
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRetracted Article: Long non-coding RNA MEG3 inhibits cell proliferation, migration, invasion and enhances apoptosis in non-small cell lung cancer cells by regulating the miR-31-5p/TIMP3 axis†
Kui Li‡
ab,
Xiaodan Wang‡a,
Zhen Huanga,
Hui Xub,
Songbai Zheng *a and
Yurong Qiu*a
*a and
Yurong Qiu*a
aDepartment of Translational Medicine Research Institute, Guangzhou Huayin Medical Laboratory Center. Ltd, The Second Floor of Life Sciences Building of Southern Medical University No. 1838, North Guangzhou Street, Guangzhou, Guangdong, China. E-mail: npivyd@163.com; Tel: +86-18520035749
bTechnical Service Department, Guangzhou Huayin Medical Institute. Ltd, Guangzhou, Guangdong, China
First published on 22nd November 2019
Abstract
Non-small cell lung cancer (NSCLC) is a malignant lung cancer and accounts for 80% of lung cancer-related deaths. Long non-coding RNA maternally expressed gene 3 (MEG3) has been identified as a tumor suppressor in multiple cancers. However, the regulatory mechanism of MEG3 in NSCLC development is still largely unknown. The expression levels of MEG3, microRNA-31-5p (miR-31-5p) and tissue inhibitor of metalloproteinase 3 (TIMP3) in NSCLC tumors and cells were measured by quantitative real time polymerase chain reaction (qRT-PCR). Cell viability, apoptosis, migration and invasion were detected by cell counting kit-8 (CCK-8), flow cytometry, western blotting and transwell assays, respectively. Xenograft mouse models were established by subcutaneously injecting NSCLC cells stably transfected with Lenti-pcDNA or Lenti-MEG3. The interaction between miR-31-5p and MEG3 or TIMP3 was validated by luciferase reporter and RNA immunoprecipitation (RIP) assays. MEG3 and TIMP3 levels were up-regulated, whereas miR-31-5p expression was down-regulated in NSCLC tumors and cells compared with normal tissues and cells. Overexpression of MEG3 repressed cell proliferation, migration and invasion, but induced apoptosis in NSCLC cells. More importantly, MEG3 effectively hindered tumor growth in vivo. Next, luciferase reporter and RIP assays confirmed the interaction between miR-31-5p and MEG3 or TIMP3. Pearson's correlation coefficient revealed that miR-31-5p was inversely correlated with MEG3 or TIMP3. Rescue experiments indicated that MEG3 regulated TIMP3 expression by sponging miR-31-5p in NSCLC cells. Thus, MEG3 inhibited cell proliferation, migration and invasion, but enhanced apoptosis in NSCLC cells through up-regulating TIMP3 expression by regulating miR-31-5p, indicating novel biomarkers for the therapy of NSCLC.
Introduction
Non-small cell lung cancer (NSCLC) is a frequently diagnosed malignant cancer and comprises more than 80% of lung cancer-related deaths.1 NSCLC is categorized as squamous cell carcinoma (SCC) and adenocarcinoma (ADC) on the basis of histologic characteristics.2 To date, there are still limited therapy strategies to effectively overcome NSCLC, because most NSCLC patients are diagnosed at an advanced stage.3 In addition, distant metastasis, inflammatory response and drug resistance also impair the therapeutic efficiency.4 In recent years, immunotherapy has developed into an effective treatment method for NSCLC. Tumor mutational burden (TMB), a standard to evaluate the mutations of a tumor genome, is closely linked with immune checkpoint inhibitor efficacy and immunotherapy outcomes.5 Therefore, it is necessary to explore the pathogenesis of NSCLC genetically to develop effective therapeutics of NSCLC.Long noncoding RNAs (lncRNAs) are conserved transcripts without protein encoding capacity, which can regulate gene expression in the form of RNA at epigenetic, transcriptional and post-transcriptional level.6 Currently, a good deal of lncRNAs have been identified, and emerging evidence has demonstrated that lncRNAs are not transcriptional noise but act as critical modulators in a variety of physiological and pathological process in many human cancers, including proliferation, differentiation, metabolism and apoptosis7–9 thus affecting the occurrence and development of cancers. Maternally expressed 3 (MEG3) mapped on chromosome 14q32.3, is involved in pathology of various diseases, such as osteoarthritis, congenital intestinal atresia and cancers.10–12 For example, deficiency of MEG3 contributed to nerve growth and neurological impairment alleviation in rats with cerebral ischemia-reperfusion injury.13 In addition, MEG3 functioned as a tumor suppressor to retard cell growth and facilitate apoptosis in oral squamous cell carcinoma in vitro and in vivo by targeting miR-548d-3p through JAK-STAT pathway.14 Similarly, MEG3 enhanced cytotoxicity and blocked autophagy induced by adenosine in hepatic carcinoma cells through inhibition of ILF3 via PI3K-AKT-mTOR and beclin-1 signaling pathways.15 In NSCLC, MEG3 was found to be down-regulated, and low MEG3 expression suggested an unfavorable prognosis of NSCLC patients as well as linked with cell tumorigenesis and drug resistant in NSCLC.16–18 Herein, we further investigated the role of MEG3 in the progression and development of NSCLC.
Increasing studies have proposed lncRNA can serve as a sponge of microRNAs (miRNAs) to regulate the expression of genes, and the widespread existence of the interaction lncRNA-miRNA-mRNA network involved in the tumorigenesis has also been identified in a variety of cancers.19,20 Therefore, in this study, we not only aimed to investigate the potential biological of MEG3, but explored the underlying molecular mechanisms of MEG3 in the tumorigenesis and progression of NSCLC.
Materials and methods
Tissue samples
37 NSCLC patients were recruited from Guangzhou Huayin Medical Laboratory Center. Ltd. All the volunteers were informed and signed informed consent. NSCLC tissues and normal tissues were harvested by surgery and stored at −80 °C. Our protocols were approved by the Ethics Committee of Guangzhou Huayin Medical Laboratory Center. Ltd.Cell culture and transfection
NSCLC cell lines H522 and A549 were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA) and human bronchial epithelial cells 16HBE were purchased from Chuan Qiu Biotechnology (Shanghai, China). All cells were cultured in Dulbecco's Modified Eagle Medium (DMEM; Gibco, Carlsbad, CA, USA) containing 10% fetal bovine serum (FBS; Gibco) and 0.05% penicillin/streptomycin.MEG3 overexpression vector (MEG3) and its negative control pcDNA, small interfering RNA (siRNA) targeting MEG3 (si-MEG3), siRNA targeting TIMP3 (si-TIMP3) and siRNA negative control (si-NC) were synthesized by Genepharma (Shanghai, China). MiR-31-5p mimics, miR-31-5p inhibitor (in-miR-31-5p), miRNA mimics negative control (miR-NC) and miRNA inhibitor negative control inhibitor (anti-NC) were purchased from RIBOBIO (Guangzhou, China). All the vectors were transfected in H522 and A549 cells using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA).
Quantitative real time polymerase chain reaction (qRT-PCR)
NSCLC tumors and cells were resuspended with TRIzol reagent (Invitrogen) to obtain total RNA. The cDNA for MEG3, miR-31-5p and TIMP3 was synthesized by All-in-One™ First-Strand cDNA Synthesis Kit (FulenGen, Guangzhou, China). After that, qRT-PCR was conducted using SYBR green (Applied Biosystems, Foster City, CA, USA). The expression levels of MEG3, miR-31-5p and TIMP3 were normalized by U6 or GAPDH. The primers for MEG3, miR-31-5p and TIMP3 were listed: MEG3, (forward, 5′-GGTCTCTCCTCAGGGATGACAT-3′; reverse, 5′-TTAAGTCTTTAGGTAAGAGGGACAGC-3′); miR-31-5p, (forward, 5′-TATTCATAGGCAAGATGCTGGC-3′; reverse, 5′-TATGGTTGTTCTCGTCTCCTTCTC-3′); TIMP3, (forward, 5′-CTGACAGGTCGCGTCTATGA-3′; reverse, 5′-GCAAGGCAGGTAGTAGCAGG-3′); U6, (forward, 5′-ACCCTGAGAAATACCCTCACAT-3′; reverse, 5′-GACGACTGAGCCCCTGATG-3′); GAPDH, (forward, 5′-TCTACATGTTCCAGTATGACTC-3′; reverse, 5′-ACTCCACGACATACTCAGCACC-3′).CCK-8 assay
Transfected H522 and A549 cells (5000 cells per well) were placed onto 96-well plate and continuously cultured for different time. After reaction with CCK-8 reagent (10 μL per well) (Beyotime, Shanghai, China) for 2 h, cell viability was calculated based on the OD value (450 nm) measured by a spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA).Transwell assay
For migration assay, transfected H522 and A549 cells were inoculated on the transwell upper chamber for 24 h. Then, the migrated cells in the lower chamber were stained with crystal violet (Sigma, St. Louis, MO, USA), and then analyzed using microscope. For invasion assay, transwell upper chamber was pre-treated with Matrigel. After that, transfected H522 and A549 cells were inoculated on the upper chamber for 24 h. The following procedure is similar to migration assay.Flow cytometry
Transfected H522 and A549 cells (2 × 105 cell per well) were seeded on 24-well plate and continuously cultured for 24 h. Then, the cells were collected, resuspended and stained with Annexin V-FITC and PI. The number of apoptotic cells was counted by BD FACS Canto II flow cytometry (Becton Dickinson).Western blot assay
Western blot assay was conducted following the standard procedure. The primary antibodies against Bax, Bcl-2, cleaved caspase 3, β-actin and TIMP3 were purchased from Abcam (Cambridge, MA, USA) and HRP-conjugated secondary antibody was purchased from Sangon (Shanghai, China).Establishment of xenograft mice models
Male nude mice (4–5 week old, N = 6) were purchased from China Medical University (Shenyang, China). Xenograft mice models were established by subcutaneously injecting of H522 cells stably transfected with Lenti-pcDNAor Lenti-MEG3. After 28 d experiments, the tumors were collected from the xenograft mice. Tumor volume was recorded every 7 d. Tumor weight was measured at day 28. Animal experiments were approved by the National Animal Care and Ethics Institution and Guangzhou Huayin Medical Laboratory Center. Ltd, and this study was performed in strict accordance with the NIH guidelines for the care and use of laboratory animals (NIH publication no. 85-23 rev. 1985).Luciferase reporter assay
The putative binding sites of between miR-31-5p and MEG3 or TIMP3 were predicted by Diana Tools (http://diana.imis.athena-innovation.gr/DianaTools/index.php), and the process of prediction was presented in the ESI 1.† Wild type (MEG3 WT, TIMP3 3′UTR WT) and mutant type (MEG3 MUT, TIMP3 3′UTR MUT) luciferase vectors were constructed and co-transfected into H522 and A549 cells with miR-31-5p or miR-NC. Luciferase activities were detected by the Dual-Luciferase Reporter Assay System (Promega Corp., Madison, WI, USA).RNA immunoprecipitation (RIP)
RIP assay for H522 and A549 cells were performed using EZ-Magna RIP™ Kit (Millipore, Billerica, MA, USA). In brief, transfected H522 and A549 cells were lysed with RIP buffer and incubated with magnetic beads conjugated with Ago2 or IgG antibody. The immunoprecipitated RNA was subjected to qRT-PCR to detect the enrichment of MEG3 or TIMP3.Statistical analysis
Our data were presented as means ± standard deviation (SD). Data analyses were carried out using SPSS software (SPSS, Chicago, Illinois, USA) and GraphPad Prism 7 (GraphPad Inc., San Diego, CA, USA). The correlation between miR-31-5p and MEG3 or TIMP3 was analyzed by Pearson's correlation coefficient. P value less than 0.05 was considered as statistically significant.Results
MEG3 was down-regulated in NSCLC
MEG3 expression in NSCLC tumor tissues and cell lines was detected by qRT-PCR. As illustrated in Fig. 1A, MEG3 level was extremely lower in NSCLC tumors than that of normal tissues. Consistently, down-regulation of MEG3 expression was observed in NSCLC cell lines (H522, A549) compared with human bronchial epithelial cells 16HBE (Fig. 1B). Therefore, we determined that MEG3 might play suppressive role in NSCLC progression. | ||
| Fig. 1 MEG3 was down-regulated in NSCLC. (A) qRT-PCR analysis of MEG3 expression in NSCLC tumors and the normal tissues. (B) qRT-PCR analysis of MEG3 expression in NSCLC cell lines (H522, A549) and human bronchial epithelial cells 16HBE. *P < 0.05. | ||
Overexpression of MEG3 restricted cell proliferation, migration and invasion, but induced apoptosis in NSCLC cells
H522 and A549 cells were transfected with MEG3 or pcDNA to further evaluate the effects of MEG3 on NSCLC cell progression. MEG3 expression was elevated in NSCLC cells transfected with MEG3, indicating the successful transfection (Fig. 2A and B). Additionally, cell growth was attenuated in MEG3 transfection cells in comparison with pcDNA group (Fig. 2C and D). Apparently, MEG3 overexpression inhibited cell migration and invasion abilities (Fig. 2E and F). Moreover, cell apoptosis was enhanced in NSCLC cells transfected with MEG3 compared with pcDNA group (Fig. 2G). At the same time, we discovered that the expression levels of apoptosis associated protein Bax and cleaved caspase 3 were increased remarkably, whereas anti-apoptosis associated protein Bcl-2 level was decreased (Fig. 2H). These findings revealed that MEG3 repressed NSCLC cell progression. | ||
| Fig. 2 MEG3 repressed cell proliferation, migration, invasion and induced apoptosis in NSCLC cells. H522 and A549 cells were transfected with MEG3 and pcDNA. (A and B) qRT-PCR analysis of MEG3 expression in transfected H522 (A) and A549 cells (B). (C and D) Cell viability of transfected H522 (C) and A549 cells (D) was analyzed using CCK-8 assay. (E and F) Cell migration (E) and invasion (F) of transfected NSCLC cells was measured using transwell assay. (G) Cell apoptosis of transfected NSCLC cells was determined using flow cytometry. (H) Protein expression of Bcl-2, Bax and cleaved caspase 3 in transfected NSCLC cells was examined by western blot. *P < 0.05. | ||
MEG3 hindered tumor growth in vivo
Xenograft mice were established by subcutaneously injecting H522 cells transfected with Lenti-MEG3 or Lenti-pcDNA to illuminate the function of MEG3 in vivo. As displayed in Fig. 3A, tumor growth was suppressed in Lenti-MEG3 transfection mice compared with Lenti-pcDNA group. Tumor weight at day 28 was lower in Lenti-MEG3 transfection mice than that of Lenti-pcDNA group (Fig. 3B). To measure MEG3 level in tumors, the mice were sacrificed at day 28 and tumors were harvested. MEG3 level was significantly enhanced in Lenti-MEG3 transfection mice (Fig. 3C). Altogether, overexpression of MEG3 suppressed tumor growth in vivo.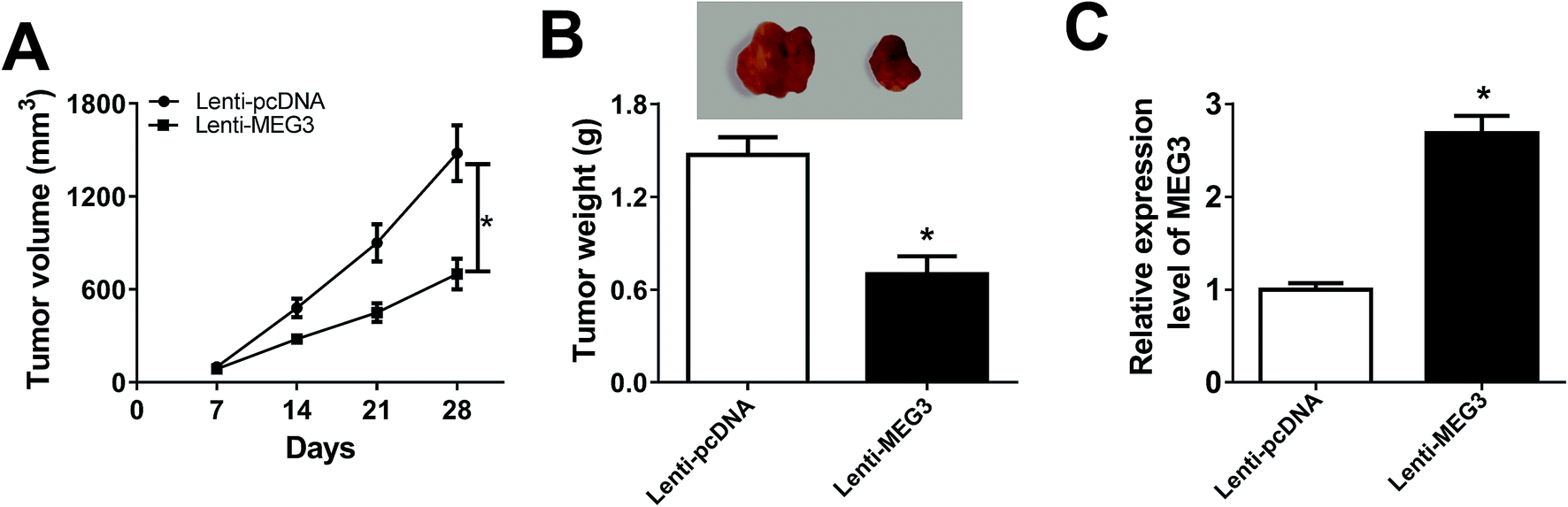 | ||
| Fig. 3 MEG3 hindered tumor growth in vivo. (A) Tumor volume was measured every 7 d. (B) Tumor weight was measured at day 28 when the mice were sacrificed. (C) MEG3 expression in tumor tissues from xenograft mice was investigated using qRT-PCR. *P < 0.05. | ||
MEG3 was a sponger of miR-31-5p
By searching from Diana Tools, we discovered that miR-31-5p had the potential to bind to MEG3 (Fig. 4A). Luciferase activity was reduced in NSCLC cells co-transfected with MEG3 WT and miR-31-5p, confirmed the interaction between MEG3 and miR-31-5p (Fig. 4B and C). Besides, RIP assay results showed that the enrichment of MEG3 was boosted in NSCLC cells transfected with miR-31-5p compared with miR-NC group (Fig. 4D). To investigate the role of miR-31-5p, the expression of miR-31-5p in NSCLC tumors and cells was measured by qRT-PCR. It was obvious that miR-31-5p expression was up-regulated in NSCLC tumors and cells compared with the corresponding normal ones (Fig. 4E and F). We further found that level of MEG3 was inversely correlated with miR-31-5p in NSCLC tissues (R = −0.9332, P < 0.0001) (Fig. 4G). Then si-MEG3 and si-NC were transfected into H522 and A549 cells with relatively high transfection efficiency (Fig. 4H). More importantly, MEG3 deletion increased miR-31-5p expression, whereas abundance of MEG3 decreased miR-31-5p expression (Fig. 4I). These results demonstrated that MEG3 could negatively regulate miR-31-5p expression in NSCLC cells.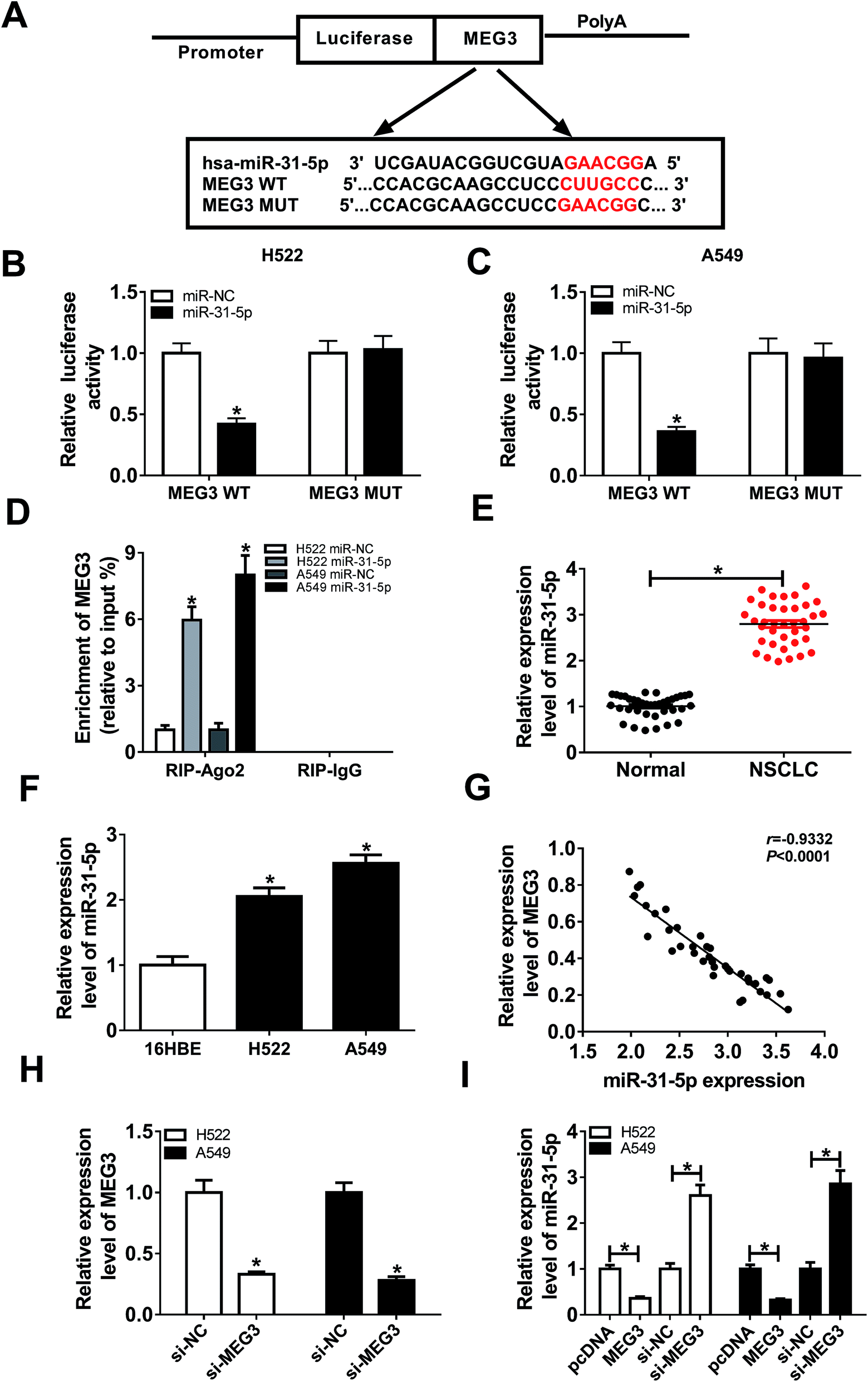 | ||
| Fig. 4 The interaction between MEG3 and miR-31-5p. (A) The potential binding sites between MEG3 and miR-31-5p were predicted by Diana Tools. (B and C) Luciferase activity of H522 (B) and A549 cells (C) co-transfected with MEG3 WT or MEG3 MUT and miR-31-5p or miR-NC was detected using Luciferase reporter assay. (D) Enrichment of MEG3 in H522 and A549 cells transfected with miR-31-5p or miR-NC after Ago2 RIP was measured using qRT-PCR. (E) qRT-PCR analysis of the expression of miR-31-5p in NSCLC tumors and the normal tissues was conducted. (F) The expression of miR-31-5p in NSCLC cell lines compared with human bronchial epithelial cells 16HBE was analyzed using qRT-PCR. (G) The correlation between MEG3 and miR-31-5p was analyzed by Pearson's correlation coefficient (R = −0.9332, P < 0.0001). (H) MEG3 expression in H522 and A549 cells transfected with si-NC or si-MEG3 was detected with qRT-PCR. (I) The expression of miR-31-5p in H522 and A549 cells transfected with pcDNA, MEG3, si-NC or si-MEG3 was determined using qRT-PCR. *P < 0.05. | ||
MEG3 regulated proliferation, migration, invasion and apoptosis by interacting with miR-31-5p in NSCLC cells
To explore the regulatory effects of MEG3/miR-31-5p axis on NSCLC cell progression, H522 and A549 cells were transfected with pcDNA, MEG3, MEG3 + miR-NC or MEG3 + miR-31-5p. As exhibited in Fig. 5A and B, miR-31-5p abolished the inhibition of MEG3 on cell viability. Consistently, cell migration and invasion were inhibited by MEG3 overexpression, whereas miR-31-5p reversed this suppression (Fig. 5C and D). Meanwhile, cell apoptosis was enhanced by MEG3, but was reduced by following miR-31-5p up-regulation (Fig. 5E). Furthermore, MEG3 boosted the expression of apoptosis associated protein Bax, cleaved caspase 3 and repressed expression of anti-apoptosis associated protein Bcl-2, while miR-31-5p up-regulation reversed protein expression trends mediated by MEG3 (Fig. 5F). Collectively, we concluded that MEG3 regulated proliferation, migration, invasion and apoptosis in NSCLC cells by targeting miR-31-5p. | ||
| Fig. 5 MiR-31-5p restored MEG3-mediated inhibition on proliferation, migration and invasion as well as promotion on apoptosis in NSCLC cells. H522 and A549 cells were transfected with pcDNA, MEG3, MEG3 + miR-NC or MEG3 + miR-31-5p. (A and B) CCK-8 analysis of cell viability in transfected H522 (A) and A549 cells (B) was performed. (C and D) Cell migration (C) and invasion (D) of transfected NSCLC cells was measured using transwell assay. (E) Cell apoptosis of transfected NSCLC cells was calculated by flow cytometry. (F) Protein expression of Bcl-2, Bax and cleaved caspase 3 in transfected NSCLC cells was analyzed using western blot. *P < 0.05. | ||
MEG3 regulated TIMP3 expression by sponging miR-31-5p
Bioinformatics analysis prediction by Diana Tools presented that miR-31-5p comprised the binding sites of 3′UTR of TIMP3 (Fig. 6A). Decreased luciferase activity in NSCLC cells co-transfected with TIMP3 3′UTR WT and miR-31-5p confirmed the interaction between TIMP3 and miR-31-5p (Fig. 6B and C). In addition, we found TIMP3 expression was down-regulated in NSCLC tumors and cells compared with normal tissues and cells (Fig. 6D and E). Person's correlation coefficient analysis results revealed that level of TIMP3 was negatively correlated with miR-31-5p (R = −0.9488, P < 0.0001) (Fig. 6F), but positively correlated with MEG3 in NSCLC tissues (R = 0.9374, P < 0.0001) (Fig. 6H). Moreover, TIMP3 protein expression in NSCLC cells was weakened by miR-31-5p (Fig. 6G). Specifically, miR-31-5p counteracted MEG3-mediated promotive effects on TIMP3 protein expression (Fig. 6I). Taken together, TIMP3 was a target of miR-31-5p. | ||
| Fig. 6 TIMP3 was a target of miR-31-5p. (A) The potential binding sites between TIMP3 and miR-31-5p analyzed by Diana Tools. (B and C) Luciferase activity of H522 (B) and A549 cells (C) co-transfected with TIMP3 3′UTR WT or TIMP3 3′UTR MUT and miR-31-5p or miR-NC was analyzed using Luciferase reporter assay. (D) qRT-PCR analysis of TIMP3 expression in NSCLC tumors and the normal tissues was conducted. (E) qRT-PCR analysis of TIMP3 expression in NSCLC cells (H522, A549) and 16HBE cells was performed. (F) The correlation betweenTIMP3 and miR-31-5p was analyzed by Pearson's correlation coefficient (R = −0.9488, P < 0.0001). (G) Western blot analysis of TIMP3 protein expression in NSCLC cells transfected with miR-31-5p or miR-NC was applied. (H) The correlation between MEG3 and TIMP3 was analyzed by Pearson's correlation coefficient (R = 0.9374, P < 0.0001). (I) Western blot analysis of TIMP3 protein expression in NSCLC cells transfected with pcDNA, MEG3, MEG3 + miR-NCorMEG3 + miR-31-5p was carried out. *P < 0.05. | ||
MiR-31-5p modulated cell proliferation, migration, invasion and apoptosis in NSCLCcells by targeting TIMP3
Since TIMP3 was a target of miR-31-5p, we speculated that miR-31-5p could affect NSCLC cell development through regulation of TIMP3. Rescue experiments were conducted to confirm the speculation. As shown in Fig. 7A and B, TIMP3 silence rescued miR-31-5p inhibitor induced suppressive effect on cell growth. Likewise, cell migration and invasion abilities were decreased by miR-31-5p inhibitor and recovered by TIMP3 silence (Fig. 7C and D). Conversely, cell apoptosis was promoted by miR-31-5p inhibitor, while TIMP3 silence reversed the promotive effect (Fig. 7E). Moreover, TIMP3 silence also reversed miR-31-5p inhibitor-mediated the promotion on Bax and cleaved caspase 3 protein expressions as well as the inhibition on Bcl-2 protein expression (Fig. 7F). Therefore, we considered that TIMP3 silence attenuated miR-31-5p inhibitor induced the inhibition on proliferation, migration, invasion as well as the promotion on apoptosis in NSCLC cells. | ||
| Fig. 7 TIMP3 silence attenuated miR-31-5p inhibitor induced suppression on proliferation, migration and invasion as well as acceleration on apoptosis in NSCLC cells. H522 and A549 cells were transfected with in-miR-NC, in-miR-31-5p, in-miR-31-5p + si-NC or in-miR-31-5p + si-TIMP3. (A and B) Cell viability of transfected H522 (A) and A549 cells (B) was detected by CCK-8 assay. (C and D) Cell migration (C) and invasion (D) of transfected NSCLC cells was measured using transwell assay. (E) Cell apoptosis of transfected NSCLC cells was determined with flow cytometry. (F) Western blot was used to determine the protein expression of Bcl-2, Bax and cleaved caspase 3 in transfected NSCLC cells. *P < 0.05. | ||
Discussion
Numerous evidences indicated that MEG3 served as a tumor suppressor and low level of MEG3 indicated poor prognosis of multiple cancer types, such as osteosarcoma, glioma and cervical cancer.21,22 Typically, MEG3 was also confirmed to be a poor prognostic factor in NSCLC, and decreased MEG3 expression predicted an unfavorable prognosis in NSCLC patients, especially in younger patients.17 Besides that, Wu et al. revealed that MEG3 overexpression interacted with miR-7-5p to up-regulate BRCA1 expression to increase cell apoptosis, thus inhibiting the progression of NSCLC.23 Lu et al. demonstrated decreased MEG3 linked with advanced pathologic stage, and tumor size, and MEG3 overexpression could suppressed cell proliferation and induced apoptosis to impede the tumorigenesis of NSCLC cells by activating p53 pathway.18 Moreover, abundance of MEG3 improved the sensitivity of NSCLC cells to paclitaxel through increasing the protein of p53.24 Therefore, it is imperative to disclose the role of MEG3 in NSCLC development.Recently, a new regulatory network has been focused on that lncRNA can crosstalk with mRNA by competing for shared miRNAs.25 In this study, bioinformatics prediction by Diana Tools revealed that miR-31-5p was a target of MEG3, and luciferase reporter assay certified the interaction between MEG3 and miR-31-5p. MiRNAs refer to small non-coding RNAs with 16–25 nucleotides in length and involve in gene regulation at post-transcriptional level by base-pairing 3′ untranslated regions (3′UTR) of the target genes.26,27 As oncogenes or tumor suppressors, they participate in cell cycle, growth, inflammation, invasion and autophagy by mRNA degradation and protein translation blockage in many diseases.28–30 For instance, miR-31-5p increased oncogenesis and development via interacting with ACOX1 and pro-migratory prostaglandin E2 in oral squamous cell carcinoma.31 Silence of miR-31-5p restricted cell progression in osteosarcoma by up-regulating AXIN1 through Wnt/β-catenin signaling.32 On the contrary, miR-31-5p was reported to be a suppressor to alleviate proliferation, migration, and invasion of hepatocellular carcinoma cells by binding to Sp1 transcription factor.33 It is apparently that ectopic expression of miR-31-5p is the major cause of different cancers. In NSCLC, miR-31-5p has been found to be increased,34 while the underlying function of miR-31-5p in NSCLC is unclear.
In this study, we investigated the regulatory mechanism of MEG3 in NSCLC cell proliferation, migration, invasion and apoptosis. Initially, we noticed that MEG3 and TIMP3 were down-regulated while miR-31-5p was up-regulated in NSCLC tumors and cells compared with normal tissues and cells. Overexpression of MEG3 was found to suppress proliferation, migration and invasion, but enhance apoptosis in NSCLC cells. More importantly, MEG3 inhibited tumor growth in vivo. In addition, luciferase reporter assay confirmed the interaction between miR-31-5p and MEG3 or TIMP3. Rescue experiments revealed that miR-31-5p attenuated MEG3-induced suppressive effect on the progression of NSCLC. Similarly, TIMP3 silence inversed miR-31-5p inhibitor-mediated promotion of NSCLC progression.
In conclusion, we determined the interaction of MEG3/miR-31-5p/TIMP3 axis in NSCLC development process. We found that MEG3 acted as a suppressor to inhibit proliferation, migration and invasion but promote apoptosis by up-regulating TIMP3 through inhibiting miR-31-5p expression, providing potential targets for the treatment of NSCLC.
Conflicts of interest
The authors have no conflict of interest to declare.References
- T. Yang, W. Zhang, L. Wang, C. Xiao, B. Guo, Y. Gong, X. Liang, D. Huang, Q. Li, Y. Nan, Y. Xiang and J. Shao, J. Int. Med. Res., 2019, 47, 481–493 CrossRef CAS PubMed.
- Y. Ma, S. Kang, X. Wu, B. Han, Z. Jin and Z. Guo, Tumori, 2018, 104, 338–343 CrossRef CAS PubMed.
- R. Zhang, Y. Xia, Z. Wang, J. Zheng, Y. Chen, X. Li, Y. Wang and H. Ming, Biochem. Biophys. Res. Commun., 2017, 490, 406–414 CrossRef CAS PubMed.
- Z. Qian, Q. Zhang, Y. Hu, T. Zhang, J. Li, Z. Liu, H. Zheng, Y. Gao, W. Jia, A. Hu, B. Li and J. Hao, Respir. Res., 2018, 19, 262 CrossRef CAS PubMed.
- H. Chang, A. Sasson, S. Srinivasan, R. Golhar, D. M. Greenawalt, W. J. Geese, G. Green, K. Zerba, S. Kirov and J. Szustakowski, Mol. Diagn. Ther., 2019, 23, 507–520 CrossRef CAS PubMed.
- T. R. Mercer, M. E. Dinger and J. S. Mattick, Nat. Rev. Genet., 2009, 10, 155–159 CrossRef CAS PubMed.
- X. Zhu, G. Yang, J. Xu and C. Zhang, Cancer Cell Int., 2019, 19, 82 CrossRef PubMed.
- X. Gao, J. Ge, W. Li, W. Zhou and L. Xu, Biol. Chem., 2018, 399, 375–386 CAS.
- Q. Jiang, J. Wang, X. Wu, R. Ma, T. Zhang, S. Jin, Z. Han, R. Tan, J. Peng, G. Liu, Y. Li and Y. Wang, Nucleic Acids Res., 2015, 43, D193–D196 CrossRef CAS.
- Z. Xia, D. Ding, N. Zhang, J. Wang, H. Yang and D. Zhang, Biomed. Pharmacother., 2019, 111, 436–442 CrossRef CAS PubMed.
- K. Chen, H. Zhu, M. Q. Zheng and Q. R. Dong, Cartilage, 2019, 1947603519855759 Search PubMed.
- B. Shen, N. Zhou, T. Hu, W. Zhao, D. Wu and S. Wang, J. Cell. Physiol., 2019, 234, 13464–13480 CrossRef CAS PubMed.
- D. You and H. You, Biomed. Pharmacother., 2019, 111, 1447–1457 CrossRef CAS PubMed.
- J. Tan, L. Xiang and G. Xu, IUBMB Life, 2019, 71, 882–890 CrossRef CAS PubMed.
- Z. Pu, L. Wu, Y. Guo, G. Li, M. Xiang, L. Liu, H. Zhan, X. Zhou and H. Tan, J. Cell. Biochem., 2019, 120, 18172–18185 CrossRef CAS PubMed.
- P. Wang, D. Chen, H. Ma and Y. Li, OncoTargets Ther., 2017, 10, 5137–5149 CrossRef PubMed.
- Z. Zhang, T. Liu, K. Wang, X. Qu, Z. Pang, S. Liu, Q. Liu and J. Du, Evidence from the GEO database, Gene, 2017, 630, 49–58 CrossRef CAS.
- K. H. Lu, W. Li, X. H. Liu, M. Sun, M. L. Zhang, W. Q. Wu, W. P. Xie and Y. Y. Hou, BMC Cancer, 2013, 13, 461 CrossRef PubMed.
- D. Liu, Y. Li, G. Luo, X. Xiao, D. Tao, X. Wu, M. Wang, C. Huang, L. Wang, F. Zeng and G. Jiang, Cancer Lett., 2017, 388, 281–291 CrossRef CAS PubMed.
- L. Zhao, T. Han, Y. Li, J. Sun, S. Zhang, Y. Liu, B. Shan, D. Zheng and J. Shi, FASEB J., 2017, 31, 893–903 CrossRef CAS.
- J. Zhang, Z. Lin, Y. Gao and T. Yao, J. Exp. Clin. Cancer Res., 2017, 36, 5 CrossRef PubMed.
- Z.-Z. Tian, X.-J. Guo, Y.-M. Zhao and Y. Fang, Int. J. Clin. Exp. Pathol., 2015, 8, 15138–15142 CAS.
- J. Wu, F. M. Meng and H. J. Li, Eur. Rev. Med. Pharmacol. Sci., 2018, 22, 5938–5945 Search PubMed.
- J. Xu, C. Su, F. Zhao, J. Tao, D. Hu, A. Shi, J. Pan and Y. Zhang, Biochem. Biophys. Res. Commun., 2018, 504, 123–128 CrossRef CAS PubMed.
- M. J. Huang, J. Y. Zhao, J. J. Xu, J. Li, Y. F. Zhuang and X. L. Zhang, Mol. Ther.--Nucleic Acids, 2019, 18, 533–545 CrossRef CAS PubMed.
- Y. Sun, D. Chen, L. Cao, R. Zhang, J. Zhou, H. Chen, Y. Li, M. Li, J. Cao and Z. Wang, Cardiovasc. Res., 2013, 100, 272–279 CrossRef CAS PubMed.
- P. Gao, J. Si, B. Yang and J. Yu, Med. Sci. Monit., 2017, 23, 881–888 CrossRef CAS PubMed.
- X. Liu, B. He, T. Xu, Y. Pan, X. Hu, X. Chen and S. Wang, J. Cancer, 2018, 9, 1218–1230 CrossRef PubMed.
- Z. Liu, F. Sun, Y. Hong, Y. Liu, M. Fen, K. Yin, X. Ge, F. Wang, X. Chen and W. Guan, Mol. Cancer, 2017, 16, 33 CrossRef PubMed.
- L. Liu, J. Wang, X. Li, J. Ma, C. Shi, H. Zhu, Q. Xi, J. Zhang, X. Zhao and M. Gu, Biochem. Biophys. Res. Commun., 2015, 457, 621–626 CrossRef CAS PubMed.
- Y. H. Lai, H. Liu, W. F. Chiang, T. W. Chen, L. J. Chu, J. S. Yu, S. J. Chen, H. C. Chen and B. C. Tan, Theranostics, 2018, 8, 486–504 CrossRef CAS PubMed.
- X. Chen, L. Zhong, X. Li, W. Liu, Y. Zhao and J. Li, Exp. Mol. Pathol., 2019, 108, 32–41 CrossRef CAS PubMed.
- G. Zhao, C. Han, Z. Zhang, L. Wang and J. Xu, Biochem. Biophys. Res. Commun., 2017, 490, 371–377 CrossRef CAS PubMed.
- C. Li, Y. Yin, X. Liu, X. Xi, W. Xue and Y. Qu, Oncotarget, 2017, 8, 24564–24578 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra07880k |
| ‡ The two authors contribute to this work equally as co-first authors. |
| This journal is © The Royal Society of Chemistry 2019 |