
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRetracted Article: Exosome-derived PTENP1 suppresses cisplatin resistance of bladder cancer (BC) by suppressing cell proliferation, migration and inducing apoptosis via the miR-103a/PDCD4 axis
Xingre Lu *,
Fengyu Chen,
Diao Yuan,
Xiang He,
Xiaowen Liu,
Yunju Zi and
Yu Lu
*,
Fengyu Chen,
Diao Yuan,
Xiang He,
Xiaowen Liu,
Yunju Zi and
Yu Lu
Department of Clinical Laboratory, The People's Hospital of Wenshan Prefecture in Yunnan Province, No. 228, Kaihua Middle Road, Wenshan, Yunnan 663000, China. E-mail: luoangedvxdf@163.com; Tel: +86 876 2124990
First published on 19th November 2019
Abstract
Bladder cancer (BC) is a lethal cancer that threatens the health of millions of people. Chemotherapy drug resistance, for example, cisplatin (DDP) resistance, is a huge limitation for BC therapy. PTEN pseudogene-1 (PTENP1) has been identified as a significant biomarker of multiple cancers. Therefore, it is essential to illuminate the molecular mechanism of PTENP1 in BC cell DDP resistance and progression. Serum exosomes were isolated using an ExoQuick precipitation kit. Serum exosomes were round-shaped vesicles of 100 ± 60 nm in size. The expression of PTENP1 was down-regulated in serum exosomes isolated from cisplatin non-responsive patients compared with responsive patients. ROC curves certified the diagnostic value of PTENP1. Apparently, PTENP1 transfection inhibited DDP-resistant BC cell proliferation, migration, cisplatin resistance and facilitated apoptosis. Next, we discovered that PTENP1 was a sponge of miR-103a, while PDCD4 was a target of miR-103a. More importantly, PTENP1 regulated DDP-resistant cell viability, migration, apoptosis and cisplatin resistance by interacting with the miR-103a/PDCD4 axis. In addition, PTENP1 hindered tumor growth of cisplatin-resistant mice. Exosome-derived PTENP1 suppressed the DDP resistance of BC by inhibiting cell proliferation, migration and promoting apoptosis through regulating the miR-103a/PDCD4 axis, representing a targeted therapy for DDP-resistant BC patients.
1. Introduction
Bladder cancer (BC) is an aggressive cancer in the genitourinary system.1 BC is capable of migrating to and invading adjacent tissues, such as sentinel lymph nodes.2 Despite early detection and conventional treatments, for example, surgical resection, chemotherapy and radiotherapy, having improved the therapeutic outcomes, the 5 year survival rate remains undesirable.3,4 Owing to chemotherapy drug resistance, for example cisplatin (DDP) resistance, many patients are still suffering from BC.5 Therefore, clarification of the mechanism of chemotherapy resistance in BC might provide novel strategies to reduce the relapse of BC patients.Long non-coding RNAs (lncRNAs) act as critical modulators in many diseases by participating in cell cycle, growth, differentiation, inflammation, metastasis, apoptosis and drug resistance.6–8 Exosomes are vesicles secreted by cells and they could transfer and exchange lncRNAs between tumors and extracellular microenvironment.9 LncRNA PTEN pseudogene-1 (PTENP1) mapped on chromosome 10 was recognized as a tumor suppressor to reduce tumorigenesis and progression of various cancers.10 It was reported that PTENP1 existed in serum exosomes of cancer patients.11 PTENP1 acted as tumor suppressor to inhibit cell cycle, proliferation in oral squamous cell carcinoma by targeting miR-21 and regulating AKT/CDK2 pathway.12 Li et al. reported that PTENP1 repressed cell proliferation, colony formation and initiated apoptosis of breast cancer by absorbing miR-20a through PI3K/AKT pathway.13 Therefore, we assumed that PTENP1 regulated DDP resistance and cell progression in BC.
MicroRNAs (miRNA) are small non-protein coding RNAs that regulate gene expression at post-transcriptional level.14 They participate in the pathological process of many diseases by regulating cell metabolism, inflammation, migration, autophagy, apoptosis and drug resistance.15–17 For example, reduced expression of miR-103a improved epilepsy induced neuron injury in rats by activating astrocytes in hippocampus through interacting with BDNF.18 Also, miR-103a was reported to restrict bone formation by acting as mechano-sensitive gene via modulating Runx2.19 In addition, miR-103a was involved in tumorigenesis of various cancers. For example, miR-103a restrained the deterioration of gastric cancer or abdominal aortic aneurysm by interacting with c-Myb or ADAM10.20,21 Therefore, we considered that PTENP1 might regulate cell proliferation, migration and drug resistance by competitively binding to miR-103a.
We attempted to reveal the molecular mechanism of exosome-derived PTENP1 in DDP-resistant BC cell proliferation, migration, apoptosis and drug resistance. Serum exosomes were isolated from cisplatin response and non-response BC patients. Rescue experiment was conducted to evaluate the regulatory effects of PTENP1, miR-103a and programmed cell death 4 (PDCD4).
2. Materials and methods
2.1. Patient samples
Cisplatin response (n = 37) and non-response (n = 34) BC patients were recruited from The People's Hospital of Wenshan Prefecture in Yunnan Province. Serum samples were collected from cisplatin response and non-response BC patients. Briefly, venous blood (5 mL) was collected from those participants by vena puncture before cisplatin treatment. Next, serum samples were centrifuged at 1600g for 10 min. Then, the samples were centrifuged at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for another 10 min to remove the residual cell debris. All experiments were performed in accordance with the Guidelines of 1964 Helsinki declaration and its later amendments or comparable ethical standards, and experiments were approved by the Ethics Committee of The People's Hospital of Wenshan Prefecture in Yunnan Province. Informed consents were obtained from human participants of this study.
000g for another 10 min to remove the residual cell debris. All experiments were performed in accordance with the Guidelines of 1964 Helsinki declaration and its later amendments or comparable ethical standards, and experiments were approved by the Ethics Committee of The People's Hospital of Wenshan Prefecture in Yunnan Province. Informed consents were obtained from human participants of this study.
2.2. Exosomes extraction
Serum exosomes were isolated from serum samples using ExoQuick precipitation kit (Systembiosciences, Mountain View, CA, USA). Firstly, the serum was centrifuged at 3000×g for 15 min to eliminate cell debris. Then, the supernatant was mixed with ExoQuick precipitation kit and centrifuged at 1500×g for 30 min, respectively. Next, the exosomes were centrifuged at 1500×g for 5 min to remove the residual liquid. Finally, the exosomes were re-suspended in phosphate-buffered saline (PBS).2.3. Transmission electron microscopy (TEM)
Serum exosome samples (20 μL) were dropped on carbon-coated copper grid for 5 min and then immersed the samples with 2% phosphotungstic acid for 2 min. After washing with PBS for 3 times, the prepared samples were captured by TEM (JEOL, Akishima, Japan).2.4. Nanoparticle tracking analysis (NTA)
NTA was applied to analyze the size and concentration of serum exosomes. In brief, the serum exosomes were re-suspended in 1 mL PBS. Next, the samples were injected in NanoSight NS300 (Malvern Panalytical, Malvern, UK). The size and concentration of serum exosomes were automatically detected by NanoSight NS300.2.5. Cell culture and transfection
T24 and 5637 cells were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). Cisplatin (DDP)-resistant T24 (T24/DDP) and 5637 (5637/DDP) cells were obtained from the Biomedical Science Cell Bank (Shanghai, China). The IC50 values for T24, T24/DDP, 5637, and 5637/DDP cells were 15.89 μM, 83.56 μM, 11.91 μM and 80.45 μM. The resistance indexes for T24/DDP and 5637/DDP cells were 5.37 and 6.75, respectively. The cells were cultured in complete DMEM medium (Gibco, Carlsbad, CA, USA). Small interfering RNA targeting PDCD4 (si-PDCD4), negative control (si-NC), PTENP1 and PDCD4 overexpression vectors were synthesized by Genepharma (Shanghai, China). MiR-103a, miR-103a inhibitor (anti-miR-103a), negative control (miR-NC) and negative control inhibitor (anti-NC) were purchased from RIBOBIO (Guangzhou, China). Cell transfection was performed using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA).2.6. Western blot
Protein CD9, CD63 and tumor susceptibility gene 101 (TSG101) were collected from serum exosomes whereas PDCD4, cleaved poly-ADP-ribose-polymerase (cleaved-PARP), cleaved caspase3 (cleaved-casp3) and cleaved caspase9 (cleaved-casp9) were isolated from T24/DDP and 5637/DDP cells. The proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, transferred to polyvinylidene difluoride membranes (Millipore, Bedford, Mass, USA) and blocked by 5% nonfat milk. Next, the membranes were incubated with primary antibodies against CD9 (ab92726, 1:5000), CD63 (ab217345, 1:2000), TSG101 (ab125011, 1:5000), PDCD4 (ab80590, 1:5000), cleaved-PARP (ab32064, 1:10000), cleaved-casp3 (ab2302, 1:10000) and cleaved-casp9 (ab2324, 1:5000) or GAPDH (ab181602, 1:10000) (Abcam, Cambridge, MA, USA) and HRP-conjugated secondary antibody (Sangon, Shanghai, China).
2.7. Quantitative real-time polymerase chain reaction (qRT-PCR)
Exosome samples and BC cells were lysed by TRIzol reagent (Invitrogen) to obtain total RNA. Next, the cDNA for PTENP1, miR-103a and PDCD4 was synthesized by All-in-One™ Kit (FulenGen, Guangzhou, China). After that, qRT-PCR was conducted using SYBR green (Applied Biosystems, Foster City, CA, USA). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and U6 were employed as internal references. The primers for PTENP1, miR-103a and PDCD4, GAPDH and U6 were listed: PTENP1, (forward, 5′-TTCGTCTTCTCCCCATTCCG-3′; reverse, 5′-TGCAGGATGGAAATGGCTCT-3′); miR-103a, (forward, 5′-ACACTCCAGCTGGGAGCAGCATTGTACAGGGC-3′; reverse, 5′-TGGTGTCGTGGAGTCG-3′); PDCD4 (forward, 5′-ATGAGCAGATACTGAATGTAAAC-3′; reverse, 5′-CTTTACTTCCTCAGTCCCAGCAT-3′); GAPDH, (forward, 5′-AGGTCGGTGTGAACGGATTTG-3′; reverse, 5′-GGGGTCGTTGATGGCAACA-3′); U6, (forward, 5′-ACCCTGAGAAATACCCTCACAT-3′; reverse, 5′-GACGACTGAGCCCCTGATG-3′).2.8. Cell counting kit-8 (CCK-8) assay
T24, T24/DDP, 5637 and 5637/DDP cells (5000 cells per well) were plated on 96-well plates overnight. Next, the cells were treated with DDP (0.005 μM, 0.05 μM, 0.5 μM, 5 μM, 50 μM, 500 μM) for 48 h and transfected with different vectors for another 48 h. For cell proliferation detection, transfected T24/DDP and 5637/DDP cells were allowed to grow for 24 h, 48 h and 72 h. Then, the cells were added with 10 μL CCK-8 (Beyotime, Shanghai, China) for 2 h. The optical density (OD) value (450 nm) was detected by a spectrophotometer. Half maximal inhibitory concentration (IC50) values were determined by GraphPad Prism 7 (San Diego, CA, USA).2.9. Transwell assay
Transfected T24/DDP and 5637/DDP cells were plated on the upper chamber of transwell (Corning, New York, NY, USA) and allowed to migrate for 48 h. After that, the cells at the lower chamber of transwell were stained with 0.1% crystal violet (Sigma, St. Louis, MO, USA). Lastly, the migration cell numbers were recorded by a microscope.2.10. Flow cytometry
Transfected T24/DDP and 5637/DDP cells were plated on 24-well plates overnight and treated with 5 μM DDP for 48 h. Then, the cells were collected and stained by fluorescein isothiocyanate tagged Annexin V (Annexin V-FITC)/propidium iodide (PI) detection kit (Invitrogen). Lastly, the apoptotic rate was analyzed by a flow cytometer.2.11. Dual-luciferase reporter assay
The interaction between miR-103a and PTENP1 or PDCD4 was proved by dual-luciferase reporter assay. Wild type PTENP1 (WT-PTENP1), PDCD4 (WT-PDCD4), mutant type PTENP1 (MUT-PTENP1) and PDCD4 (MUT-PDCD4) luciferase vectors were constructed. Those vectors were co-transfected in T24/DDP and 5637/DDP cells with miR-103a or miR-NC. Luciferase activities were determined using a luminometer.2.12. Establishment of xenograft mice
Male nude mice (4–5 weeks) were purchased from Jinan Pengyue Animal Center (Jinan, China). The mice were randomly divided into 4 groups (n = 6): Vector, PTENP1, Vector + DDP and PTENP1 + DDP. Animal models were constructed by subcutaneously injecting T24/DDP cells transfected with Vector and PTENP1. After a week, the mice were intraperitoneally injected with cisplatin (5 mg kg−1) or PBS. The mice were sacrificed after 4 weeks and tumors were collected. Animal experiments were approved by Ethics Committee of The People's Hospital of Wenshan Prefecture in Yunnan Province and followed NIH Guidelines for the Care and Use of Laboratory Animals (NIH publication no. 85-23 rev. 1985) and was approved by the Institutional Animal Care and Use Committee of National Tissue Engineering Center (Shanghai, China).2.13. Statistical analysis
The data were presented as means ± standard deviation (SD). Statistical analysis was performed by GraphPad Prism 7. Two-tailed t-tests were applied to compare the differences among multiple groups. Receiver operating characteristic (ROC) curves were plotted to evaluate the diagnostic value of PTENP1. P-value less than 0.05 (P < 0.05) was considered statistically significant.3. Results
3.1. Downregulation of PTENP1 in serum exosomes from cisplatin-resistant BC patients
Serum exosomes were extracted from cisplatin-resistant and non-resistant BC patients to explore the potential role of exosomes-derived PTENP1. TEM image of exosomes showed the morphology (round-shaped vesicles) and size (100 ± 60 nm) of exosomes (Fig. 1A). Nanoparticle trafficking results exhibited that the concentration of exosomes was approximately 5.3 × 107 particles per mL while the diameters were ranged from 50 nm to 160 nm (Fig. 1B). In addition, the expression of exosomal markers CD9, CD63 and TSG101 was enhanced in serum exosomes compared with supernatant (Fig. 1C). Next, we measured the expression of PTENP1 in serum exosomes isolated from cisplatin response and non-response BC patients by qRT-PCR. As illustrated in Fig. 1D, PTENP1 was low expressed in cisplatin non-response patients. What's more, ROC curves revealed that the area under the ROC curve (AUC) was 0.769. The sensitivity and specificity of PTENP1 indicated the existence of BC (Fig. 1E). Besides, cisplatin-resistant patients possessed low level of PTENP1 whereas cisplatin-response patients comprised high level of PTENP1 (Fig. 1F). Taken together, PTENP1 expression was closely linked with cisplatin resistance of BC patients.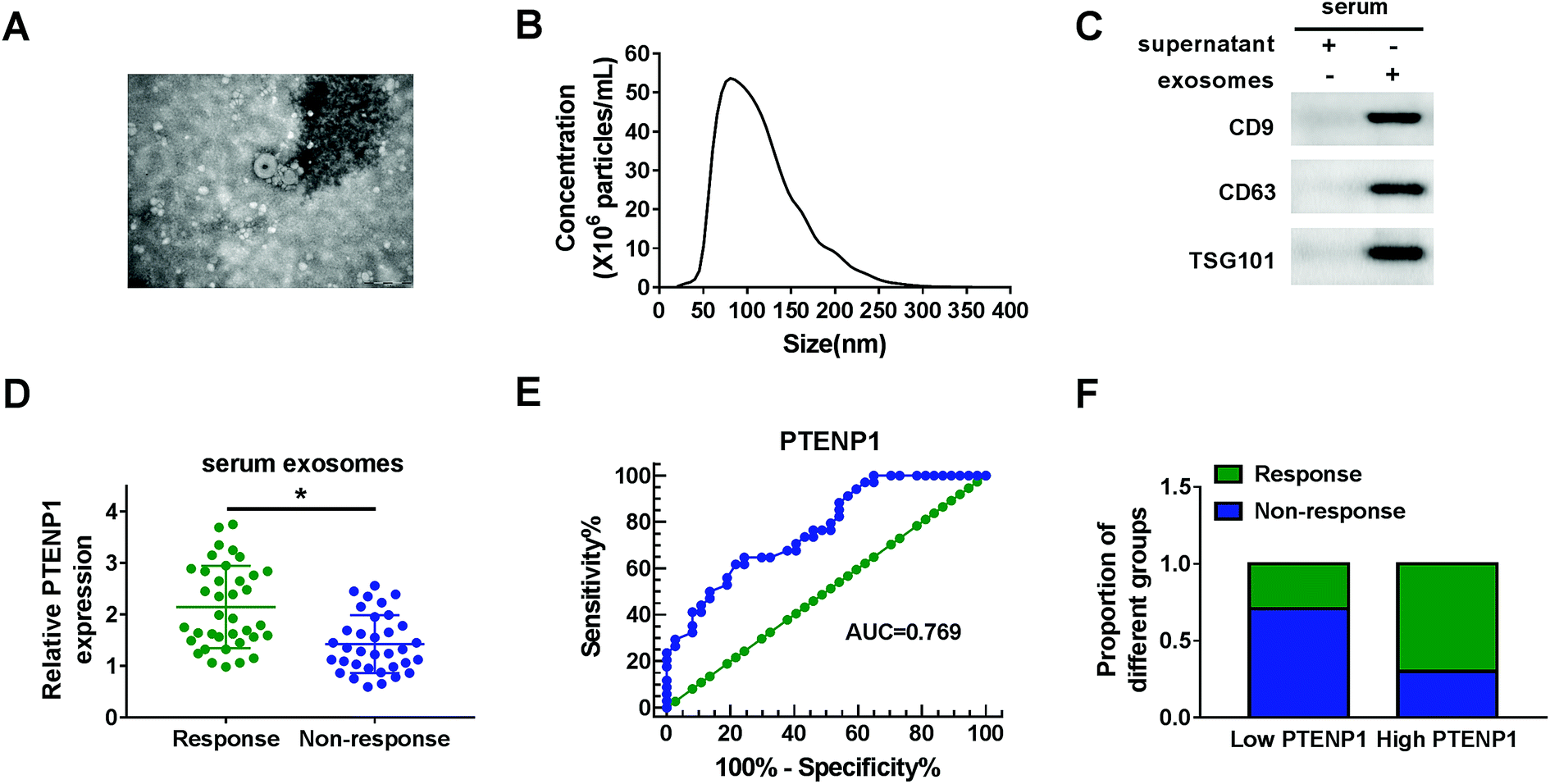 | ||
| Fig. 1 PTENP1 expression was down-regulated in serum exosomes from cisplatin-resistant BC patients. (A) The image of exosomes was captured by TEM. (B) Size and concentration of exosomes were analyzed by nanoparticle trafficking. (C) The expression of exosomal markers CD9, CD63 and TSG101 was detected by western blot. (D) PTENP1 expression in serum exosomes from cisplatin response (n = 37) and non-response (n = 34) BC patients was measured by qRT-PCR. (E) ROC curves were plotted to determine the diagnostic value of PTENP1 in serum exosomes. (F) The proportion of response and non-response to cisplatin in BC patients with high and low expression of PTENP1. *P < 0.05. | ||
3.2. Overexpression of PTENP1 suppressed cell proliferation, migration, cisplatin resistance and induced apoptosis of cisplatin-resistant BC cells
The influence of PTENP1 on T24/DDP and 5637/DDP cells was further investigated to illuminate the function of PTENP1 in cisplatin-resistant BC cells. CCK-8 assay was conducted to evaluate cisplatin-induced cytotoxicity of T24, T24/DDP, 5637 and 5637/DDP cells. As displayed in Fig. 2A, cell viability was distinctly inhibited by cisplatin in T24 and 5637 cells compared with DDP-resistant cells. Besides, DDP IC50 was higher in DDP-resistant cells than that of normal BC cells. We also observed that PTENP1 expression was down-regulated in T24/DDP and 5637/DDP cells in comparison with T24 and 5637 cells (Fig. 2B). Then, T24/DDP and 5637/DDP cells were transfected with Vector and PTENP1 to investigate the effects of PTENP1 on DDP-resistant cell growth, migration, apoptosis and cisplatin resistance. The transfection efficiency was confirmed by qRT-PCR (Fig. 2C). CCK-8 assay result exhibited that DDP IC50 was decreased by PTENP1 transfection, indicating PTENP1 inhibited cisplatin resistance of T24/DDP and 5637/DDP cells (Fig. 2D). Consistently, cell proliferation and migration of DDP-resistant cells were attenuated by PTENP1 (Fig. 2E and F). Conversely, PTENP1 transfection improved cell apoptosis of DDP-resistant cells (Fig. 2G). Meanwhile, the expression of pro-apoptotic protein cleaved-PARP, cleaved-casp3 and cleaved-casp9 in T24/DDP and 5637/DDP cells was boosted by PTENP1 compared with Vector group (Fig. 2H and I). These findings demonstrated that PTENP1 could repress cell progression and cisplatin resistance of DDP-resistant BC cells.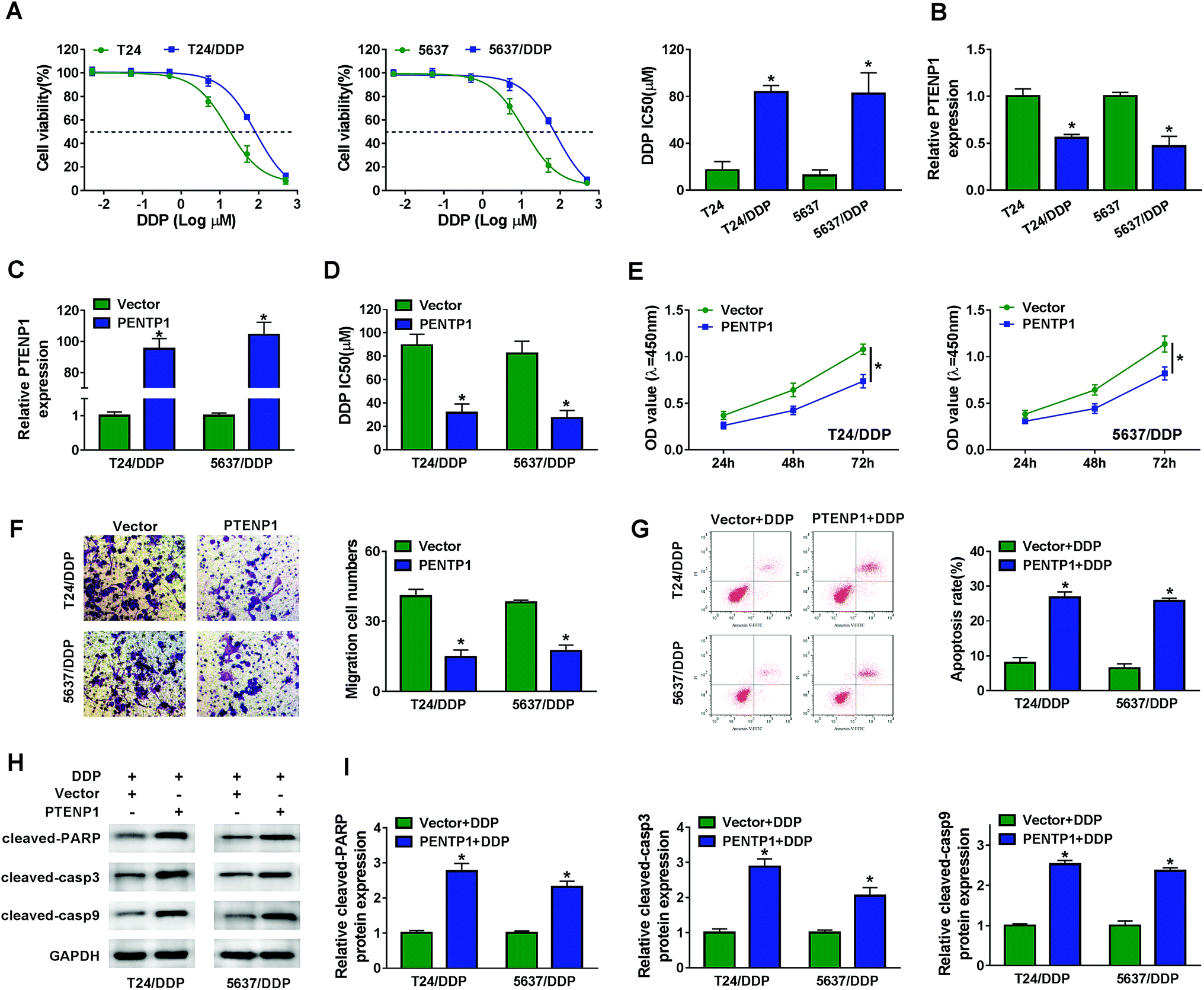 | ||
| Fig. 2 PTENP1 inhibited cell proliferation, migration, cisplatin resistance and stimulated cell apoptosis in BC. T24/DDP and 5637/DDP cells were transfected with Vector and PTENP1. (A) Cell viability of T24, T24/DDP, 5637 and 5637/DDP cells treated with cisplatin (DDP) (0.005 μM, 0.05 μM, 0.5 μM, 5 μM, 50 μM, 500 μM) for 48 h was evaluated by CCK-8 assay. (B) PTENP1 expression in T24, T24/DDP, 5637 and 5637/DDP cells was measured by qRT-PCR. (C) Analysis of PTENP1 expression in cells using qRT-PCR was performed. (D) IC50 of DDP was determined by CCK-8 assay. (E and F) Cell viability and migration were detected by CCK-8 and transwell assay, respectively. (G) Cell apoptosis of T24/DDP and 5637/DDP cells transfected with Vector or PTENP1 and treated with 5 μM DDP was assessed by flow cytometry. (H and I) Protein expression of cleaved-PARP, cleaved-casp3 and cleaved-casp9 was assessed by western blot. *P < 0.05. | ||
3.3. PTENP1 modulated cisplatin-resistant BC cell proliferation, migration, apoptosis and drug resistance by interacting with miR-103a
Bioinformatics analysis by MiRcode indicated that PTENP1 could specifically bind to miR-103a (Fig. 3A). Declined luciferase activity in T24/DDP and 5637/DDP cells co-transfected with WT-PTENP1 and miR-103a validated the interaction between PTENP1 and miR-103a (Fig. 3B). In addition, miR-103a expression in T24/DDP and 5637/DDP cells was reduced by PTENP1 (Fig. 3C). Synchronously, we discovered that miR-103a expression was relatively higher in T24/DDP and 5637/DDP cells than that of T24 and 5637 cells (Fig. 3D). To explore the regulatory effects of miR-103a on cell progression, T24/DDP and 5637/DDP cells were transfected with miR-NC, miR-103a, anti-NC and anti-miR-103a. As displayed in Fig. 3E, miR-103a expression was elevated by miR-103a transfection and lowered by anti-miR-103a transfection. Reduced DDP IC50 in cells transfected with anti-miR-103a manifested that miR-103a inhibitor could decrease cisplatin resistance (Fig. 3F). More importantly, miR-103a inhibitor attenuated cell proliferation (Fig. 3G), migration (Fig. 3H) and initiated apoptosis (Fig. 3I) of T24/DDP and 5637/DDP cells. As expected, miR-103a inhibitor boosted the expression of protein cleaved-PARP, cleaved-casp3 and cleaved-casp9 (Fig. 3J–K). Collectively, PTENP1 acted as a sponge of miR-103a to regulate cell progression of DDP-resistant BC cells.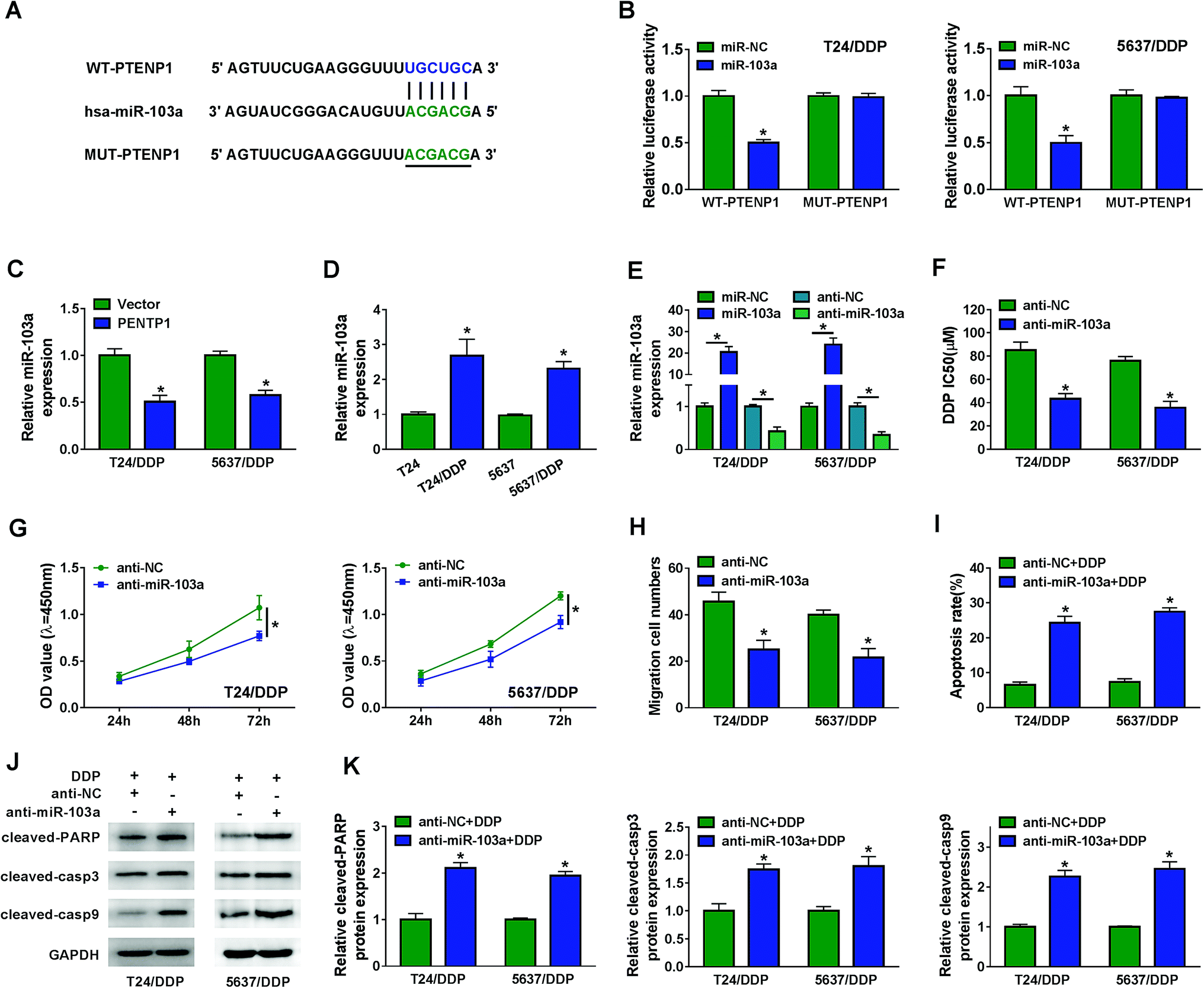 | ||
| Fig. 3 PTENP1 acted as a sponge of miR-103a. (A) The putative binding sites between PTENP1 and miR-103a were predicted by MiRcode. (B) Luciferase activity of T24/DDP and 5637/DDP cells co-transfected with WT-PTENP1 or MUT-PTENP1 and miR-103a or miR-NC was examined by dual-luciferase reporter assay. (C) The expression of miR-103a in T24/DDP and 5637/DDP cells transfected with Vector and PTENP1 was detected by qRT-PCR. (D) The expression of miR-103a in T24, T24/DDP, 5637 and 5637/DDP cells was examined by qRT-PCR. (E) The expression of miR-103a in T24/DDP and 5637/DDP cells transfected with miR-NC, miR-103a, anti-NC and anti-miR-103a was measured by qRT-PCR. (F) IC50 of DDP in T24/DDP and 5637/DDP cells transfected with anti-NC and anti-miR-103a was analyzed by CCK-8 assay. (G and H) Cell viability and migration were evaluated by CCK-8 and transwell assay. (I) Cell apoptosis of T24/DDP and 5637/DDP cells transfected with anti-NC or anti-miR-103a and treated with 5 μM DDP was detected by flow cytometry. (J and K) Protein expression of cleaved-PARP, cleaved-casp3 and cleaved-casp9 was analyzed by western blot. *P < 0.05. | ||
3.4. PDCD4 acted as a target of miR-103a to regulate cisplatin resistance, cell viability, migration and apoptosis in BC
As predicted by starBase, miR-103a contained the binding sites of PDCD4 (Fig. 4A). Luciferase activity was decreased in T24/DDP and 5637/DDP cells co-transfected with WT-PDCD4 and miR-103a, revealing that PDCD4 interacted with miR-103a (Fig. 4B). We also noticed that PDCD4 protein expression in T24/DDP and 5637/DDP cells was boosted by miR-103a inhibitor (Fig. 4C). Interestingly, PDCD4 protein expression was down-regulated in T24/DDP and 5637/DDP cells compared with T24 and 5637 cells (Fig. 4D). Enhanced protein expression of PDCD4 in DDP-resistant cells transfected with PDCD4 indicated cell transfection was performed successfully (Fig. 4E). In addition, DDP IC50 was decreased in T24/DDP and 5637/DDP cells transfected with PDCD4, revealing that cisplatin resistance was repressed by PDCD4 (Fig. 4F). As expected, we discovered that overexpression of PDCD4 reduced proliferation (Fig. 4G), migration (Fig. 4H) and enhanced apoptosis (Fig. 4I) of DDP-resistant cells. Upregulation of protein cleaved-PARP, cleaved-casp3 and cleaved-casp9 further clarified that PDCD4 induced DDP-resistant cell apoptosis (Fig. 4J–K). Altogether, miR-103a regulated DDP-resistant cell viability, migration, apoptosis and drug resistance by targeting PDCD4.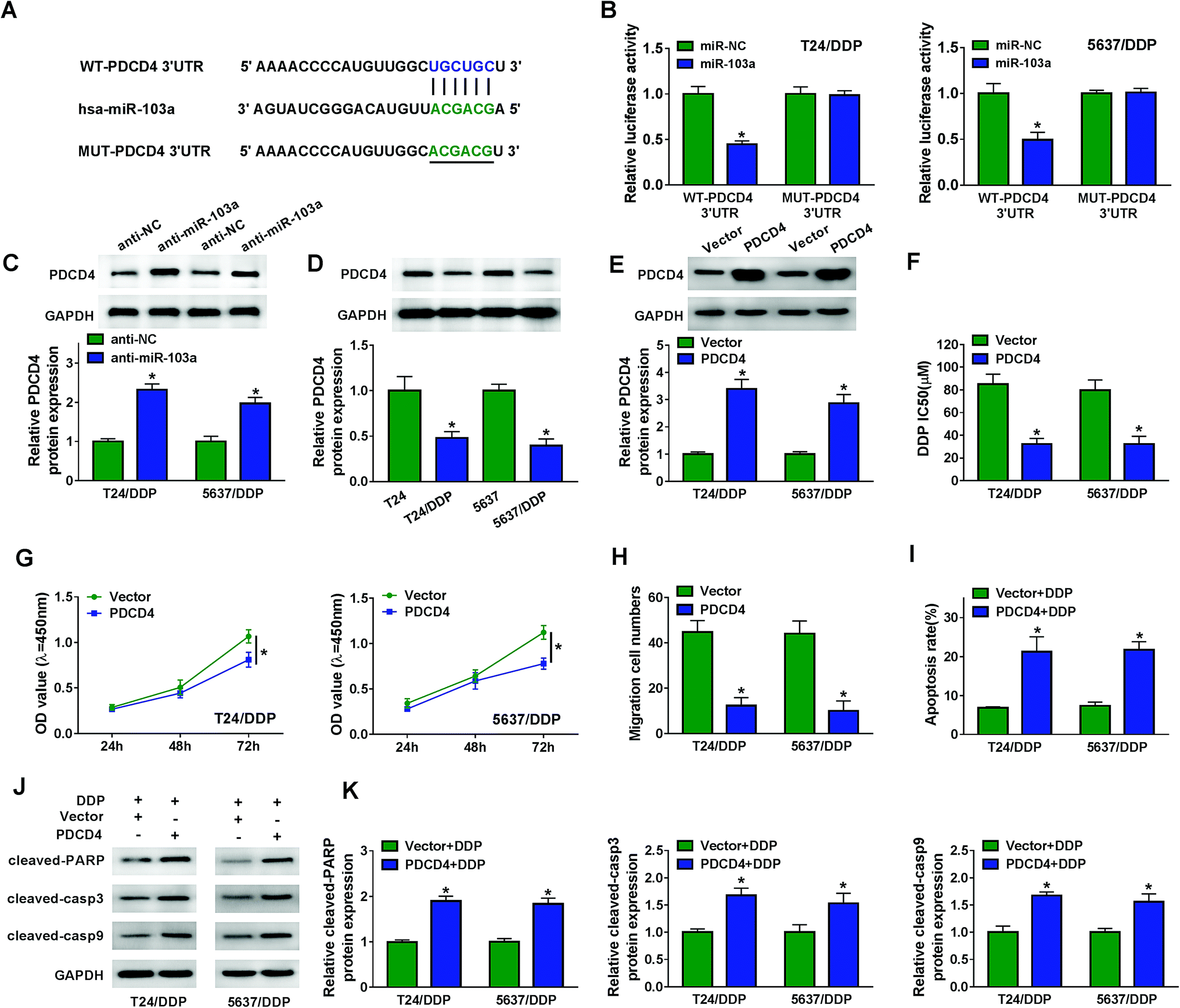 | ||
| Fig. 4 PDCD4 was a target of miR-103a. (A) The putative binding sites between PDCD4 and miR-103a were analyzed by starBase. (B) Luciferase activity of T24/DDP and 5637/DDP cells co-transfected with WT-PDCD4 or MUT-PDCD4 and miR-103a or miR-NC was evaluated by dual-luciferase reporter assay. (C) PDCD4 protein expression in T24/DDP and 5637/DDP cells transfected with anti-NC and anti-miR-103a was assessed by western blot. (D) PDCD4 protein expression in T24, T24/DDP, 5637 and 5637/DDP cells was analyzed by western blot. (E) PDCD4 protein expression in T24/DDP and 5637/DDP cells transfected with Vector and PDCD4 was determined by western blot. (F) IC50 of DDP was evaluated by CCK-8 assay. (G and H) Cell viability and migration were determined by CCK-8 and transwell assay. (I) Cell apoptosis of T24/DDP and 5637/DDP cells transfected with Vector or PDCD4 and treated with 5 μM DDP was examined by flow cytometry. (J and K) Protein expression of cleaved-PARP, cleaved-casp3 and cleaved-casp9 was analyzed by western blot. *P < 0.05. | ||
3.5. PTENP1 participated in DDP-resistant cell progression by regulating PDCD4 expression via sponging miR-103a
To expound the molecular mechanism of DDP-resistant cell progression, T24/DDP and 5637/DDP cells were transfected with Vector, PTENP1, PTENP1 + miR-NC, PTENP1 + miR-103a, PTENP1 + si-NC and PTENP1 + si-PDCD4. As illustrated in Fig. 5A, PDCD4 protein expression was elevated by PTENP1 whereas decreased by miR-103a and PDCD4 knockdown. Significantly, DDP IC50 was reduced by PTENP1 and the reduction was attenuated by miR-103a and PDCD4 silencing in T24/DDP and 5637/DDP cells (Fig. 5B). Meanwhile, miR-103a and PDCD4 silencing counteracted PTENP-mediated suppression on proliferation (Fig. 5C), migration (Fig. 5D) and promotion on apoptosis (Fig. 5E) of T24/DDP and 5637/DDP cells. Besides, PTENP promoted protein expression of cleaved-PARP, cleaved-casp3 and cleaved-casp9. However, the promotion of PTENP on apoptosis-associated protein expression was reversed by miR-103a and PDCD4 silencing (Fig. 5F and G). These results demonstrated that PTENP could interact with miR-103a/PDCD4 axis to regulate cell progression and cisplatin resistance.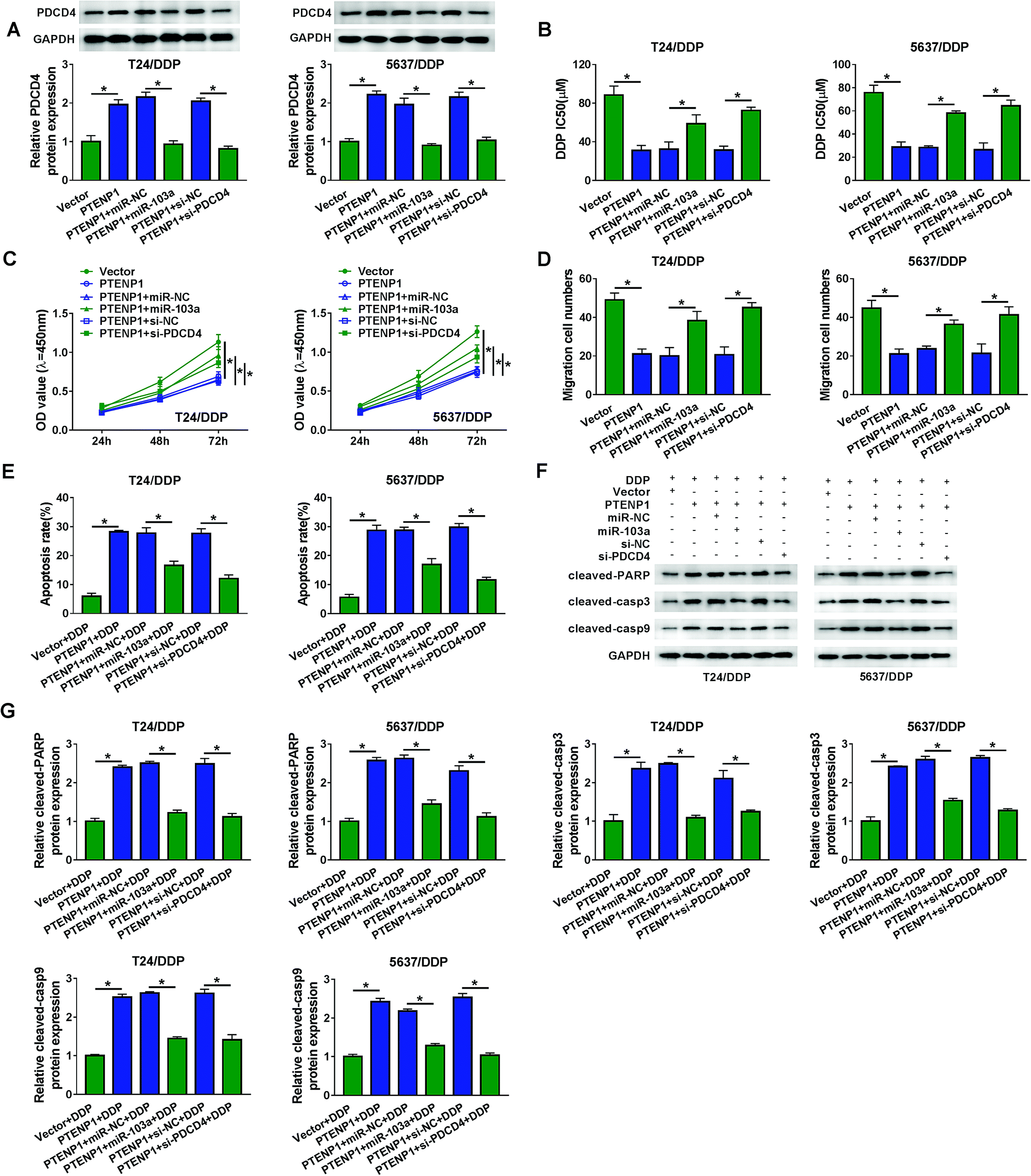 | ||
| Fig. 5 PTENP1 regulated cell proliferation, migration, apoptosis and cisplatin resistance in BC by miR-103a/PDCD4 axis. T24/DDP and 5637/DDP cells were transfected with Vector, PTENP1, PTENP1 + miR-NC, PTENP1 + miR-103a, PTENP1 + si-NC and PTENP1 + si-PDCD4. (A) PDCD4 protein expression was evaluated by western blot. (B) IC50 of DDP was assessed by CCK-8 assay. (C and D) Cell proliferation and migration were detected by CCK-8 and transwell assay. (E) Evaluation of cell apoptosis by flow cytometry. (F and G) Western blot was performed to analyze protein expression of cleaved-PARP, cleaved-casp3 and cleaved-casp9. *P < 0.05. | ||
3.6. PTENP1 hindered tumor growth of DDP-resistant mice
We further investigated the regulatory effects of PTENP1 on xenograft mice. As shown in Fig. 6A–C, PTENP1 suppressed tumor volume and weight of DDP-resistant mice. In addition, the expression of PTENP1 was increased whereas miR-103a was decreased in tumor tissues collected from DDP-resistant mice (Fig. 6D). Meanwhile, the expression of PDCD4 protein in DDP-resistant mice tumors was boosted by PTENP1 (Fig. 6E). Therefore, PTENP1 restrained tumor growth in DDP-resistant mice tumor by up-regulating PDCD4 via sponging miR-103a.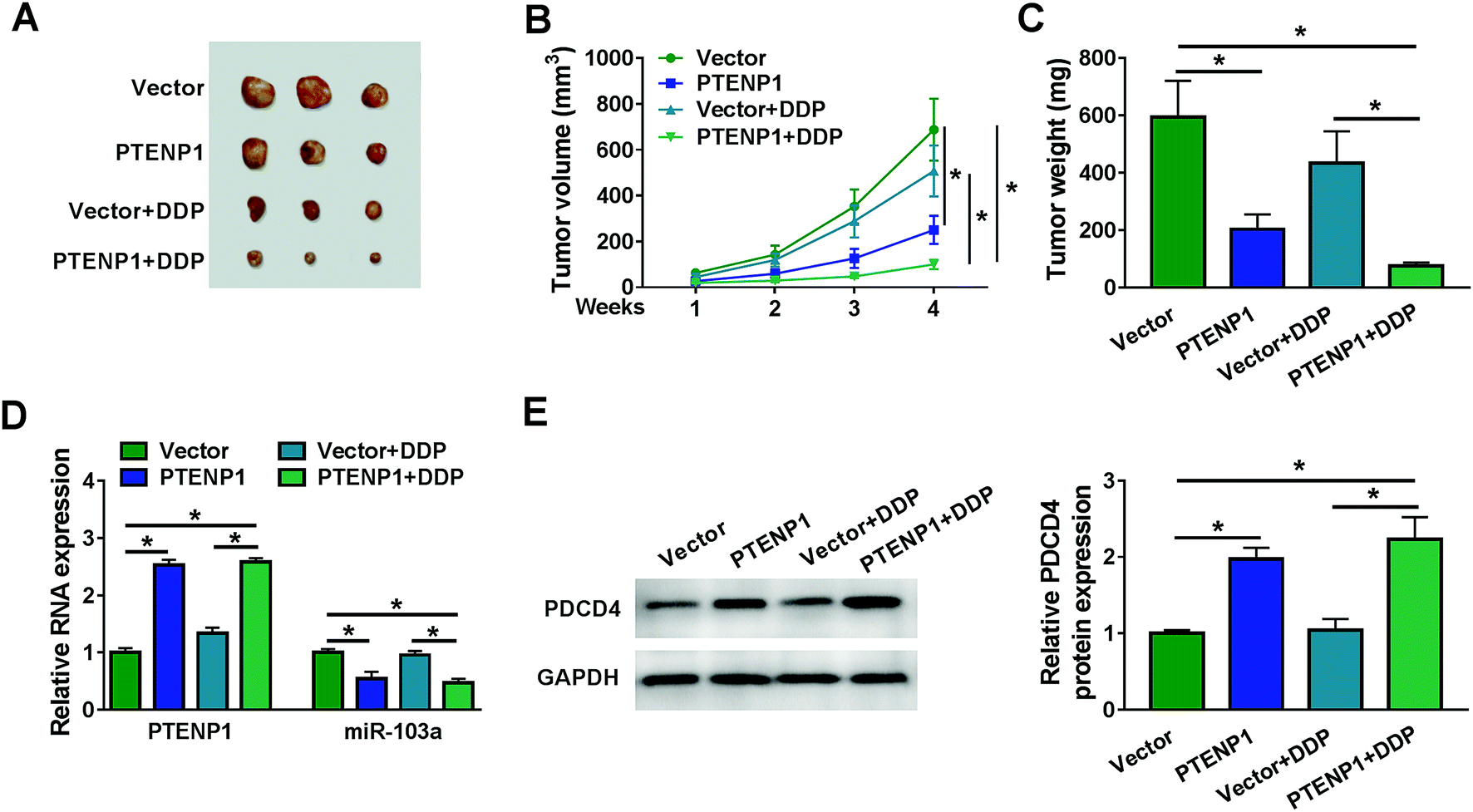 | ||
| Fig. 6 PTENP1 inhibited DDP-resistant tumor growth in vivo. (A) Images of tumors collected from the xenograft mice were captured. (B) Tumor volume was measured every week. (C) Tumor weight was measured after 4 weeks. (D) The expression of PTENP1 and miR-103a in tumors collected from the xenograft mice was detected by qRT-PCR. (E) Protein expression of PDCD4 was evaluated by western blot. *P < 0.05. | ||
4. Discussion
Growing evidences have validated that PTENP1 was involved in the progression of a variety of cancers.22–24 For example, PTENP1 alleviated the malignancy of hepatocellular carcinoma in vivo by inducing cell apoptosis via sponging miR-193a-3p and regulating PTEN/AKT pathway.25 Consistently, PTENP1 was reported to attenuate cell survival, metastasis and accelerate cell apoptosis in gastric cancer by competitively binding to miR-106b as well as miR-93 to regulate PTEN expression.26 Overexpression of PTENP1 attenuated cell survival and migration by regulating AKT/MAPK pathway in breast cancer.27 In addition, enhanced expression of PTENP1 inhibited cell growth, migration in esophageal squamous cell carcinoma or breast cancer by regulating SOCS6 or miR-19b, respectively.28,29 Therefore, it is imperative to disclose the role of PTENP1 in cisplatin resistance of BC cells.As bioinformatics analysis by online database MiRcode, PTENP1 could specifically bind to miR-103a. We also confirmed that PTENP1 regulated BC cell viability, migration, apoptosis and DDP resistance by interacting with miR-103a. Previous studies have clarified that miR-103a was a crucial biomarker of different diseases, such as acute myocardial infarction and cancer.30,31 For example, miR-103a was reported to improve the cancerogenic effects of macrophages in hypoxic lung cancer by interacting with PTEN.32 Moreover, miR-103a participated in cell progression in colorectal carcinoma or gastric cancer by regulating Wnt pathway or ATF7.33,34 Therefore, it is of great clinical significance to reveal the role of miR-103a in DDP-resistant BC cells.
In conclusion, we demonstrated the regulatory effects of PTENP1 in DDP-resistant BC cells. Down-regulation of PTENP1 expression in cisplatin non-response exosomes indicated that PTENP1 might inhibit cisplatin resistance. Functional experiment confirmed that PTENP1 attenuated cell viability, migration and cisplatin resistance in DDP-resistant cells. What's more, we discovered that PTENP1 could regulate cell proliferation, migration, apoptosis and cisplatin resistance in DDP-resistant BC cells by sponging miR-103a to alter PDCD4 expression. Animal experiments revealed that overexpression of PTENP1 hindered DDP-resistant tumor growth in vivo. In short, exosome-derived PTENP1 attenuated cisplatin resistance and cell progression of BC through miR-103a/PDCD4 axis, providing prospective therapy of DDP-resistant BC patients.
Funding
This study was supported by “Two Kinds of Talents” funding project of Yunnan Province (Grant No. 2016HB050).Conflicts of interest
There is no conflict of interest regarding the publication of this paper.References
- E. Laloglu, H. Aksoy, Y. Aksoy, F. Ozkaya and F. Akcay, Ann. Clin. Biochem., 2016, 53, 647–653 CrossRef CAS PubMed.
- W. Usuba, F. Urabe, Y. Yamamoto, J. Matsuzaki, H. Sasaki, M. Ichikawa, S. Takizawa, Y. Aoki, S. Niida, K. Kato, S. Egawa, T. Chikaraishi, H. Fujimoto and T. Ochiya, Cancer Sci., 2019, 110, 408–419 CAS.
- J. Li, Y. Li, F. Meng, L. Fu and C. Kong, Biosci. Rep., 2018, 38 DOI:10.1042/bsr20171701.
- W. S. Tan, W. P. Tan, M. Y. Tan, P. Khetrapal, L. Dong, P. deWinter, A. Feber and J. D. Kelly, Cancer Treat. Rev., 2018, 69, 39–52 CrossRef CAS PubMed.
- P. Li, X. Yang, Y. Cheng, X. Zhang, C. Yang, X. Deng, P. Li, J. Tao, H. Yang, J. Wei, J. Tang, W. Yuan, Q. Lu, X. Xu and M. Gu, Cell. Physiol. Biochem., 2017, 41, 921–932 CrossRef CAS PubMed.
- L. Chen, H. Yang, Z. Yi, L. Jiang, Y. Li, Q. Han, Y. Yang, Q. Zhang, Z. Yang, Y. Kuang and Y. Zhu, J. Cancer Res. Clin. Oncol., 2019, 145, 637–652 CrossRef CAS PubMed.
- F. Fei, Y. He, S. He, Z. He, Y. Wang, G. Wu and M. Li, Biosci. Rep., 2018, 38 DOI:10.1042/bsr20180420.
- X. Hu, D. Ding, J. Zhang and J. Cui, Biosci. Rep., 2019, 39 DOI:10.1042/bsr20181038.
- N. A. Patel, L. D. Moss, J. Y. Lee, N. Tajiri, S. Acosta, C. Hudson, S. Parag, D. R. Cooper, C. V. Borlongan and P. C. Bickford, J. Neuroinflammation, 2018, 15, 204 CrossRef PubMed.
- R. K. Li, J. Gao, L. H. Guo, G. Q. Huang and W. H. Luo, Cancer Gene Ther., 2017, 24, 309–315 CrossRef CAS PubMed.
- R. Zheng, M. Du, X. Wang, W. Xu, J. Liang, W. Wang, Q. Lv, C. Qin, H. Chu, M. Wang, L. Yuan, J. Qian and Z. Zhang, Mol. Cancer, 2018, 17, 143 CrossRef PubMed.
- L. Gao, W. Ren, L. Zhang, S. Li, X. Kong, H. Zhang, J. Dong, G. Cai, C. Jin, D. Zheng and K. Zhi, Mol. Carcinog., 2017, 56, 1322–1334 CrossRef CAS PubMed.
- X. Gao, T. Qin, J. Mao, J. Zhang, S. Fan, Y. Lu, Z. Sun, Q. Zhang, B. Song and L. Li, J. Exp. Clin. Cancer Res., 2019, 38, 256 CrossRef PubMed.
- X. Liang, Q. Zhao, T. Geng, S. Luo and Q. He, J. Biochem. Mol. Toxicol., 2018, 32, e22169 CrossRef PubMed.
- G. Wang, S. Wang and C. Li, Tumour Biol., 2017, 39, 1010428317703825 Search PubMed.
- Y. Zhang, Y. Zhao, L. Liu, H. Su, D. Dong, J. Wang, Y. Zhang, Q. Chen and C. Li, Technol. Cancer Res. Treat., 2018, 17, 1533033818793652 Search PubMed.
- L. Wu, Z. Chen and Y. Xing, Cell Biol. Int., 2018 DOI:10.1002/cbin.11041.
- P. Zheng, H. Bin and W. Chen, Cancer Cell Int., 2019, 19, 109 CrossRef PubMed.
- B. Zuo, J. Zhu, J. Li, C. Wang, X. Zhao, G. Cai, Z. Li, J. Peng, P. Wang, C. Shen, Y. Huang, J. Xu, X. Zhang and X. Chen, J. Bone Miner. Res., 2015, 30, 330–345 CrossRef CAS PubMed.
- J. Liang, X. Liu, H. Xue, B. Qiu, B. Wei and K. Sun, Cell Proliferation, 2015, 48, 78–85 CrossRef CAS PubMed.
- T. Jiao, Y. Yao, B. Zhang, D. C. Hao, Q. F. Sun, J. B. Li, C. Yuan, B. Jing, Y. P. Wang and H. Y. Wang, BioMed Res. Int., 2017, 2017, 9645874 Search PubMed.
- Y. Zhang and C. Xu, J. Cell. Biochem., 2019, 120, 19738–19748 CrossRef CAS PubMed.
- G. Yu, W. Yao, K. Gumireddy, A. Li, J. Wang, W. Xiao, K. Chen, H. Xiao, H. Li, K. Tang, Z. Ye, Q. Huang and H. Xu, Mol. Cancer Ther., 2014, 13, 3086–3097 CrossRef CAS PubMed.
- N. Haddadi, Y. Lin, G. Travis, A. M. Simpson, N. T. Nassif and E. M. McGowan, Mol. Cancer, 2018, 17, 37 CrossRef PubMed.
- Y. Y. Qian, K. Li, Q. Y. Liu and Z. S. Liu, Oncotarget, 2017, 8, 107859–107869 Search PubMed.
- R. Zhang, Y. Guo, Z. Ma, G. Ma, Q. Xue, F. Li and L. Liu, Oncotarget, 2017, 8, 26079–26089 Search PubMed.
- S. Chen, Y. Wang, J. H. Zhang, Q. J. Xia, Q. Sun, Z. K. Li, J. G. Zhang, M. S. Tang and M. S. Dong, Oncol. Lett., 2017, 14, 4659–4662 CrossRef PubMed.
- T. Gong, S. Zheng, S. Huang, S. Fu, X. Zhang, S. Pan, T. Yang, Y. Sun, Y. Wang, B. Hui, J. Guo and X. Zhang, Mol. Carcinog., 2017, 56, 2610–2619 CrossRef CAS PubMed.
- X. Shi, X. Tang and L. Su, Oncol. Res., 2018, 26, 869–878 CrossRef PubMed.
- A. R. Scheffer, S. Holdenrieder, G. Kristiansen, A. von Ruecker, S. C. Muller and J. Ellinger, World J. Urol., 2014, 32, 353–358 CrossRef CAS PubMed.
- L. Huang, L. Li, X. Chen, H. Zhang and Z. Shi, Cardiol. J., 2016, 23, 556–562 Search PubMed.
- Y. L. Hsu, J. Y. Hung, W. A. Chang, S. F. Jian, Y. S. Lin, Y. C. Pan, C. Y. Wu and P. L. Kuo, Mol. Ther., 2018, 26, 568–581 CrossRef CAS PubMed.
- X. Hu, J. Miao, M. Zhang, X. Wang, Z. Wang, J. Han, D. Tong and C. Huang, Mol. Cells, 2018, 41, 390–400 CAS.
- A. Fasihi, B. M. Soltani, A. Atashi and S. Nasiri, J. Cell. Biochem., 2018, 119, 5104–5117 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |