride recombination to afford the silylated product, identical to the reaction pathway suggested by the group of Hou.9b The free energy barrier of path A is 29.1 kcal mol−1. However, we found a new pathway (path B) that is more reasonable than path A. In path B, the initial step is proton-abstraction by a second N,N-dimethylaniline. The subsequent H2 generation is the rate-determining step with a free energy barrier of 23.5 kcal mol−1. This H2 generation transition state is in consistent with the observation by Rieger and co-workers.26 The process of path C is similar to that of path B, but the corresponding proton shuttle is the silylated product. The free energy barrier of path C is 27.2 kcal mol−1. Among these three pathways, pathway B is more favourable than paths A and C. We recalculated the three pathways using the wb97xd functional and the related reaction barriers were 27.1, 21.9 and 24.3 kcal mol−1, respectively. It is clear that the results obtained using the wb97xd and M062X functionals are in good agreement, and thus our calculations are reasonable. | ||
| Fig. 3 Free-energy profiles for B(C6F5)3-catalyzed dehydrogenative silylation of N,N-dimethylaniline with PhSiH3 (pathway B), along with the optimized structures of the stationary points. Key bond lengths are given in Å. | ||
3.3 Comparison of C–H and N–H silylation of PhNHMe
Generally, if the aniline bears para-phenyl C–H and N–H bonds, both C–H and N–H silylation reactions will occur. Therefore, we chose PhNHMe as the substrate to investigate its cross-dehydrocoupling reaction catalyzed by B(C6F5)3 with PhSiH3.First, we studied the N–H silylation of PhNHMe. One possible pathway is comparable to that proposed by Stephan and co-workers14b (see Fig. 4(A) and S8, ESI†), including a nucleophilic attack and H2 liberation. The second step is the rate-limiting step with the free energy barrier of 28.0 kcal mol−1 (25.9 kcal mol−1 according to the wb97xd functional calculations). However, we also obtained an alternative pathway in which a second PhNHMe served as a proton shuttle to assist the proton migration (the related free energy profile and geometries of the structures are shown in Fig. 4(B) and S8, ESI†). Our proposed pathway includes a nucleophilic attack (ΔG≠ = 17.4 kcal mol−1), proton abstraction (ΔG≠ = 10.7 kcal mol−1) and H2 generation (rate-limiting step, ΔG≠ = 27.1 kcal mol−1, 25.0 kcal mol−1 for the wb97xd functional calculations). Therefore, both of the pathways are possible. The two pathways of N–H silylation of N-methylaniline are both different from the mechanism of N–H silylation of indole proposed by the group of Paradies.14c
 | ||
| Fig. 4 Free-energy profiles for N–H silylation of PhNHMe with PhSiH3 catalyzed by B(C6F5)3, along with the optimized structures of the stationary points. Key bond lengths are given in Å. | ||
Next, we studied the C–H silylation of PhNHMe and found that this reaction is comparable to that of N,N-dimethylaniline. The most favourable pathway is identical to path B in C–H silylation of N,N-dimethylaniline and requires the overcoming of a free energy barrier of 36.6 kcal mol−1 (the related free energy profile and geometries of the structures are shown in Fig. S9 and S10, ESI†). It is clear that N–H silylation of PhNHMe is more facile than the C–H silylation. The transformation from C–H to C–Si bonds and N–H to N–Si bonds are endothermic by 43.8 and 20.2 kcal mol−1, respectively. So the N–H silylated product is more stable than the C–H silylated product, thereby lowering the corresponding reaction barrier. Since PhNHMe was not used in the N–H silylation experiments, we choose PhNH2 and Ph2NH that were used as substrates in experiments to study the related N–H silylation reactions with Ph2MeSiH in dichloromethane. The related free energy barriers of the rate-determining steps are 22.6 and 16.9 kcal mol−1, respectively, which are reasonable barriers for experimental conditions.14c
3.4 Other possible Lewis acids and substrates
The group of Zhang reported that Lewis acid Al(C6F5)3 can also catalyzed the C–H silylation of N-methylindole with Ph2SiH2.12 We studied the mechanisms of the reaction. There are three possible reaction pathways and the related free energy barriers are 38.0, 26.5 and 31.8, kcal mol−1 (Table 1), respectively. The N-methylindoline promoted pathway (path B) is most favourable for C–H silylation of N-methylindole catalyzed by Al(C6F5)3, which is different from that catalyzed by B(C6F5)3. Furthermore, we test other two Lewis acids (Ga(C6F5)3 and B(2,6-Cl2C6H3)(p-HC6F4)2)27 to catalyze the same reaction like Al(C6F5)3. The most favorable pathways catalyzed by the two Lewis acids are same with that catalyzed by Al(C6F5)3. The related free energy barriers of the rate-determining steps are 21.7 and 21.8 kcal mol−1, respectively. The geometric structures of the related transition states are shown Fig. S11 in the ESI.† It seems that all of these Lewis acids could promote the silylation of N-methylindole.| B(C6F5)3 | Al(C6F5)3 | Ga(C6F5)3 | B(2,6-Cl2C6H3)(p-HC6F4)2 | |
|---|---|---|---|---|
| Barriers (path A) | 24.8 | 38.0 | 35.4 | 23.8 |
| Barriers (path B) | 26.8 | 26.5 | 21.7 | 21.8 |
| Barriers (path C) | 27.9 | 31.8 | 35.4 | 25.3 |
The substrates in the silylation reactions are thiophenes, pyridines, indoles and anilines in experiments. We want to extend the reaction to other types of reaction substrates. So we selected six other heterocyclic compounds to study their C–H silylation reaction with PhSiH3 catalyzed by B(C6F5)3. These substrates are shown in Table 2. The most favourable pathways for the C–H silylation of these substrates are identical to path C in C–H silylation of N-methylindole. The related free energy barriers of the rate-determining steps are 21.3, 17.9, 16.0, 19.8, 51.2 and 63.7 kcal mol−1 (Table 2), respectively. The geometric structures of the related transition states are shown Fig. S12 in the ESI.† These results suggest that entries 1–4 would undergo C–H silylation reactions under moderate conditions while entries 6 and 7 can't.
| Substrate | ΔG (TS) | Substrate | ΔG (TS) | Substrate | ΔG (TS) | |||
|---|---|---|---|---|---|---|---|---|
| 1 |  |
21.3 | 2 |  |
17.9 | 3 |  |
16.0 |
| 4 |  |
19.8 | 5 |  |
51.2 | 6 |  |
63.7 |
Similarly, we chose six other different types of substrates (entries 1–6 in Table 3) to study their N–H silylation reaction with PhSiH3 catalyzed by B(C6F5)3. The related free energy barriers of the rate-determining steps are 17.7, 27.7, 28.7, 33.3, 32.6 and 36.4 kcal mol−1 (Table 3), respectively. The geometric structures of the related transition states are shown Fig. S13 in the ESI.† The results suggest that entries 1–3 would undergo N–H silylation reaction while entries 4–6 would not undergo the same reaction. Entries 4–6 can form stable Lewis adducts with Lewis acid B(C6F5)3. The free energies of their Lewis adducts are 22.3, 26.3 and 24.9 kcal mol−1 lower than the related free Lewis acids and bases. These stable Lewis adducts inhibit the corresponding N–H silylation reaction.
| Substrate | ΔG (TS) | Substrate | ΔG (TS) | Substrate | ΔG (TS) | |||
|---|---|---|---|---|---|---|---|---|
| 1 |  |
17.7 | 2 |  |
27.7 | 3 |  |
28.7 |
| 4 |  |
33.3 | 5 |  |
32.6 | 6 |  |
36.4 |
4 Conclusions
Transition-metal-catalyzed C–H silylation of arenes is an attractive synthetic approach since it was known as efficient, atom-economical and superior selectivity. The protocol of B(C6F5)3 catalytic C–H and N–H silylation of aromatic compounds features environmental conditions, high regioselectivity, and no requirement for removing residual metal catalysts. Efforts have been made to deepen the understanding of the reaction mechanisms of these reactions to enhance the activity of the catalysts. So we performed detailed density functional theory calculations on the mechanism of the dehydrogenative silylation of N-methylindole, N,N-dimethylaniline and N-methylaniline. The main conclusions are summarized as follows:(1) The most favourable pathways for the silylation of N-methylindole and N,N-dimethylaniline are similar except for the last step (Scheme 5A and B). The first two steps are the same: nucleophilic attack and proton abstraction, and the last step is hydride migration for N-methylindole and H2 liberation for N,N-dimethylaniline. In the two reactions, aniline and indole not only serve as Lewis bases to attack the B–Si complex but also as a Brønsted base to assist proton transfer, leading to the lowering of the reaction barrier.
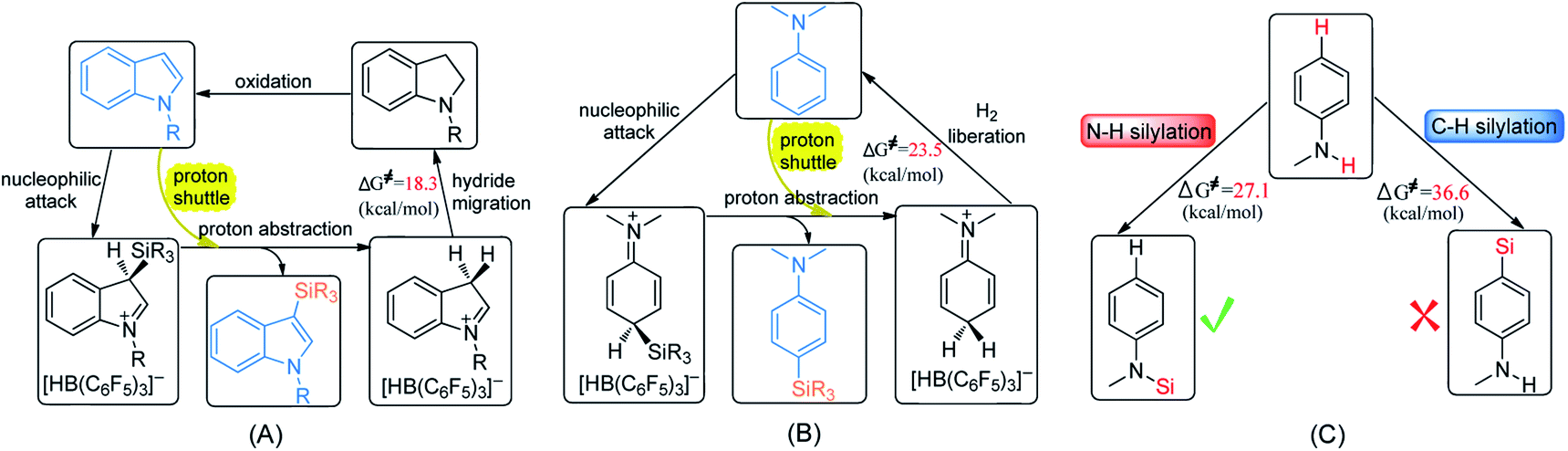 | ||
| Scheme 5 Proposed mechanisms for (A) C–H silylation of N-methylindole, (B) C–H silylation of N,N-dimethylaniline and (C) selectivity of silylation of N-methylaniline. | ||
(2) For the conversion of the resulting indoline to indole, we found two compete pathways where indoline and indole both can act as proton-shuttle to facilitate the reaction. Our proposed mechanism can explain the experimental observations that an ion pair was formed at room temperature while N-methylindole and H2 was generated at 120 °C.
(3) With respect to the dehydrogenative silylation of PhNHMe that bears both N–H and para-phenyl C–H bonds, the N–H silylated product is more stable than the C–H silylated product, so N–H silylation of PhNHMe is more favourable than the C–H silylation (Scheme 5C). N–H silylation of PhNHMe includes nucleophilic attack, proton abstraction and H2 liberation.
(4) The C–H silylation of N-methylindole catalyzed by Al(C6F5)3 follows the pathway that promoted by N-methylindoline, which is different from that catalyzed by B(C6F5)3. Lewis acids (Ga(C6F5)3, and B(2,6-Cl2C6H3)(p-HC6F4)2) can also catalyze the same reaction like Al(C6F5)3. Moreover, we identified four other substrates that would undergo C–H silylation and three other substrates that would undergo N–H silylation with hydrosilanes using B(C6F5)3 as the catalyst.
Of course, our designed reactions need to be examined by experiments. We anticipate this investigation can be helpful for obtaining other metal-free catalysts to promote silylation of other substrates.
Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported by The Natural Science Foundation of the Jiangsu Higher Education Institutions of China (grant no. 18KJB150010, 19KJB150034), Foundation of Nanjing Xiaozhuang University (2018NXY24), the Program of Jiangsu Key Laboratory of New Power Batteries.Notes and references
- (a) L. H. Chan, R. H. Lee, C. F. Hsieh, H. C. Yeh and C. T. Chen, J. Am. Chem. Soc., 2002, 124, 6469–6479 CrossRef CAS PubMed; (b) W. Wei, P. I. Djurovich and M. E. Thompson, Chem. Mater., 2010, 22, 1724–1731 CrossRef CAS.
- R. Ramesh and D. S. Reddy, J. Med. Chem., 2018, 61, 3779–3798 CrossRef CAS PubMed.
- (a) H. F. Sore, W. R. J. D. Galloway and D. R. Spring, Chem. Soc. Rev., 2012, 41, 1845–1866 RSC; (b) M. Gimferrer, Y. Minami, Y. Noguchi, T. Hiyama and A. Poater, Organometallics, 2018, 37, 1456–1461 CrossRef CAS.
- (a) R. Sharma, R. Kumar, I. Kumar, B. Singh and U. Sharma, Synthesis, 2015, 47, 2347–2366 CrossRef CAS; (b) C. Cheng and J. F. Hartwig, Chem. Rev., 2015, 115, 8946–8975 CrossRef CAS PubMed; (c) Z. Xu, W.-S. Huang, J. Zhang and L.-W. Xu, Synthesis, 2015, 47, 3645–3668 CrossRef CAS; (d) S. Bähr and M. Oestreich, Angew. Chem., Int. Ed., 2017, 56, 52–59 CrossRef PubMed; (e) Y. Qin, L. Zhu and S. Luo, Chem. Rev., 2017, 117, 9433–9520 CrossRef CAS PubMed; (f) J. Zhang, S. Park and S. Chang, Chem. Commun., 2018, 54, 7243–7246 RSC; (g) P. Wedi and M. van Gemmeren, Angew. Chem., Int. Ed., 2018, 57, 13016–13027 CrossRef CAS PubMed.
- For selected examples, see: (a) H. F. T. Klare, M. Oestreich, J. Ito, H. Nishiyama, Y. Ohki and K. Tatsumi, J. Am. Chem. Soc., 2011, 133, 3312–3315 CrossRef CAS PubMed; (b) J. Oyamada, M. Nishiura and Z. Hou, Angew. Chem., Int. Ed., 2011, 50, 10720–10723 CrossRef CAS PubMed; (c) C. Cheng and J. F. Hartwig, Science, 2014, 343, 853–857 CrossRef CAS PubMed; (d) C. Cheng and J. F. Hartwig, J. Am. Chem. Soc., 2015, 137, 592–595 CrossRef CAS PubMed; (e) B. J. Truscott, A. M. Z. Slawin and S. P. Nolan, Dalton Trans., 2013, 42, 270–276 RSC; (f) B. Marciniec, S. Kostera, B. Wyrzykiewicza and P. Pawluć, Dalton Trans., 2015, 44, 782–786 RSC; (g) W.-B. Liu, D. P. Schuman, Y.-F. Yang, A. A. Toutov, Y. Liang, H. F. T. Klare, N. Nesnas, M. Oestreich, D. G. Blackmond, S. C. Virgil, S. Banerjee, R. N. Zare, R. H. Grubbs, K. N. Houk and B. M. Stoltz, J. Am. Chem. Soc., 2017, 139, 6867–6879 CrossRef CAS PubMed; (h) T. Yoshida, L. Ilies and E. Nakamura, Org. Lett., 2018, 20, 2844–2847 CrossRef CAS PubMed; (i) T. He, L.-C. Liu, L. Guo, B. Li, Q.-W. Zhang and W. He, Angew. Chem., Int. Ed., 2018, 57, 10868–10872 CrossRef CAS PubMed; (j) M. Skrodzki, S. Witomska and P. Pawluć, Dalton Trans., 2018, 47, 5948–5951 RSC.
- S. Furukawa, J. Kobayashi and T. Kawashima, J. Am. Chem. Soc., 2009, 131, 14192–14193 CrossRef CAS PubMed.
- L. D. Curless and M. J. Ingleson, Organometallics, 2014, 33, 7241–7246 CrossRef CAS.
- L. D. Curless, E. R. Clark, J. J. Dunsford and M. J. Ingleson, Chem. Commun., 2014, 50, 5270–5272 RSC.
- (a) Q. Yin, H. F. T. Klare and M. Oestreich, Angew. Chem., Int. Ed., 2016, 55, 3204–3207 CrossRef CAS PubMed; (b) Y. H. Ma, B. L. Wang, L. Zhang and Z. M. Hou, J. Am. Chem. Soc., 2016, 138, 3663–3666 CrossRef CAS PubMed; (c) N. Gandhamsetty, S. Park and S. Chang, J. Am. Chem. Soc., 2015, 137(48), 15176–15184 CrossRef CAS PubMed.
- Q.-A. Chen, H. F. T. Klare and M. Oestreich, J. Am. Chem. Soc., 2016, 138, 7868–7871 CrossRef CAS PubMed.
- Y. Han, S. Zhang, J. He and Y. Zhang, J. Am. Chem. Soc., 2017, 139, 7399–7407 CrossRef CAS PubMed.
- Y. Han, S. Zhang, J. He and Y. Zhang, ACS Catal., 2018, 8, 8765–8773 CrossRef CAS.
- J. Zhang, S. Park and S. Chang, J. Am. Chem. Soc., 2018, 140, 13209–13213 CrossRef CAS PubMed.
- (a) J. Hermeke, M. Mewald and M. Oestreich, J. Am. Chem. Soc., 2013, 135, 17537–17546 CrossRef CAS PubMed; (b) M. Pérez, C. B. Caputo, R. Dobrovetsky and D. W. Stephan, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 10917–10921 CrossRef PubMed; (c) L. Greb, S. Tamke and J. Paradies, Chem. Commun., 2014, 50, 2318–2320 RSC.
- (a) T. Stahl, P. Hrobarik, C. D. F. Konigs, Y. Ohki, K. Tatsumi, S. Kemper, M. Kaupp, H. F. T. Klare and M. Oestreich, Chem. Sci., 2015, 6, 4324–4334 RSC; (b) M. Arun, G. Srimanta, F. Sheng, D. Amit, S. V. Kumar, L. Peng and M. Debabrata, Angew. Chem., Int. Ed., 2017, 56, 14903–14907 CrossRef PubMed; (c) T. Lee and J. F. Hartwig, J. Am. Chem. Soc., 2017, 139, 4879–4886 CrossRef CAS PubMed; (d) L. Rubio-Pérez, M. Iglesias, J. Munárriz, V. Polo, V. Passarelli, J. J. Pérez-Torrentea and L. A. Oro, Chem. Sci., 2017, 8, 4811–4822 RSC; (e) Y. Ma, S. Lou, G. Luo, Y. Luo, G. Zhan, M. Nishiura, Y. Luo and Z. Hou, Angew. Chem., Int. Ed., 2018, 57, 15222–15226 CrossRef CAS PubMed; (f) R. Sakamoto, B.-N. Nguyen and K. Maruoka, Asian J. Org. Chem., 2018, 7, 1085–1088 CrossRef CAS.
- (a) D. J. Parks, J. M. Blakwell and W. E. Piers, J. Org. Chem., 2000, 65, 3090–3098 CrossRef CAS PubMed; (b) S. Rendler and M. Oestreich, Angew. Chem., Int. Ed., 2008, 47, 5997–6000 CrossRef CAS PubMed; (c) K. Sakata and H. Fujimoto, J. Org. Chem., 2013, 78, 12505–12512 CrossRef CAS PubMed; (d) M. Wen, F. Huang, G. Lu and Z.-X. Wang, Inorg. Chem., 2013, 52, 12098–12107 CrossRef CAS PubMed; (e) A. Y. Houghton, J. Hurmalainen, A. Mansikkamäki, W. E. Piers and H. M. Tuononen, Nat. Chem., 2014, 6, 983–988 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- (a) Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed; (b) Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS PubMed; (c) J. Miao, S. Hua and S. Li, Chem. Phys. Lett., 2012, 541, 7–11 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS PubMed.
- (a) K. Fukui, Acc. Chem. Res., 1981, 14, 363–368 CrossRef CAS; (b) H. P. Hratchian and H. B. Schlegel, Theory and Applications of Computational Chemistry: The First 40 Years, ed. C. E. Dykstra, Elsevier, Amsterdam, 2005, pp. 195−249 Search PubMed.
- C. Y. Legault, CYLview, 1.0b, Université de Sherbrooke, 2009, http://www.cylview.org Search PubMed.
- (a) S. W. Benson, The Foundations of Chemical Kinetics, McGraw-Hill, New York, 1960 Search PubMed; (b) Q. Liu, Y. Lan, J. Liu, G. Li, Y.-D. Wu and A. Lei, J. Am. Chem. Soc., 2009, 131, 10201–10210 CrossRef CAS PubMed; (c) F. Schoenebeck and K. N. Houk, J. Am. Chem. Soc., 2010, 132, 2496–2497 CrossRef CAS PubMed; (d) B. Liu, M. Gao, L. Dang, H. Zhao, T. B. Marder and Z. Lin, Organometallics, 2012, 31, 3410–3425 CrossRef CAS; (e) Q. Zhou and Y. Li, J. Am. Chem. Soc., 2015, 137, 10182–10189 CrossRef CAS PubMed.
- (a) A. F. G. Maier, S. Tussing, T. Schneider, U. Flörke, Z.-W. Qu, S. Grimme and J. Paradies, Angew. Chem., Int. Ed., 2016, 55, 12219–12223 CrossRef CAS PubMed; (b) M. Kojima and M. Kanai, Angew. Chem., Int. Ed., 2016, 55, 12224–12227 CrossRef CAS PubMed.
- S. Zhang, Y. Han, J. He and Y. Zhang, J. Org. Chem., 2018, 83(3), 1377–1386 CrossRef CAS PubMed.
- V. Sumerin, F. Schulz, M. Nieger, M. Leskelä, T. Repo and B. Rieger, Angew. Chem., Int. Ed., 2008, 47, 6001–6003 CrossRef CAS PubMed.
- (a) Á. Gyömöre, M. Bakos, T. Földes, I. Pápai, A. Domján and T. Soós, ACS Catal., 2015, 59, 5366–5372 CrossRef; (b) Y. Hoshimoto, T. Kinoshita, S. Hazra, M. Ohashi and S. Ogoshi, J. Am. Chem. Soc., 2018, 140(23), 7292–7300 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Free energy profiles, optimized geometries, corrected free energies, imaginary frequency and Cartesian coordinates. See DOI: 10.1039/c9ra07985h |
| ‡ P. Du and J. Zhao contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |