
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of novel polycarbonyl Schiff bases by ring-opening reaction of ethyl 5-acyl-4-pyrone-2-carboxylates with primary mono- and diamines†
D. L. Obydennov *,
L. R. Khammatova,
V. D. Steben'kov and
V. Y. Sosnovskikh
*,
L. R. Khammatova,
V. D. Steben'kov and
V. Y. Sosnovskikh
Institute of Natural Sciences and Mathematics, Ural Federal University, 51 Lenina Ave., 620000 Ekaterinburg, Russian Federation. E-mail: dobydennov@mail.ru
First published on 3rd December 2019
Abstract
An approach for the introduction of the tricarbonyl moiety into aromatic, heterocyclic, and aliphatic amines with the use of acylpyrones has been developed for the synthesis and the design of novel polycarbonyl Schiff base ligands, including salphen structures. This Michael addition–ring-opening reaction proceeds under mild conditions (stirring at 0–20 °C) via the attack at the C-6 position of the pyrone ring in good to high yields (up to 99%) with excellent selectivity. The products can be easily isolated by crystallization without the use of chromatography. The scope of the reaction, tautomeric equilibrium of open-chain products, and their cyclization into pyridone structures were investigated.
Introduction
Polycarbonyl compounds are widely used in various fields of materials chemistry, analytical and inorganic chemistry due to their multifarious chemical properties1 and complex-formation ability.2 Functional derivatives of 1,3-diketones, such as Schiff bases, play an especially important role as N,N′,O,O′-ligands and can give stable metal-chelate complexes with various metal ions.3 These coordination compounds are actively used as catalytic systems for diverse organic reactions (synthesis of heterocycles4a–e and polymerization4f), as well as chemosensors,5 materials for OLEDs,6 OFEDs,7 and solar cells.8To date, although a large number of metal salen complexes have been studied, methods for further functionalization of the ligand structure remain important. The introduction of additional coordinating centers for the synthesis of multinucleating salphen complexes makes it possible to improve their catalytic, magnetic, optical properties and ion sensing.9
It is well known that the main method for the preparation of Schiff bases involves the condensation of carbonyl compounds with aromatic or aliphatic amines,10 whereas the use of Michael reaction as an effective synthetic tool remains limited for the construction of ligands.11 Unlike the 1,3-dicarbonyl motif, the introduction of the polycarbonyl moiety into the molecule based on 1,3,5-triketones and other open-chain polyketones has some drawback, such as side reactions (the retro-Claisen cleavage, the intramolecular cyclization), a low selectivity, and low chemical reactivity of these substrates due to the presence of the acidic protons involved in the formation of the intramolecular hydrogen bonds.12 In the literature there are only a few examples of salen ligand synthesis based on 2,4,6-hexanetriones.13 Using acyl-4-pyrones as highly reactive synthetic equivalents of 1,3,5-triones, which can undergo Michael reaction under the action of amines as a key stage to circumvent the above difficulties. In this case, the reaction proceeds under mild conditions to avoid the formation of heterocyclization products and with high chemoselectivity. The ability of 4-pyrones to react with various N-nucleophiles can allow the introduction of the polycarbonyl moiety in wide scope of substrates.
5-Acylcomanoates 1 (ref. 14) are convenient building blocks for the construction of heterocyclic systems via a nucleophilic attack at the C-6 position due to the presence of the electron-withdrawing substituent at the C-5 position followed by pyrone ring-opening (Fig. 1). Usually, 4-pyrones react with N-nucleophiles via the ANRORC (Addition of the Nucleophile, Ring Opening, and Ring Closure) mechanism and give more stable azaheterocycles.14a,b Earlier,15 for the reaction of 2,5-dicarbethoxy-4-pyrone and ethyl 5-benzoylcomanoate with o-phenylenediamine, the formation of polyfunctional open-chain polycarbonyl substrates only as intermediates in the synthesis of polycyclic pyridones have been shown.
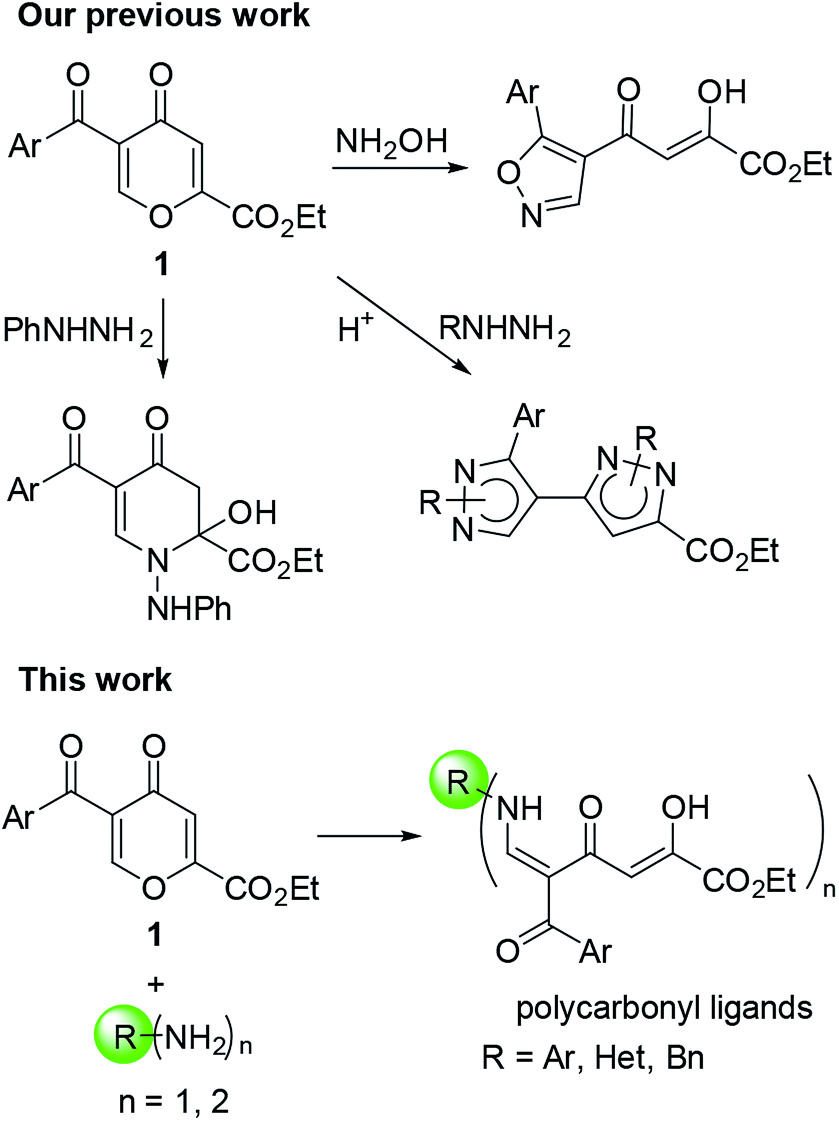 | ||
| Fig. 1 Reactions of acylpyrones 1 with primary amines. | ||
In addition, in organic chemistry close attention is paid to the development of highly efficient synthetic methods, such as “Click Chemistry”. This term describes reactions that are high yielding, wide in scope, stereospecific, simple to perform, high atom-economic, include facile the isolation of a product without the use of chromatography and can be carried out in easily removable or benign solvents.16 The search for such transformations is actively conducted, and Michael reaction and ring-opening reaction belong to a number of click-reactions.16 Pyrones can undergo the tandem of Michael reaction/pyrone ring-opening, therefore, they are of interest for the development of new synthetic click-approaches for the construction of polycarbonyl structures.
This work describes the general approach for the introduction of the 1,3,5-triketone moiety into the molecules bearing the amino group with the use of 5-acyl-4-pyrone-2-carboxylic acid esters to obtain and design new polycarbonyl Schiff base ligands, including salphen structures, as well as to study their tautomeric equilibrium and the substrate scope of this method.
Results and discussion
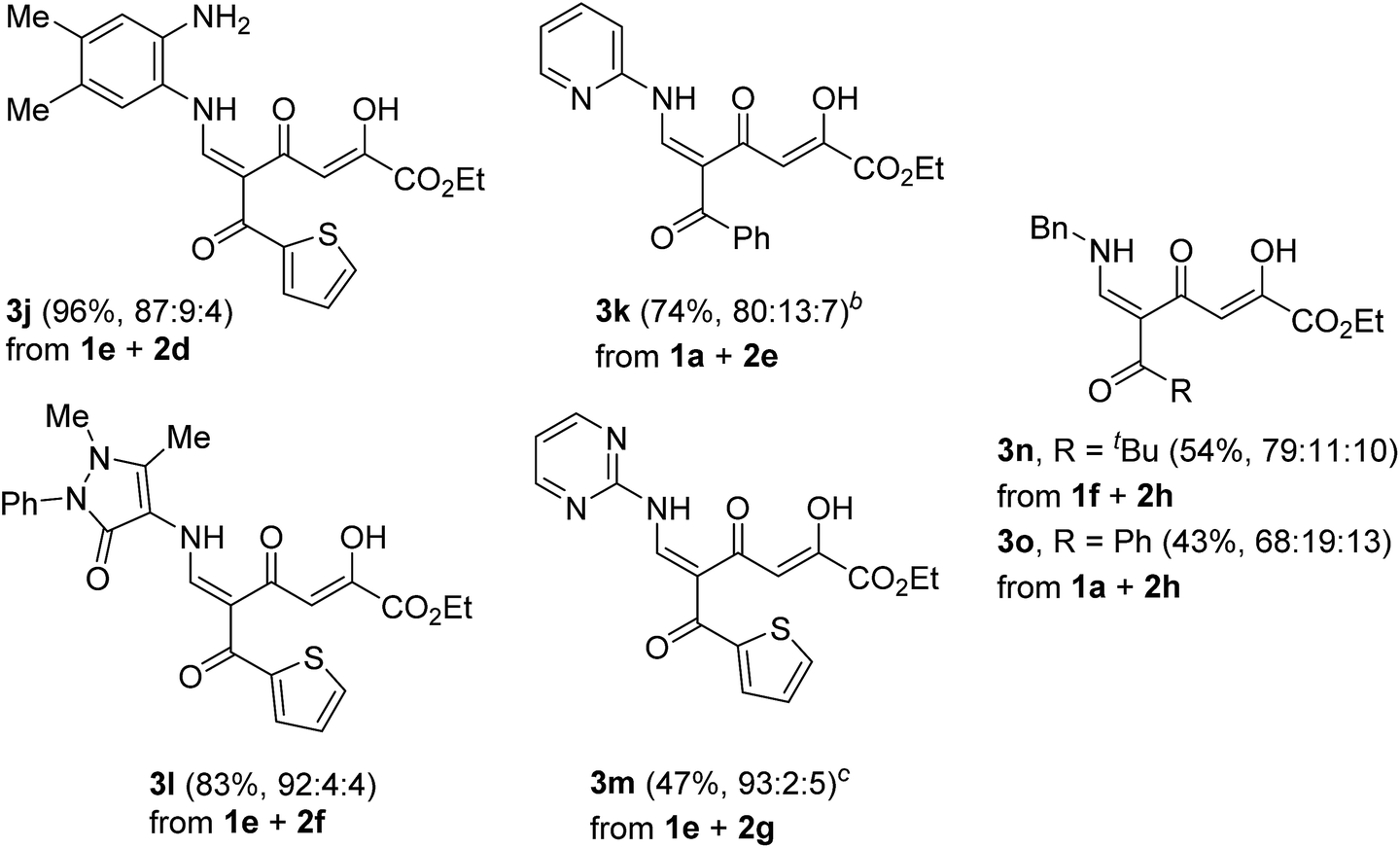
We started the study with the reaction of 5-acylcomanoates 1 with various aromatic mono- and diamines 2 in EtOH at 0 °C or 20 °C (Scheme 1 and Table 1). It was found that pyrones 1 gave tetracarbonyl enaminones 3a–o in medium to high yields (47–96%). The nature of the substituents in the aromatic amine has a slight effect on the outcome of the reaction though unreactive amines bearing the electron-withdrawing group, such as 2-aminopyridine and 2-aminopyrimidine, gave enaminones 3k and 3m at 20 °C in good yields (74% and 47%, respectively). The transformation was found to be tolerant to steric hindrance of a bulky acyl group and, for pivaloylpyrone 1f bearing the bulky tert-butyl group, product 3i was obtained in 61% yield.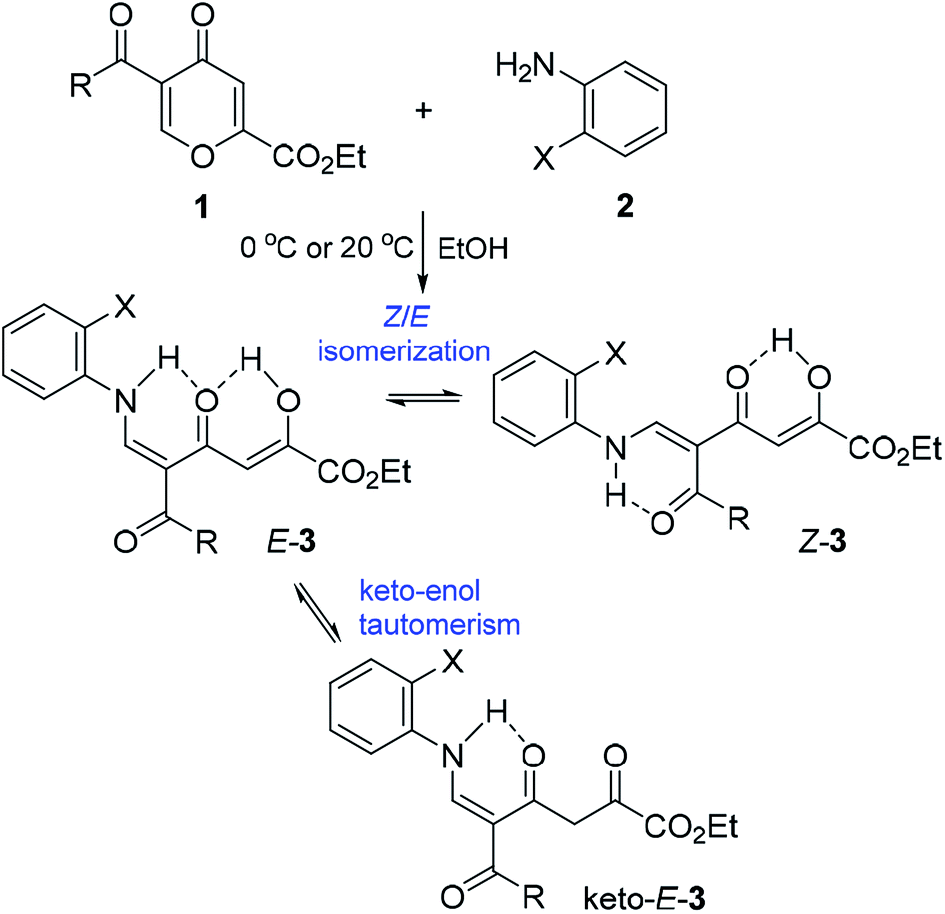 | ||
| Scheme 1 Reaction of pyrones 1 with amines. | ||
| Adduct | Pyrone | Amine | R | X | Yield, % | E-3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Z-3:keto-E-3 :Z-3:keto-E-3 |
|---|---|---|---|---|---|---|
| a Pyrone 1 (1.11 mmol) and amine 2 (1.11 mmol) were stirred in EtOH (6 mL) at 0 °C for 1 h.b The reaction was performed at 20 °C for 2 h.c The reaction was performed at 20 °C for 4 days, 2.22 mmol of the amine was used. | ||||||
| 3a | 1a | 2a | Ph | H | 87 | 82:13:5 |
| 3b | 1b | 2a | 4-MeOC6H4 | H | 85 | 95:2:3 |
| 3c | 1a | 2b | Ph | OH | 68 | 76:17:7 |
| 3d | 1c | 2b | 4-ClC6H4 | OH | 81 | 76:17:7 |
| 3e | 1b | 2c | 4-MeOC6H4 | NH2 | 86 | 79:15:6 |
| 3f | 1c | 2c | 4-ClC6H4 | NH2 | 78 | 78:16:6 |
| 3g | 1d | 2c | 2-Naph | NH2 | 90 | 79:13:8 |
| 3h | 1e | 2c | 2-Th | NH2 | 90 | 89:8:3 |
| 3i | 1f | 2c | t-Bu | NH2 | 61 | 79:14:7 |
 |
||||||
It was found that the polycarbonyl moiety can be introduced into the molecule of aliphatic amine, such as benzylamine (2h). Indeed, enaminones 3n,o were obtained in lower yield (43–54%) compared to aromatic amines. This result can be explained by easier intramolecular cyclization of enaminone 3 into stable pyridones because of higher nucleophilicity of the alkylamine.
Products 3 are multifunctionalized structures bearing the enaminodione and diketone moieties, which can undergo keto–enol tautomerism and Z/E isomerization as the result of the push–pull nature of the double bond. Two geometric isomers Z-3 and E-3 and also tautomer keto-E-3, which exist in equilibrium in the solution of CDCl3, were detected by NMR spectroscopy (Scheme 1 and Table 1).
A clear influence of the substituents on the tautomeric equilibrium is observed, which can be explained by the stability of the hydrogen bond formed and steric interactions. The introduction of the electron-donating MeO group into the aroyl substituent of N-phenyl-substituted enaminones 3a increases the content of the most stable isomer E-3 from 82% to 95%. Although a change in the structure of the amine has an effect on the observed equilibrium, the introduction of the ortho substituent reduces the content of major tautomer E-3 to 76–79%. Enaminones 3h,j,l,m, which bear the thenoyl substituent, are especially distinguished in this series because the content of the major isomer E-3 reaches 87–93%.
The structure of tautomer E-3 was confirmed on the basis of the coupling constants extracted from the 1H-coupled 13C NMR spectrum of 3b, along with the analysis of 1H–13C HMBC spectrum of 3b. It is known17 that, in the case of enones, coupling constant 3JH,C between the β-olefinic protons and the carbonyl group strongly depends on the dihedral angle and is a good criterion for establishing the configuration of the double bond when other experiments cannot be applied. For NC-substituted enaminones, the CH proton of the N–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) moiety has 3JH,CN coupling with the cyano group in the range of 4.6–5.8 Hz and 9.6–10.8 Hz for their cis- and trans-orientation, respectively.18 Also in the literature the 3JH,CO coupling has been measured19 for similar structures, alkoxymethylidenediones (cis-3JH,CO = 2.6–4.0 Hz, trans-3JH,CO = 7.7–10.1 Hz) and the magnitudes of these 3JH,CO coupling constants are convenient parameters for establishment of enaminodione configuration in our case. It was found that for enaminodione 3b the carbonyl group of the aroyl substituent (C-4′) is a quartet at δ 193.0 ppm with 3JH,C = 3.7 Hz, which indicates the cis-arrangement of the H-6 proton relative to C-4′ (Fig. 2). The signal for the carbonyl group of the diketone moiety (C-4) in the spectrum of 3b appears as a doublet of doublets and exhibits a 3JC–H coupling of 7.8 Hz with the H-6 proton and a small 2JC–H coupling of 3.7 Hz, which is in consistent with the magnitude of the coupling constant 2JC–H in the CH–CO moiety (2.3–3.3 Hz).20 The value of the first (7.8 Hz) is more than 3JH,C for C-4′ (3.7 Hz) and is in agreement with the trans-arrangement of H-6 and C-4, which corresponds structure E-3b.
moiety has 3JH,CN coupling with the cyano group in the range of 4.6–5.8 Hz and 9.6–10.8 Hz for their cis- and trans-orientation, respectively.18 Also in the literature the 3JH,CO coupling has been measured19 for similar structures, alkoxymethylidenediones (cis-3JH,CO = 2.6–4.0 Hz, trans-3JH,CO = 7.7–10.1 Hz) and the magnitudes of these 3JH,CO coupling constants are convenient parameters for establishment of enaminodione configuration in our case. It was found that for enaminodione 3b the carbonyl group of the aroyl substituent (C-4′) is a quartet at δ 193.0 ppm with 3JH,C = 3.7 Hz, which indicates the cis-arrangement of the H-6 proton relative to C-4′ (Fig. 2). The signal for the carbonyl group of the diketone moiety (C-4) in the spectrum of 3b appears as a doublet of doublets and exhibits a 3JC–H coupling of 7.8 Hz with the H-6 proton and a small 2JC–H coupling of 3.7 Hz, which is in consistent with the magnitude of the coupling constant 2JC–H in the CH–CO moiety (2.3–3.3 Hz).20 The value of the first (7.8 Hz) is more than 3JH,C for C-4′ (3.7 Hz) and is in agreement with the trans-arrangement of H-6 and C-4, which corresponds structure E-3b.
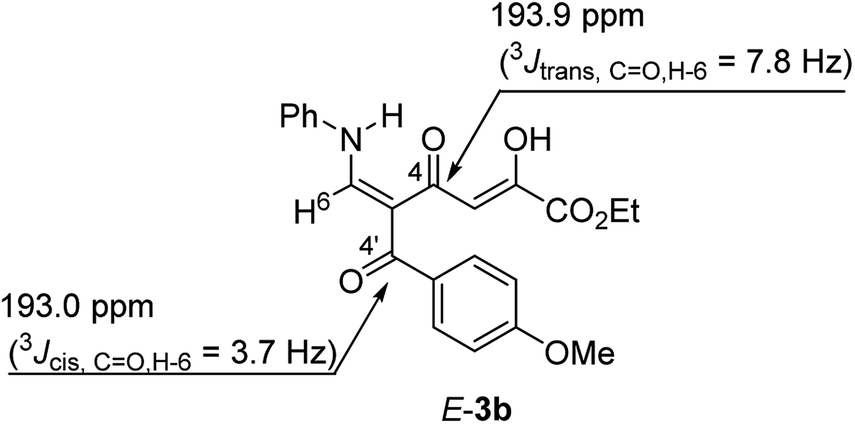 | ||
| Fig. 2 Selected 13C NMR signals of E-3b. | ||
The 1H NMR spectra of polycarbonyl compounds (het)aryl substituted 3a–m feature a main set of signals of tautomer E-3 with the 5E-configuration of the double bond which consists of a doublet of the NH proton at δ 12.07–12.60 ppm (3J = 12.4–13.6 Hz) and a downfield singlet for the OH group (δ 14.27–14.50 ppm), apparently involved in a strong intramolecular hydrogen bonds, a doublet of the CHN proton at δ 7.86–8.81 ppm (3J = 12.4–13.6 Hz), and a vinylic proton singlet at δ 6.59–7.15 ppm. For Bn-substituted 3n,o, the signals of both the CHN proton and the NH proton are shifted upfield compared with those of (het)aryl substituted 3a–m and appeared at δ 7.49–7.69 and 10.95 ppm, respectively.
The characteristic signals of minor tautomer Z-3 with 5Z-configuration include a doublet of the CHN proton (δ 8.27–9.30 ppm, 3J = 12.8–14.0 Hz), which is shifted downfield by ca. 0.5 ppm compared with the same proton of E-3, a singlet for the CHCO proton (δ 5.84–6.34 ppm) is shifted upfield by 0.7–1.2 ppm compared to E-3 isomer due to a shielding influence of the aromatic ring of the acyl moiety (Fig. 3). Such a difference in the chemical shifts of these protons was not observed for pivaloyl-substituted enaminones 3i,n (only 0.01–0.2 ppm). The OH proton and the NH proton appear as broadened signals at δ 15.0–15.4 ppm and δ 11.19–12.60 ppm (for aryl substituted 3a–m, δ 10.04–10.95 ppm for benzyl substituted 3n,o), respectively, as a result of the formation of the intramolecular hydrogen bonds. It is important to note that the OH proton signal is shifted downfield by ca. 0.8 ppm in comparison with the same proton of E-3 isomer, which indicates the stronger intramolecular hydrogen bond (OH⋯O) in the Z-3 isomer and is consistent with the proposed structures.
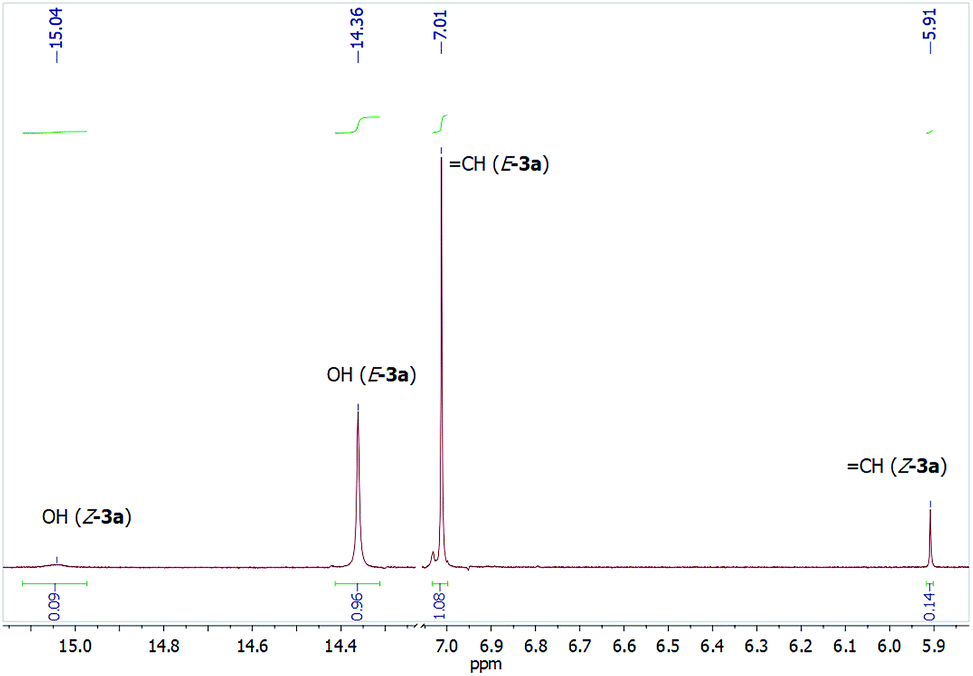 | ||
| Fig. 3 Fragment of the 1H NMR spectrum of 3a in CDCl3. | ||
It is well known14b,d that diketobutanoates predominantly exist as the enol form and contain some quantity of the diketo form (0–10%). Therefore, the structure of keto-E-3 was established on the basis of the relative content of the tautomer (0–13%). In 1H NMR spectra the feature signals of the tautomer are a doublet (3J ≈ 13.0 Hz) of the downfield NH proton at δ 12.22–12.60 ppm (for keto-E-3a–m) or δ 11.23–11.25 ppm (for keto-E-3n–o) and a singlet of the CH2 group at δ 4.07–4.40 ppm. The signal of the NH proton of the tautomer is shifted downfield by 0.2–0.6 ppm compared to the same proton of E-3 isomer because of stronger electron-withdrawing properties of the non-conjugated carbonyl group of the diketone moiety.
Enaminones 3 are of interest as chemically active substances for further transformations. When substrates 3 bearing the amino group react with pyrone 1 in EtOH at room temperature for 24 h, they give polycarbonyl ligands 4a–e in 64–79% yields (Method A, Scheme 2 and Table 2), which are yellow or red precipitates and stable during storage in the solid state. Polycarbonyl ligands 4 can also be obtained directly from o-phenylenediamines 2 by the reaction with two equivalents of 4-pyrones 1 in 31–63% yields (Method B).
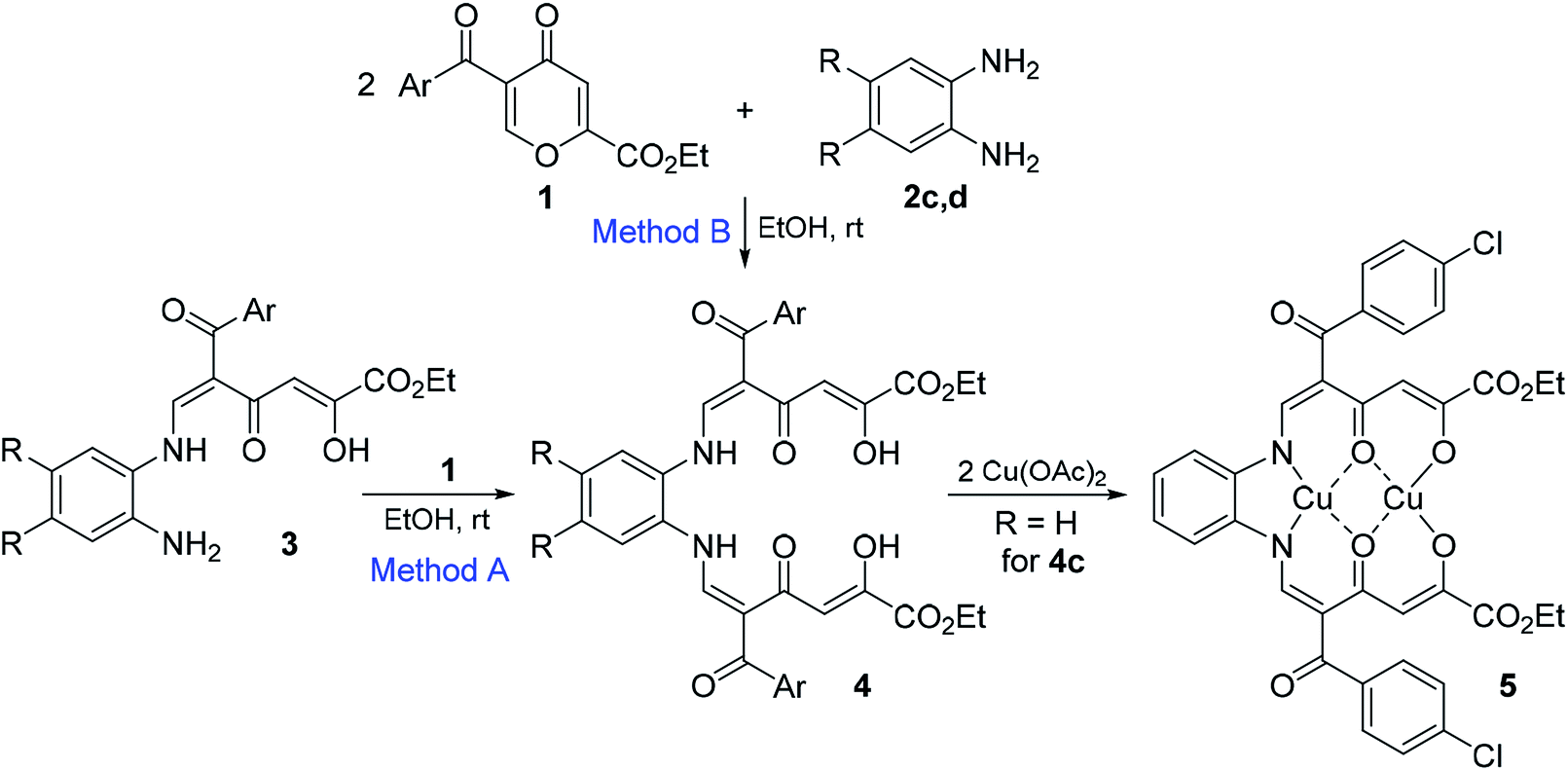 | ||
| Scheme 2 Reactions of pyrones 1 with diamines 2. | ||
| Bis-adduct | Ar | R | Yield from enaminone 3, % (Method A) | Yield from diamine, % (Method B) | 5E,5′E-4:5E,5′Z-4 |
|---|---|---|---|---|---|
| a Method A: pyrone 1 (0.26 mmol) and enaminone 3 (0.26 mmol) were stirred in EtOH (2 mL) at 20 °C for 24 h. Method B: pyrone 1 (0.72 mmol) and diamine 2 (0.36 mmol) were stirred in EtOH (2 mL) at 0 °C for 1 h and then at 20 °C for 24 h. | |||||
| 4a | Ph | H | 64 | 57 | 50:17 |
| 4b | 4-MeOC6H4 | H | 73 | 38 | 69:17 |
| 4c | 4-ClC6H4 | H | 65 | 75 | 39:26 |
| 4d | 2-Th | H | 75 | 31 | 71:14 |
| 4e | 2-Th | Me | 79 | 63 | 74:10 |
Theoretically, compounds 4 can be represented as ten open-chain tautomeric structures, but in the 1H NMR spectra it is possible to detect and assign some signals only for 5E,5′E-4 and 5E,5′Z-4 tautomers by comparing NMR signals of parent polycarbonyl compounds 3.
The structure of compounds 4 was confirmed on the basis of elemental analysis data, IR and NMR spectra. In 1H NMR spectra in CDCl3 major isomer 5E,5′E-4 appeared as a singlet of the H-3 proton at δ 6.82–6.95 ppm, doublets of the H-6 proton and the NH group at δ 7.92–8.09 ppm and 12.41–12.58 ppm (3J = 12.4–12.6 Hz), respectively. The characteristic signals of minor isomer 5E,5′Z-4 include two singlets at δ 5.90–6.36 and 6.41–6.45 ppm attributed to the vinylic protons of the diketone motifs and two doublets of the CHN protons at δ 7.99–8.12 ppm and 8.36–8.53 ppm.
It was found that the content of the most stable 5E,5′E-4 tautomer is equal to 39–74%. The nature of the substituents in the aroyl group has a strong influence on the content of this tautomer. In the case of electron-donating substituents, the content of 5E,5′E-isomer is the highest (69–74% for 4b,d,e), while the introduction of the electron-withdrawing substituent (Ar = 4-ClC6H4) leads to a decrease in the content of the main isomer to 39%. These data are also consistent with the amount of minor 5E,5′Z-isomer (10–26%), the largest content (26%) was found for 4c (Ar = 4-ClC6H4).
Compounds 4 are polydentate salphen structures, which makes them promising ligands for the formation of binuclear complex compounds with various metal ions, which have been recently received much attention for various application from catalysis to materials chemistry.9 It was found that compound 4c upon treatment with copper(II) acetate gives binuclear chelate complex 5, whose structure is in agreement with the literature data13 and is confirmed by elemental analysis, ESI-MS data, NMR and IR21 spectroscopies. 1H NMR spectra for Cu(II)-coordinated compounds are usually not informative as the result of strong signal peak broadening and unpredictable chemicals shifts due to paramagnetic properties of Cu(II) compounds.22 In contrast, binuclear complexes can produce narrow 1H NMR resonance because of magnetic interaction of two paramagnetic copper(II) centers.22 The 1H NMR spectrum of compound 5 appears to be useful and features a single set of signals, which consists of a broadened triplet for the Me group at δ 1.10 ppm, broadened quartet at δ 3.76 ppm, a broadened singlet for CH proton at δ 5.74 ppm, and a multiplet for the aromatic protons at δ 7.24–7.80 ppm.
It is interesting to note that the reaction of benzoylpyrone 1a (2 equiv.) with p-phenylenediamine (2i, 1 equiv.) proceeds very readily to form compound 6 in almost quantitative yield (99%) (Scheme 3). Such a large difference in reactivity compared to o-phenylenediamine can be explained by less steric hindrances during the formation of the polycarbonyl ligands. In contrast to aromatic diamines, the reaction of pyrone 1a with ethylenediamine led to a product, which exists in solution as a complex mixture of tautomers.
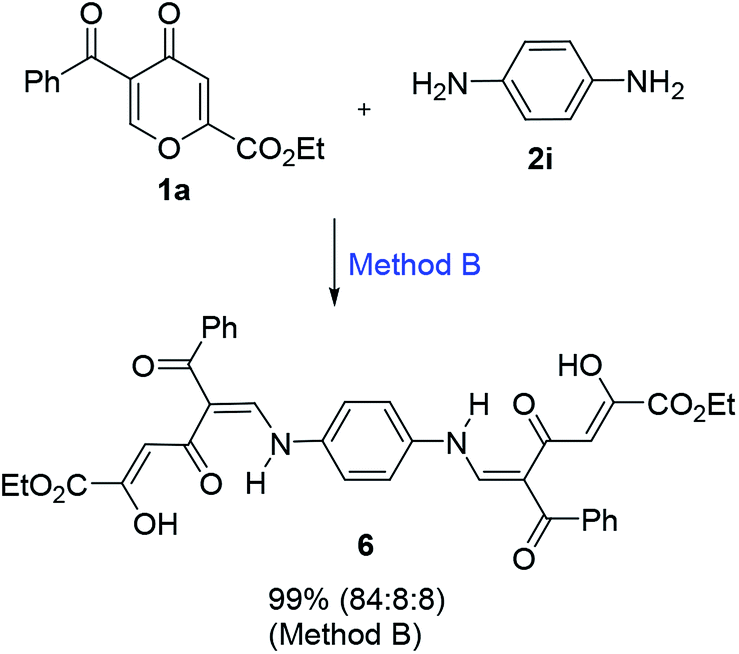 | ||
| Scheme 3 Reaction of pyrone 1a with p-phenylenediamine. | ||
The main drawback of the polycarbonyl Schiff base ligands bearing the triketone moiety is their ability to cyclize into pyridones, which may limit their application. Therefore, further we decided to study the chemical properties of the polycarbonyl compounds 3, which are connected with their stability and the possibility of further modification for the design of new ligands.
Enaminones 3e,f,h,j bearing the amino group in the ortho position can undergo intramolecular cyclization to form more stable polycyclic pyridones 7 and 8 (Scheme 4 and Table 3). It was found that reflux of compounds 3 in EtOH without the addition of acid leads to the formation of dihydropyridones 7 in 72–89%. Although quinoxalines 8 were detected in the 1H NMR spectra of products 7 (the content reached 24%), structures 7 are proved by the data of elemental analysis and the IR spectra of the obtained precipitates 7 exhibit the absence of the characteristic bands of the carbonyl groups of pyridones 8 at 1683–1710 cm−1. The obtained result probably shows that the formation of compounds 8 occurs upon the dissolution of dihydropyridone 7 in DMSO-d6. Recrystallization of compounds 7 from acetic acid or boiling in EtOH with the addition of hydrochloric acid lead to the formation of dehydration products 8 in 80–92%.
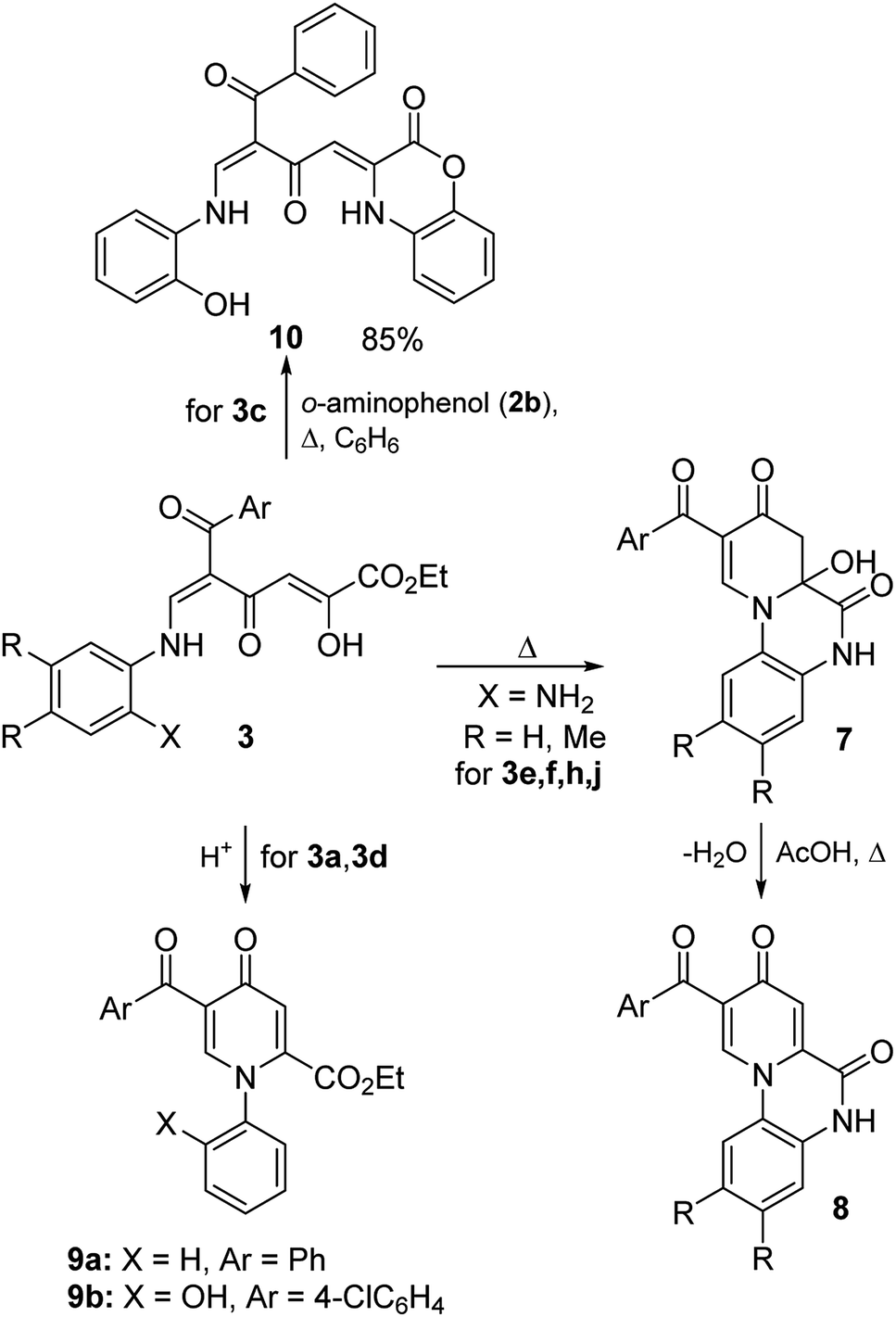 | ||
| Scheme 4 Cyclization reactions of polycarbonyl compounds 3. | ||
| Starting enaminone | Ar | R | Product 7, 8 | Yield 7, % | Yield 8, % | 7:8 (in DMSO-d6) |
|---|---|---|---|---|---|---|
| 3e | 4-MeOC6H4 | H | a | 72 | 89 | 81:19 |
| 3f | 4-ClC6H4 | H | b | 89 | 89 | 76:24 |
| 3h | 2-Th | H | c | 86 | 92 | 93:7 |
| 3j | 2-Th | Me | d | 85 | 80 | 97:3 |
Enaminones 3a,d, which were obtained from aniline and o-aminophenol, undergo cyclization in the presence of acids gave thermodynamically more stable pyridones 9 in 78–79% (Scheme 4). On the other hand, it was important to find ways for the modification of such polycarbonyl ligands into structures containing heterocyclic substituents by reactions on the active diketobutanoic moiety. Using enaminone 3c as a representative example, it was shown that this compound is able to react with o-aminophenol (2b) under reflux in benzene to form benzoxazine 10 in 85% yield.
The reaction of thenoylpyrone 1e with 2,3-diaminopyridine (2j) currently led to dihydropyridone 11 (the content of dehydratation product 12 is 8% in DMSO-d6) in 91% yield without the isolation of polycarbonyl intermediate 3 (Scheme 5). Compound 11 gave pyrido[1,2-a]quinoxaline-6,8-dione 12 in 81% yield under heating in AcOH. The structure of products 11,12 was supported by the literature data23 regarding reactions of 2,3-diaminopyridones with polycarbonyl compounds.
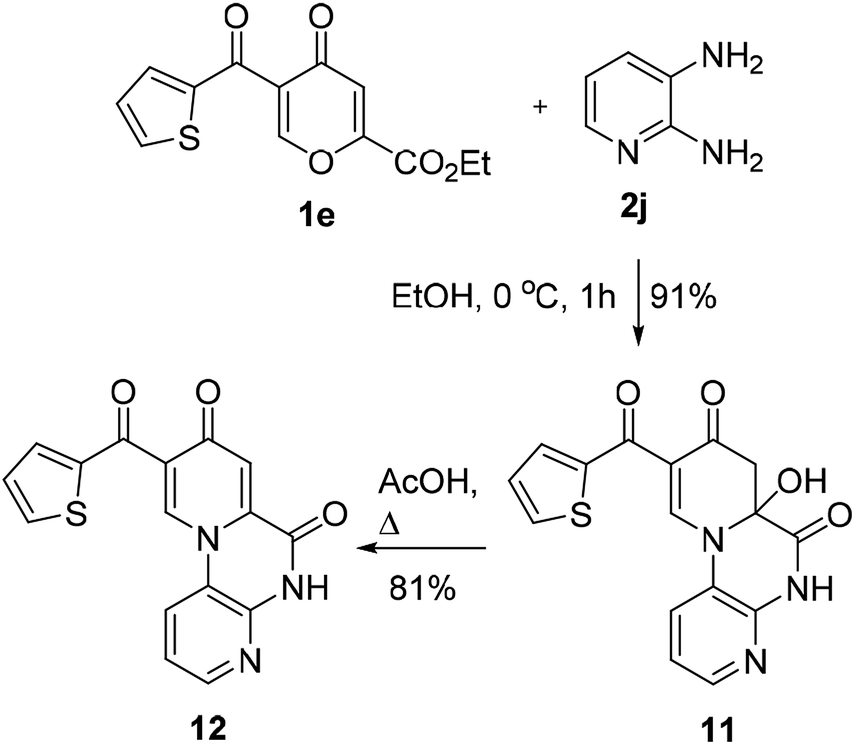 | ||
| Scheme 5 Reaction of pyrone 1e with 2,3-diaminopyridine. | ||
Conclusions
In summary, we have found a convenient, convergent, and effective approach for the introduction of a triketone moiety into various amines using ethyl esters of 5-acyl-4-pyrone-2-carboxylic acids, which allows the synthesis of a wide range of new polycarbonyl Schiff base ligands and salphen structures. Important advantages of this reaction include its wide substrate scope, catalyst free, atom economy, high yields, mild conditions and easy performing and isolation of products without the use of chromatography. Obtained polycarbonyl compounds in the solution of CDCl3 exist as several tautomers, the major tautomer has 5E-configuration of the double bond. The found strategy for the introduction of the triketone moiety can be used further for the deep functionalization of polymers and biopolymers containing a free amino group for the creation of novel catalysts or materials.Experimental
General
NMR spectra were recorded on Bruker DRX-400 (1H – 400 MHz and 13C – 100 MHz) and Bruker Avance III-500 (1H – 500 MHz and 13C – 126 MHz) spectrometers in DMSO-d6 or CDCl3. Chemical shifts are reported relative to TMS, CHCl3 (δ = 7.26 ppm, 1H NMR), DMSO-d6 (δ = 2.50 ppm, 1H NMR), CDCl3 (δ = 77.06 ppm, 13C NMR) and DMSO-d6 (δ = 39.52 ppm, 13C NMR) as internal standards. IR spectra were recorded on a Shimadzu IRSpirit-T (QATR-S) instrument (FTIR mode, ZnSe crystal). Elemental analysis was performed on a PerkinElmer PE 2400 automatic analyzer. The mass spectrum (ESI-MS) of compound 5 was measured with a Waters Xevo QTof instrument. All solvents used were dried and distilled per standard procedures. Melting points were determined on a Stuart SMP40 apparatus. Pyrones 1 were prepared according the literature procedure.24General approach for the preparation of enaminodiones 3
Pyrone 1 (1.11 mmol) was added to EtOH (6 mL) containing amine 2 (1.11 mmol) (for 3m – 2.22 mmol) under cooling on an ice bath. The resulted mixture was stirred at 0 °C for 1 h (for 3k – at 20 °C for 2 h, or 3m – at 20 °C for 1 day). The precipitate was filtered and washed with EtOH (for 3i, the reaction mixture was additionally maintained at −20 °C for 12 h).:Z-3a:keto-3a = 82:13:5. 1H NMR (CDCl3, ppm) δ: E-3a (82%) – 1.31 (t, J = 7.1 Hz, 3H, Me), 4.30 (q, J = 7.1 Hz, 2H, OCH2), 7.01 (s, 1H, CH), 7.10 (d, J = 7.7 Hz, 2H, H-2, H-6 Ph), 7.22 (tt, J = 7.4 Hz, J = 1.1 Hz, 1H, H-4 Ph), 7.38 (dd, J = 7.7 Hz, J = 7.4 Hz, 2H, H-3, H-5 Ph), 7.48 (dd, J = 7.8 Hz, J = 7.2 Hz, 2H, H-3, H-5 Ph′), 7.57 (tt, J = 7.8 Hz, J = 1.2 Hz, 1H, H-4 Ph′), 7.73 (dd, J = 7.2 Hz, J = 1.4 Hz, 1H, H-2, H-6 Ph′), 8.08 (d, J = 13.4 Hz, 1H, CHN), 12.41 (d, J = 13.4 Hz, 1H, NH), 14.36 (s, 1H, OH); 13C NMR (CDCl3, ppm) δ 14.1, 62.1, 103.9, 111.5, 117.9, 126.2, 128.6, 129.3, 130.1, 132.2, 138.5, 139.8, 153.3, 160.1, 162.6, 194.08, 194.12. Anal. calcd for C21H19NO5: C, 69.03; H, 5.24; N, 3.83. Found: C, 69.08; H, 5.13; N, 4.04.:Z-3b:keto-3b = 95:2:3. 1H NMR (CDCl3, ppm) δ: E-3b (95%) – 1.31 (t, J = 7.1 Hz, 3H, Me), 3.89 (s, 3H, MeO), 4.29 (q, J = 7.1 Hz, 2H, OCH2), 6.90 (s, 1H, CH), 6.97 (d, AA′BB′, J = 8.9 Hz, 2H, H-3 H-5 Ar), 7.12 (d, J = 7.7 Hz, 2H, H-2, H-6 Ph), 7.21 (tt, J = 7.4 Hz, J = 0.9 Hz, 1H, H-4 Ph), 7.39 (dd, J = 7.9 Hz, J = 7.4 Hz, 2H, H-3, H-5 Ph), 7.74 (d, AA′BB′, J = 8.9 Hz, 2H, H-2, H-6 Ar), 8.05 (d, J = 13.3 Hz, 1H, CHN), 12.34 (d, J = 13.3 Hz, 1H, NH), 14.38 (s, 1H, OH); 13C NMR (CDCl3, ppm) δ 14.0 (Me), 55.5 (MeO), 62.1 (CH2), 103.9 (C-3), 111.7 (C-5), 113.8 (C-3, C-5 Ar), 117.8 (C-2, C-6 Ph), 126.0 (C-4 Ph), 130.0 (C-3, C-5 Ph), 131.8 (C-2, C-6 Ar), 132.1 (C-1 Ar), 138.6 (C-1 Ph), 152.4 (C-6), 159.9 (C-2), 162.6 (C-1), 163.1 (C-4 Ar), 193.0 (Ar-C-4′O), 193.9 (C-4O). Anal. calcd for C22H21NO6: C, 66.83; H, 5.35; N, 3.54. Found: C, 66.61; H, 5.27; N, 3.86.:Z-3c:keto-3c = 76:17:7. 1H NMR (CDCl3, ppm) δ: E-3c (76%) – 1.31 (t, J = 7.1 Hz, 3H, Me), 4.30 (q, J = 7.1 Hz, 2H, OCH2), 6.79 (br, 1H, HOAr), 6.86–6.93 (m, 1H, Ar), 6.95–7.10 (m, 4H, Ar, CH), 7.48 (t, J = 7.4 Hz, 2H, H-3, H-5 Ph), 7.57 (t, J = 7.2 Hz, 1H, H-4 Ph), 7.73 (d, J = 7.3 Hz, 2H, H-2, H-6 Ph), 8.13 (d, J = 13.3 Hz, 1H, CHN), 12.60 (d, J = 13.3 Hz, 1H, NH), 14.80 (s, 1H, OH). Anal. calcd for C21H19NO6: C, 66.14; H, 5.02; N, 3.67. Found: C, 66.32; H, 4.91; N, 3.38.:Z-3d:keto-3d = 76:17:7. 1H NMR (CDCl3, ppm) δ: E-3d (76%) – 1.31 (t, J = 7.1 Hz, 3H, Me), 4.30 (q, J = 7.1 Hz, 2H, OCH2), 6.10 (br, 1H, HOAr), 6.88–7.12 (m, 5H, Ar, CH), 7.45 (d, AA′BB′, J = 8.4 Hz, 2H, H-3, H-5 Ar′), 7.68 (d, AA′BB′, J = 8.4 Hz, 2H, H-2, H-6 Ar′), 8.10 (d, J = 13.6 Hz, 1H, CHN), 12.59 (d, J = 13.6 Hz, 1H, NH), 14.49 (s, 1H, OH). Anal. calcd for C21H18ClNO6: C, 60.66; H, 4.36; N, 3.37. Found: C, 60.61; H, 4.10; N, 3.30.:Z-3e:keto-3e = 79:15:6. 1H NMR (CDCl3, ppm) δ: E-3e (79%) – 1.31 (t, J = 7.1 Hz, 3H, Me), 3.77 (br, 2H, NH2), 3.88 (s, 3H, MeO), 4.29 (q, J = 7.1 Hz, 2H, OCH2), 6.78–6.87 (m, 2H, Ar), 6.92–7.00 (m, 4H, CH, Ar, Ar′), 7.07 (td, J = 7.6 Hz, J = 0.9 Hz, 1H, H-4 Ar), 7.74 (d, AA′BB′, J = 8.7 Hz, 2H, H-2, H-6 Ar′), 7.95 (d, J = 13.1 Hz, 1H, CHN), 12.24 (d, J = 13.1 Hz, 1H, NH), 14.33 (s, 1H, OH). Anal. calcd for C22H22N2O6: C, 64.38; H, 5.40; N, 6.83. Found: C, 64.59; H, 5.60; N, 6.64.:Z-3f:keto-3f = 78:16:6. 1H NMR (CDCl3, ppm) δ: E-3f (78%) – 1.34 (t, J = 7.1 Hz, 3H, Me), 3.79 (br, 2H, NH2), 4.32 (q, J = 7.1 Hz, 2H, OCH2), 6.81–6.90 (m, 2H, Ar), 6.98 (d, J = 7.9 Hz, 1H, H-6 Ar), 7.00 (s, 1H, CH), 7.07 (td, J = 7.7 Hz, J = 0.9 Hz, 1H, H-4 Ar), 7.47 (d, AA′BB′, J = 8.4 Hz, 2H, H-3, H-5 Ar′), 7.69 (d, AA′BB′, J = 8.4 Hz, 2H, H-2, H-6 Ar′), 7.97 (d, J = 13.2 Hz, 1H, CHN), 12.33 (d, J = 13.2 Hz, 1H, NH), 14.28 (s, 1H, OH). Anal. calcd for C21H19ClN2O5: C, 60.80; H, 4.62; N, 6.72. Found: C, 60.78; H, 4.62; N, 6.67.:Z-3g:keto-3g = 79:13:8. 1H NMR (CDCl3, ppm) δ: E-3g (79%) – 1.25 (t, J = 7.1 Hz, 3H, OCH2Me), 3.79 (br, 2H, NH2), 4.27 (q, J = 7.1 Hz, 2H, OCH2), 6.79 (td, J = 7.4 Hz, J = 0.9 Hz, 1H, H-5 Ar), 6.85 (dd, J = 8.0 Hz, J = 0.9 Hz, 1H, H-3 Ar), 6.95 (d, J = 7.5 Hz, 1H, H-6 Ar), 7.08 (td, J = 7.7 Hz, J = 0.9 Hz, 1H, H-4 Ar), 7.10 (s, 1H, CH), 7.51–7.68 (m, 2H, Naph), 7.84 (dd, J = 8.5 Hz, J = 1.5 Hz, 1H, Naph), 7.89–8.00 (m, 3H, Naph), 8.07 (d, J = 13.1 Hz, 1H, CHN), 8.24 (d, J = 0.7 Hz, 1H, H-1 Naph), 12.38 (d, J = 13.1 Hz, 1H, NH), 14.34 (s, 1H, OH). Anal. calcd for C25H22N2O5: C, 69.76; H, 5.15; N, 6.51. Found: C, 69.88; H, 5.14; N, 6.30.:Z-3h:keto-3h = 89:8:3. 1H NMR (CDCl3, ppm) δ: E-3h (89%) – 1.33 (t, J = 7.1 Hz, 3H, Me), 3.79 (br, 2H, NH2), 4.31 (q, J = 7.1 Hz, 2H, OCH2), 6.82–6.88 (m, 2H, H-5, H-3 Ar), 7.04 (dd, J = 7.2 Hz, J = 1.0 Hz, 1H, H-6 Ar), 7.04 (s, 1H, CH), 7.09 (td, J = 7.9 Hz, J = 1.1 Hz, 1H, H-4 Ar), 7.14 (dd, J = 5.0 Hz, J = 3.8 Hz, 1H, H-4 Th), 7.55 (dd, J = 3.8 Hz, J =1.0 Hz, 1H, H-3 Th), 7.67 (dd, J = 5.0 Hz, J =1.0 Hz, 1H, H-5 Th), 8.14 (d, J = 13.1 Hz, 1H, CHN), 12.22 (d, J = 13.1 Hz, 1H, NH), 14.28 (s, 1H, OH). Anal. calcd for C19H18N2O5S: C, 59.06; H, 4.70; N, 7.25. Found: C, 58.88; H, 4.88; N, 7.49.:Z-3i:keto-3i = 79:14:7. 1H NMR (CDCl3, ppm) δ: E-3i (79%) – 1.35 (s, 9H, t-Bu), 1.38 (t, J = 7.1 Hz, 3H, Me), 3.75 (br, 2H, NH2), 4.36 (q, J = 7.1 Hz, 2H, OCH2), 6.59 (s, 1H, CH), 6.83–6.92 (m, 2H, Ar), 7.05 (dd, J = 7.8 Hz, J = 0.9 Hz, 1H, H-6 Ar), 7.11 (td, J = 7.6 Hz, J = 1.1 Hz, 1H, H-4 Ar), 7.86 (d, J = 12.4 Hz, 1H, CHN), 11.95 (d, J = 12.4 Hz, 1H, NH), 14.36 (s, 1H, OH). Anal. calcd for C19H24N2O5: C, 63.22; H, 6.71; N, 7.77. Found: C, 63.53; H, 6.58; N, 7.89.:Z-3j:keto-3j = 87:9:4. 1H NMR (CDCl3, ppm) δ: E-3j (87%) – 1.33 (t, J = 7.1 Hz, 3H, OCH2Me), 2.16 (s, 3H, Me), 2.19 (s, 3H, Me), 3.59 (br, 2H, NH2), 4.31 (q, J = 7.1 Hz, 2H, OCH2), 6.65 (s, 1H, Ar), 6.80 (s, 1H, Ar), 7.02 (s, 1H, CH), 7.14 (dd, 1H, J = 5.0 Hz, J = 3.8 Hz, H-4 Th), 7.54 (dd, 1H, J = 3.8 Hz, J = 1.1 Hz, H-3 Th), 7.67 (dd, 1H, J = 5.0 Hz, J = 1.1 Hz, H-5 Th), 8.11 (d, J = 13.2 Hz, 1H, CHN), 12.23 (d, J = 13.2 Hz, 1H, NH), 14.32 (s, 1H, OH); 13C NMR (CDCl3, ppm) δ 14.1, 19.0, 19.4, 62.1, 103.5, 111.6, 119.5, 120.1, 124.8, 127.8, 128.7, 132.9, 133.3, 135.7, 136.3, 145.3, 153.9, 159.7, 162.7, 185.3, 193.0. Anal. calcd for C21H22N2O5S: C, 60.85; H, 5.35; N, 6.76. Found: C, 60.51; H, 5.65; N, 6.65.:Z-3k:keto-3k = 80:13:7. 1H NMR (CDCl3, ppm) δ: E-3k (80%) – 1.35 (t, J = 7.1 Hz, 3H, Me), 4.34 (q, J = 7.1 Hz, 2H, OCH2), 7.00 (d, J = 8.1 Hz, 1H, Py), 7.10 (dd, J = 7.3 Hz, J = 4.8 Hz, 1H, Py), 7.12 (s, 1H, CH), 7.51 (dd, J = 7.7 Hz, J = 7.3 Hz, 2H, H-3, H-5 Ph), 7.61 (tt, J = 7.7 Hz, J = 1.4 Hz, 1H, H-4 Ph), 7.73 (td, J = 7.8 Hz, J = 1.3 Hz, 1H, Py), 7.79 (2H, dd, J = 7.9 Hz, J = 1.4 Hz, H-2, H-6 Ph), 8.30 (dd, J = 4.8 Hz, J = 1.3 Hz, 1H, Py), 8.81 (d, J = 12.5 Hz, 1H, CHN), 12.41 (d, J = 12.5 Hz, 1H, NH), 14.44 (s, 1H, OH); 13C NMR (CDCl3, ppm) δ 14.1, 62.2, 103.8, 112.3, 112.9, 120.7, 128.6, 129.6, 132.3, 138.8, 139.4, 149.0, 149.9, 151.2, 160.8, 162.5, 194.4, 194.8. Anal. calcd for C20H18N2O5: C, 65.57; H, 4.95; N, 7.65. Found: C, 65.62; H, 4.76; N, 7.94.:Z-3l:keto-3l = 92:4:4. 1H NMR (CDCl3, ppm) δ: E-3l (92%) – 1.34 (t, J = 7.1 Hz, 3H, OCH2Me), 2.38 (s, 3H, Me), 3.11 (s, 3H, Me), 4.32 (q, J = 7.1 Hz, 2H, OCH2), 7.12 (dd, J = 5.0 Hz, J = 3.9 Hz, 1H, H-4 Th), 7.15 (s, 1H, CH), 7.33–7.38 (m, 3H, Ph), 7.48 (dd, J = 8.0 Hz, J = 7.4 Hz, 2H, H-3, H-5 Ph), 7.60 (dd, J = 5.0 Hz, J = 0.8 Hz, 1H, H-5 Th), 7.64 (dd, J = 3.9 Hz, J = 0.8 Hz, 1H, H-3, Th), 9.03 (d, J = 12.6 Hz, 1H, CHN), 12.52 (d, J = 12.6 Hz, 1H, NH), 14.27 (s, 1H, OH). Anal. calcd for C24H23N3O6S: C, 59.86; H, 4.81; N, 8.73. Found: C, 59.94; H, 4.70; N, 8.72.:Z-3m:keto-3m = 93:2:5. 1H NMR (CDCl3, ppm) δ: E-3m (93%) – 1.35 (t, 3H, J = 7.1 Hz, Me), 4.34 (q, J = 7.1 Hz, 2H, OCH2), 6.99 (s, 1H, CH), 7.07 (t, J = 4.8 Hz, 1H, H-5 pyrimidine), 7.18 (dd, J = 4.9 Hz, J = 3.8 Hz, 1H, H-4 Th), 7.60 (dd, J = 3.8 Hz, J = 0.8 Hz, 1H, H-3 Th), 7.73 (dd, J = 4.9 Hz, J = 0.8 Hz, 1H, H-5 Th), 8.57 (d, J = 4.8 Hz, 2H, H-4, H-6 pyrimidine), 8.88 (d, J = 12.6 Hz, 1H, CHN), 12.07 (d, J = 12.6 Hz, 1H, NH), 14.50 (s, 1H, OH). Anal. calcd for C17H15N3O5S: C, 54.68; H, 4.05; N, 11.25. Found: C, 54.79; H, 4.06; N, 11.04.:Z-3n:keto-3n = 79:11:10. 1H NMR (CDCl3, ppm) δ: E-3m (79%) – 1.20 (s, 9H, t-Bu), 1.34 (t, J = 7.1 Hz, 3H, Me), 4.32 (q, J = 7.1 Hz, 2H, OCH2), 4.54 (d, J = 6.0 Hz, 2H, CH2Ph), 6.63 (s, 1H, CH), 7.28 (d, J = 7.1 Hz, 2H, H-2, H-6 Ph), 7.32–7.44 (3H, m, Ph), 7.69 (d, J = 13.3 Hz, 1H, CHN), 10.67 (dt, J = 13.3 Hz, J = 6.0 Hz, 1H, NH), 14.43 (s, 1H, OH). Anal. calcd for C20H25NO5: C, 66.84; H, 7.01; N, 3.90. Found: C, 67.00; H, 6.91; N, 4.11.:Z-3o:keto-3o = 68:19:13. 1H NMR (CDCl3, ppm) δ: E-3o (68%) – 1.31 (t, J = 7.1 Hz, 3H, Me), 4.29 (q, J = 7.1 Hz, 2H, OCH2), 4.54 (d, J = 6.0 Hz, 2H, CH2Ph), 7.01 (s, 1H, CH), 7.27 (dd, J = 6.9 Hz, J = 1.3 Hz, 2H, H-2, H-6 PhCH2), 7.31–7.52 (5H, m, PhCH2, Ph), 7.53 (t, J = 7.5 Hz, 1H, H-4 Ph), 7.63 (dd, J = 7.8 Hz, J = 1.4 Hz, 1H, H-2, H-6 Ph), 7.69 (d, J = 13.7 Hz, 1H, CHN), 10.95 (dt, J = 13.7 Hz, J = 6.0 Hz, 1H, NH), 14.43 (s, 1H, OH). Anal. calcd for C22H21NO5: C, 69.64; H, 5.58; N, 3.69. Found: C, 69.48; H, 5.61; N, 3.97.General method for the preparation of polycarbonyl compounds 4 and 6
CH), 7.13–7.22 (m, 1H, Ar), 7.24–7.33 (m, 1H, Ar), 7.50 (2H, dd, J = 7.9 Hz, J = 7.2 Hz, H-3, H-5 Ph), 7.55–7.66 (m, 1H, H-4 Ph), 7.75–7.81 (m, 2H, H-2, H-6 Ph), 7.98 (d, J = 12.5 Hz, 1H, CHN), 12.58 (d, J = 12.5 Hz, 1H, NH), 14.18 (s, 1H, OH); 13C NMR (DMSO-d6, ppm) δ 14.0, 62.1, 103.7, 113.2, 120.3, 127.2, 129.1, 129.4, 131.5, 132.4, 139.4, 154.6, 160.8, 162.3, 193.9, 194.4. Anal. calcd for C36H32N2O10: C, 66.25; H, 4.94; N, 4.29. Found: C, 66.38; H, 4.86; N, 4.29.CH), 6.96 (d, AA′BB′, J = 8.8 Hz, 2H, H-3, H-5 Ar′), 7.14–7.30 (m, 2H, Ar), 7.77 (d, J = 8.8 Hz, AA′BB′, 2H, H-2, H-6 Ar′), 7.92 (d, J = 12.5 Hz, 1H, CHN), 12.49 (d, J = 12.5 Hz, 1H, NH), 14.18 (s, 1H, OH); 13C NMR (DMSO-d6, ppm) δ 14.0, 55.5, 62.1, 103.7, 113.9, 120.2, 127.5, 131.5, 131.9, 132.2, 153.6, 156.8, 160.6, 162.4, 163.3, 192.7, 194.2. Anal. calcd for C30H36N2O12: C, 64.04; H, 5.09; N, 3.93. Found: C, 63.78; H, 4.92; N, 4.05.CH), 7.15–7.40 (m, 2H, Ar), 7.39–7.50 (m, 2H, Ar′), 7.70 (d, J = 8.5 Hz, AA′BB′, 2H, H-3, H-5 Ar′), 7.93 (d, J = 12.6 Hz, 1H, CHN), 12.55 (d, J = 12.6 Hz, 1H, NH), 14.09 (s, 1H, OH). 13C NMR (CDCl3, ppm) δ 14.0, 62.2, 103.6, 113.0, 120.3, 127.9, 129.0, 129.8, 130.8, 137.6, 138.8, 154.5, 160.9, 162.2, 192.5, 194.1. Anal. calcd for C36H30Cl2N2O10: C, 59.93; H, 4.19; N, 3.88. Found: C, 59.64; H, 3.82; N, 4.15.CH), 7.15 (dd, J = 4.8 Hz, J =3.8 Hz, 1H, H-4 Th), 7.23–7.33 (m, 2H, Ar), 7.59 (dd, J = 3.8 Hz, J = 0.8 Hz, 1H, H-3 Th), 7.70 (dd, J = 4.8 Hz, J = 0.8 Hz, 1H, H-5 Th), 8.09 (d, J = 12.4 Hz, 1H, CHN), 12.48 (d, J = 12.4 Hz, 1H, NH), 14.12 (s, 1H, OH); 13C NMR (CDCl3, ppm) δ 14.1, 62.2, 103.2, 113.5, 120.4, 127.6, 128.1, 131.5, 133.4, 134.0, 144.7, 153.0, 160.8, 162.3, 185.0, 193.7. Anal. calcd for C32H28N2O10S2: C, 57.82; H, 4.25; N, 4.21. Found: C, 57.81; H, 4.20; N, 4.12.CH), 7.00 (br, 1H, Ar), 7.14 (dd, J = 5.0 Hz, J = 3.8 Hz, 1H, H-4 Th), 7.57 (dd, J = 3.8 Hz, J = 0.9 Hz, 1H, H-3 Th), 7.69 (dd, J = 5.0 Hz, J = 0.9 Hz, 1H, H-5 Th), 8.05 (d, J = 12.6 Hz, 1H, CHN), 12.41 (d, J = 12.6 Hz, 1H, NH), 14.14 (s, 1H, OH); 13C NMR (CDCl3, ppm) δ 14.0, 19.5, 62.1, 103.3, 113.0, 121.5, 128.0, 129.1, 133.4, 133.9, 136.9, 144.9, 153.4, 160.5, 162.4, 185.0, 193.5. Anal. calcd for C34H32N2O10S2: C, 58.95; H, 4.66; N, 4.04. Found: C, 58.90; H, 4.77; N, 4.05.Binuclear copper(II) complex 5
Cu(OAc)2 (0.028 g, 0.15 mmol) was added to solution of polycarbonyl compounds 4c (0.050 g, 0.069 mmol) in DMF (1 mL). The reaction mixture was stirred for 10 min at room temperature. The precipitate that formed was filtered and washed with EtOH. Yield 87% (0.051 g), red-brown powder, mp > 330 °C. 1H NMR (DMSO-d6, ppm) (selected signals) δ 1.10 (br, 3H, Me), 3.76 (br, 2H, CH2), 5.74 (br, 1H,CH), 7.24–7.80 (m, 6H, Ar). IR (ATR): 2982, 1733, 1723, 1713, 1631, 1569, 1338, 1630, 1393, 1340, 1247, 995, 911, 816, 749 cm−1. Anal. calcd for C36H26Cl2Cu2N2O10: C, 51.19; H, 3.10; N, 3.32. Found: C, 51.06; H, 3.11; N, 3.62. HRMS (ESI) calcd for C36H27N2O10Cl2Cu2 [M + H]+ 842.9635, found 842.9622.
:(5E,5′Z)-6:keto-(5E,5′E)-6 = 84:8:8. 1H NMR (CDCl3, ppm) δ: (5E,5′E)-6 (84%) – 1.33 (3H, t, J = 7.1 Hz, Me), 4.31 (2H, q, J = 7.1 Hz, OCH2), 6.94 (1H, s, CH), 7.15 (2H, s, Ar), 7.50 (dd, J = 7.8 Hz, J = 7.3 Hz, 2H, H-3, H-5 Ph), 7.57–7.60 (m, 1H, H-4 Ph), 7.75 (d, 2H, J = 7.3 Hz, H-2, H-6 Ph), 8.02 (d, J = 13.2 Hz, 1H, CHN), 12.46 (d, J = 13.2 Hz, 1H, NH), 14.27 (s, 1H, OH). Anal. calcd for C36H32N2O10: C, 66.25; H, 4.94; N, 4.29. Found: C, 66.14; H, 4.81; N, 4.45.General approach for the preparation of 6a-hydroxy-9-acyl-6a,7-dihydro-5H-pyrido[1,2-a]quinoxaline-6,8-diones 7
Enaminone 3e,f,h,j (0.259 mmol) was refluxed in EtOH (2 mL) for 20 min. The precipitate that formed was filtered and washed with EtOH.General approach for the preparation of 9-acyl-5H-pyrido[1,2-a]quinoxaline-6,8-diones 8
Compound 7 (0.147 mmol) was refluxed in AcOH (2 mL) for 20 min. After cooling, the reaction mixture was diluted with H2O (3 mL). The precipitate that formed was filtered and washed with cold EtOH. The product was dried at 100 °C.CH), 6.68 (t, J = 7.6 Hz, 1H, Ar), 6.80–6.87 (m, 2H, Ar), 6.93 (t, J = 6.9 Hz, 1H, Ar), 6.97 (d, J = 8.1 Hz, 1H, Ar), 7.22 (t, J = 7.7 Hz, 1H, Ar), 7.42 (d, J = 7.8 Hz, 1H, Ar), 7.44–7.51 (m, 3H, Ar, Ph), 7.59 (t, J = 7.3 Hz, 1H, H-4 Ph), 7.77 (s, 1H, CHN), 7.83 (2H, d, J = 7.7 Hz, H-2, H-6 Ph), 9.56 (br, 1H, NH), 9.70 (s, 1H, OH), 10.25 (br, 1H, NH). Anal. calcd for C25H18N2O5: C, 70.42; H, 4.25; N, 6.57. Found: C, 70.09; H, 4.09; N, 6.57.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Russian Science Foundation (Grant 18-13-00186). Analytical studies were carried out using equipment of the Center for Joint Use “Spectroscopy and Analysis of Organic Compounds” at the Postovsky Institute of Organic Synthesis of the Russian Academy of Sciences (Ural Branch) and the Laboratory of Complex Investigations and Expert Evaluation of Organic Materials of the Center for Joint Use at the Ural Federal University. We also thank both reviewers for helpful suggestions.Notes and references
-
(a) P. R. Christensen, A. M. Scheuermann, K. E. Loeffler and B. A. Helms, Nat. Chem., 2019, 11, 442 CrossRef CAS PubMed
; (b) E. A. Shokova, J. K. Kim and V. V. Kovalev, Russ. J. Org. Chem., 2015, 51, 755 CrossRef CAS
-
(a) N. V. Zolotareva and V. V. Semenov, Russ. Chem. Rev., 2013, 82, 964 CrossRef
- See reviews:
(a) K. C. Gupta and A. K. Sutar, Coord. Chem. Rev., 2008, 252, 1420 CrossRef CAS
-
(a) Y. Ren, J. Chen, C. Qi and H. Jiang, ChemCatChem, 2015, 7, 1535 CrossRef CAS
-
(a) S. J. Wezenberg, E. C. Escudero-Adán, J. Benet-Buchholz and A. W. Kleij, Org. Lett., 2008, 10, 3311 CrossRef CAS PubMed
- C. Bizzarri, E. Spuling, D. M. Knoll, D. Volz and S. Bräse, Coord. Chem. Rev., 2018, 373, 49 CrossRef CAS
- A. K. Asatkar, S. P. Senanayak, A. Bedi, S. Panda, K. S. Narayan and S. S. Zade, Chem. Commun., 2014, 50, 7036 RSC
- G. Salassa, J. W. Ryan, E. C. Escudero-Adána and A. W. Kleij, Dalton Trans., 2014, 43, 210 RSC
-
(a) R. M. Clarke and T. Storr, Dalton Trans., 2014, 43, 9380 RSC
- K. T. Mahmudov and A. J. L. Pombeiro, Chem.–Eur. J., 2016, 22, 16356 CrossRef CAS
-
(a) Y. S. Kudyakova, D. N. Bazhin, M. V. Goryaeva, Y. V. Burgart and V. I. Saloutin, Russ. Chem. Rev., 2014, 83, 120 CrossRef
-
(a) P. P. Mukovoz, P. A. Slepukhin, E. A. Danilova, O. P. Aysuvakova and A. P. Glinushkin, Russ. J. Gen. Chem., 2018, 88, 1363 CrossRef CAS
-
(a) D. E. Fenton and S. E. Gayda, J. Chem. Soc., Dalton Trans., 1977, 2095 RSC
-
(a) D. L. Obydennov, L. R. Khammatova, O. S. Eltsov and V. Y. Sosnovskikh, Org. Biomol. Chem., 2018, 16, 1692 RSC
- D. L. Obydennov and V. Y. Sosnovskikh, Chem. Heterocycl. Compd., 2014, 50, 579 CrossRef CAS
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS
-
(a) V. S. Berseneva, A. V. Tkachev, Y. Yu. Morzherin, W. Dehaen, I. Luyten, S. Toppet and V. A. Bakulev, J. Chem. Soc., Perkin Trans. 1, 1998, 2133 RSC
-
(a) J. Saloň, V. Milata, A. Gatial, N. Prónayová, J. Leško, P. Černuchová, Z. Rappoport, G. Vo-Thanh and A. Loupy, Eur. J. Org. Chem., 2005, 4870 CrossRef
- C. Hametner, P. Černuchová, V. Milata, G. Vo-Thanh and A. Loupy, Org. Magn. Reson., 2005, 43, 171 CrossRef CAS PubMed
- A. Brbot-Šaranović, G. Pavlović and M. Cindrić, Struct. Chem., 2000, 1, 65 CrossRef
-
(a) A. V. Pestov, L. A. Khamidullina, V. Y. Sosnovskikh, P. A. Slepukhin and I. S. Pusyrev, Polyhedron, 2016, 106, 75 CrossRef CAS
-
(a) J. H. Satcher and A. L. Balch, Inorg. Chem., 1995, 34, 3371 CrossRef CAS
-
(a) V. Y. Sosnovskikh, A. V. Safrygin, R. A. Irgashev, M. A. Ezhikova and M. I. Kodess, RSC Adv., 2016, 6, 30056 RSC
- D. L. Obydennov, A. O. Goncharov and V. Y. Sosnovskikh, Russ. Chem. Bull., 2016, 65, 2233 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra07653k |
| This journal is © The Royal Society of Chemistry 2019 |