
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and reactivity of 2-thionoester pyrroles: a route to 2-formyl pyrroles†
Min Joon
Kim
,
Sophie M.
Gaube
,
Michael H. R.
Beh
,
Craig D.
Smith
and
Alison
Thompson
 *
*
Department of Chemistry, Dalhousie University, P. O. Box 15000, Halifax, NS B3H 4R2, Canada. E-mail: Alison.Thompson@dal.ca
First published on 7th October 2019
Abstract
2-Functionalised pyrroles exhibit considerable synthetic utility. Herein, the synthesis and reactivity of 2-thionoester (–C(S)OR) pyrroles is reported. 2-Thionoester pyrroles were synthesised using a Knorr-type approach from aliphatic starting materials. 2-Thionoester pyrroles were reduced to the corresponding 2-formyl pyrroles, or the deuterated formyl variant, in one step using RANEY® nickel, thereby removing the need for the much-utilised hydrolysis/decarboxylation/formylation steps that are typically required to convert Knorr-type 2-carboxylate pyrroles into 2-formyl pyrroles. 2-Thionoester pyrroles proved tolerant of typical functional group interconversions for which the parent 2-carboxylate pyrroles have become known.
2-Formyl pyrroles are critical to the synthesis of (poly)pyrroles such as porphyrins and BODIPYs courtesy of their availability from widely used Knorr-type syntheses, and their robust and reliable reactivity.1 However, attempts to reduce the 2-carboxylate functionality of Knorr-type pyrroles with H-DIBAL are unfruitful, which is unsurprising considering the doubly-vinylogus carbamate character of the ester (Fig. 1, top). Instead, transformation to 2-formyl pyrroles typically entails hydrolysis of the 2-carboxylate functionality,2,3 followed by decarboxylation and finally formylation.4–7 Alternatively, reduction of the 2-carboxylate functionality to the corresponding alcohol, followed by oxidation back to the desired aldehyde,8 also provides the requisite 2-formyl pyrrole. In addition to the inevitably poor atom economy that results from such circuitousness, these reactions can be low yielding and necessitate harsh conditions. The potential of utilising new reactivity at the 2-position of pyrroles opens new synthetic opportunities in pyrrole chemistry that build on traditional approaches.9 Herein, we report the one-step reduction of 2-thionoester pyrroles to provide 2-formyl pyrroles, and explore the stability of 2-thionoester pyrroles under conditions typically used for the synthetic manipulation of Knorr-type pyrroles.
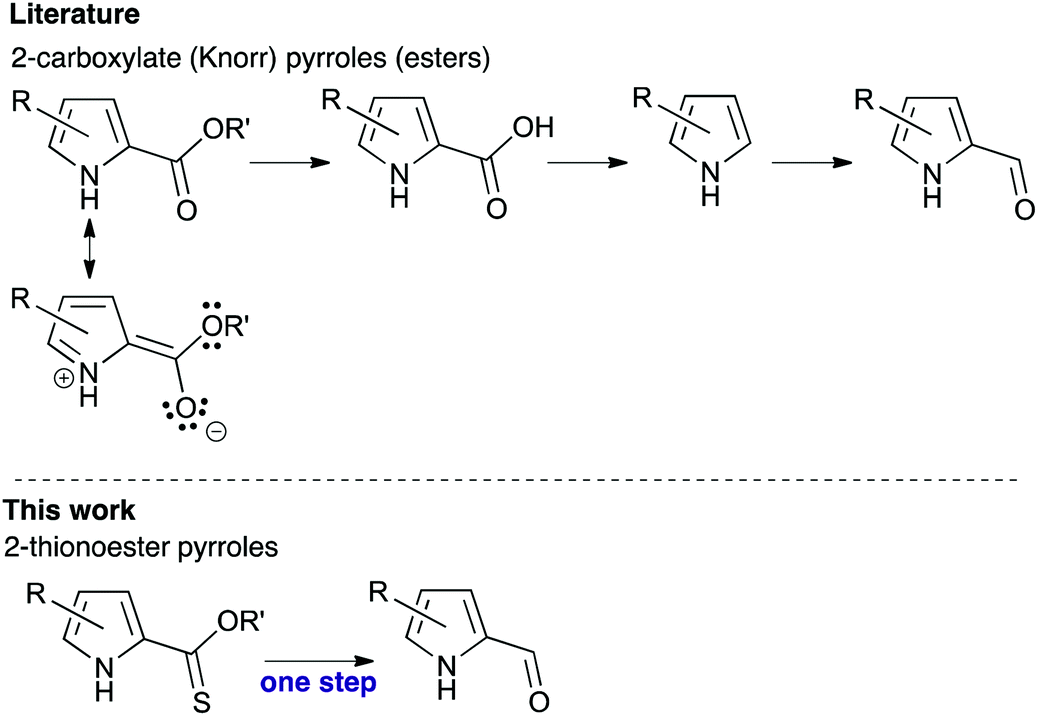 | ||
| Fig. 1 Conversion of Knorr-type pyrroles to 2-formyl pyrroles. | ||
2-Thionoester pyrroles, which bear the –C(S)OR functional group at the 2-position, have not been studied in detail despite the fact that thionoesters are more electrophilic compared to the corresponding “all-oxygen ester” functional groups (–CO2R).10,11 In contrast, 2-thioester pyrroles (–C(O)SR) have been thoroughly evaluated as such species are common secondary metabolites in marine bacteria and plants.9,12,13 In particular, we noticed a role for thiophilic reagents in the reduction of 2-thioester pyrroles14–17 induced by RANEY® nickel.14,15,18
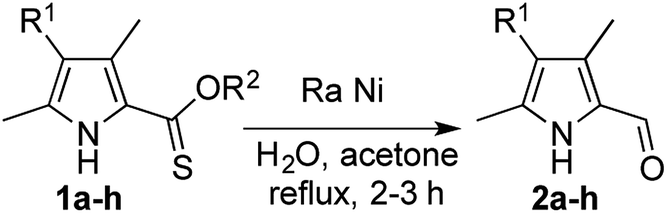
We thus explored the reactivity of 2-thionoester pyrroles with RANEY® nickel, taking advantage of the availability of RANEY® nickel as a slurry in water. O-Ethyl 2-thionoester pyrrole 1a, initially prepared via treatment of the corresponding 2-carboxylate pyrrole with Lawesson's reagent,9 was reacted with excess 2800 RANEY® nickel to provide the 2-formyl pyrrole 2a in 67% isolated yield (Table 1, entry 1). To commence the reaction, a slurry of RANEY® nickel in water and acetone was activated via heating at reflux temperature for an hour before 1a was added. Although the purpose of the activation step is unclear, this procedure is documented alongside the success of W2 RANEY® nickel in the desulfurisation of a 2-thioester pyrrole.19 We found that this activation step was essential for success of our desired reduction using currently commercially available 2800 RANEY® nickel, which is different from W2 RANEY® nickel. After the starting material has been fully consumed, the reaction mixture was filtered through a short pad of silica to remove the spent RANEY® nickel and thus enable isolation of the desired aldehyde. Execution of the reaction under an atmosphere of hydrogen did not improve the yield,18,20–22 supporting the synergy of the thiophilicity and hydride content of the RANEY® nickel, and we were unable to reduce the stoichiometry without significant detriment to yield (see ESI†). Similarly, dissolution of the substrate in acetone was key to success, as the use of other water-miscible solvents (methanol, ethanol, tetrahydrofuran and isopropanol),23 all previously reported as suitable for reactions involving RANEY® nickel, resulted only in isolation of starting material. Furthermore, treatment of 1a with nickel boride returned only starting material.24
Although yields were moderate (Table 1), the one-step desulfurative reduction of 2-thionoester pyrroles to 2-formyl pyrroles is more streamlined than the common hydrolysis/decarboxylation/formylation route shown in Fig. 1. The one-step synthesis of 2-formyl pyrroles from O-ethyl 2-thionoester pyrroles in reasonable yield was successful for substrates bearing unsubstituted positions (1a, Table 1, entry 1) as well as those with alkyl (entries 2 and 3) and aryl (entry 4) substituents. Similarly, the incorporation of a conjugated keto-substituent was tolerated (entry 5). The reduction of O-benzyl 2-thionoester pyrroles was also successful (entries 6–9). Long-chain substituents were tolerated (entry 7), but incorporation of an alkyl ester functionality served to reduce the efficacy of the desired thionoester reduction using RANEY® nickel (entry 8).
|
|
||||
|---|---|---|---|---|
| Entry | Substrate | R1 | R2 | Yield of 2 (%)a |
| a Isolated yield. b 15% starting material recovered. c Deuterated 2e′ using RANEY® nickel in D2O slurry. d Expected product is 2b. | ||||
| 1 | 1a | –H | Et | 67 |
| 2 | 1b | –Me | Et | 52 |
| 3 | 1c | –Et | Et | 63 |
| 4 | 1d | –Ph | Et | 45 |
| 5 | 1e | –C(O)CH3 | Et | 44b (46c) |
| 6 | 1f | –Me | Bn | 45d |
| 7 | 1g | –(CH2)4CH3 | Bn | 70 |
| 8 | 1h | –(CH2)2C(O)OCH3 | Bn | 12 |
Although a complete mechanism for desulfurisation by RANEY® nickel remains unclear,25,26 we explored a number of avenues to try to understand the atom efficiency of the reaction, as well as the source of the formyl hydrogen atom. First, although we did not attempt to dry the pyrophoric reductant, water from the commercially available RANEY® nickel slurry was manually removed by pipette. Acetone was then added, and the slurry shaken. After being allowed to settle, the liquid was removed once again. This process was repeated several times so as to produce a slurry of RANEY® nickel in wet acetone. The reaction of pyrrole 1e with this acetone-based slurry resulted in only trace amounts of 2e suggesting that the water within the slurry is critical to the formation of the aldehyde. To investigate whether deuterium27,28 could be introduced into the 2-formyl moiety, a slurry of RANEY® nickel in D2O was prepared through removal/addition as above, with the added sonication step after each addition. The solvent removal/addition and sonicator wash cycle was repeated 10 times to ensure efficient exchange. Reaction of the deuterated slurry with pyrrole 1e, in acetone, resulted in formation of the deuterated aldehyde i.e. R–C(O)D, 2e′ in moderate yield (Table 1, entry 6). Interestingly, without the use of sonication a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of the deuterated and undeuterated aldehydes was recovered, again indicating the critical role of the slurry water to the reductive capability of the RANEY® nickel.
:1 mixture of the deuterated and undeuterated aldehydes was recovered, again indicating the critical role of the slurry water to the reductive capability of the RANEY® nickel.
The manner by which RANEY® nickel effects desulfurative reduction has been long debated.25,26,29 Assuming a radical mechanism, we used two radical clocks/traps in the hopes of intercepting radical intermediate species (Fig. 2).30,31 However, in neither case was trapping observed with reduction of 1a serving to return the 2-formyl pyrrole 2a (50–60% yield) no matter whether large excess of a cyclopropane were present or not. However, in light of the large excess of RANEY® nickel used, we maintain that a radical mechanism is most likely for the reduction. In contrast, reaction of 1a with RANEY® nickel in the presence of 50 eq. TEMPO resulted in the isolation of only 7% 2a. However, no trapped products could be identified via either mass spectrometry or NMR spectroscopy.32,33
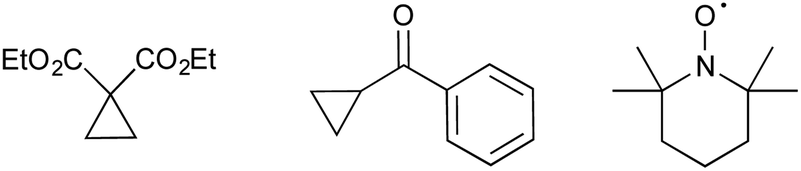 | ||
| Fig. 2 Cyclopropane radical clocks and TEMPO. | ||
With the one-step reduction to form 2-formyl pyrroles in hand, we sought a practical synthesis of 2-thionoester pyrroles that did not entail thionation of 2-carboxylate pyrroles. We thus turned our attention to the preparation of 2-thionoester pyrroles from aliphatic materials, modelling the Knorr synthesis by incorporation of thionoester functionality. Reaction of potassium ethyl xanthogenate and chloroacetone, following modified literature methods,34,35 gave 3 (Scheme 1), and then extrusion of sulfur produced the desired acetothioacetate 4. Treatment with sodium nitrite gave the oxime 5, which could be isolated and characterised although the typical procedure entailed treating the crude material with 2,4-pentanedione and zinc powder to provide O-ethyl 2-thionoester pyrrole 1e. As with traditional Knorr-type syntheses,36 by-products accompanied the desired pyrrole: 6 is the ester (–CO2R) variant of 1e; and 1a is produced as a consequence of deacylation. As such, the basic Knorr conditions were modified using various stoichiometries of sodium carbonate, tripotassium phosphate and sodium acetate salts to assert that the use of sodium carbonate resulted in the highest yield (33%) of the desired 2-thionoester pyrrole 1e. Cognisant that a Knorr-type synthesis of 2-carboxylate pyrroles yields 30–60% of the desired product on the same scale, this synthetic approach to 2-thionoester pyrroles is in keeping with the most prevalent of the routes to functionalised pyrroles.37,38 The corresponding benzyl thionoester pyrrole was synthesised following a similar approach, but originating with the reaction of CS2 with benzyl alcohol in the presence of potassium hydroxide to produce the required potassium O-benzyl carbonodithioate starting material.
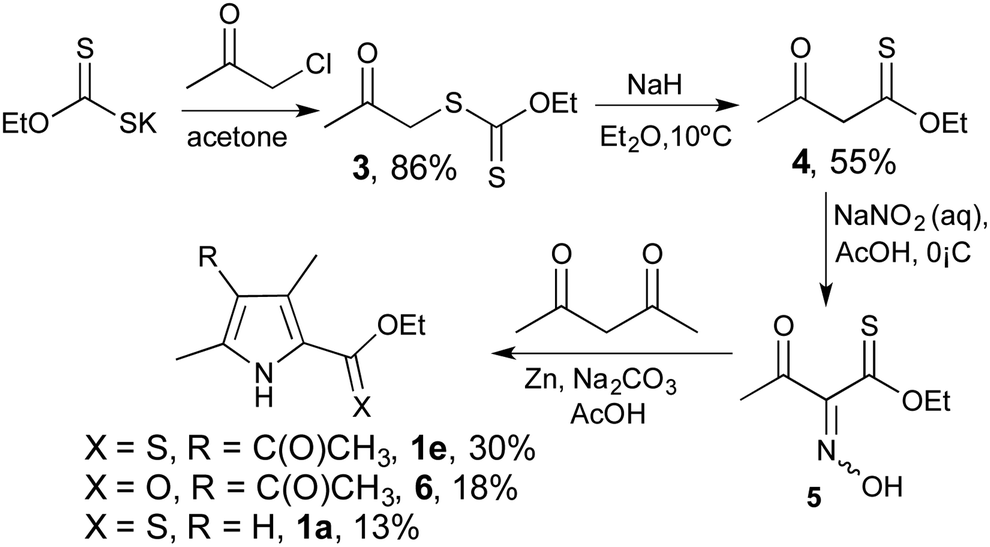 | ||
| Scheme 1 Knorr-type synthesis of o-ethyl 2-thionoester pyrrole. | ||
In order to probe the utility of 2-thionoester pyrroles beyond their conversion to 2-formyl pyrroles, we explored the potential of 1e as a substrate for the most common reactions that utilise 2-carboxylate pyrrole analogues. As shown in Scheme 2, treatment of 1e with borane cleanly reduced the acyl group to give the corresponding ethyl derivative 1c, no matter whether borane were generated in situ (Method A) or used as reagent (Method B). 2-Thionoester 1c mirrored the reactivity of 2-carboxylate pyrroles in the preparation of dipyrrins, thereby providing 7. Similarly, treatment of 1e with base effected hydrolysis and decarboxylation to give 8, and treatment with acid resulted in deacylation to render 1a. However, reaction of pyrrole 1e with CAN or Pb(OAc)4 returned the corresponding ester (–CO2R) with no sign of the desired oxidation of the 5-methyl group, clearly presenting a limitation as none of the desired 5-formyl species was produced.
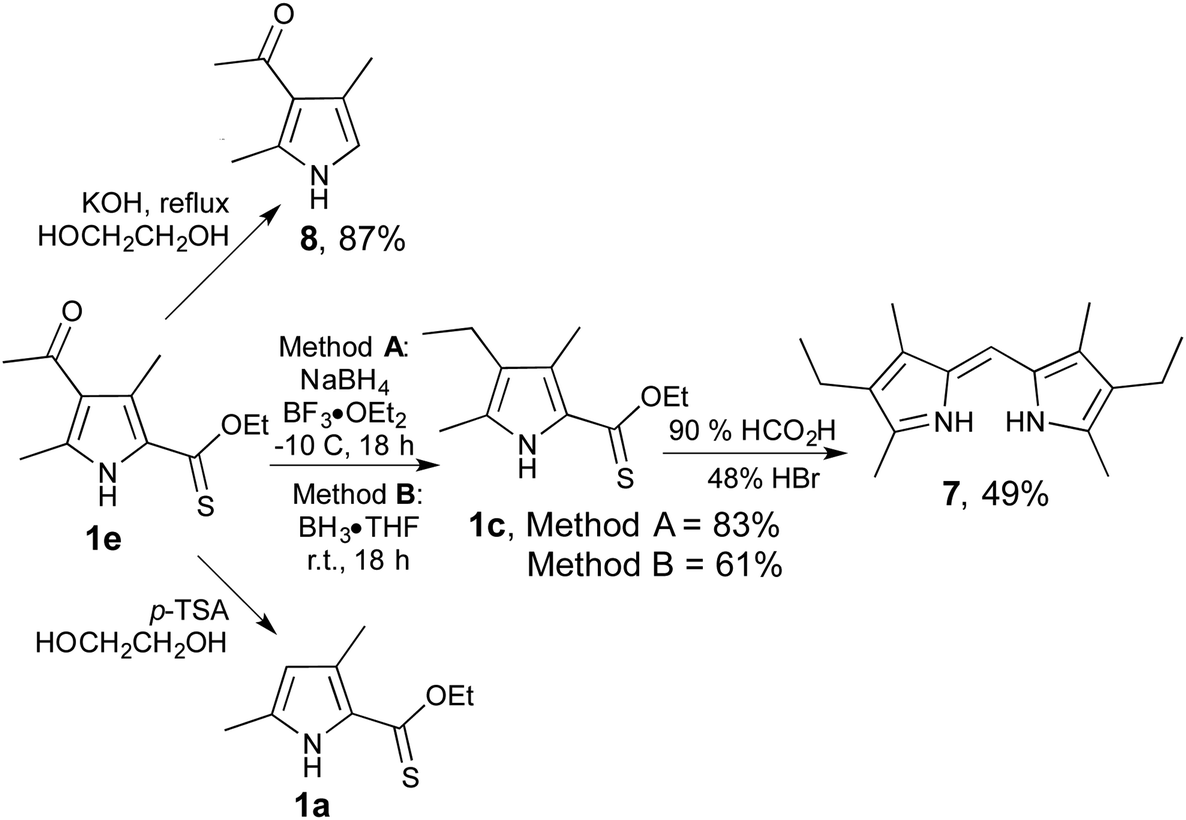 | ||
| Scheme 2 Reactions of 2-thionoester pyrrole 1e. | ||
In conclusion, an alternative route to 2-formyl pyrroles has been achieved via development of a modified Knorr procedure to produce 2-thionoester pyrroles, followed by the use of RANEY® nickel to promote reduction of the thionoester moiety to an aldehyde. The tolerance and utility of 2-thionoester pyrroles in reactions that are in widespread use within dipyrrin and porphyrin chemistry were investigated. Given the circuitousness nature of the synthesis of 2-formyl pyrroles of this genre and for these applications, the synthesis and use of 2-thioester pyrroles is presented as a viable alternative to the specialised manipulation of traditional Knorr-type 2-carboxylate pyrroles.
Experimental section
General information
Anhydrous solvents, silica 40–63 μm 60 Å and reagents were purchased and used as received. NMR spectra were obtained using 300 or 500 MHz instruments using CDCl3 or DMSO as solvents and are reported in part per million (ppm). Internal solvents were referenced at 7.26 ppm for 1H and at 77.16 ppm for 13C when using CDCl3, and at 2.50 ppm for 1H when using DMSO-d6. Coupling constants (J) are given in Hertz (Hz). Mass spectra were obtained using a time-of-flight ion trap instrument operating in ESI+/− mode. Flash chromatography was performed using Silicycle ultra-pure silica (230–400 mm). Pyrroles 1a, 1b, 1e, and 1f were prepared according to literature procedure involving Lawesson's reagent.9 Compound 3 was prepared according to the literature method.39 RANEY® nickel used was W. R. Grace Grade 2800 (CAS# 7440-02-0), purchased in the form of a slurry in water with the nickel composition stated as equal to or greater than 89%. Before each use, the bottle containing the RANEY® nickel slurry was thoroughly shaken before aliquots were drawn out quickly with a pipette in order to enable efficient transfer of the slurried material to a reaction flask.O-Ethyl 4-ethyl-3,5-dimethyl-1H-pyrrole-2-carbothioate (1c)
According to the literature method,40 the title compound was obtained as a yellow solid (0.63 g, 60%). Mp 66–68 °C; 1H-NMR (500 MHz, CDCl3): δ 9.08 (broad s, 1H), 4.66 (q, 2H, J = 7.0 Hz), 2.36 (q, 2H, J = 7.5 Hz), 2.27 (s, 3H), 2.21 (s, 3H), 1.46 (t, 3H, J = 7.0 Hz), 1.04 (t, 3H, J = 7.5 Hz); 13C NMR (125 MHz, CDCl3): δ 199.5, 134.2, 128.7, 126.0, 66.5, 17.2, 15.4, 14.3, 12.0, 11.9, one carbon signal missing; HRMS-ESI (m/z): [M + Na]+ calculated for C11H17NNaOS, 234.0923; found 234.0931.O-Ethyl 3,5-dimethyl-4-phenyl-1H-pyrrole-2-carbothioate (1d)
According to the literature method,40 the title compound was obtained as a yellow solid (0.76 g, 39%). Mp 89–92 °C; 1H-NMR (300 MHz, CDCl3): δ 9.31 (broad s, 1H), 7.39–7.45 (m, 2H), 7.29–7.34 (m, 1H), 7.22–7.25 (m, 2H), 4.71 (q, 2H, J = 6.9 Hz), 2.30 (s, 3H), 2.26 (s, 3H), 1.49 (t, 3H, J = 6.9 Hz); 13C NMR (125 MHz, CDCl3): δ 200.0, 134.5, 134.3, 130.2, 129.0, 128.4, 126.6, 124.8, 66.8, 14.3, 12.9, 12.5, one carbon signal missing; HRMS-ESI (m/z): [M + Na]+ calculated for C15H17NNaOS, 282.0923; found 282.0923.O-Benzyl 3,5-dimethyl-4-pentyl-1H-pyrrole-2-carbothioate (1g)
According to the literature method,40 the title compound was obtained as a yellow oil (0.53 g, 50%). 1H-NMR (300 MHz, CDCl3): δ 9.13 (broad s, 1H), 7.35–7.49 (m, 5H), 5.70 (s, 2H), 2.34 (t, 2H, J = 7.2 Hz), 2.24 (s, 3H), 2.22 (s, 3H), 1.42 (quin., 2H, J = 7.2 Hz), 1.28–1.35 (m, 4H), 0.91 (t, 3H, J = 7.2 Hz); 13C NMR (125 MHz, CDCl3): δ 198.7, 136.2, 135.0, 129.1, 128.6, 128.4, 128.2, 127.0, 124.7, 72.0, 31.7, 30.5, 23.9, 22.7, 14.2, 12.4, 12.0; HRMS-ESI (m/z): [M + Na]+ calculated for C19H25NNaOS, 338.1549; found 338.1545.O-Benzyl 3,5-dimethyl-4-methylpropionic-1H-pyrrole-2-carbothioate (1h)
According to the literature method,40 the title compound was obtained as a yellow solid (0.45 g, 43%). Mp 98–100 °C; 1H-NMR (500 MHz, CDCl3): δ 9.11 (broad s, 1H), 7.33–7.45 (m, 5H), 5.67 (s, 2H), 3.65 (s, 3H), 2.69 (t, 2H, J = 7.5 Hz), 2.41 (t, 2H, J = 7.5 Hz), 2.23 (s, 3H), 2.23 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 198.9, 173.4, 136.1, 135.0, 128.6, 128.4, 128.3, 126.1, 122.2, 72.2, 51.7, 34.8, 19.5, 12.3, 11.9, one carbon signal missing; HRMS-ESI (m/z): [M + Na]+ calculated for C18H21NNaO3S, 354.1134; found 354.1127.General procedure for synthesising 2-formyl pyrroles from 2-thionoester pyrroles (GP1)
Slurried RANEY® nickel (2 mL containing water and RANEY® nickel) was suspended in acetone, and the mixture was heated at reflux temperature for 1 hour. 2-Thionoester pyrrole 1 (0.47 mmol) was dissolved in acetone (10 mL), and the resulting solution was added to the reaction mixture containing RANEY® nickel in acetone held at reflux temperature. The reaction mixture was heated at reflux temperature for 2 hours, with stirring under nitrogen gas. Analysis using thin layer chromatography showed the complete consumption of starting material and the appearance of the product at lower Rf. The reaction mixture was allowed to cool to room temperature and then filtered through a short pad of silica, washing with acetone (500 mL). The filtrate was concentrated under vacuum, diluted with ethyl acetate and washed with brine. The organic layer was then dried using anhydrous sodium sulfate, and the solvent was removed through use of a rotary evaporator to yield a dark greenish brown solid. The crude product was purified via column chromatography over silica, eluting with 40% ethyl acetate/hexanes.General procedure for radical studies (GP2)
Following GP1, the radical clock or TEMPO was added along with 2-thionoester pyrrole 1a. Each reaction was run in parallel with a reference reaction prepared by GP1 using the same batch of RANEY® nickel. Cyclopropyl phenyl ketone: 500 equivalencies of the radical clock were added with 1a, and the product 2a was isolated in 55% (in absence of a radical clock: 53%). Diethyl cyclopropane-1,1-dicarboxylate: 200 equivalencies of the radical clock were added with 1a, and the product 2a was isolated in 64% (in absence of a radical clock: 58%). TEMPO: 50 equivalencies of TEMPO were added with 1a, and the product 2a was isolated in 7% (in absence of a radical trap: 37%).2-Formyl-3,5-dimethylpyrrole (2a)
Following GP1, the title compound was obtained as a beige solid (67%). Mp 87–88 °C; 1H-NMR (300 MHz, CDCl3): δ 10.16 (broad s, 1H), 9.43 (s, 1H), 5.82 (m, 1H), 2.29 (s, 3H), 2.27 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 176.0, 138.7, 134.9, 128.9, 112.1, 13.3, 10.7; HRMS-ESI (m/z): [M + Na]+ calculated for C7H9NNaO, 146.0576; found 146.0576.2-Formyl-3,4,5-trimethylpyrrole (2b) from 1b
Following GP1, the title compound was obtained as a beige solid (52%). Mp 142–145 °C; 1H-NMR (300 MHz, CDCl3): δ 9.99 (broad s, 1H), 9.44 (s, 1H), 2.24 (s, 6H), 1.91 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 175.6, 136.2, 132.9, 127.9, 118.3, 11.7, 9.0, 8.5; HRMS-ESI (m/z): [M + Na]+ calculated for C8H11NNaO, 160.0733; found 160.0731.4-Ethyl-2-formyl-3,5-dimethylpyrrole (2c)
Following GP1, the title compound was obtained as a beige solid (63%). Mp 98–100 °C; 1H-NMR (300 MHz, CDCl3): δ 10.03 (broad s, 1H), 9.45 (s, 1H), 2.38 (q, 2H, J = 7.5 Hz), 2.27 (s, 3H), 2.26 (s, 3H), 1.06 (t, 3H, J = 7.5 Hz); 13C NMR (125 MHz, CDCl3): δ 175.8, 136.0, 132.4, 128.1, 125.1, 17.2, 15.3, 11.7, 9.0; HRMS-ESI (m/z): [M + Na]+ calculated for C9H13NNaO, 174.0889; found 174.0889.2-Formyl-3,5,-dimethyl-4-phenylpyrrole (2d)
Following GP1, the title compound was obtained as a beige solid (45%). Mp 148–150 °C; 1H-NMR (300 MHz, DMSO-d6): δ 11.77 (br s, 1H), 9.56 (s, 1H), 7.39–7.44 (t, 2H), 7.25–7.31 (m, 3H), 2.25 (s, 3H), 2.21 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 176.6, 135.6, 134.3, 131.7, 130.0, 128.5, 128.4, 126.7, 125.6, 12.4, 9.6; HRMS-ESI (m/z): [M + Na]+ calculated for C13H13NNaO, 222.0889; found 222.0887.4-Acetyl-2-formyl-3,5-dimethylpyrrole (2e)
Following GP1, the title was compound was prepared as a beige solid (44%). Mp 168–170 °C; 1H-NMR (300 MHz, CDCl3): δ 9.89 (br s, 1H), 9.65 (s, 1H), 2.58 (s, 3H), 2.57 (s, 3H), 2.46 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 195.0, 177.7, 142.6, 134.2, 128.4, 123.9, 31.4, 15.4, 11.5; HRMS-ESI (m/z): [M + Na]+ calculated for C9H11NNaO2, 188.0682; found 188.0681.4-Acetyl-2-formyl-d-3,5-dimethylpyrrole (2e′)
To a slurry of RANEY® nickel (2 mL containing water and RANEY® nickel), deuterium oxide (5 mL portion) was added. The reaction vessel was stirred and sonicated at room temperature for 30 seconds, and the solvent was decanted. The exchange/wash cycle was repeated 10 times. After a final wash, deuterium oxide (2 mL) and acetone (10 mL) were added. GP1 was followed to obtain the title compound as a beige solid (46%). Mp 1H-NMR (300 MHz, CDCl3): δ 9.69 (br s, 1H), 2.58 (s, 3H), 2.57 (s, 3H), 2.47 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 195.0, 177.4 (t), 142.6, 134.2, 128.3, 123.9, 31.4, 15.5, 11.6; HRMS-ESI (m/z): [M + Na]+ calculated for C9H10DNNaO2, 189.0745; found 189.0736.2-Formyl-3,5-dimethylpyrrole (2b) from 1f
Following GP1, the title compound was prepared (45%). 1H-NMR (300 MHz, CDCl3): δ 9.86 (broad s, 1H), 9.42 (s, 1H), 2.23 (s, 6H), 1.91 (s, 3H) matching the data given above for this compound.2-Formyl-3,5-dimethyl-4-pentylpyrrole (2g)
Following GP1, the title compound was obtained as a beige solid (70%). Mp 70–71 °C; 1H-NMR (300 MHz, CDCl3): δ 9.87 (br s, 1H), 9.47 (s, 1H), 2.37 (t, 2H, J = 7.5 Hz), 2.28 (s, 3H), 2.27 (s, 3H), 1.45 (quin., 2H, J = 7.5. Hz), 1.28–1.38 (m, 4H), 0.92 (t, 3H, J = 7.0 Hz); 13C NMR (125 MHz, CDCl3): δ 175.8, 135.6, 132.3, 128.0, 123.6, 31.8, 30.4, 23.9, 22.7, 14.2, 11.8, 9.0; HRMS-ESI (m/z): [M + Na]+ calculated for C12H19NNaO, 216.1359; found 216.1362.2-Formyl-3,5-dimethyl-4-methylpropionic pyrrole (2h)
Following GP1, the title was compound was obtained as a beige solid (12%). Mp 128–129 °C; 1H-NMR (500 MHz, CDCl3): δ 9.82 (br s, 1H), 9.45 (s, 1H), 3.66 (s, 3H), 2.72 (t, 2H, J = 7.0 Hz), 2.44 (t, 2H, J = 7.5 Hz), 2.27 (s, 3H), 2.26 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 176.0, 173.4, 135.9, 132.3, 128.1, 121.1, 51.8, 34.7, 19.4, 11.7, 8.9; HRMS-ESI (m/z): [M + Na]+ calculated for C11H15NNaO3, 232.0944; found 232.0946.O-Ethyl acetothioacetate (4)
The title compound was synthesised following a modified literature procedure.34 A solution of S-acetonyl O-ethyl dithiocarbonate 339 (31.4 g, 176 mmol) in anhydrous Et2O (70 mL) was added drop-wise over 20 minutes to a suspension of NaH (60% in oil, 10.7 g, 268 mmol) in anhydrous Et2O (350 mL) at 10 °C, with stirring under nitrogen. After 10 minutes, the mixture was poured into aqueous K2HPO4 solution (0.91 M, 390 mL) and stirred. The ethereal layer was separated and the aqueous layer was extracted with Et2O (3 × 200 mL). The combined organic extracts were washed with brine (300 mL), dried over anhydrous MgSO4 and concentrated in vacuo. The crude product was purified via fractional vacuum distillation (heated at 140–150 °C), using a water-fed aspirator (∼25–60 mmHg), and the desired product was collected at 84 °C as a yellow liquid (14.1 g, 55%). The product was present in both enol and keto forms with an approximate 3:1 enol–keto ratio, according to the 1H NMR spectrum. 1H-NMR (300 MHz, CDCl3): δ 13.75 (s, 1H), 5.67 (s, 1H), 4.54 (q, J = 7.1 Hz, 0.7H), 4.47 (q, J = 7.1 Hz, 2H), 3.89 (s, 0.7H), 2.26 (s, 1H), 2.03 (s, 3H), 1.41 (t, J = 7.1 Hz, 1H), 1.38 (t, J = 7.1 Hz, 3H), in accordance with literature.34 However, the literature34 reports a 4:1 enol–keto ratio.
O-Ethyl 2-oximinoacetothioacetate (5)
The title compound was synthesised following a modified literature procedure.41 The resulting product mixture was poured into distilled water (25 mL) and extracted into CH2Cl2 (3 × 10 mL). The combined organic fractions were washed with saturated NaHCO3, dried over anhydrous sodium sulfate and concentrated in vacuo to yield the product as a bright yellow solid (277 mg, 23%). Mp 75–77 °C; 1H-NMR (300 MHz, CDCl3): δ 8.13 (s, 1H), 4.66 (q, J = 7.1 Hz, 2H), 2.42 (s, 3H), 1.45 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3): δ 204.6, 193.5, 155.0, 68.5, 26.0, 13.6; HRMS-ESI (m/z): [M + Na]+ calculated for C6H9NO3SNa, 198.0195; found 198.0189.O-Ethyl 4-acetyl-3,5-dimethyl-1H-pyrrole-2-carbothioate (1e)
The title compound was synthesised following a modified literature procedure typically used for Knorr-type synthesis of 2-carboxylate pyrroles.41 A solution of sodium nitrite (530 mg, 7.67 mmol) in distilled water (1.9 mL) was added drop-wise to a solution of O-ethyl acetothioacetate (4) (1.02 g, 6.98 mmol) in AcOH (1.6 mL) at 0 °C. The reaction mixture was stirred at room temperature and monitored via thin layer chromatography until the starting material was consumed. After 45 minutes, the resulting mixture containing crude O-ethyl 2-oximinoacetothioacetate (5) was transferred to a dropping funnel and added drop-wise to a slurry of 2,4-pentanedione (0.71 mL, 6.98 mmol), Na2CO3 (1.63 g, 15.3 mmol) and AcOH (10 mL). At the same time, Zn dust (0.958 g, 14.6 mmol) was slowly added to the slurry. The rate of addition and an ice-bath were used to keep the temperature below 30 °C. After the addition was complete, the reaction mixture was stirred at room temperature and monitored via thin layer chromatography. After 30 minutes the resulting mixture was poured into distilled water (150 mL) and then filtered through Celite, washing with CH2Cl2 until the washings were colorless.40 The filtrate was extracted with CH2Cl2 (3 × 100 mL). The combined organic fractions were washed with saturated NaHCO3 (150 mL) and brine (150 mL), then dried over anhydrous Na2SO4 and the solvent evaporated in vacuo. The resulting residue was purified via column chromatography on silica, eluting with a gradient of 5–20% EtOAc/hexanes, yielding the desired product as a yellow solid (0.53 g, 33%). Mp 109–111 °C; 1H-NMR (500 MHz, CDCl3): δ 9.41 (br s, 1H), 4.68 (q, J = 7.1 Hz, 2H), 2.58 (s, 3H), 2.52 (s, 3H), 2.45 (s, 3H), 1.49 (t, J = 7.1 Hz, 3H), in accordance with literature.93-Acetyl-2,4-dimethyl-1H-pyrrole (8) from 1e
To a solution of ground KOH (252 mg, 4.50 mmol) in ethylene glycol (1.6 mL) was added 1e (507 mg, 2.25 mmol). The reaction mixture was heated at 160 °C, with stirring, for 1 hour, monitoring via thin layer chromatography (40% EtOAc/hexanes). Water (5 mL) was then added and the mixture was extracted into CH2Cl2 (3 × 5 mL). The combined organic layers were dried over anhydrous Na2SO4 and concentrated in vacuo to yield the desired product as an off-white solid (269 mg, 87%). 1H-NMR (500 MHz, CDCl3): δ 7.93 (br s, 1H), 6.36 (s, 1H), 2.50 (s, 3H), 2.42 (s, 3H), 2.27 (s, 3H), in accordance with literature.41O-Ethyl 4-ethyl-3,5-dimethyl-1H-pyrrole-2-carbothioate (1c) from 1e
1,3,7,9-Tetramethyl-2,8-diethyl-5-H-4,6-dipyrrin hydrobromide (7)
Pyrrole 1c (64 mg, 0.3 mmol) was added to formic acid (0.29 mL) and the resulting mixture heated to reflux temperature before HBr (0.096 mL, 0.84 mmol) was added. The reaction mixture was stirred for an hour at reflux temperature. Then, the mixture was allowed to cool down to room temperature and was kept in the fridge for 2 days. The precipitate was collected and washed with ether to obtain a red-orange solid (49 mg, 49%). 1H-NMR (300 MHz, CDCl3): δ 12.92 (br s, 2H), 7.02 (s, 1H), 2.66 (s, 6H), 2.42 (q, J = 7.6 Hz, 4H), 2.26 (s, 6H), 1.07 (t, J = 7.6 Hz, 6H), in accordance with literature.42O-Ethyl 3,5-dimethyl-1H-pyrrole-2-carbothioate (1a) from 1e
The title compound was synthesised following a modified literature procedure.43 Ethylene glycol (1.5 mL), para-toluene sulfonic acid (16 mg, 0.08 mmol) and 1e (174 mg, 0.77 mmol) were added to a 2 mL microwave vial equipped with a stir bar. The vial was sealed and the mixture was pre-stirred for 1 minute then heated for 2 minutes at 100 °C in a laboratory microwave. Once allowed to cool to room temperature, the vial was unsealed. Water (5 mL) was added and the mixture was extracted into CH2Cl2 (3 × 5 mL). The combined organic fractions were dried over anhydrous Na2SO4 and then concentrated in vacuo. The crude mixture was purified via column chromatography on silica, eluting with 5% EtOAc/hexanes to yield the desired product as a yellow solid (95 mg, 67%). 1H-NMR (300 MHz, CDCl3): δ 9.09 (br s, 1H), 5.85 (s, 1H), 4.65 (q, J = 7.1 Hz, 2H), 2.30 (s, 3H), 2.25 (s, 3H), 1.46 (t, J = 7.1 Hz, 3H), in accordance with literature.9Potassium O-benzyl carbonodithioate
The title compound was synthesised following a modified literature procedure.25 Carbon disulfide (17.4 mL, 289 mmol) and benzyl alcohol (25 mL, 242 mmol) were added to a suspension of KOH (13.5 g, 242 mmol) in diethyl ether (375 mL). The suspension was stirred at room temperature for 3 hours. The product was collected via suction filtration and washed with Et2O to yield the desired product as a pale yellow solid (34.3 g, 64%). 1H-NMR (300 MHz, DMSO-d6): δ 7.35–7.27 (m, 5H), 5.33 (s, 2H), in accordance with literature.35O-Benzyl acetothioacetate
The title compound was synthesised following a modified literature procedure.39 A solution of chloroacetone (5.4 mL, 67.4 mmol) in acetone (34 mL) was added drop-wise to a solution of potassium O-benzyl carbonodithioate in acetone (84 mL) at 0 °C over a period of 10 minutes with stirring. After 1 hour, the solvent was removed in vacuo. The resulting mixture containing S-acetonyl O-benzyl dithiocarbonate was dissolved in dry Et2O (27 mL) and was added drop-wise over 20 minutes to a suspension of NaH (60% in oil, 3.2 g, 81.0 mmol) in dry Et2O (135 mL) under nitrogen at 10 °C. After 10 minutes, the reaction mixture was poured into aqueous KH2PO4 solution (1.2 M, 200 mL) and stirred at room temperature. The ethereal layer was separated and the aqueous layer was extracted with ether (3 × 100 mL). The combined organic fractions were washed with brine (150 mL), dried with anhydrous MgSO4 and concentrated in vacuo. The resulting residue was purified via column chromatography on silica, eluting with 2% ethyl acetate/hexanes to isolate the desired product as a yellow liquid (1.9 g, 13%). The product was isolated as a mixture containing enol and keto forms with an approximate 3:1 enol–keto ratio according to the 1H NMR spectrum. 1H-NMR (500 MHz, CDCl3): δ 13.70 (s, 1H), 7.40–7.35 (m, 6.9H), 5.76 (s, 1H), 5.51 (s, 0.7H), 5.46 (s, 2H), 3.95 (s, 0.7H), 2.24 (s, 1H), 2.04 (s, 3H); 13C-NMR{1H} (125 MHz, CDCl3): δ 213.1, 208.1, 177.8, 135.4, 134.7, 128.9, 128.8, 128.75, 128.72, 128.6, 128.5, 103.3, 74.7, 70.9, 61.7, 31.1, 29.9, 23.3; HRMS-ESI (m/z): [M + Na]+ calculated for C11H12O2SNa, 231.0450; found 231.0460.
O-Benzyl 4-acetyl-3,5-dimethyl-1H-pyrrole-2-carbothioate
The title compound was synthesised following a modified literature procedure.41 A solution of NaNO2 (235 mg, 3.41 mmol) in distilled water (852 μL) was added drop-wise to a solution of O-benzyl acetothioacetate (645 mg, 3.10 mmol) in AcOH (774 μL) at 0 °C. The reaction mixture was stirred at room temperature and monitored via thin layer chromatography until the starting material was consumed. The reaction mixture was then transferred to a dropping funnel and added drop-wise to a slurry of 2,4-pentanedione (316 μL, 3.10 mmol), Na2CO3 (361 mg, 3.41 mmol) and AcOH (2 mL). At the same time, Zn dust (11.4 g, 173.9 mmol) was slowly added to the slurry with the rate of addition and an ice-bath was used to keep the temperature below 30 °C. After full addition, the reaction mixture was stirred at room temperature. The resulting mixture was poured into distilled water (25 mL) and then filtered through Celite, washing with CH2Cl2. The filtrate was extracted with CH2Cl2 (3 × 10 mL). The combined organic fractions were washed with saturated NaHCO3 (10 mL) and brine (10 mL), then dried over anhydrous Na2SO4 and evaporated in vacuo. The resulting residue was purified via column chromatography on silica, eluting with a gradient of 5–20% ethyl acetate/hexanes, then washed with pentane (3 × 25 mL), yielding the desired product as a yellow solid (168 mg, 19%). Mp 122–124 °C; 1H-NMR (500 MHz, CDCl3): δ 9.43 (br s, 1H), 7.45–7.36 (m, 5H), 5.68 (s, 2H), 2.54 (s, 3H), 2.51 (s, 3H), 2.43 (s, 3H); 13C-NMR{1H} NMR (125 MHz, CDCl3): δ 200.7, 195.9, 141.0, 135.4, 128.8, 128.62, 128.58, 128.56, 127.1, 125.0, 73.1, 31.6, 15.5, 14.0; HRMS-ESI (m/z): [M + Na]+ calculated for C16H17NO2SNa, 310.0872; found 310.0869.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the Natural Sciences and Engineering Research Council of Canada (NSERC) courtesy of a Discovery Grant (AT) and the CREATE Training Program in BioActives (510963). NSERC (MJK, MHRB), Nova Scotia Graduate (MJK, MHRB) and Laing Undergraduate Research Award (SMG) scholarships are acknowledged.References
- J. M. Wood, D. P. Furkert and M. A. Brimble, Nat. Prod. Rep., 2019, 36, 289–306 RSC.
- L. Knorr, Justus Liebigs Ann. Chem., 1886, 236, 290–314 CrossRef.
- J. B. Paine III and D. Dolphin, J. Org. Chem., 1985, 50, 5598–5604 CrossRef.
- G. Anguera, W. Y. Cha, M. D. Moore, J. T. Brewster, M. Y. Zhao, V. D. Lynch, D. Kim and J. L. Sessler, Angew. Chem., Int. Ed., 2018, 57, 2575–2579 CrossRef CAS.
- S. Neya, T. Yoneda, H. Omori, T. Hoshino, A. T. Kawaguchi and M. Suzuki, Tetrahedron, 2017, 73, 6780–6785 CrossRef CAS.
- S. Shah, C. Lee, H. Choi, J. Gautam, H. Jang, G. J. Kim, Y.-J. Lee, C. L. Chaudhary, S. W. Park, T.-g. Nam, J.-A. Kim and B.-S. Jeong, Org. Biomol. Chem., 2016, 14, 4829–4841 RSC.
- Y. Yang, S. Chen, L. Liu, S. Li, Q. Zeng, X. Zhao, H. Li, Z. Zhang, L.-S. Bouchard, M. Liu and X. Zhou, ACS Appl. Mater. Interfaces, 2017, 9, 23400–23408 CrossRef CAS PubMed.
- A. Tsotinis, P. A. Afroudakis, K. Davidson, A. Prashar and D. Sugden, J. Med. Chem., 2007, 50, 6436–6440 CrossRef CAS.
- B. R. Groves, D. A. Smithen, T. S. Cameron and A. Thompson, RSC Adv., 2016, 6, 69691–69697 RSC.
- M. A. Shalaby and H. Rapoport, J. Org. Chem., 1999, 64, 1065–1070 CrossRef CAS.
- T. L. Cottrell, The strengths of chemical bonds, Butterworths Scientific Publications, London, England, 2nd edn, 1958 Search PubMed.
- A. El Gamal, V. Agarwal, S. Diethelm, I. Rahman, M. A. Schorn, J. M. Sneed, G. V. Louie, K. E. Whalen, T. J. Mincer, J. P. Noel, V. J. Paul and B. S. Moore, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 3797–3802 CrossRef CAS PubMed.
- C. T. Walsh, S. Garneau-Tsodikova and A. R. Howard-Jones, Nat. Prod. Rep., 2006, 23, 517–531 RSC.
- T. M. Cresp and M. V. Sargent, J. Chem. Soc., Perkin Trans. 1, 1973, 2961–2971 RSC.
- C. E. Loader and H. J. Anderson, Can. J. Chem., 1969, 46, 3879–3885 Search PubMed.
- B. Föhlisch, D. Abu Bakr and P. Fischer, J. Org. Chem., 2002, 67, 3682–3686 CrossRef.
- C. Savarin, J. Srogl and L. S. Liebeskind, Org. Lett., 2000, 2, 3229–3231 CrossRef CAS PubMed.
- R. Mozingo, D. E. Wolf, S. A. Harris and K. Folkers, J. Am. Chem. Soc., 1943, 65, 1013–1016 CrossRef CAS.
- E. Bullock, T.-S. Chen and C. E. Loader, Can. J. Chem., 1966, 44, 1007–1011 CrossRef CAS.
- L. Birry and A. Lasia, J. Appl. Electrochem., 2004, 34, 735–749 CrossRef CAS.
- I. Pogorelić, M. Filipan-Litvić, S. Merkaš, G. Ljubić, I. Cepanec and M. Litvić, J. Mol. Catal. A: Chem., 2007, 274, 202–207 CrossRef.
- B. Klenke and I. H. Gilbert, J. Org. Chem., 2001, 66, 2480–2483 CrossRef CAS PubMed.
- X. Wang and R. Rinaldi, ChemSusChem, 2012, 5, 1455–1466 CrossRef CAS.
- J. M. Khurana and R. Arora, Synthesis, 2009, 1127–1130 CrossRef CAS.
- H. Hauptmann and W. F. Walter, Chem. Rev., 1962, 62, 347–404 CrossRef CAS.
- J. Rentner, M. Kljajic, L. Offner and R. Breinbauer, Tetrahedron, 2014, 70, 8983–9027 CrossRef CAS.
- S. S. Bokatzian-Johnson, M. L. Maier, R. H. Bell, K. E. Alston, B. Y. Le and E. A. Cioffi, J. Labelled Compd. Radiopharm., 2007, 50, 380–383 CrossRef CAS.
- R. J. Heys, J. Labelled Compd. Radiopharm., 2010, 53, 716–721 CrossRef.
- G. R. Pettit and R. E. Kadunce, J. Org. Chem., 1962, 27, 4566–4570 CrossRef CAS.
- A. Call, C. Casadevall, F. Acuna-Pares, A. Casitas and J. Lloret-Fillol, Chem. Sci., 2017, 8, 4669–5218 RSC.
- W. Liu, L. Li and C.-J. Li, Nat. Commun., 2015, 6, 6526 CrossRef CAS.
- P. J. Wright and A. M. English, J. Am. Chem. Soc., 2003, 125, 8655–8665 CrossRef CAS.
- X. Zhang, H. Wang and Y. Guo, Rapid Commun. Mass Spectrom., 2006, 20, 1877–1882 CrossRef CAS.
- A. J. Bridges and G. H. Whitham, J. Chem. Soc., Perkin Trans. 1, 1975, 1603–1606 RSC.
- F. Carta, A. Akdemir, A. Scozzafava, E. Masini and C. T. Supuran, J. Med. Chem., 2013, 56, 4691–4700 CrossRef CAS.
- L. Knorr, Ber. Dtsch. Chem. Ges., 1884, 17, 1635 CrossRef.
- E. V. Antina, G. B. Guseva, A. E. Loginova, A. S. Semeikin and A. I. V'yugin, Russ. J. Gen. Chem., 2010, 80, 2374–2381 CrossRef CAS.
- H. Fischer, Org. Synth., 1935, 15, 17 CrossRef.
- M. D. Brown, D. W. Gillon, G. D. Meakins and G. H. Whitham, J. Chem. Soc., Perkin Trans. 1, 1985, 1623–1626 RSC.
- B. Groves, D. Smithen, S. Cameron and A. Thompson, RSC Adv., 2016, 6, 69691–69697 RSC.
- S. Mula, A. K. Ray, M. Banerjee, T. Chaudhuri, K. Dasgupta and S. Chattopadhyay, J. Org. Chem., 2008, 73, 2146–2154 CrossRef CAS.
- T. E. Wood, B. Berno, C. S. Beshara and A. Thompson, J. Org. Chem., 2006, 71, 2964–2971 CrossRef CAS.
- J. Regourd, I. M. Comeau, C. S. Beshara and A. Thompson, J. Heterocycl. Chem., 2006, 43, 1709–1714 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details including synthetic procedures and NMR spectra. See DOI: 10.1039/c9ra07527e |
| This journal is © The Royal Society of Chemistry 2019 |