
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceKinetically-controlled mechanism-based isolation of metabolic serine hydrolases in active form from complex proteomes: butyrylcholinesterase as a case study†
Hui Liu,
Huimin Zhou,
Huaqiao Du,
Qiaoling Xiao and
Marco Pistolozzi *
*
School of Biology & Biological Engineering, South China University of Technology, Higher Education Mega Center, Guangzhou, People's Republic of China. E-mail: marco_pistolozzi@scut.edu.cn
First published on 26th November 2019
Abstract
In this work an activity-based probe containing a carbamate group was designed to isolate human butyrylcholinesterase (hBChE), a metabolic serine hydrolase (mSH), from complex proteomes. The method took advantage of the native interaction mechanism of mSHs with carbamate pseudo-substrates for temporarily capturing the enzyme on a resin functionalized with the carbamate probe and releasing the enzyme in active form after removal of the contaminating proteins. The isolation relied on the possibility of manipulating the carbamylation and decarbamylation kinetics favoring the former during the capture and wash steps and the latter in the release step. The designed probe captured and released all the active hBChE isoenzymes present in plasma with high selectivity (up to ∼2000-fold purification) and reasonable yields (17% to 36%). The parameters affecting the performance were the incubation time used in the load and elution steps, the plasma to resin volumetric ratio, the elution temperature and the nature and concentration of the eluting agent. The carbamate resin could be prepared either by coupling a fully synthesized probe with an activated resin or by building the probe onto the resin by a step-by-step procedure, without major differences in performance between the two routes. The prepared resins allowed to process up to about 8.5 mL of plasma per g of resin with constant performance. Since the method was based on the general catalytic cycle of mSHs, we expect this approach to be applicable to other enzymes of the family, by selecting a suitable target-selective feature to link to the carbamate group.
1. Introduction
Serine hydrolases (SHs) constitute a large class of enzymes that catalyze the hydrolysis of esters, amides, or thioester groups of small molecules, peptides, and proteins. In mammals, this class comprises more than 200 enzymes including serine proteases (e.g. trypsin, chimotrypsin, subtilisin enzymes) and other enzymes, collectively termed “metabolic SHs” (mSHs), consisting of peptidases, lipases, esterases, thioesterases and amidases. mSHs are known for their involvement in a wide range of (patho)physiological pathways, however, despite their importance, a large part of them has yet not been fully characterized.1–4 mSHs share a similar catalytic cycle in which a characteristic nucleophilic serine reacts with the substrate, forming an acyl-enzyme ester intermediate that is quickly hydrolized by water.2 In addition to the natural substrates, other molecules bearing carbamate and organophosphate groups can act as mSH substrates4–6 (Fig. 1a), albeit with low efficiency, and for this reason are regarded as pseudo-substrates. In fact, since the hydrolysis of the acyl-enzyme is much slower when the acyl group is a carbamate or a phosphonate rather than an ester, the reaction of mSHs with pseudo-substrates results in the formation of adducts with significantly longer lifespan than those formed with the natural substrates.7,8 In the case of recombinant enzymes, the use of a His-tag enables a simple, often single-step, purification of active mSHs through immobilized metal affinity chromatography (IMAC) or metal oxides (MOAC)9,10 and other similar methods based on magnetic micro- and nanoparticles.11,12 Very promising purification strategies based on cleavable self-aggregating tags13 have recently emerged, and might replace IMAC and MOAC strategies in the future. However, the isolation of untagged mSHs from their native complex proteomes is still a very challenging task and several techniques are often necessary to obtain a pure enzyme preparation,14–16 making mSHs isolation time-consuming, labor-intensive and rather expensive. In virtue of their pseudo-substrate behaviour, organophosphonates have been used to design activity-based probes that could be used to monitor mSHs activity in complex proteomes using a strategy known as activity-based protein profiling (ABPP).1,4,17–20 In the frame of mSHs, this chemoproteomic strategy employs a probe containing a fluorophosphonate function at one end that targets the active site of mSHs forming irreversible adducts. The other end of the probe is functionalized with a reporter tag (e.g., a fluorescent molecule such as rhodamine) that allows selective detection of the targeted mSHs after SDS-PAGE separation. This approach has been used to study quantitative differences of mSHs activity in different conditions21 or, in a competitive format, to find new ligands targeting the active site of mSHs and test their specificity,22 and to find off-targets of environmental pesticides.23,24 In addition to their use for monitoring purposes, if opportunely functionalized (e.g. with an affinity or a clickable tag), such probes can be used for the enrichment of mSHs for subsequent identification by MS analysis. However, since these probes bind irreversibly to the active site, the enzymes are isolated as the inactive probe-conjugates. In other works in which the phosphonate probe was directly linked to a chromatographic material, the target enzyme could not be recovered,25 or required the use of strong nucleophilic reagents to restore the native active site.26,27 Therefore such probes cannot be easily used if an active enzyme is desired. Due to these limitations, we explored the possibility to isolate selected mSHs from complex proteomes using activity-based carbamate probes. The proposed approach consists in substituting one of the N-alkyl groups of the carbamate moiety of a suitable enzyme pseudo-substrate with a spacing arm that is covalently attached to a solid surface (Fig. 1). Upon incubation of the proteome with the functionalized solid, the enzyme reacts with the carbamate moiety and a certain amount, which depends on the carbamylation and decarbamylation rates, is constantly attached to the solid surface as a carbamate adduct (i.e., with a covalent bond) allowing the removal of unbound or reversibly bound contaminating proteins by rapidly washing with large volumes of a suitable buffer. Then, in virtue of the native capacity of the enzyme to slowly hydrolyze the covalent carbamyl–enzyme bond, it would be possible to recover the active enzyme by simply modifying the equilibrium between carbamylation and decarbamylation. To test the principle of this approach we selected human butyrylcholinesterase (EC 3.1.1.8, hBChE) as the model mSH and human plasma as the model proteome. | ||
| Fig. 1 (a) Simplified schematic representation of the interaction between carbamates and metabolic serine hydrolases (mSHs). The formation of the enzyme-pseudo-substrate reversible complex and the following first-order carbamylation step are implicitally included in the carbamylation step. (b) Schematic representation of the method for the isolation of mSHs: in the load and wash steps the decarbamylation is the rate limiting step, therefore hBChE accumulates on the resin as a carbamate adduct. In the elution step, the carbamylation rate is reduced therefore the decarbamylation prevails. The sepharose bead is indicated with the letter “S”. (c) The structure of the probe used in this study. 6a and 6b were obtained following the syntheses depicted in Scheme 1 and S1 (ESI†), respectively. | ||
2. Results and discussion
2.1. Probe design
The design of the carbamate probe was based on the structure of bambuterol monocarbamate (MONO), a product of the bioconversion of the BChE inhibitor bambuterol (BMB).7,28 MONO can react with ChEs following the general pseudo-substrate mechanism with high potency and selectivity towards hBChE,7,29 thus representing a suitable lead structure to characterize this activity-based method. Since the decarbamylation rate constant (k3) of carbamyl-ChEs is modulated by the number and nature of the substituents on the carbamic nitrogen, and in general monoalkylated carbamates are hydrolyzed faster than dialkylated ones,30–32 one of the N-methyl groups of MONO was removed to increase the decarbamylation kinetics which, according to the working hypothesis, is a key parameter modulating the elution rate of the enzyme from the resin. The other N-methyl group was substituted with a flexible spacing arm which was then linked to sepharose (Fig. 1c).2.2. Synthesis of the probe and kinetic characterization of its interaction with hBChE
The synthesis of the probe involved two activation steps and N,N′-carbonyldiimidazole (CDI) was used as the activating reagent for both of them (Strategy 1, Scheme 1). First it was used as a zero-length crosslinker to form an amide bond between Boc-6-aminohexanoic acid (Boc-6Ahx) and 4,7,10-trioxa-1,13-tridecanediamine (TTD), then it was used as a one-carbon spacer to form a carbamate bond between the amino-end of Boc-6Ahx-TTD (1) and one of the phenol groups of terbutaline (TBT) to form compound 3. The imidazolyl urea intermediate (compound 2) was isolated for characterization, although the reaction could be performed one-pot.33 The kinetic characterization of compound 3 indicated that the introduction of the spacing arm in the structure of MONO did not significantly modify the bimolecular carbamylation rate constant (kI), while the removal of one of the N-methyl groups increased the k3, as expected (Fig. 2 and Table 1). In addition, since a linear correlation was obtained by plotting the pseudo-first order kinetic constants (kobs) against the inhibitor concentrations up to 2 μM, the analysis indicated that the probe has a relatively low affinity (high KD) for hBChE, likely above 20 μM. | ||
| Scheme 1 Synthesis of sepharose-6Ahx-TTD-TBT 6a (Strategy 1). Boc-6Ahx, Boc-6-aminohexanoic acid; CDI N,N′-carbonyldiimidazole; TTD, 4,7,10-trioxa-1,13-tridecanediamine; TBT, rac-terbutaline hemisulfate; 4NPCF, p-nitrophenyl chloroformate; DMAP, 4-(dimethylamino)pyridine. | ||
 | ||
| Fig. 2 (a) Time- and concentration-dependent inhibition of hBChE after pre-incubation with various concentrations of compound 3. The one-phase exponential fit of the measured residual activity (solid lines) provided the kobs values. (b) Linear correlation of the kobs values vs. the concentration of compound 3. The slope of the linear regression corresponds to the bimolecular carbamylation rate constant, kI. (c) Secondary plot obtained from the analysis the area under the inhibition–time curves (AUIC). The slope of the linear regression corresponds to the decarbamylation rate constant, k3. (d) Recovery of hBChE activity after inhibition by compound 3 and dilution. | ||
| Compound | kI (M−1 min−1) | k3 (×10−3, min−1) |
|---|---|---|
| 3 | (7.7 ± 0.1) × 105 | 57 ± 3 |
| MONO29 | (3.6 ± 0.2) × 105 | 10.2 ± 0.6 |
2.3. Synthesis and characterization of the carbamate resins 6a (Strategy 1) and 6b (Strategy 2)
Compound 3 was deprotected to form compound 4, which was linked to a sepharose resin containing p-nitrophenyl carbonate groups (5) to obtain the final resin 6a (Strategy 1, Scheme 1 and Fig. 1). An alternative strategy (Strategy 2) involved the sequential addition of the building blocks starting from 5 to form a carbamate resin containing the same probe (6b) by using a different route (ESI, Scheme S1†). In this simplified procedure 6-aminohexanoic acid (6Ahx) was coupled to 5 to obtain a sepharose-6Ahx derivatized resin (7). The addition of TTD and N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (ECD) afforded sepharose-6Ahx-TTD (8) which was then activated with CDI and reacted with TBT to form the final carbamate resin 6b.The amount of carbamate probe immobilized on resin 6a was determined by HPLC analysis of the TBT released from the resin upon alkaline treatment and resulted 5.5 nmol mg−1 of wet (spin filtered) resin. The same approach gave only an approximate estimation of the carbamate immobilized on resin 6b because we found that CDI was able to activate the hydroxyl groups present on the sepharose matrix and a large amount of TBT was coupled directly to the matrix. Based on the amount of TBT released from 6b and the amount released from a test resin prepared by reacting sepharose, CDI and TBT (i.e. without the presence of the spacing arm), we estimated the amount to be in the range of 0.2–0.4 nmol mg−1 of wet (spin filtered) resin.
2.4. Characterization of the retention and release mechanisms
The capability of the prepared carbamate resins to retain and release hBChE was tested by a series of single-step purification experiments in which the resins were placed on spin filters, incubated with plasma and then quickly washed with large volumes of a wash buffer. The enzyme was eluted in three fractions obtained by incubation with an elution buffer. The eluted proteins were washed and concentrated by ultracentrifugation, then an aliquot of the eluate was tested for hBChE activity to determine the yield. Another aliquot was loaded in duplicate on the left and right sides of polyacrylamide gels to evaluate the purity. The gel electrophoresis was performed in non-denaturing conditions, then the gels were cut in half and one half was developed by silver staining, the other half by BChE activity staining. Bradford assay was used for protein quantification of selected samples to determine the specific activity of hBChE and the purification factors. The results obtained with resins 6a and 6b were essentially similar. The results reported in this paper were obtained using resin 6a, unless otherwise specified.The incubation of plasma with the carbamate resins resulted in the expected retention and release of active hBChE (Fig. 3a). In preliminary experiments we found that (1) the incubation of plasma for 30–60 min was sufficient to bind significant amounts of hBChE, (2) most of the protein contaminants could be removed by washing the resins with a phosphate buffer at pH 7 and (3) active hBChE could be eluted using tetramethylammonium chloride (4MAC), a weak hBChE activator/inhibitor.34 We used this information to characterize the retention and release mechanisms of hBChE.
 | ||
| Fig. 3 (a) Left panel, non-denaturing PAGE showing the effect of active site inhibition (lane 2) and acid denaturation (lane 3) (before the load step), and the effect of the removal of the probe from the resin (lane 4) on the retention of hBChE; lane 1, untreated plasma loaded on resin containing the probe (6a); right panel, hBChE activity of the corresponding eluates; FT, flow-through fraction. (b) Non-denaturing PAGE showing the effect of denaturation (after load and wash steps) on the release of hBChE; the arrows indicate the position of hBChE tetramers. | ||
The pre-incubation of plasma with BMB or the denaturation by acid treatment as well as the removal of the carbamate moieties from the resin (by alkaline treatment, before the incubation with plasma), prevented the binding of hBChE (Fig. 3a), demonstrating that (1) the retention mechanism involved the active site, (2) only active hBChE was retained and (3) the presence of the carbamate was essential for the retention.
Temperature-, acid- and solvent-induced denaturation of the enzyme bound to the resin prevented its release (Fig. 3b), consistently with the proposed retention mechanism involving a covalent attachment of the enzyme to the resin.
The fact that the protein contaminants were not retained when the probe was chemically stripped from the resin (Fig. 3a) is indicative of a certain binding specificity of the contaminants for the probe. However, the acid treatment had no effect on the retention and release of the contaminants indicating that either these protein are stable at low pH or that their binding to the probe is not particularly dependent on their folding. The fact that, with the exception of the denaturation by heat shock at pH 7, denaturation conditions impaired, and, in the case of acetonitrile (ACN) and acid shock, completely suppressed, also the release of most of the contaminants (Fig. 3b), suggested that also the binding of the contaminants might be covalent in nature. However, further research is required to determine the exact nature of the interaction between the contaminant proteins and the probe.
2.5. Proteomic analysis of the eluate
In addition to hBChE, other five major bands were observed in the silver stained gel (Fig. 3a). Proteomic analysis of the tryptic digest of the eluate indicated the presence of three main proteins in addition to hBChE (Table 2): serum albumin, and the immunoglobulins IgG and IgM. Based on this information we could tentatively assign the slowest band(s) to IgM (∼950 kDa and ∼1140 kDa for pentamer and hexamer, respectively), which is known to remain in the stacking gel unless reduced.35 The other bands were attributed to hBChE tetramers (the main form of hBChE in plasma, ∼340 kDa), Ig heavy chain μ (IGHM, ∼80 kDa), serum albumin (HSA, ∼65 kDa), Ig heavy chain γ (IGHG, ∼55 kDa) and Ig light chain λ (IGLC, ∼25 kDa) (Fig. 3a). The activity-stained gel revealed the presence of small amounts of trimers, dimers, albumin adducts and monomers of hBChE in the eluate (Fig. 3a), indicating that the carbamate resins were able to retain and release all the active isoforms of hBChE normally present in plasma,36 apparently without affecting their natural composition. This is a major difference compared with other hBChE isolation methods,37 including the most recent and best performing affinity chromatographic (AC) method based on Hupresin resin,38 which were reported to retain only the tetramers.| UniProt accession | Description | Score | Average massb | Matched products | Matched peptides | Sequence coverage (%) | Top3 peptides intensity sum | Relative mol abundance (%) |
|---|---|---|---|---|---|---|---|---|
| a The sample analyzed was obtained by pooling aliquots of samples 4 and 5, (Fig. 7), as detailed in the (ESI, S6).b The average masses indicated in the table are those reported in the UniProt database and include signal peptides whereas do not include glycosylations and other post-translational modifications. Butyrylcholinesterase and immunoglobulin heavy chains are normally glycosylated. | ||||||||
| P06276 | Butyrylcholinesterase | 207 | 68![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 988 988 |
33 | 9 | 10.1 | 93615 |
22.8 |
| P0CG04 | Immunoglobulin lambda-1, constant | 708 | 11519 |
23 | 3 | 31.1 | 32170 |
7.8 |
| P01860 | Immunoglobulin gamma-3 heavy chain | 217 | 42313 |
30 | 7 | 16.7 | 27375 |
6.7 |
| P01871 | Immunoglobulin mu heavy chain | 337 | 50124 |
43 | 13 | 23.2 | 26994 |
6.6 |
| P02768 | Serum albumin | 1136 | 71363 |
226 | 42 | 52.2 | 231080 |
56.2 |
According to the semi-quantitative data obtained from this proteomic analysis (Table 2) and the normal concentrations of hBChE, HSA and immunoglobulins in plasma (i.e. 10 μg L−1,8 35–50 g L−1,39 14 g L−1,35 1.5 g L−1,35 for hBChE, HSA, IgG and IgM, respectively), the amount of HSA and Igs retained was below 0.05% of their initial amount. These results highlighted the high selectivity of the method since only three proteins were present in significant amounts in the eluates and only in a small fraction compared to their initial amount. The selectivity compared well with that obtained with Hupresin, which was reported to retain a much higher number of protein contaminants when used in a similar format,38 although the total amount of contaminants was comparable.
hBChE and plasma platelet-activating factor acetylhydrolase (PLA2G7) are the only mSHs known to be present in the plasma of healthy individuals,2,36 although other mSHs might be present in traces. Therefore the selectivity of the probe for mSHs cannot be inferred from data obtained with plasma and should be established by using other tissues.
2.6. Effect of experimental parameters on the performance of the carbamate resins
In order to find the best conditions to isolate hBChE, the effects of several experimental parameters of each step of the isolation (i.e. load, wash and elution) on the isolation performance were systematically investigated. | ||
| Fig. 4 (a) Non-denaturing PAGE showing the effect of plasma incubation time on protein retention; (b) non-denaturing PAGE showing the effect of different concentration of 4MAC on protein release; (inset) hBChE activity of the isolates versus the concentration of 4MAC employed for the elution (apparent KD = 36 mM); (c) non-denaturing PAGE showing the effect of 4MAC incubation time on protein release; (inset) comparison of the theoretical (solid line) and experimental (red circles) kinetics of the release of hBChE from the resin. The arrows indicate the position of hBChE tetramers. | ||
| Variables investigated | Condition | Wash volume (mL) | Effect on yielda (%) | Effect on puritya (%) |
|---|---|---|---|---|
| a The effect is reported in percentage normalized to the yield and apparent purity obtained in the reference condition. | ||||
| pH and temperature | pH 7, 37 °C | 5 | Reference | Reference |
| pH 5, 37 °C | 5 | +20 | −62 | |
| pH 7, 4 °C | 5 | +18 | −62 | |
| pH 5, 4 °C | 5 | +21 | −73 | |
| Presence of NaCl and rinse volume | +NaCl (pH 7, 37 °C) | 5 | +11 | +11 |
| 10 | +4 | +6 | ||
| 20 | −14 | −11 | ||
| Pre-elution agent and conditionsa | Pre-elution repeats | Elution agent and conditionsb | Effect on yieldc (%) | Effect on purityc (%) |
|---|---|---|---|---|
| a The pre-elution step was performed at 4 °C. Hx, hexanoic acid 3 mM; IBU, rac-ibuprofen 3 mM; SAL, salicylic acid 3 mM; 4MAC, tetramethylammonium chloride 500 mM.b The elution step was performed at 37 °C, 3 repeats; 4MAC, tetramethylammonium chloride 500 mM; NaCl 500 mM; TBT, rac-terbutaline 500 mM; 3, Boc-6Ahx-TTD-TBT 500 μM.c The effect is reported in percentage normalized to the yield and apparent purity obtained in the reference condition. | ||||
| — | — | 4MAC, pH 7 | Reference | Reference |
| — | — | 4MAC, pH 5 | −8 | −20 |
| — | — | NaCl, pH 7 | −77 | −67 |
| — | — | TBT, pH 7 | −26 | −7 |
| — | — | 3, pH 7 | −51 | +25 |
| Hx, pH 7 | 1 | 4MAC, pH 7 | −5 | +13 |
| IBU, pH 7 | 1 | 4MAC, pH 7 | +3 | 0 |
| SAL, pH 7 | 1 | 4MAC, pH 7 | +9 | +6 |
| 4MAC, pH 5 | 1 | 4MAC, pH 7 | −3 | +5 |
| 4MAC, pH 7 | 1 | 4MAC, pH 7 | −14 | +11 |
| 4MAC, pH 5 | 3 | 4MAC, pH 7 | −9 | 0 |
| 4MAC, pH 7 | 3 | 4MAC, pH 7 | −9 | −17 |
The incubation time was a major parameter affecting hBChE elution (Fig. 4c). In agreement with the proposed mechanism, the release of hBChE reached a plateau after ∼45 min of incubation with 4MAC, and followed the same profile that was calculated on the basis of the k3 measured for compound 3 (Fig. 4c, inset). This evidence provided a further proof of the retention mechanism and indicated that the immobilization of the probe on the resin did not affect the decarbamylation kinetics.
Temperature was another major parameter affecting the elution as showed by the little loss in hBChE yield observed when additional pre-incubation steps with 4MAC were performed at 4 °C (at either pH 5 and pH 7) before the usual elution with 4MAC at 37 °C (Table 4 and Fig. 5). Consistently with the effect of temperature on catalytic rate constants, the decarbamylation rate of hBChE was reduced at low temperature, thus preventing the competitive effect of 4MAC. This was a further indication that hBChE retention involved a covalent interaction with the resin. The low temperature also prevented the release of the contaminants and the same amount of contaminants were obtained with or without the additional pre-incubation steps with 4MAC at 4 °C (Fig. 5). We interpreted this result as a further indication that also the other proteins might be retained through the formation of covalent bonds.
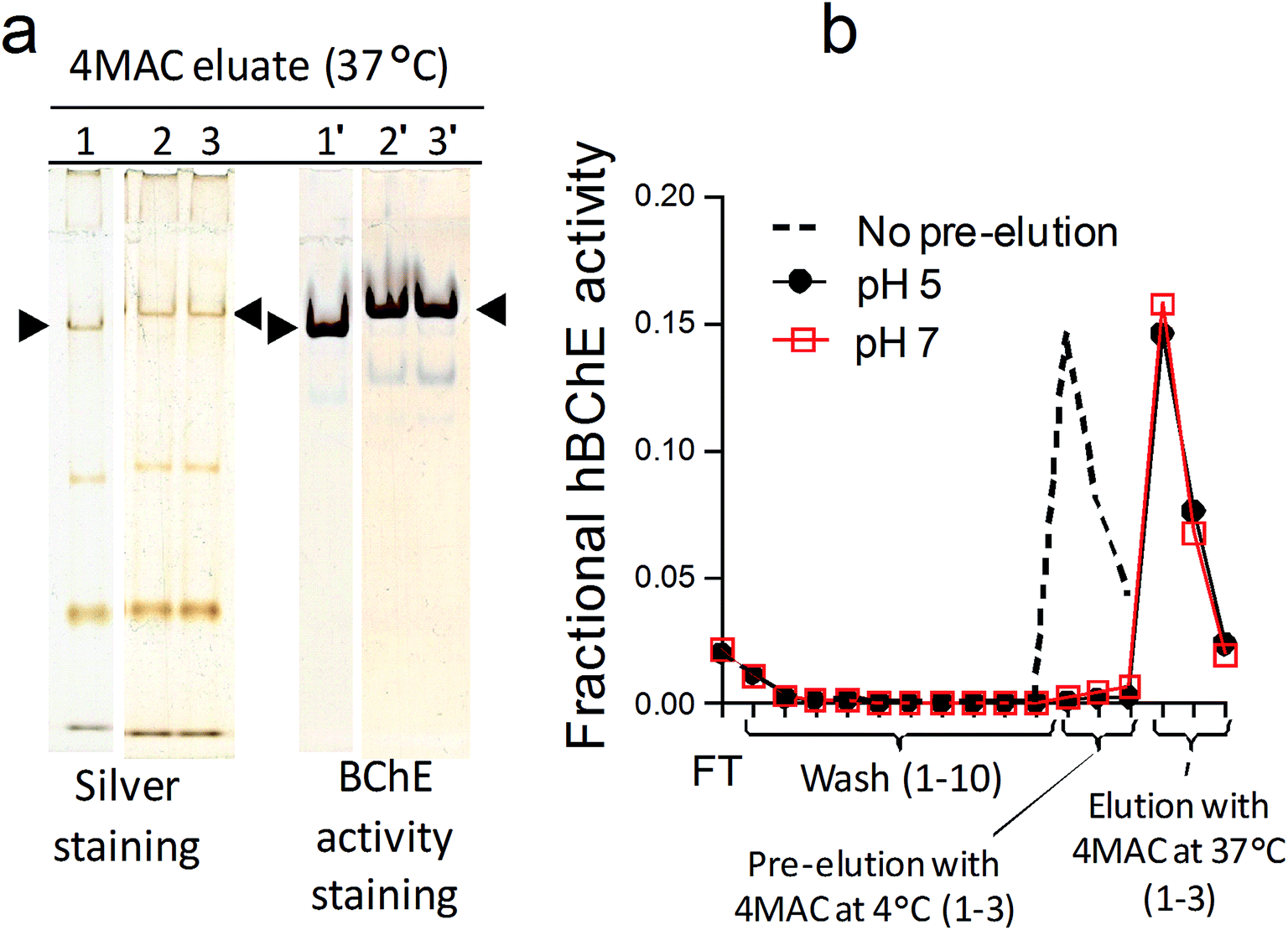 | ||
| Fig. 5 (a) Non-denaturing PAGE showing the lack of effect of additional pre-elution steps performed using 4MAC at 4 °C on the amount of proteins eluted with 4MAC at 37 °C; lane 1 and 1′, elution with 4MAC, pH 7 at 37 °C (no pre-elution); lane 2 and 2′, pre-elution with 4MAC, pH 5 at 4 °C followed by elution with 4MAC, pH 7 at 37 °C; lane 3 and 3′, pre-elution with 4MAC, pH 7 at 4 °C followed by elution with 4MAC, pH 7 at 37 °C. (b) hBChE activity of the eluates obtained in the three conditions; FT, flow-through fraction. | ||
The yields of hBChE obtained with 4MAC ranged from 17% to 36% of the loaded activity when fresh resin was used. The other eluting agents tested (i.e., compound 3, TBT, a weak hBChE inhibitor,43 and NaCl, which was reported to reduce the carbamylation but not the decarbamylation rate44), were less efficient than 4MAC (Table 4).
 | ||
| Fig. 6 Effect of repeated use of the resin on the yield and purity of hBChE isolated from plasma. The effect of repeated use on the isolation performance was evaluated by: (samples A1–A3) repeating three times the whole isolation protocol (i.e., each plasma incubation was followed by wash and elution steps); (sample B) repeating three times only the plasma incubation and the wash step followed by a single final elution (×3); (sample C) repeating three times only the plasma incubation followed by a single wash step (×10) and a single elution step (×3). (a) Non-denaturing gel electrophoresis of the eluates obtained with the three protocols; samples B and C were diluted 3-fold to match the amount of plasma loaded in samples A1–A3 (b) hBChE activity of the eluates obtained with the three protocols. | ||
 | ||
| Fig. 7 Effect of plasma to resin volumetric ratio on the yield and purity of hBChE isolated from plasma. Non-denaturing gel electrophoresis of the eluates obtained the different conditions described in Table 5; the samples in lanes 4 and 5 were diluted 2.3-fold and 3.2-fold, respectively, to match the amount of plasma loaded in the other samples. | ||
| Resin | Sample (μL) | Yield (%) | Specific activity (U mg−1) | Purification factor |
|---|---|---|---|---|
| — | Original plasma | — | 0.03 | — |
| 1 × 70 | 36.3 | 28.2 | 853 | |
| 6a | 4 × 70 | 24.4 | 38.2 | 1157 |
| 8 × 70 | 7.3 | 42.3 | 1282 | |
| 1 × 70 | 29.1 | 28.7 | 837 | |
| 6b | 4 × 70 | 20.1 | 56.5 | 1649 |
| 8 × 70 | 13.6 | 84.6 | 2470 |
3. Experimental
3.1 Materials
A full list of chemicals, reagents, solvents and their providers is given as (ESI, S1†).3.2 Synthesis of compounds 1, 2, 3 and 4 (Strategy 1)
The general methods used for the synthesis and characterization of the newly synthesized compounds is reported as (ESI, S2†). 1H-NMR, 13C-NMR and HRMS spectra recorded for characterization are reported as (ESI†), as indicated.TLC: Rf 0.26; 1HNMR (400 MHz, CDCl3, TMS) (Fig. S1†) δ 6.56 (1H, s), 4.69 (1H, s), 3.68–3.53 (12H, m), 3.36 (2H, q, J = 6.0 Hz), 3.11 (2H, q, J = 6.5 Hz), 2.82 (2H, t, J = 7.6 Hz), 1.57–1.39 (11H, m), 1.39–1.29 (2H, m). 1HNMR (400 MHz, CDCl3, D2O, TMS) (Fig. S2†) δ 4.78 (2H, s), 3.87–3.50 (12H, m), 3.35 (2H, t, J = 6.2 Hz), 3.10 (2H, t, J = 7.1 Hz), 2.78 (2H, t, J = 6.7 Hz), 2.15 (2H, t, J = 7.5 Hz), 1.84–1.68 (4H, m), 1.64 (2H, p, J = 7.5 Hz), 1.56–1.38 (11H, m), 1.38–1.26 (2H, m). 13CNMR (101 MHz, CDCl3) (Fig. S3†) δ 173.1, 156.1, 79.0, 70.5, 70.4, 70.07, 70.05, 69.9, 69.6, 40.4, 39.6, 37.6, 36.5, 32.6, 29.7, 28.9, 28.4, 26.4, 25.4; HRMS (ESI-TOF) (Fig. S10†) m/z 434.3233 (434.3225 calcd for C21H44N3O6, [M + H]+).
TLC: Rf 0.42; HPLC (C18, UV shoulder 230 nm) k′ 3.98 (Rt 7.0 min). 1HNMR (400 MHz, CDCl3) (Fig. S4†) δ 8.21 (1H, t, J = 1.1 Hz), 7.56–7.46 (2H, m), 7.04 (1H, t, J = 1.2 Hz), 6.41 (1H, s), 4.67 (1H, s), 3.70–3.62 (8H, m), 3.59–3.52 (4H, m), 3.50 (2H, t, J = 5.8 Hz), 3.32 (2H, q, J = 6.2 Hz), 3.10 (2H, q, J = 6.7 Hz), 2.16 (2H, t, J = 7.5 Hz), 1.91 (2H, dt, J = 12.0, 5.6 Hz), 1.73 (2H, p, J = 6.2 Hz), 1.63 (2H, p, J = 7.6 Hz), 1.53–1.39 (11H, m), 1.38–1.27 (2H, m); 13CNMR (101 MHz, CDCl3) (Fig. S5†) δ 173.2, 156.1, 149.0, 135.9, 129.8, 116.5, 79.1, 70.4, 70.3, 70.2, 70.1, 69.9, 69.6, 40.4, 39.8, 37.5, 36.5, 29.8, 29.1, 28.6, 28.4, 26.4, 25.4; HRMS (ESI-TOF) (Fig. S11†) m/z 528.3403 (528.3392 calcd for C25H46N5O7, [M + H]+).
TLC: Rf 0.23; HPLC (C18, UV max 274–277 nm, 243 nm) k′ 2.48 (Rt 4.89 min). 1HNMR (400 MHz, CD3OD) (Fig. S6†) δ 6.76 (1H, t, J = 1.8 Hz), 6.67 (1H, t, J = 1.8 Hz), 6.49 (1H, t, J = 2.2 Hz), 4.83 (1H, dd, J = 9.9, 3.1 Hz), 3.68–3.55 (10H, m), 3.51 (2H, t, J = 6.2 Hz), 3.26 (4H, dt, J = 13.5, 6.8 Hz), 3.16 (1H, dd, J = 12.4, 3.2 Hz), 3.06–2.97 (3H, m), 2.17 (2H, t, J = 7.5 Hz), 1.86–1.69 (4H, m), 1.61 (2H, p, J = 7.5 Hz), 1.53–1.40 (11H, m), 1.40–1.24 (11H, m); 13CNMR (101 MHz, CD3OD) (Fig. S7†) δ 178.6, 176.1, 159.8, 157.1, 153.7, 144.9, 111.3, 111.0, 109.8, 79.8, 71.5, 70.21, 70.19, 70.3, 69.9, 69.8, 58.0, 49.9, 49.6, 41.2, 39.4, 37.8, 37.0, 30.7, 30.4, 28.8, 27.4, 26.7, 25.8, 23.1; HRMS (ESI-TOF) (Fig. S12†) m/z 685.4385 (685.4382 calcd for C34H61N4O10, [M + H]+).
TLC: Rf 0.03; HPLC (C18, UV max 276–277 nm, 243 nm) k′ 0.04 (Rt 1.47 min). 1HNMR (400 MHz, CD3OD) (Fig. S8†) δ 6.76 (1H, s), 6.67 (1H, s), 6.50 (1H, t, J = 2.2 Hz), 4.85 (2H, dd, J = 9.9, 2.9 Hz), 3.66–3.55 (10H, m), 3.51 (2H, t, J = 6.1 Hz), 3.26 (4H, dt, J = 11.4, 6.8 Hz), 3.21–3.13 (1H, m), 3.06–2.96 (1H, m), 2.91 (2H, t, J = 7.7 Hz), 2.21 (2H, t, J = 7.4 Hz), 1.85–1.71 (4H, m), 1.71–1.58 (4H, m), 1.45–1.31 (11H, m); 13CNMR (101 MHz, CD3OD) (Fig. S9†) δ 175.8, 159.9, 157.1, 153.7, 144.9, 111.4, 111.0, 109.8, 71.5, 71.20, 71.18, 70.3, 69.84, 69.75, 58.2, 49.9, 49.5, 40.5, 39.4, 37.8, 36.6, 30.7, 30.4, 28.2, 26.9, 26.3, 25.7; HRMS (ESI-TOF) (Fig. S13†) m/z 585.3866 (585.3858 calcd for C29H53N4O8, [M + H]+).
3.3 Synthesis of the sepharose-supported probes
:20. The resin (8) was stored in NaH2PO4 0.1 M/EtOH 80:20 at 4 °C.:20. The resin (6b) was stored in NaH2PO4 0.1 M/EtOH 80:20 at 4 °C.3.4. Methods for the characterization of the solid-supported probes
380 M−1 cm−1.47 A detailed description of the protocol is provided as (ESI, S4†). The amount of active groups immobilized on the resin used for Strategy 1 and Strategy 2 were 41 μmol g−1 and 31 μmol g−1 of wet (spin filtered) resin, respectively.:20. Standard TBT was analyzed in the same conditions (calibration curve (10–50 μM): y = 2639.7x + 2425.6, r2 = 1). The same strategy was used to determine the content of compounds 3 and 4 in their respective preparations.3.5 Measurement of hBChE activity
hBChE activity was measured in 96-well plates by an EnSpire Multimode 2300 plate reader (PerkinElmer, Waltham, MA) using a modified Ellman's spectrophotometric assay.48A detailed description of the protocol is provided as (ESI, S5†).
3.6 Characterization of the kinetics of the interaction between compound 3 (Boc-6Ahx-TTD-TBT) and hBChE
:300 in assay buffer (200 μL) were pre-incubated at 37 °C with solutions of compound 3 (25 μL) at different concentrations (final concentrations, 29.5–1777 nM, in water). At set times (2–60 min), the inhibition was stopped by adding the substrate working solution (25 μL) and the residual hBChE activity (νi) was spectrophotometrically determined as described in paragraph 3.5. A sample pre-incubated with water instead of compound 3 was measured in parallel to determine the maximum enzyme activity (ν0).:240) and this solution was kept at 37 °C. At set time points 225 μL of this sample were added onto a well of a 96-well plate containing 25 μL of substrate working solution. The hBChE activity (νi) was spectrophotometrically determined at 37 °C as described in the (ESI, S5†). A sample pre-incubated with water in place of compound 3 was analyzed in parallel to measure the maximum enzyme activity (ν0).3.7. Investigation of the parameters affecting the isolation of hBChE from plasma
000×g, 12 min, 4 °C). A 12 μL aliquot of this concentrated preparation was analyzed by non-denaturing gel electrophoresis to evaluate the purity.For sepharose-6Ahx-TTD-TBT (6a, Strategy 1), the effect on yield and purity of various protocol parameters was systematically investigated. In particular, the effect of resin amount (10 mg, 25 mg, 50 mg), plasma volume (42 μL, 45 μL, 95 μL, 135 μL), incubation time in the load step (10 min, 20 min, 30 min, 60 min), nature of the wash buffer (PB, NaOAc (50 mM, pH 5), NaCl (200 mM, 500 mM, 1 M, 1.5 M) in PB), temperature of the wash buffer (4 °C or 37 °C, tested with both PB and NaOAc (50 mM, pH 5)), volume of the wash buffer (500 μL × 10, 500 μL × 20, 500 μL × 40), tested with NaCl 1.5 M in PB at 37 °C, nature of the elution buffer (rac-terbutaline hemisulfate (TBT) 500 mM in PB, NaCl 500 mM in PB, hexanoic acid (Hx) 3 mM in PB; ibuprofen (IBU) 3 mM in PB, salicylic acid (SAL) 3 mM in PB, compound 3 500 μM in PB, 4MAC 500 mM in NaOAc (50 mM, pH 5); 4MAC (0.5 mM, 1 mM, 5 mM, 20 mM, 100 mM, 500 mM, 1000 mM) in PB), incubation time of the elution buffer (10–60 min, tested with 4MAC 500 mM in PB), temperature of the elution buffer (4 °C or 37 °C, tested with 4MAC 500 mM at pH 5 and pH 7).
In the experiment aimed at demonstrating that if denaturated, the hBChE bound to sepharose-6Ahx-TTD-TBT (6a) cannot be eluted (Fig. 3b), aliquots of plasma (45 μL) were first loaded on aliquots of resin (50 mg) and incubated (30 min, 37 °C), washed with PB (500 μL × 10 at 4 °C) then the resin samples were spin-dried and treated with: 100 μL of PB or NaOAc pH 5 and heated (95 °C, 5 min); 100 μL of water/ACN 30/70 v/v (37 °C, 30 min); 95 μL of PB/HCl 4 M 90/5 v/v, neutralized with 5 μL NaOH 4 M after 15 min of incubation at 37 °C. An untreated sample was prepared in parallel for reference.
In the experiments aimed at demonstrating the effect of temperature in the elution step (Table 4 and Fig. 5), samples of resin incubated with plasma and washed according to the general protocol, were pre-incubated 45 min (×1 or ×3) with 100 μL of 4MAC at 4 °C, pH 5 or pH 7. The eluate obtained from this pre-incubation was collected for analysis. The resin samples were then incubated and eluted with 4MAC 500 mM, pH 7 at 37 °C, as in the general protocol.
In order to understand if the yield was limited by overloading effects, we evaluated the effect of three repeated incubations of 70 μL of plasma on the isolation performance, using three slightly different protocols (Fig. 6): (sample A) repeating the plasma incubation-wash-elution steps three times; (sample B) repeating three times only the plasma incubation and the wash steps followed by a single elution step; (sample C) repeating three times only the plasma incubation followed by a single wash step and a single elution step. Resin amount, 25 mg; plasma incubation step, 10 min; wash step, 500 μL PB × 10, at 37 °C; elution step, 100 μL 4MAC 500 mM × 3, at 37 °C.
In order to evaluate the possibility of using the resin more than three times, three samples (25 mg) of the sepharose-6Ahx-TTD-TBT under study (6a, Strategy 1, or 6b, Strategy 2) were incubated with one, four or eight aliquots of human plasma (each aliquot, 70 μL), respectively. Each incubation lasted for 10 min, and was performed at 37 °C. After the last incubations, the resin samples were washed with PB (500 μL × 10, 500 μL × 40 and 500 μL × 80, respectively, at 37 °C) and incubated with 4MAC (100 μL × 3) for 45 min at 37 °C. The eluates were concentrated and washed with PB (400 μL × 10) through ultrafiltration units (Amicon® Ultra-0.5 mL, 30 KDa MWCO, 14000×g, 15 min, 4 °C), then they were characterized for their total protein content (paragraph 3.8) and hBChE activity (paragraph 3.5) to calculate the specific activities of hBChE and the purification factors (Table 6).
3.8. Methods for the characterization of the isolated hBChE
The electrophoresis was at run at constant voltage (80 V) using a 0.05 M Tris-0.38 M glycine buffer (pH 8.3) at 4 °C and was stopped just before the bromophenol blue could elute from the gel (∼1.5 h). The gel was then cut in half: one half was stained for BChE activity by the method of Karnovsky and Roots50 using S-butyrylcholine iodide as the substrate; the other half was developed by silver staining (short silver nitrate protocol).51 The optical density of the bands developed by silver staining was used to evaluate differences in purity (apparent purity) among the samples separated in the same gel.
The yield was calculated as the ratio between the hBChE activity in the eluate and the hBChE activity measured for the loaded plasma. The specific activity was calculated as the ratio between the hBChE activity and the total protein content of the sample. The purification factor was obtained as the ratio between the specific activity of hBChE in the eluate and that in plasma.
3.8.3.1 Sample preparation. Aliquots of the ultrafiltrates obtained with resin 6a showed in Fig. 7, lanes 4 and 5 were prepared for proteomic analysis by using a previously published filter-assisted sample preparation (FASP) protocol, with minor modifications.52 A detailed description of the protocol is provided as (ESI, S6†). The resulting solution was analyzed by UPLC-HDMSE.
3.8.3.2 UPLC. The tryptic digest peptides were separated on a UPLC system (I class, waters) equipped with a Acquity UPLC BEH C18, 1.7 μm (100 mm × 2.1 mm i.d.), 130 Å. Mobile phase A was 0.1% formic acid in water, mobile phase B was 0.1% formic acid in ACN. The peptides were eluted with a 3.04 B%/CV (CV, column volume) linear gradient as follows (time, flow rate, B%): initial, 100 μL min−1, 2%; 50 min, 100 μL min−1, 40%; 51 min, 600 μL min−1, 80%; 55 min, 600 μL min−1, 80%; 56 min, 600 μL min−1, 2%; 58.5 min, 600 μL min−1, 2%; 58.6 min, 100 μL min−1, 2%; 60 min, 100 μL min−1, 2%. Column temperature, 55 °C; sample temperature, 10 °C. The lockmass compound leucine enkephalin 200 pg μL−1 (m/z 556.2771) in water/ACN (50/50, 0.1% formic acid) was delivered to the lockspray source of the mass spectrometer at 5 μL min−1 using the fluidics of the MS apparatus.
3.8.3.3 Mass spectrometry. The mass spectrometry of the tryptic peptides separated by chromatography was performed on-line by a Q-TOF mass spectrometer (Synapt G2-S HDMS, Waters) equipped with an ESI source. The mass spectrometer was operated in positive and resolution modes. Data were acquired with the instrument operating in MSE mode with ion mobility enabled (HDMSE).52 Source settings: source (capillary 3.00 kV, cone 35 kV offset 80 kV), temperature (source 100 °C, desolvation 350 °C), gas flows (cone gas 50 L h−1, desolvation gas 500 L h−1, nebulizer 6.0 bar), lockspray capillary 2.50 kV. Quad profile (mass, dwell time, ramp time): 1 (400, 20, 20), 2 (500, 20, 40), 3 (1000). Data were acquired collecting spectra in the 50–2000 m/z range every 0.6 s (data format: continuum) using the following ion mobility-dependent energy profile: bin 1–20, constant 17 eV; bin 20–110 ramp from 17 eV to 45 eV; bin 110 to 200 ramp from 45 eV to 60 eV.52 The lockspray signal was measured by averaging 3 × 0.6 s scans every 30 s, within a mass window of ±0.5 Da.
3.8.3.4 Data processing, protein identification and semi-quantification. The LC-HDMSE data were processed using ProteinLynx Global SERVER (PLGS) v2.5.2 (Waters), using the following parameters: UniprotKB Homo sapiens proteome dataset (UP000005640, reviewed, 20
416 entries, 30-04-2019); peptide and fragment tolerance, automatic (the software set 5 ppm and 13 ppm, respectively); maximum trypsin missed cleavages, 1; maximum protein mass 500 kDa; fixed modification, carbamidomethylation at Cys; variable modifications, oxidation at Met; false positive rate, 4%. Minimum fragment ion matches per peptide, 1; minimum fragment ion matches per protein, 3; minimum peptide matches per protein, 1. Processing parameters: low energy threshold, 10 counts; elevated energy threshold, 5 counts; intensity threshold, 500 counts. A protein score above 5 was set post-processing (100 is a typical accepted threshold for reliable identification). Protein semi-quantification (relative abundance) was based on the integrated peptide ion intensities according to the Top3 method.53
4. Conclusions
In summary, the results demonstrated that the designed probe captured and released hBChE from plasma with high selectivity and reasonable yields. The best purification factor obtained was similar to that obtained with Hupresin, the best AC method for the purification of hBChE from plasma currently available (∼2000-fold).38 The yield was probably limited by the release of small quantities of probe from the resin. The main parameters affecting the performance were the incubation time used in the load and the elution steps, the P/R, the elution temperature and the nature and concentration of the eluting agent whereas all the other parameters investigated had limited effect. The carbamate resin could be prepared using simple and inexpensive synthetic steps either by coupling a fully synthesized probe with an activated resin or by building the probe on the resin by a step-by-step procedure, without major differences in performance between the two routes. The prepared resins allowed to process up to about 8.5 mL of plasma per g of resin with constant performance.In conclusion, this work demonstrated the feasibility of using mechanism-based probes bearing carbamate moieties for the selective and kinetically-controlled capture and release of active hBChE from complex proteomes. The method is based on the manipulation of the kinetic equilibrium between the two reactions catalyzed by the target enzyme and does not require the use of reactivating reagents. Overall the method is simple and fast and its use is similar to that of traditional AC resins. In contrast to traditional AC methods, this approach enabled to retain all the active hBChE isoenzymes. Since the method is based on the general catalytic cycle of mSHs, we expect this approach to be applicable to other enzymes of the family, by selecting a suitable target-selective feature to link to the carbamate group. Further research is required to establish the relationship between probe-enzyme reaction kinetics and purification performance, to fully understand the reasons underlying the limited yield and reusability and to explore other possible setups.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Fundamental Research Fund for the Central Universities, China [Grant 2014ZM0053] and the Research Start-up Funds of South China University of Technology [Grants x2sw/D6191290 and j2rs/D6173430].Notes and references
- G. M. Simon and B. F. Cravatt, J. Biol. Chem., 2010, 285, 11051–11055 CrossRef CAS PubMed
.
- J. Z. Long and B. F. Cravatt, Chem. Rev., 2011, 111, 6022–6063 CrossRef CAS PubMed
- T. Fukami and T. Yokoi, Drug Metab. Pharmacokinet., 2012, 27, 466–477 CrossRef CAS PubMed
- B. Chen, S.-S. Ge, Y.-C. Zhao, C. Chen and S. Yang, RSC Adv., 2016, 6, 113327–113343 RSC
- S. R. Feaster and D. M. Quinn, Methods Enzymol., 1997, 286, 231–252 CAS
- D. A. Bachovchin, T. Ji, W. Li, G. M. Simon, J. L. Blankman, A. Adibekian, H. Hoover, S. Niessen and B. F. Cravatt, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20941–20946 CrossRef CAS PubMed
- M. Pistolozzi, H. Du, H. Wei and W. Tan, Drug Metab. Dispos., 2015, 43, 344–352 CrossRef PubMed
- H. Zhou, Q. Xiao, W. Tan, Y. Zhan and M. Pistolozzi, J. Pharm. Biomed. Anal., 2017, 144, 167–174 CrossRef CAS PubMed
- M. P. Patricelli, H. A. Lashuel, D. K. Giang, J. W. Kelly and B. F. Cravatt, Biochemistry, 1998, 37, 15177–15187 CrossRef CAS PubMed
- Y. Ben Ali, H. Chahinian, S. Petry, G. Muller, F. Carrière, R. Verger and A. Abousalham, Biochemistry, 2004, 43, 9298–9306 CrossRef CAS PubMed
- Z. Rashid, H. Naeimi, A.-H. Zarnani, M. Nazari, M.-R. Nejadmoghaddam and R. Ghahremanzadeh, RSC Adv., 2016, 6, 36840–36848 RSC
- K. Salimi, D. D. Usta, İ. Koçer, E. Çelik and A. Tuncel, RSC Adv., 2017, 7, 8718–8726 RSC
- X. Yang, M. Pistolozzi and Z. Lin, Biotechnol. Lett., 2018, 40, 745–753 CrossRef CAS PubMed
- T. Tsujita, H. Ninomiya and H. Okuda, J. Lipid Res., 1989, 30, 997–1004 CAS
- P. Masson and O. Lockridge, Arch. Biochem. Biophys., 2010, 494, 107–120 CrossRef CAS PubMed
- J. F. Breyer, C. Schmidt, B. Mizaikoff, F. Worek and H. Thiermann, Drug Test. Anal., 2015, 7, 947–956 CrossRef PubMed
- Y. Liu, M. P. Patricelli and B. F. Cravatt, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 14694–14699 CrossRef CAS PubMed
- D. Kidd, Y. Liu and B. F. Cravatt, Biochemistry, 2001, 40, 4005–4015 CrossRef CAS PubMed
- B. F. Cravatt, A. T. Wright and J. W. Kozarich, Annu. Rev. Biochem., 2008, 77, 383–414 CrossRef CAS PubMed
- J. L. Counihan, B. Ford and D. K. Nomura, Curr. Opin. Chem. Biol., 2016, 30, 68–76 CrossRef CAS PubMed
- D. K. Nomura, J. Z. Long, S. Niessen, H. S. Hoover, S. W. Ng and B. F. Cravatt, Cell, 2010, 140, 49–61 CrossRef CAS PubMed
- D. A. Bachovchin, T. Ji, W. Li, G. M. Simon, J. L. Blankman, A. Adibekian, H. Hoover, S. Niessen and B. F. Cravatt, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20941–20946 CrossRef CAS PubMed
- D. K. Nomura and J. E. Casida, J. Agric. Food Chem., 2011, 59, 2808–2815 CrossRef CAS PubMed
- D. Medina-Cleghorn, A. Heslin, P. J. Morris, M. M. Mulvihill and D. K. Nomura, ACS Chem. Biol., 2014, 9, 423–432 CrossRef CAS PubMed
- V. Delorme, B. Raux, R. Puppo, J. Leclaire, J. F. Cavalier, S. Marc, P. K. Kamarajugadda, G. Buono, F. Fotiadu, S. Canaan and F. Carrière, Biochimie, 2014, 107(Pt A), 124 CrossRef CAS PubMed
- Y. Ashani and I. B. Wilson, Biochim. Biophys. Acta, 1972, 276, 317–322 CrossRef CAS
- H. F. Voss, Y. Ashani and I. B. Wilson, Methods Enzymol., 1974, 34, 581–591 CAS
- A. Tunek and L. A. Svensson, Drug Metab. Dispos., 1988, 16, 759–764 CAS
- Q. Xiao, H. Zhou, H. Wei, H. Du, W. Tan, Y. Zhan and M. Pistolozzi, J. Pharm. Biomed. Anal., 2017, 144, 175–182 CrossRef CAS PubMed
- G. Voss, Arch. Toxicol., 1976, 36, 117–120 CAS
- M. Rotenberg and S. Almog, Clin. Chim. Acta, 1995, 240, 107–116 CrossRef CAS
- E. Groner, Y. Ashani, D. Schorer-Apelbaum, J. Sterling, Y. Herzig and M. Weinstock, Mol. Pharmacol., 2007, 71, 1610–1617 CrossRef CAS PubMed
- K. J. Padiya, S. Gavade, B. Kardile, M. Tiwari, S. Bajare, M. Mane, V. Gaware, S. Varghese, D. Harel and S. Kurhade, Org. Lett., 2012, 14, 2814–2817 CrossRef CAS PubMed
- J. Stojan, M. Golicnik, M. T. Froment, F. Estour and P. Masson, Eur. J. Biochem., 2002, 269, 1154–1161 CrossRef CAS PubMed
- T. L. Rižne, Biochem. Mol. Biol. Educ., 2014, 42, 152–159 CrossRef PubMed
- B. Li, M. Sedlacek, I. Manoharan, R. Boopathy, E. G. Duysen, P. Masson and O. Lockridge, Biochem. Pharmacol., 2005, 70, 1673–1684 CrossRef CAS PubMed
- O. Lockridge, L. M. Schopfer, G. Winger and J. H. Woods, J. Med. Chem. Biol. Radiol. Def., 2005, 3, nihms5095 Search PubMed
- S. Onder, E. David, O. Tacal, L. M. Schopfer and O. Lockridge, Front. Pharmacol., 2017, 8, 713 CrossRef PubMed
- N. L. Anderson and N. G. Anderson, Mol. Cell. Proteomics, 2002, 1, 845–867 CrossRef CAS PubMed
- E. Chosson, S. Uzan, F. Gimenez, I. W. Wainer and R. Farinotti, Chirality, 1993, 5, 71–77 CrossRef CAS PubMed
- C. Bertucci, M. Pistolozzi, G. Felix and U. H. Danielson, J. Sep. Sci., 2009, 32, 1625–1631 CrossRef CAS PubMed
- O. Lockridge, E. David, L. M. Schopfer, P. Masson, X. Brazzolotto and F. Nachon, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2018, 1102–1103, 109–115 CrossRef CAS PubMed
- Z. Kovarik and V. Simeon-Rudolf, J. Enzyme Inhib. Med. Chem., 2004, 19, 113–117 CrossRef CAS PubMed
- V. Tõugu and T. Kesvatera, Biochim. Biophys. Acta, 1996, 1298, 12–30 CrossRef
- W. H. Scouten, Curr. Protoc. Protein Sci., 2001, ch.
9, unit 9.3 Search PubMed
- T. Miron and M. Wilcheck, Methods Enzymol., 1987, 135, 84–90 CAS
- G. N. Bowers Jr, R. B. McComb, R. C. Christensen and R. Schaffer, Clin. Chem., 1980, 26, 724–729 CAS
- G. L. Ellman, K. D. Courtney, V. Jr Ander and R. M. Featherstone, Biochem. Pharmacol., 1961, 7, 88–95 CrossRef CAS PubMed
- S. Bevc, J. Konc, J. Stojan, M. Hodošček, M. Penca, M. Praprotnik and D. Janežič, PLoS One, 2011, 6, e22265 CrossRef CAS PubMed
- M. J. Karnovsky and L. Roots, J. Histochem. Cytochem., 1964, 12, 219–221 CrossRef CAS PubMed
- M. Chevallet, S. Luche and T. Rabilloud, Nat. Protoc., 2006, 1, 1852–1858 CrossRef CAS PubMed
- U. Distler, J. Kuharev, P. Navarro and S. Tenzer, Nat. Protoc., 2016, 11, 795–812 CrossRef CAS PubMed
- J. C. Silva, M. V. Gorenstein, G.-Z. Li, J. P. C. Vissers and S. J. Geromanos, Mol. Cell. Proteomics, 2006, 5, 144–156 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Chemicals, reagents and solvents; general methods for the chemical synthesis; synthesis of the solid-supported probes; determination of the amount of active carbonate groups on the resins; measurement of hBChE activity; sample preparation for the proteomic analysis; Scheme S1; 1H-NMR, 13C-NMR and HRMS spectra. See DOI: 10.1039/c9ra07583f |
| This journal is © The Royal Society of Chemistry 2019 |