DOI:
10.1039/C9RA07737E
(Paper)
RSC Adv., 2019,
9, 38902-38911
Three cobalt-based coordination polymers with tripodal carboxylate and imidazole-containing ligands: syntheses, structures, properties and DFT studies†
Received
24th September 2019
, Accepted 20th November 2019
First published on 26th November 2019
Abstract
Three cobalt-based coordination polymers [Co(Htatb)(1,3-bimyb)] (1), [Co(Htatb)(bimbp)]·DMF (2), and [Co(Htatb)(1,4-bimyb)]·H2O (3) [H3tatb = 4,4′,4′′-s-triazine-2,4,6-tribenzoic acid, 1,3-bimyb = 1,3-bis(imidazole-1-ylmethyl)benzene, bimbp = 4,4′-bis(imidazolyl)biphenyl, 1,4-bimyb = 1,4-bis (imidazole-1-ylmethyl)benzene] were synthesized by hydrothermal reactions and characterized by single-crystal X-ray diffraction, thermogravimetric analyses, IR spectroscopy, UV-vis spectroscopy and elemental analysis. Compound 1 shows a double-strand chain structure, due to the intermolecular O–H⋯O hydrogen bonds and aromatic π–π stacking interactions, the adjacent chains are connected to produce a 3D supramolecular structure. Compound 2 shows a 2D structure with a 1D channel. Compound 3 displays a 2D layer structure, furthermore these layers are joined by O–H⋯O hydrogen bonding to generate a four-fold interpenetrating 3D architecture. The fluorescence properties of 1–3 and the magnetic behavior of 1 and 2 have also been investigated. Based on their crystal structures, compounds 1 and 2 were investigated using hybrid DFT methods at the B3LYP/6-31G (d) level. The DFT-BS approach was applied to study the magnetic coupling behavior. The results reveal that the calculated exchange coupling constants J were in good agreement with the experimental data.
1 Introduction
Metal–organic coordination polymers of mixed-ligand assembly have become a very attractive research subject.1–4 This is not only due to their intriguing structural diversity but also to the intriguing potential applications of functional materials, such as selective molecular recognition and separation, gas storage and separation, absorption, luminescence, molecular magnetism, ion-exchange and heterogeneous catalysis.5–8 In this context, the predesign and assembly of novel crystalline materials by structure-directing factors, such as central metal ions, organic ligands, the metal–ligand ratio, solvents, temperature, pH value, and other factors, have been validated and summarized.9–12 The mixed-ligand strategy by the judicious choice of various organic linkers has been proven to be highly-efficient for the construction of metal–organic coordination polymers. Among such systems, most outstanding is the incorporation of polycarboxylates and N-donor co-ligands,13–15 which has successfully been utilized to generate diverse and interesting polymeric networks with potential properties and contributes to refining our knowledge of self-assembly processes. Within polycarboxylate ligands, aromatic polycarboxyl compounds have extensively been documented as multifunctional tectons, owing to their versatile linking capability in virtue of both covalent bonding and supramolecular interactions.16–18
In our strategy, multidentate O- or N-donor ligands have also been employed in the construction of coordination polymers. Among the family of organic carboxylate, tripodal carboxylate shows more superiority,19–21 the ligand 4,4′,4′′-s-triazine-2,4,6-tribenzoic acid (H3tatb) as a class example of tripodal ligands has been utilized, and some metal–organic coordination polymers based on H3tatb have also been investigated.22–28 However, metal–organic coordination polymers based on H3tatb and N-donor ligands have only been investigated scarcely.29 To explore the influence of N-donor ligands on achieving different dimensional and topological structures based on tripodal carboxylate ligand, we also employ 1,3-bis(imidazole-1-ylmethyl)benzene(1,3-bimyb), 4,4′-bis(imidazolyl)biphenyl(bimbp), and 1,4-bis(imidazole-1-ylmethyl)benzene(1,4-bimyb) with different conformations as co-ligands. Three cobalt-based coordination polymers [Co(Htatb)(1,3-bimyb)] (1), [Co(Htatb)(bimbp)]·DMF (2), and [Co(Htatb)(1,4-bimyb)]·H2O (3) were synthesized and characterized. In addition, their fluorescent properties and magnetic behavior were also investigated in this paper.
2 Experimental sections
2.1 Materials and chemical analysis
The H3tatb, 1,3-bimyb, bimbp and 1,4-bimyb ligands were purchased in the Jinan Henghua Sci. & Tec. Co., Ltd.; all other reagents and solvents employed were commercially available and used without further purification. Elemental analyses were performed with a PerkinElmer 2400 CHN Elemental analyzer. Infrared spectra on KBr pellets were recorded on a Nicolet 170SX FT-IR spectrophotometer in the range 400–4000 cm−1. UV-vis spectra were recorded on a Shimadzu UV-3600 spectrophotometer in the region 200–700 nm. TG analyses were conducted with a Nietzsch STA 449C micro analyzer under atmosphere at a heating rate of 5 °C min−1. X-ray powder diffraction (PXRD) patterns were recorded on a Shimadzu XRD-7000 diffractometer analyzer. The magnetic susceptibilities were obtained on crystalline samples using a Quantum Design MPMS SQUID magnetometer. The fluorescence spectra were studied using a Hitachi F-7100 fluorescence spectrophotometer at room temperature.
2.2 Computational details
All calculations have been processed in Gaussian 03 package.30 The magnetic isotropic shielding tensors were carried out with the hybrid DFT method on the basis of B3LYP functional.31 The experimentally determined geometries for the complete structures of compounds 1–3 were used for the calculation of the magnetic exchange coupling constants.32 Neither variation of the geometrical parameters nor the geometry optimization was attempted in this calculation because a small variation in the geometry can have a big effect on the calculated magnetic interaction parameters.
2.3 Synthesis of [Co(Htatb)(1,3-bimyb)] (1)
A mixture of CoCl2·6H2O (0.1 mmol, 0.024 g), H3tatb (0.1 mmol, 0.026 g), 1,3-bimyb (0.1 mmol, 0.024 g) and 8 mL DMF-H2O (V![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :V = 1:1) was stirred for 30 min in air. The mixture was then transferred to a 20 ml airtight glass reactor and kept at 100 °C for 5 days under autogenous pressure, and then cooled to room temperature at a rate of 5 °C h−1. Red crystals of 1 were obtained and washed with DMF and dried in the air (yield: 53% based on Co). Elemental analysis (%). Calcd for C38H27N7O6Co: C, 61.96; H, 3.69; N, 13.31%. Found: C, 61.64; H, 3.78; N, 13.56%. IR data (KBr cm−1): 3412 (s), 2358 (w), 1711 (s), 1652 (s), 1547 (m), 1502 (m), 1406 (m), 1134 (w), 1072 (w), 837 (m), 765 (m), 672 (w), 585 (w).
:V = 1:1) was stirred for 30 min in air. The mixture was then transferred to a 20 ml airtight glass reactor and kept at 100 °C for 5 days under autogenous pressure, and then cooled to room temperature at a rate of 5 °C h−1. Red crystals of 1 were obtained and washed with DMF and dried in the air (yield: 53% based on Co). Elemental analysis (%). Calcd for C38H27N7O6Co: C, 61.96; H, 3.69; N, 13.31%. Found: C, 61.64; H, 3.78; N, 13.56%. IR data (KBr cm−1): 3412 (s), 2358 (w), 1711 (s), 1652 (s), 1547 (m), 1502 (m), 1406 (m), 1134 (w), 1072 (w), 837 (m), 765 (m), 672 (w), 585 (w).
2.4 Synthesis of [Co(Htatb)(bimbp)]·DMF (2)
Complex 2 was prepared as for 1 by using bimbp ligand (0.1 mmol, 0.029 g) instead of 1,3-bimyb. Red crystals of 2 were obtained and washed with DMF and dried in the air (yield: 56% based on Co). Elemental analysis. Calcd for C45H34N8O7Co: C, 63.01; H, 3.99; N, 13.06%. Found: C, 63.17; H, 3.87; N, 13.27%. IR data (KBr cm−1): 3429 (s), 2107 (m), 1714 (s), 1648 (s), 1521 (m), 1454 (s), 1402 (s), 1154 (m), 1018 (w), 872 (m), 765 (m), 669 (m).
2.5 Synthesis of [Co(Htatb)(1,4-bimyb)]·H2O (3)
The procedure is similar to that of 1, except that the 1,3-bimyb ligand was replaced by 1,3-bimyb ligand (0.1 mmol, 0.024 g), and the mixture was heated at 100 °C for 5 days, and then it was cooled to room temperature at 5 °C h−1. Purple crystals of 3 were obtained and washed with DMF and dried in the air (yield: 54% based on Co). Elemental analysis (%). Calcd for C38H29N7O7Co: C 60.48, H 3.87, N 12.99; found: C 60.75; H 3.64; N 12.76; IR data (KBr cm−1): 3421 (s), 2374 (w), 1718 (s), 1654 (s), 1560 (m), 1508 (m), 1396 (m), 1234 (w), 1083 (w), 825 (m), 771 (m), 653 (w), 572 (w).
2.6 X-ray crystallographic studies
Diffraction intensities for the three complexes were collected at 293 K on a Bruker SMART 1000 CCD diffractometer employing graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å). A semi-empirical absorption correction was applied using the SADABS program.33 The structures were solved by direct methods and refined by full-matrix least-squares on F2 using the SHELXS 2014 and SHELXL 2014 programs, respectively.34,35 Non-hydrogen atoms were refined anisotropically and hydrogen atoms were placed in geometrically calculated positions. The crystallographic data for compounds 1–3 are listed in Table 1, and selected bond lengths and angles are listed in Table S1.†
Table 1 Crystal data and structural refinement summary of compounds 1–3
| Identification code |
1 |
2 |
3 |
| Empirical formula |
C38H27N7O6Co |
C45H34N8O7Co |
C38H29N7O7Co |
| Formula weight |
736.59 |
857.73 |
754.61 |
| Crystal system |
Triclinic |
Triclinic |
Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P |
| a (Å) |
9.0115(15) |
10.004(2) |
8.571(2) |
| b (Å) |
10.9375(17) |
14.689(4) |
11.968(3) |
| c (Å) |
17.301(3) |
15.139(4) |
16.874(4) |
| α (°) |
94.820(3) |
71.253(4) |
91.273(3) |
| β (°) |
100.924(3) |
87.999(4) |
103.657(3) |
| γ (°) |
92.663(3) |
89.725(4) |
95.169(3) |
| V (Å3) |
1665.0(5) |
2105.2(9) |
1673.4(7) |
| Dc (mg m−3) |
1.469 |
1.353 |
1.498 |
| Z |
2 |
2 |
2 |
| μ (mm−1) |
0.576 |
0.469 |
0.577 |
| Reflections collected/unique |
8424/5817 [R(int) = 0.0139] |
10365/7331 [R(int) = 0.0129] |
11540/6022 [R(int) = 0.0268] |
| Goodness-of-fit (GOF) on F2 |
1.062 |
1.036 |
1.033 |
| Final R indices [I > 2σ(I)] |
R1 = 0.0397 wR2 = 0.1113 |
R1 = 0.0438 wR2 = 0.1098 |
R1 = 0.0498 wR2 = 0.1302 |
| Largest difference in peak and hole (e Å−3) |
0.372 and −0.373 |
0.797 and −0.445 |
0.714 and −0.567 |
3 Results and discussion
3.1 Crystal structures of [Co(Htatb)(1,3-bimyb)] (1)
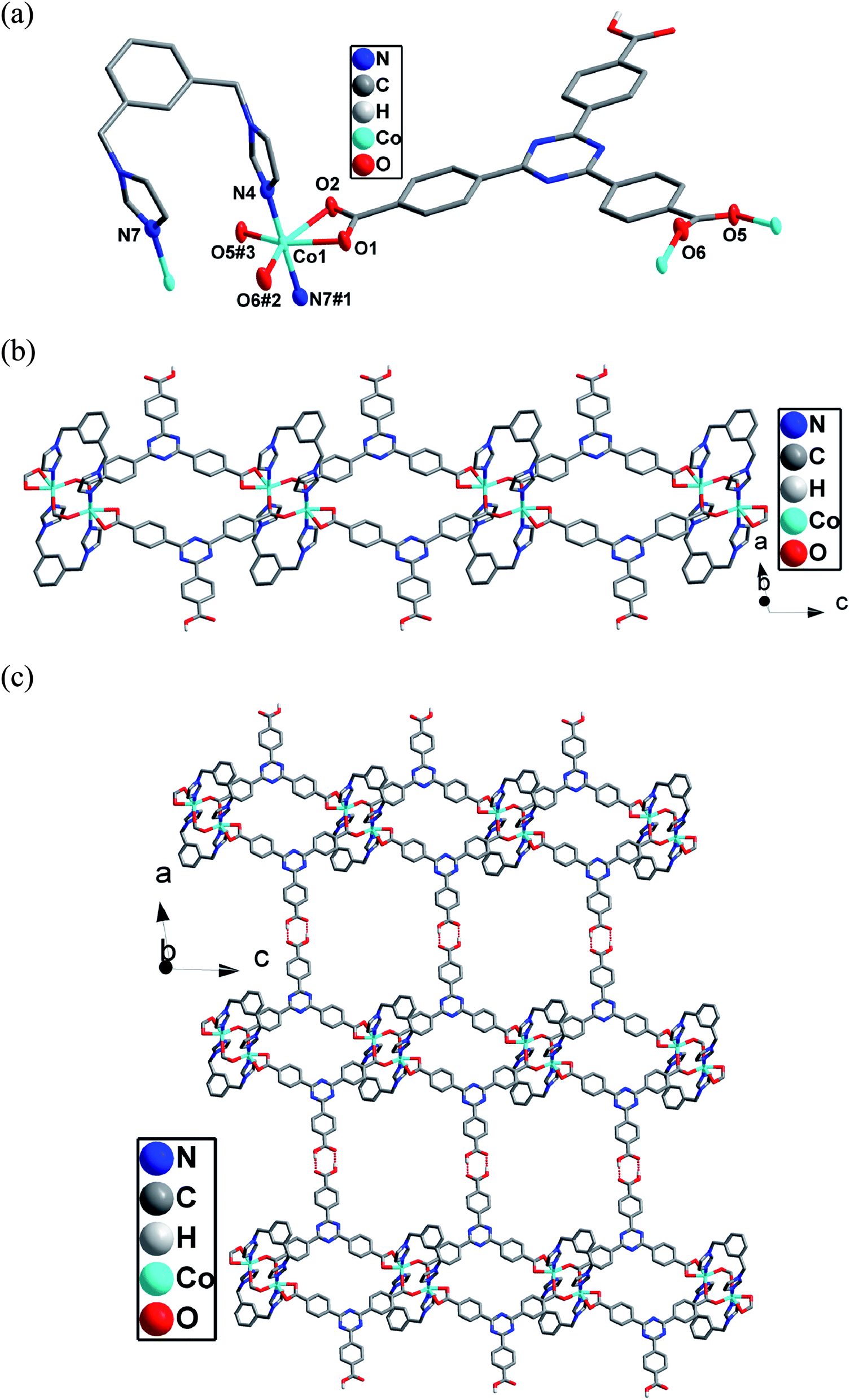
Single-crystal X-ray analysis reveals that complex 1 shows a double-strand 1D chain structure. The asymmetric unit of 1 consists of one independent CoII ion, one Htatb ligand, one 1,3-bimyb ligand. Each CoII ion coordinates to two nitrogen atoms from two 1,3-bimyb ligand, four carboxylate oxygen atoms from three Htatb ligands, forming a CoN2O4 distorted octahedral geometry (see Fig. 1a). The bond lengths of Co–O/N are in the range of 2.0104(18) Å to 2.0045(17) Å, the O/N–Co–O/N bond angles cover the range of 59.41(6)–177.70(8)°. In compound 1, the tripodal carboxylate ligands are partly deprotonated and one carboxylate group adopts μ2–η1–η1 bridging mode to link two CoII ions, and another one adopts μ1–η1–η1 chelating mode to link one CoII ion, resulting in a double-strand chain structure (Fig. 1b). The flexible 1,3-bimyb ligand joins the adjoining two CoII ions to form a ring on both sides of a 1D chain, with the Co⋯Co separation is 4.176 Å. The adjacent chains are joined to generate a 2D layer structure through intermolecular O–H⋯O hydrogen bonds [O3–H3A⋯O4 distance: 2.6478(18) Å, angle: 176.95(4)] (Fig. 1c). Further, through aromatic π–π stacking interactions between phenyl rings of two Htatb ligands (centroid-to-centroid distance: 3.681(3) Å), the adjacent layers generate a 3D supramolecular structure (Fig. S1 in the ESI†).
 |
| | Fig. 1 (a) The coordination environments of Co(II) ion in compound 1. Hydrogen atoms are omitted for clarity except the Htatb ligand. Displacement ellipsoids are drawn at the 50% probability level (symmetry code: #1 −x + 1, −y + 1, −z + 1, #2 x, y, z − 1, #3 −x + 1, −y + 1, −z) (b) The double-strand chain structure of 1. (c) The 2D layer structure of 1. | |
3.2 Crystal structures of [Co(Htatb)(bimbp)]·DMF (2)
Compound 2 shows a 2D structure with 1D channel, and compound 2 is made up of the CoII ion, Htatb ligand, bimbp ligand and free DMF molecule. Each CoII ion is located in a distorted octahedral geometry and is coordinated to four oxygen atoms of two Htatb ligand and two nitrogen atoms of two bimbp ligands, as shown in Fig. 2a. The bond lengths of Co–O/N are in the range of 2.0394(19)–2.2578(19) Å, the O/N–Co–O/N bond angles cover the range of 59.20(7)–178.24(8)°. As compound 1 is same, the carboxylic groups of Htatb ligand have two coordination modes: one carboxylate group adopts μ2–η1–η1 bridging mode to link two CoII ions, and another one adopts μ1–η1–η1 chelating mode to link one CoII ion, resulting in a double-strand chain structure (Fig. S2 in the ESI†). Through bimbp ligands bridging, the adjacent chains are connected to generate a 2D structure with 1D channel (See Fig. 2b and S3†). The potential volume accessible for DMF, determined by PLATON calculation, is 410.3 Å3 per unit cell volume (2105.2 Å3), which represents 19.5% void per unit volume for 2.
 |
| | Fig. 2 (a) The coordination environments of Co(II) ion in 2. Hydrogen atoms are omitted for clarity except the Htatb ligand. (symmetry code: #1 −x − 1, −y + 1, −z + 2, #2 x, y + 1, z − 1, #3 x − 1, y, z + 1). (b) The 2D structure with 1D channel of 2. | |
3.3 Crystal structures of [Co(Htatb)(1,4-bimyb)]·H2O (3)
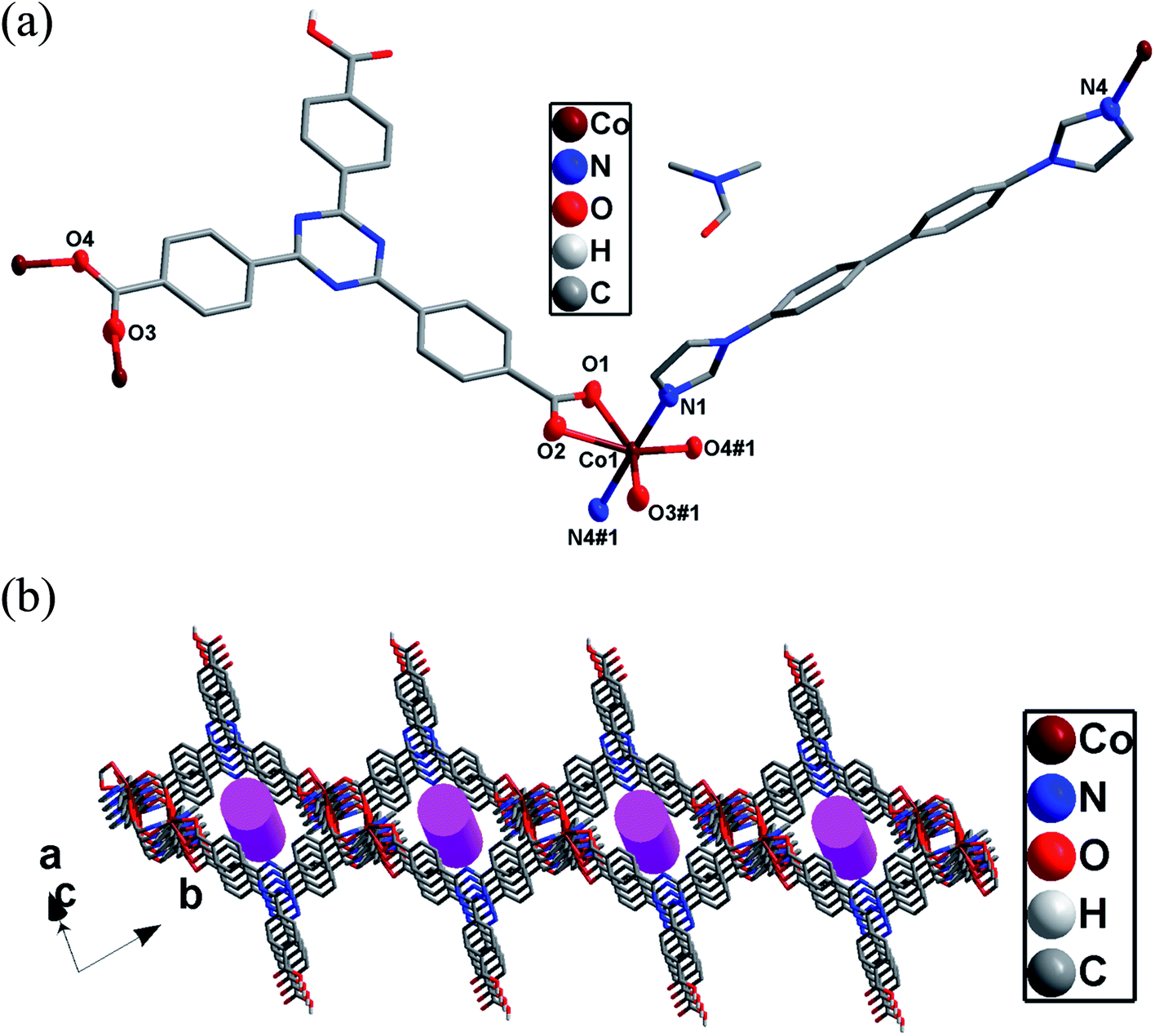
Complex 3 exhibits a 4-fold interpenetrating 3D structure. The asymmetric unit of 3 comprises one CoII ion, one Htatb ligand, one 1,4-bimyb ligand and one free water molecule, which is disposed about a centre of inversion. Each four-coordinated CoII center is surrounded by two nitrogen atoms coming from two 1,4-bimyb ligands, and two oxygen atoms from two Htatb ligands, taking a distorted tetrahedral geometry (Fig. 3a). The bond lengths of Co–O and Co–N are in the range of 1.973(2)–2.040(3) Å. Compared with complexes 1 and 2, the carboxylic groups of 3 adopts μ1–η1–η0 and μ1–η1–η0 bridging mode to link two CoII ions, resulting in a 1D chain structure, through 1,4-bimyb ligand bridging, the adjacent chains are connected to generate a 2D layer structure (Fig. S4†). These 2D layers are further joined by O–H⋯O hydrogen bonding [O5–H5⋯O2 distance: 2.592(3) Å, angle: 176.95(4)] to produce a 3D architecture (Fig. 3b). In order to simplify the 3D framework, we considered the Htatb anion as a 3-connected node and CoII ion as a 5-connected node, 1,4-bimyb ligand as linkers, the whole framework can be simplified as a 3,5-connected 2-nodal net topology with a point symbol of (3·72)(32·75·83)(Fig. 3c). However, due to the absence of large guest molecules to fill the void space, the potential voids are filled via mutual interpenetration of three independent equivalent frameworks, generating a four-fold interpenetrating 3D architecture (Fig. 3d).
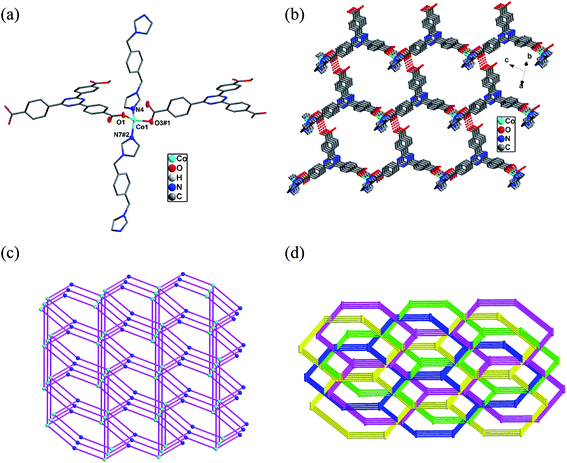 |
| | Fig. 3 (a) The coordination environments of Co(II) ion in 3. Hydrogen atoms are omitted for clarity except the Htatb ligand. (symmetry code: #1 x + 1, y, z + 1, #2 x − 1, y − 1, z). (b) The 3D architecture of 3. (c) The 3,5-connected net topology of 3. (d) The four-fold interpenetrating 3D architecture of 3. | |
3.4 Structural diversity of 1–3
It is noteworthy that a variety of framework structures can be achieved on the basis of the choice of H3tatb and imidazole-containing ligands. From Htatb containing compounds 1–3, Htatb ligand shows two types of coordination modes: one adopts μ2–η1–η1 and μ1–η1–η1 bridging mode for 1 and 2, another adopts μ1–η1–η0 and μ1–η1–η0 bridging mode for 3, and then the different N-containing co-ligands were employed, resulting in three different dimensional frameworks (compound 1 for 1D, compound 2 for 2D, compound 3 from 2D to 3D). The different imidazole-containing ligands may affect the structure of compounds, when a flexible ligand (1,3-bimyb) is employed to generate 1D structure for 1, when a rigid ligand (bimbp) is attended, the 2D structure of 2 are obtained, when a semi-rigid ligand is used, a 4-fold interpenetrating 3D architecture for 3 is obtained. By comparing of the structures of compounds 1–3, the H3tatb and imidazole-containing ligands may tune the final structural features.
3.5 Thermogravimetric analysis
To study the thermal stability of 1–3, thermogravimetric (TG) analyses were performed on polycrystalline samples under a nitrogen atmosphere with a heating rate of 10 °C min−1 (see Fig. S5–S7†). The TG curve of 1 reveals that no weight loss occurs from 20 °C until about 220 °C, above which, a significant weight loss of 92.4% is observed and ended at about 460 °C indicating the release the organic ligands (calcd. 92.0%). The TG curve of 2 shows two step weight losses, the first weight loss in the range 50–140 °C (obsd 8.7%, calcd. 8.5%) was assignable to the loss of DMF molecules. The second weight loss of 85.3% in the temperature range 210 to 470 °C corresponds to the release of the Htatb and bimbp ligands (calcd. 84.6%). TG curve of 3 reveals that the first weight loss of 2.6% from 40 °C to 120 °C corresponds to the loss of the lattice water molecules (calcd. 2.4%), and then the larger weight loss(obsd 90.4%) occurred in the range of 230–500 °C, corresponding to the decomposition of the Htatb and 1,4-bimyb ligands (calcd. 89.8%). The final decomposition products of 1–3 were confirmed to be CoO, which have also been further confirmed by PXRD patterns of compounds.
3.6 Infrared spectra of complexes 1–3
IR spectra of complexes 1–3 show features attributable to compositions of the coordination polymers. The observed strong characteristic peaks appearing around 3412, 3429 and 3421 cm−1 in spectra are attributed to the O–H stretching vibrations, respectively.36 Due to partial deprotonation of carboxylate in complexes 1–3, the absorptions about 1711, 1714 and 1718 cm−1 can be attributed to the stretching vibrations of the vCOOH in the carboxylate.37,38 The presence of the characteristic bands at 1652, 1648 and 1654 cm−1 for 1–3 suggest the v–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N– stretching vibrations of Htatb ligand. The intense characteristic peaks appearing around 1547 and 1502, 1406 cm−1 for 1, 1521 and 1454, 1348 cm−1 for 2, 1560 and 1508, 1396 cm−1 for 3 in the IR spectra correspond to asymmetric and symmetric stretching vibrations of carboxylic groups, respectively.39,40 The presence of the characteristic bands at 1134, 1154 and 1234 cm−1 for 1–3 suggest the vC–O stretching vibrations. The presence of the characteristic bands at 1072 cm−1 for 1, 1018 cm−1 for 2, 1083 cm−1 for 3 suggest the vC–N stretching vibrations of the imidazole ring.41 The presence of the characteristic bands at 1072 cm−1 for 1, 1018 cm−1 for 2, 1083 cm−1 for 3 suggest the vC–N stretching vibrations of the imidazole ring.42 The absorptions about 650–880 cm−1 of 1–3 can be attributed to the vC–H bending vibrations of phenyl ring.
N– stretching vibrations of Htatb ligand. The intense characteristic peaks appearing around 1547 and 1502, 1406 cm−1 for 1, 1521 and 1454, 1348 cm−1 for 2, 1560 and 1508, 1396 cm−1 for 3 in the IR spectra correspond to asymmetric and symmetric stretching vibrations of carboxylic groups, respectively.39,40 The presence of the characteristic bands at 1134, 1154 and 1234 cm−1 for 1–3 suggest the vC–O stretching vibrations. The presence of the characteristic bands at 1072 cm−1 for 1, 1018 cm−1 for 2, 1083 cm−1 for 3 suggest the vC–N stretching vibrations of the imidazole ring.41 The presence of the characteristic bands at 1072 cm−1 for 1, 1018 cm−1 for 2, 1083 cm−1 for 3 suggest the vC–N stretching vibrations of the imidazole ring.42 The absorptions about 650–880 cm−1 of 1–3 can be attributed to the vC–H bending vibrations of phenyl ring.
3.7 UV-vis spectroscopy
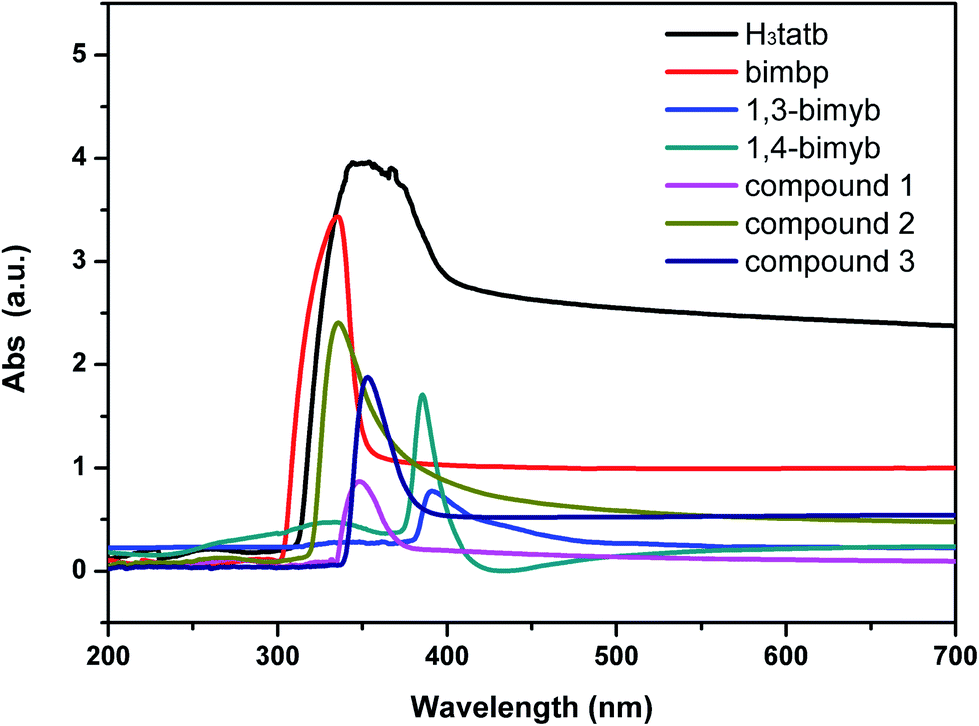
The UV-vis spectrum of the free ligands and complexes 1–3 in DMF-H2O (V:V = 1:1) is described in Fig. 4. In the spectra of the H3tatb ligand, one broad and strong absorption band at 354 nm occurred in the range of 300–400 nm, and the absorption bands of the bimbp, 1,3-bimyb and 1,4-bimyb ligands, found at 335 nm, 391 nm and 386 nm, can be assigned to the π → π* and n → π* transitions of the these ligands. In the spectra of complexes 1 and 3, the maximum of absorbance are observed at 348 nm (1) and 351 nm (3), can be assigned to the n → π* transitions of the ligands and exhibit slight blue shifted compared to the H3tatb ligand (354 nm), these shifts reflect coordination of the ligands to the Co(II) atom. The absorption bands of complex 2 was observed at 336 nm (2), which can be attributed to the intraligand π → π* and n → π* transitions of the coordinated ligands.
 |
| | Fig. 4 The UV-vis spectrum of the free ligands and complexes 1–3 in DMF-H2O (V:V = 1:1). | |
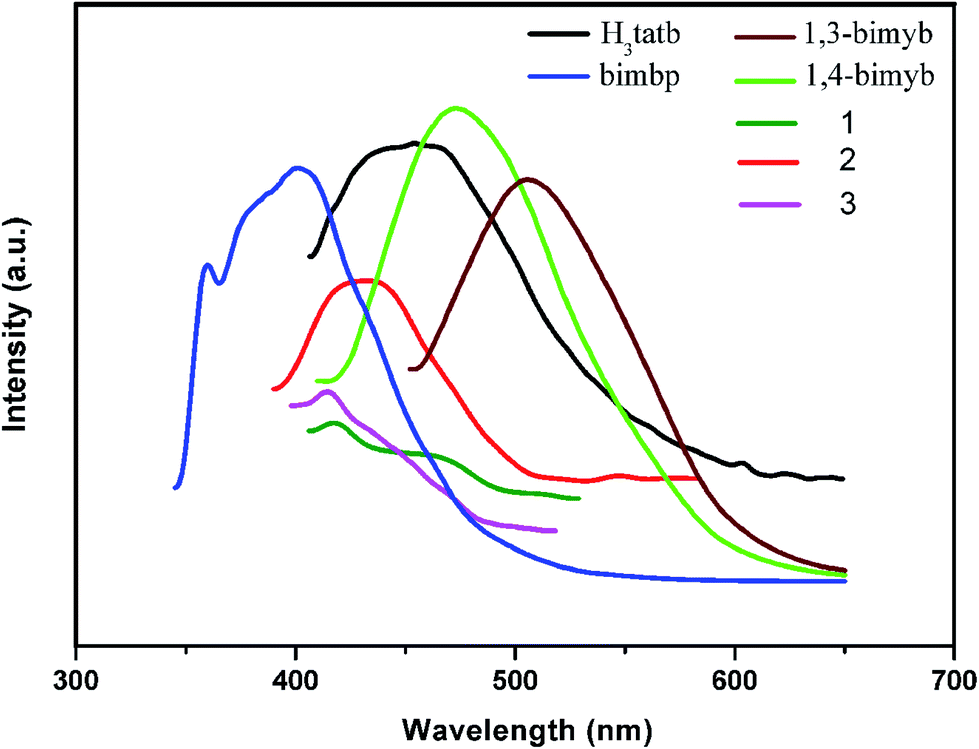
3.8 Photoluminescence properties
The luminescent emission spectra of 1–3 were examined in the solid state at room temperature as is shown in Fig. 5. The main emission peak of the free ligand H3tatb appears at 454 nm (λex = 364 nm) and can be assigned to the intra-ligand π*–π transitions.43 Complexes 1 and 3 show similar weaker emission peaks at 414 nm and 417 nm (λex = 366 nm), however, the intense emission of 1,3-bimyb and 1,4-bimyb ligand were observed at 506 nm (λex = 433 nm) and 473 nm (λex = 400 nm), respectively. Relative to their ligands, complexes 1 and 3 show a blue shift, probably owing to ligand to metal charge transfer (LMCT).44,45 Complex 2 show an emission peaks at 432 nm (λex = 345 nm), in comparison with that of free H3tatb and bimbp ligand (an intense emission at 401 nm (λex = 340 nm)), which are attributed to H3tatb or bimbp ligand-based charge transfer.46,47
 |
| | Fig. 5 The solid state luminescent emissions of complexes 1–3 and ligands. | |
3.9 Magnetic property
The magnetic susceptibilities (χM) of 1 and 2 were measured in the temperature range of 2–300 K with a field of 1000 Oe. To simulate the experimental magnetic behavior, we analyzed the experimental χMT data of 1 and 2 by the dinuclear models as expressed by eqn (1).48| |
 | (1) |
N, g, β, k and T are the Avogadro number, Zeeman factor, Bohr magneton, Boltzmann constant and temperature in kelvin, respectively. J is the exchange coupling constant between adjacent Co(II) ions.
The magnetic results of complex 1 are illustrated in Fig. 6a. As observed, the experimental χMT value at 300 K is 3.796 cm3 ·K mol−1, which is somewhat larger than the expected value (3.75 cm3 ·K mol−1) of the two isolated spin-only Co(II) ions (S = 3/2). The distance between two closest Co(II) centers aligned in the 1D chain is 4.176 Å. When the temperature decreases, the χMT descends gradually until 75 K and then rapidly drops to 2 K. The temperature dependence of the reciprocal susceptibility (1/χM) obeys the Curie–Weiss law above 30 K with θ = −8.44 K, C = 3.69 cm3 K mol−1 and R = 6.24 ×10−4 (R = ∑[(χM)obs − (χM)calc]2/∑[(χM)obs]2). The best fitting for the experimental data was found with J = −7.27 cm−1, g = 2.24 and R = 4.13 × 10−4. The values of J and θ indicate the antiferromagnetic interactions between adjacent Co(II) (S = 3/2) ions.
 |
| | Fig. 6 Thermal variation of χM and χMT for 1 (a) and 2 (b) (○, χM experimental values, □, χMT experimental values and the solid line represents the best fit obtained from the Hamiltonian given in the text.). | |
The experimental χMT values of 2 is 3.713 cm3 K mol−1 at room temperature (Fig. 6b), which are very close to the expected value of 3.75 cm3 K mol−1 for two isolated spin-only Co(II) ions with S = 3/2. As the temperature decreases, the χMT value gradually decreases till 50 K to reach a value of 2.73 cm3 K mol−1 after which an obvious fall was appeared to reach a value of 0.03 cm3 K mol−1 at 2 K, manifesting a significant antiferromagnetic exchange between the magnetic centres in Co2 dimer. The temperature dependence of the reciprocal susceptibility (1/χM) obeys the Curie–Weiss law above 30 K with θ = −7.21 K, C = 3.73 cm3 K mol−1 and R = 5.17 ×10−4. The best-fit parameters for the experimental data gives, J = −9.42 cm−1, g = 2.28 and R = 1.14 × 10−4. In 2, the magnetic coupling between two Co(II) centers is transmitted through two carboxylate bridges, the Co⋯Co distance of 4.156 Å is responsible for the antiferromagnetic coupling. In these compounds, this behavior indicates a dominant antiferromagnetic interaction between the Co(II) ions for 1 and 2. However, complex 3 features a 3D supramolecular structure, the Co⋯Co distances through the Htatb and 1,4-bimyb ligands bridging are 17.026 and 14.079 Å, due to the large distance between adjacent Co(II) ions, we did not discuss the magnetic behavior of 3.
3.10 Magnetic properties of DFT calculations
The DFT calculations have been widely proved to be one of the most efficient tools to investigate magnetic structure of transition metal complexes.49–51 We used a phenomenological Heisenberg Hamiltonian Ĥ = −JŜ1Ŝ2 to describe the exchange coupling in a dinuclear compound, the coupling constant J can be related to the energy difference between the lowest and highest spin states. For the case in which S1 = S2, the coupling constant may be obtained by using the following eqn (3).52| | |
EHS − ELS = −2JSi(Si + 1/2)
| (2) |
where EHS is the energy that corresponds to the state with the highest total spin, ELS corresponds to the state with the lowest total spin (S = 0), and Si is the total spin on each metal atom.
When using DFT-based wave functions, a reasonable estimate of the energy corresponding to the low spin state, ELS can be obtained directly from the energy of a broken-symmetry solution EBS. In this case, compounds 1 and 2 are 1D chain and 2D layer, and the theoretical calculations would be a difficult task for such a large periodical system. Here, the monomer [Co2(Htatb)2(1,3-bimyb)2] of 1 [Co2(Htatb)2(bimbp)2] of 2 were intercepted, we arrive to the following expressions for J:
The calculated coupling constant J and related quantities are listed in Table 2. We have used via eqn (3) to estimate the magnetic coupling constants (J) of compounds 1 and 2. The computed J values (J = −9.04 cm−1 for 1 and J = −11.63 cm−1 for 2) predict antiferromagnetic, the results were agreed with the experimental data. We have used the non-spin projected, giving a better agreement for compounds 1 and 2. This is due to the strong localization of the wavefunction at the metal centers both computational techniques produce results that are in remarkable agreement with the experimental value. Full molecule calculation using hybrid B3LYP functional to reproduce BS–HS energy gap of compounds is not only found to be successful in describing the magnetic behavior of the compounds correctly in this study but also yielded J that are in excellent agreement with the experimental value. So the full molecule DFT based calculation based on X-ray structural data could be successfully used to rationalize the magnetic behavior of this class of compounds.
Table 2 Calculated energy (au.) and magnetic exchange constant J (cm−1) for 1 and 2
| Compound |
EBS/(au.) |
EHS/(au.) |
Calculation, J/(cm−1) |
Experiment, J/(cm−1) |
| 1 |
−9684.132164 |
−9684.131917 |
−9.04 |
−7.27 |
| 2 |
−10160.989084 |
−10160.988766 |
−11.63 |
−9.42 |
4 Conclusions
In summary, three cobalt-based coordination polymers have been synthesized by the self-assembly of Co(II) salts with H3tatb and imidazole-containing Ligands. Assemblies of these compounds generate three types of diverse frameworks: one 1D chain, one 2D layer and 2D to 3D structure. By comparing of the structures of 1–3, the N-donor ligand can also slightly tune the final structural features. Moreover, the fluorescent properties of 1–3 and magnetic behavior of 1 and 2 have also been investigated. According to the crystal structures, the DFT-BS approach was applied to study the magnetic coupling behavior for 1 and 2, the result reveals that the calculated exchange coupling constants J were in good agreement with the experimental data.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21503183, 21573189, 21663031 and 21763028). The authors thank Professor Wen-Liang Wang (Shaanxi Normal University) for his help.
References
- Y. P. Wu, W. Zhou, J. Zhao, W. W. Dong, Y. Q. Lan, D. S. Li, C. H. Sun and X. H. Bu, Angew. Chem., Int. Ed., 2017, 56, 13001–13005 CrossRef CAS PubMed.
- Q. G. Zhai, X. H. Bu, X. Zhao, D. S. Li and P. Y. Feng, Acc. Chem. Res., 2017, 50, 407–417 CrossRef CAS PubMed.
- Y. Zhao, X. G. Yang, X. M. Lu, C. D. Yang, N. N. Fan, Z. T. Yang, L. Y. Wang and L. F. Ma, Inorg. Chem., 2019, 58, 6215–6221 CrossRef CAS PubMed.
- G. Wang, Q. Sun, Y. Liu, B. Huang, Y. Dai, X. Zhang and X. Qin, Chem.–Eur. J., 2015, 21, 2364–2367 CrossRef CAS PubMed.
- Y. L. Wang, J. H. Fu, J. J. Wei, X. Xu, X. F. Li and Q. Y. Liu, Cryst. Growth Des., 2012, 12, 4663–4668 CrossRef CAS.
- D. S. Li, J. Zhao, Y. P. Wu, B. Liu, L. Bai, K. Zou and M. Du, Inorg. Chem., 2013, 52, 8091–8098 CrossRef CAS PubMed.
- Y. Zhao, L. Wang, N. N. Fan, M. L. Han, G. P. Yang and L. F. Ma, Cryst. Growth Des., 2018, 18, 7114–7121 CrossRef CAS.
- H. R. Fu, N. Wang, J. H. Qin, M. L. Han, L. F. Ma and F. Wang, Chem. Commun., 2018, 54, 11645–11648 RSC.
- X. Y. Zhang, B. Li and J. P. Zhang, Inorg. Chem., 2016, 55, 3378–3383 CrossRef CAS PubMed.
- I. H. Park, R. Medishetty, J. Y. Kim, S. S. Lee and J. J. Vittal, Angew. Chem., Int. Ed., 2014, 53, 5591–5595 CrossRef CAS PubMed.
- S. Q. Chen, Q. G. Zhai, S. N. Li, Y. C. Jiang and M. C. Hu, Inorg. Chem., 2015, 54, 10–12 CrossRef CAS PubMed.
- Y. He, W. Zhou, G. Qian and B. Chen, Chem. Soc. Rev., 2014, 43, 5657–5658 RSC.
- M. G. Campbell, D. Sheberla, S. F. Liu, T. M. Swager and M. Dinca, Angew. Chem., Int. Ed., 2015, 54, 4349–4352 CrossRef CAS PubMed.
- K. Manna, T. Zhang, F. X. Greene and W. Lin, J. Am. Chem. Soc., 2015, 137, 2665–2673 CrossRef CAS PubMed.
- L. L. Shen, L. Yang, Y. Fan and L. Wang, CrystEngComm, 2015, 17, 9363–9369 RSC.
- W. Lin, Y. Cui, Y. Yu, Q. Hu and G. Qian, Dalton Trans., 2018, 47, 15882–15887 RSC.
- Y. J. Cheng, R. Wang, S. Wang, X. J. Xi, L. F. Ma and S. Q. Zang, Chem. Commun., 2018, 54, 13563–13566 RSC.
- Z. J. Lin, J. Lv, M. C. Hong and R. Cao, Chem. Soc. Rev., 2014, 43, 5867–5895 RSC.
- Y. W. Li, H. Ma, Y. Q. Chen, K. H. He, Z. X. Li and X. H. Bu, Cryst. Growth Des., 2012, 12, 189–196 CrossRef CAS.
- J. P. Zhang, Y. B. Zhang, J. B. Lin and X. M. Chen, Chem. Rev., 2012, 112, 1001–1033 CrossRef CAS PubMed.
- M. P. Suh, H. J. Park, T. K. Prasad and D. W. Lim, Chem. Rev., 2012, 112, 782–835 CrossRef CAS PubMed.
- S. Q. Ma, D. F. Sun, M. Ambrogio, J. A. Fillinger, S. Parkin and H. C. Zhou, J. Am. Chem. Soc., 2007, 129, 1858–1859 CrossRef CAS PubMed.
- S. Q. Ma, X. S. Wang, D. Q. Yuan and H. C. Zhou, Angew. Chem., Int. Ed., 2008, 47, 4130–4133 CrossRef CAS PubMed.
- W. Gao, F. F. Xing, D. Zhou, M. Shao and S. R. Zhu, Inorg. Chem. Commun., 2011, 14, 601–605 CrossRef CAS.
- H. B. Zhang, N. Li, C. B. Tian, T. F. Liu, F. L. Du, P. Lin, Z. H. Li and S. W. Du, Cryst. Growth Des., 2012, 12, 670–678 CrossRef CAS.
- J. W. Rong, W. W. Zhang and J. F. Bai, CrystEngComm, 2016, 18, 7728–7736 RSC.
- C. S. Liu, Z. H. Zhang, M. Chen, H. Zhao, F. H. Duan, D. M. Chen, M. H. Wang, S. Zhang and M. Du, Chem. Commun., 2017, 53, 3941–3944 RSC.
- X. Duan, R. Lv, Z. G. Ji, B. Li, Y. J. Cui, Y. Yang and G. D. Qian, Inorg. Chem. Front., 2018, 5, 1193–1198 RSC.
- X. B. Liu, Z. Y. Xiao, A. Huang, W. Wang, L. L. Zhang, R. M. Wang and D. F. Sun, Z. Anorg. Allg. Chem., 2016, 642, 31–35 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, Jr, J. A. Montgomery, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, Gaussian(version 6.1), Inc., Wallingford CT, 2004 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1997, 107, 8554–8560 CrossRef CAS.
- E. Ruiz, A. Rodríguez-Fortea, J. Tercero, T. Cauchy and C. J. Massobrio, Chem. Phys., 2005, 123, 074102 Search PubMed.
- G. M. Sheldrick, SADABS, A Program for Empirical Absorption Correction of Area Detector Data, University of Göttingen, Germany, 1997 Search PubMed.
- G. M. Sheldrick, SHELXS-2014/7, Program for Crystal Structure Solution, University of Göttingen, Germany 2014 Search PubMed.
- G. M. Sheldrick, SHELXL-2014/7, Program for Crystal Structure Refinement, University of Göttingen, Germany 2014 Search PubMed.
- P. S. Kalsi, Spectroscopy of Organic Compounds, New Age International, New Delhi, 2008 Search PubMed.
- I. Mohammed-Ziegler and A. Grün, Spectrochim. Acta, Part A, 2005, 62, 506–517 CrossRef PubMed.
- I. Mohammed-Ziegler, A. Hamdi, R. Abidi and J. Vincens, Supramol. Chem., 2006, 18, 219–234 CrossRef CAS.
- B. Dolenský, R. Konvalinka, M. Jakubek and V. Král, J. Mol. Struct., 2013, 1035, 124–128 CrossRef.
- S. Mandal, G. Das and H. Askari, Struct. Chem., 2014, 25, 43–51 CrossRef CAS.
- L. J. Bellamy, The Infrared Spectra of Complex Molecules, Wiley, New York, 1958 Search PubMed.
- G. Świderski, A. Z. Wilczewska, R. Świsłocka, M. Kalinowska and W. Lewandowski, J. Therm. Anal. Calorim., 2018, 134, 513–525 CrossRef.
- Y. Gong, Z. Hao, J. L. Sun, H. F. Shi, P. G. Jiang and J. H. Lin, Dalton Trans., 2013, 42, 13241–13250 RSC.
- Y. Q. Xu, D. Q. Yuan, B. L. Wu, L. Han, M. Y. Wu, F. L. Jiang and M. C. Hong, Cryst. Growth Des., 2006, 6, 1168–1174 CrossRef CAS.
- Y. J. Cui, Y. F. Yue, G. D. Qian and B. L. Chen, Chem. Rev., 2012, 112, 1126–1162 Search PubMed.
- X. P. Yang, S. Q. Wang, L. J. Zhang, S. M. Huang, Z. P. Li, C. R. Wang, T. Zhu and L. Bo, J. Mater. Chem. C, 2016, 4, 1589–1593 RSC.
- B. C. Yuan, F. Wang, J. B. Tao, M. Li and X. P. Yang, Inorg. Chim. Acta, 2019, 490, 24–28 CrossRef CAS.
- O. Kahn, Molecular Magnetism, VCH Publishers Inc, New York, 1993 Search PubMed.
- L. Tang, F. Fu, J. J. Wang, L. J. Gao, D. D. Chao and Z. Wang, Polyhedron, 2015, 88, 116–124 CrossRef CAS.
- C. Beghidja, G. Rogez, J. Kortus, M. Wesolek and R. Welter, J. Am. Chem. Soc., 2006, 128, 3140–3141 CrossRef CAS PubMed.
- E. Ruiz, P. Alemany, S. Alvarez and J. Cano, Inorg. Chem., 1997, 36, 3683–3688 CrossRef CAS PubMed.
- A. Rodríguez-Fortea, P. Alemany, S. Alvarez and E. Ruiz, Inorg. Chem., 2001, 40, 5868–5877 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray crystallographic files in CIF format for 1–3, the details of selected bond lengths/angles (Table S1), and hydrogen bonds for 1 and 3 (Table S2), the 3D supramolecular structure of 1 (Fig. S1), the 1D double-strand chain of 2 view along [010] direction (Fig. S2), the 2D layer structure of 2 (Fig. S3), the 2D layer structure of 3 (Fig. S4), and the TG curves of compounds 1–3 (Fig S5–S7). CCDC 1954874–1954876 contains the supplementary crystallographic data for 1–3. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra07737e |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Huan-Huan Wang,
Yu-Hao Fu,
Yi-Tong Wang,
JiJiang Wang
*,
Huan-Huan Wang,
Yu-Hao Fu,
Yi-Tong Wang,
JiJiang Wang