
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA novel ratiometric fluorescent probe for rapid detection of hydrogen peroxide in living cells†
Linan Huac,
Jiayi Liua,
Jie Zhanga,
Hailiang Zhangb,
Pengfei Xu *b,
Zhu Chen*a and
Enhua Xiao*a
*b,
Zhu Chen*a and
Enhua Xiao*a
aDepartments of Radiology, The Second Xiangya Hospital, Central South University, Changsha, Hunan 410011, P. R. China
bInstitute of Clinical Pharmacy & Pharmacology, Jining First People's Hospital, Jining Medical University, Jining 272000, P. R. China
cDepartments of Radiology, Zhuzhou Central Hospital, Zhuzhou, Hunan 412000, P. R. China
First published on 2nd December 2019
Abstract
In this work, we present a new ratiometric fluorescent probe JNY-1 for rapid and convenient detection of H2O2. The probe could selectively and sensitively respond to H2O2 within 10 min. In addition, this probe was successfully applied for monitoring and imaging of H2O2 in liver cancer HepG2 cells under physiological conditions.
Introduction
Reactive oxygen species (ROS) is a collective term that describes the chemical species that are formed upon incomplete reduction of oxygen and includes the hypochlorous acid/hypochlorite ions (HOCl/−OCl), hydrogen peroxide (H2O2), superoxide anion (O2−) and the hydroxyl radical (HO˙). ROS in the body are generated endogenously from oxygen mainly through the mitochondrial respiration process as well as exogenously by exposure to deleterious conditions including UV light, xenobiotics and infectious agents.1 ROS are classically known as indicators of oxidative stress, which is a fundamental process that operates in a diverse array of human diseases, including Alzheimer's diseases, cardiovascular disorders, and cancer.2–4 Hydrogen peroxide (H2O2) is one of the most important ROS. Cellular hydrogen peroxide (H2O2) arises primarily as a ubiquitous dismutation product of superoxide. Mounting evidence suggests that H2O2 production drives a broad spectrum of cellular processes. For example, when H2O2 is generated at low concentrations (<0.7 μM) in a regulated fashion, it functions as a ubiquitous intracellular second messenger, regulating a multitude of physiological and pathological processes in cell.5 Under conditions of stress or stimulation by exogenous chemicals, H2O2 can be generated aberrantly and can be converted to dramatically more “damaging” ROS such as hypochlorous acid (HOCl) and hydroxyl radical (HO˙) by myeloperoxidase and Fenton reagents, respectively. The resulting ROS can attack cellular structures or biomolecules such as proteins, liposomes, and DNA, which has been associated with aging, Alzheimer's disease, and cancer.6,7 However, the transient increase of intracellular H2O2 may be necessary for mitosis.8 Therefore, development of a rapid and highly selective method for monitoring H2O2 level change H2O2 in living cells is of great significance.However, it is challenging to capture and detect H2O2 in living cells. This is mainly due to H2O2 in the cells possessing the following characteristics. At first, H2O2 can readily moves out of cells through free diffusion. So, it's difficult to detect intracellular H2O2 in situ. Second, H2O2 easily reacts with biosubstances to form other ROS. Therefore, in order to accurately detect H2O2, the methods require a fast response time. Last but most important, there are many ROS with high reactivity coexisting in the living system which can interfer the detection of H2O2. Thus, high selectivity is also expected to be achieved for detection of H2O2. Therefore, detection of H2O2 with spatial-temporal accuracy is of great significance for mechanistic studies of H2O2-related biology.
Fluorescence imaging based on small-molecule fluorescent probes has emerged as an efficient methodology to visualize the spatial and temporal distribution of biomolecules in biological specimens due to its real-time, sensitive, and noninvasive characteristics.9–11 Up to now, numerous fluorescent probes for endogenous H2O2 detection that have been elaborated,12–16 and the detection mechanisms of these H2O2 probes are mainly based on boronate oxidation17 or Baeyer–Villiger-type reactions.18 To some extent, these probes always have some minor defects. Several boronate-based probes react more slowly with H2O2 compared to other reactive species (e.g., peroxynitrite and hypochlorite).19–21 Some of them have been used to detect peroxynitrite22–24 or benzoyl peroxide25 instead of H2O2. On the other hand, the vast majority of these probes are intensity-based turn-on fluorescent probes.26 However, interpretation of the results obtained with turn-on probes could be complex. As the fluorescence intensity is very sensitive to environmental factors such as probe concentration, temperature, medium polarity, and pH and also instrumental set-ups, such intensity-based probes are not suitable for quantitative analysis. In this context, ratiometric probes that have an internal correction capability are in great demand. Ratiometric probes provided two separate signals for the probe itself and for its reaction product with the analyte or its analyte bound form, which can normalize the interference. Thus, ratiometric probes with high response rate, excellent selectivity, and good sensitivity should be developed for fulfilling the stringent issues of biological examination.
To address this issue, we herein report a novel ratiometric fluorescent probe JNY-1 (Scheme 1) for detection of H2O2 with high selectivity and sensitivity. In this work, we choose an integration of fluorescein and coumarin fluorophores as the fluorogen because of its' tunable fluorescent properties which could be used for design ratiometric response probes. The pentafluorobenzenesulfonyl group was selected as the reaction unit. As we all known, it is more stable to hydrolysis than ester bond, and the pentafluorobenzene ring enhances the reactivity of the sulfonates toward H2O2, which can contribute to enhance the selectivity and sensitivity of probe towards to H2O2.27. As shown in Fig. 1, in the absent of H2O2, probe JNY-1 is present in spirocyclic form only with coumarin-like emission. After selectively deprotecting the pentafluorobenzenesulfonyl group by H2O2, probe JNY-1 is present in its strongly fluorescent quinoid form and the fluorescence shift from 440 nm (coumarin-like emission) to 540 nm (fluorescein-like emission).
 | ||
| Scheme 1 The synthsize route of probe JNY-1. | ||
 | ||
| Fig. 1 Fluorescence (a) and UV/Vis absorption (b) spectra of JNY-1 (10 μM) in the absence and presence of H2O2 (100 μM). | ||
Results and discussion
The synthetic route of probe JNY-1 is shown in Scheme 1. First, an aldehyde-functionalized fluorescein was prepared according to previous reports.28,29 The further reaction of fluorescein monoaldehyde with diethyl malonate afforded a coumarin hybrid 1, which can be treated with 2,3,4,5,6-pentafluorobenzene-1-sulfonyl chloride, eventually resulting in the formation of probe JNY-1, which was characterized by 1H, 13C NMR and mass spectra.With the probe JNY-1 in hand, we then tested its spectroscopic properties. The fluorescence and absorption spectra were measured in DMF/phosphate buffer (30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :70 v/v, 10 mM, pH 7.4) system at room temperature (25 °C). As displayed in Fig. 1b, the probe JNY-1 itself showed the maximal absorbance centered at 340 nm. With the excitation at 340 nm, the probe JNY-1 offered fluorescence emission centered at 440 nm, which is belong to coumarin-like emission. Upon the addition of H2O2, the reaction of JNY-1 and H2O2 happened and resulted the dissociation of the pentafluorobenzenesulfonyl group from JNY-1. Obviously, this detection process is not reversible. Thus, a new absorbance centered at 480 nm emerged. Correspondingly, the fluorescein-like emission centered at 540 nm was observed.
:70 v/v, 10 mM, pH 7.4) system at room temperature (25 °C). As displayed in Fig. 1b, the probe JNY-1 itself showed the maximal absorbance centered at 340 nm. With the excitation at 340 nm, the probe JNY-1 offered fluorescence emission centered at 440 nm, which is belong to coumarin-like emission. Upon the addition of H2O2, the reaction of JNY-1 and H2O2 happened and resulted the dissociation of the pentafluorobenzenesulfonyl group from JNY-1. Obviously, this detection process is not reversible. Thus, a new absorbance centered at 480 nm emerged. Correspondingly, the fluorescein-like emission centered at 540 nm was observed.
As shown in Fig. 2, the time course of the fluorescent emission of probe JNY-1 (10 μM) at 540 nm in the presence of H2O2 (10 equiv.) was studied. By examining the kinetic data for H2O2, it is apparent that it would take no more than 10 minutes to reach the equilibrium. Generally, it needs 30 min or more to reach the reaction equilibrium between arylboronate based probes and H2O2. JNY-1 showed faster response to H2O2 mainly due to the pentafluorobenzene ring enhances the reactivity of the sulfonates toward H2O2. The rapid response time is favorable for detection and imaging of H2O2 in the complex biological systems.
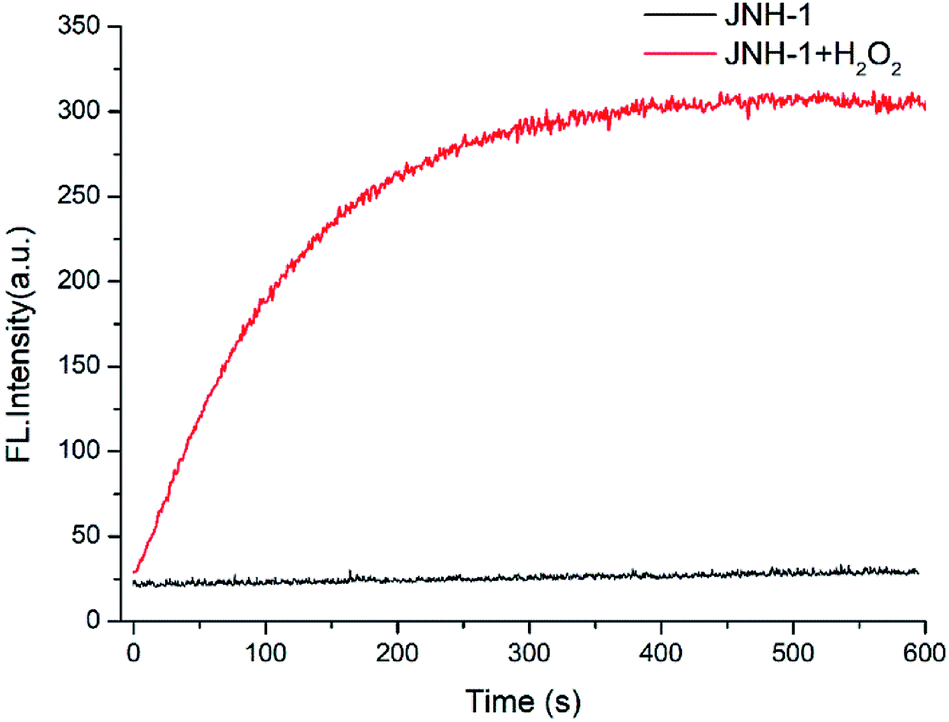 | ||
| Fig. 2 The time course of the emission intensity of JNY-1 (10 μM) at 540 nm in the presence and absence of H2O2 (100 μM) with λex = 340 nm. | ||
Next, the detailed emission titration experiments of probe JNY-1 (10 μM) with various concentrations of H2O2 were also carried out in the DMF/phosphate buffer (30:70 v/v, 10 mM, pH 7.4) system. As shown in Fig. 3, when the probe JNY-1 was treated with different concentrations of H2O2 (0–200 μM), the emission band at 440 nm decreased progressively, while a new emission peak around 540 nm was observed. Importantly, as shown in Fig. 4, the ratio of emission intensities (I540 nm:I440 nm) increased by nearly 180-fold and exhibited good linear correlation with the amount of H2O2 (0–100 μM, R2 = 0.98563). The detection limit was calculated to be 0.08 μM (3S/m, in which S is the standard deviation of blank measurements, n = 11, and m is the slope of the linear equation). As shown in Fig. S4,† with addition of H2O2 (0–200 μM), the initial absorption peak centered at 340 nm decreased slightly, along with a simultaneous emergence of the red shifted new absorption peaks at 500 nm. Correspondingly, JNY-1 displayed distinct color changes from colorless to orange, which could be easily observed by the naked eye (Fig. S6†).
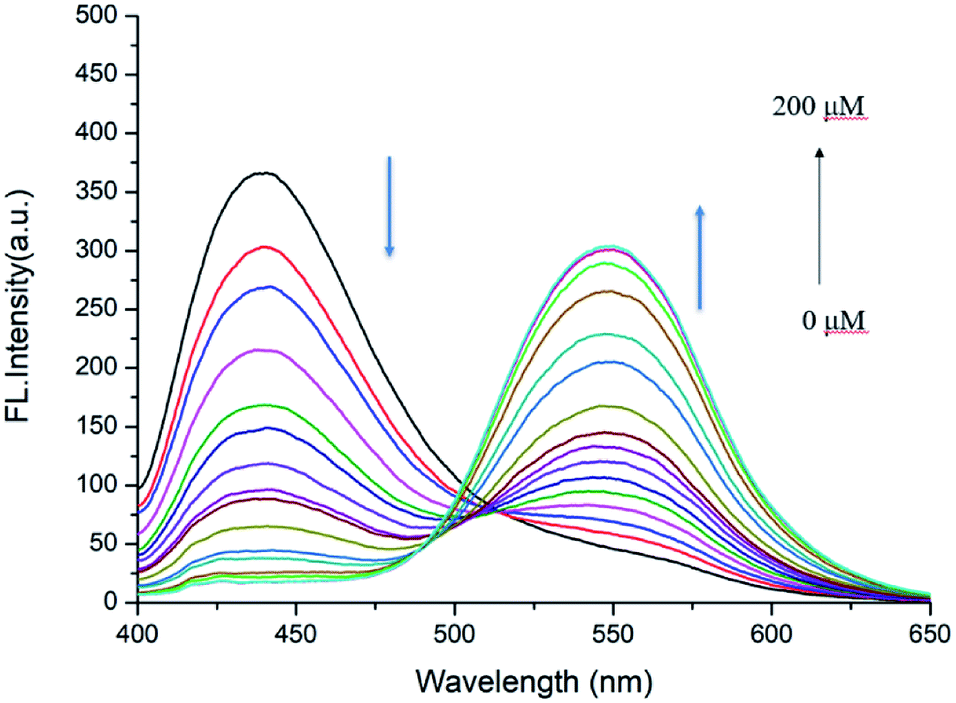 | ||
| Fig. 3 Fluorescence responses of probe JNY-1 (10 μM) toward different concentrations of H2O2 (0–200 μM) with λex = 380 nm. | ||
 | ||
| Fig. 4 Fluorescence intensity ratio (I540 nm:I440 nm) of probe JNY-1 versus H2O2 concentration (0–100 μM) with λex = 380 nm, error bars, SD (n = 3). | ||
To verify the selectivity of JNY-1 for H2O2, we also measured the ratiometric signals change upon addition of a panel of ROS, reactive nitrogen species (RNS), and the representative biological species. As shown in Fig. 5, only H2O2 could trigger a large ratiometric response. However, the ratiometric signals showed no or very minor changes for other species, including 1O2, ONOO−, NO, ClO−, tert-butyl hydroperoxide (TBHP), ·OH, O2˙−. GSH, ascorbic acid, and glucose. This result indicated that probe JNY-1 exhibited remarkably high selectivity for H2O2.
 | ||
| Fig. 5 Fluorescence responses I540 nm:I440 nm of JNY-1 (10 μM) with various analytes. (1) Free probe; (2) H2O2; (3) 1O2; (4) ONOO−; (5) NO; (6) ClO−; (7) tert-butyl hydroperoxide (TBHP); (8) ·OH; (9) O2˙−; (10) GSH; (11) ascorbic acid and (12) glucose. Each spectrum was recorded at 10 min after the addition, error bars, SD (n = 3). | ||
To further evaluate the ability of probe JNY-1 for detection of H2O2 in living cells, we conducted the cell imaging experiments. As shown in Fig. 6, the liver cancer HepG2 cells in group A and B were incubated with 10 μM of JNY-1 at 37 °C for 15 min before fluorescence imaging. Group A was the control and group B was added 100 μM of H2O2. As described in Fig. 6A, HepG2 cells incubated with 10.0 μM JNY-1 show fluorescence in blue channel, but there is negligible fluorescence signal through green channel. On the contrary, the probe-loaded cells appear clear cellular profiles with bright green fluorescence when treated with 100 μM H2O2 for 10 min, the original fluorescence signal in the blue channel weakened accordingly. These imaging results indicate that JNY-1 shows pleasurable cell-permeability and could be applied in dual-color fluorescence imaging of intracellular H2O2.
 | ||
| Fig. 6 Confocal fluorescence images of the liver cancer HepG2 cells with probe JNH-1. Fluorescence images were obtained before and after incubation for 10 min without (A) or with (B) 100 μM H2O2. The images were collected at 425–465 nm (blue channel) and 520–560 nm (green channel) upon excitation at 405 nm. Scale bar: 20 μm. | ||
Conclusions
In summary, we have described the design and synthesis of a new ratiometric fluorescent probe JNY-1 for detection of H2O2. The probe could selectively and sensitively respond to H2O2 within 10 min without interferences of other reactive oxygen species (ROS), reactive nitrogen species (RNS), and biologically relevant species. This probe was successfully applied for monitoring and imaging of H2O2 in liver cancer HepG2 cells under physiological conditions. The combination of ratiometric capability, sensitivity, specificity, and rapid response property makes the probe a candidate for various potential applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Nature Science Foundation of China (No. 81701754 and 81601471), Provincial Natural Science Foundation of Hunan (No. 2019JJ40434) and Scientific Research Project of Hunan Health and Family Planning Commission (No. B20180048).Notes and references
- X. Chen, F. Wang, J. Y. Hyun, T. Wei, J. Qiang, X. Ren, I. Shin and J. Yoon, Chem. Soc. Rev., 2016, 45, 2976–3016 RSC.
- W. Ahmad, B. Ijaz, K. Shabbiri, F. Ahmed and S. Rehman, J. Biomed. Sci., 2017, 24, 76 CrossRef PubMed.
- H. K. Seitz and F. Stickel, Nat. Rev. Cancer, 2007, 7, 599–612 CrossRef CAS PubMed.
- S. Prasad, S. C. Gupta and A. K. Tyagi, Cancer Lett., 2017, 387, 95–105 CrossRef CAS PubMed.
- S. G. Rhee, Science, 2006, 312, 1882–1883 CrossRef PubMed.
- I. Levitan, S. Volkov and P. V. Subbaiah, Antioxid. Redox Signaling, 2010, 13, 39–75 CrossRef CAS PubMed.
- S. Kanvah, J. Joseph, G. B. Schuster, R. N. Barnett, C. L. Cleveland and U. Landman, Acc. Chem. Res., 2010, 43, 280–287 CrossRef CAS PubMed.
- S. G. Rhee, T. S. Chang, Y. S. Bae, S. R. Lee and S. W. Kang, J. Am. Soc. Nephrol., 2003, 14, S211–S215 CrossRef CAS PubMed.
- J. Chan, S. C. Dodani and C. J. Chang, Nat. Chem., 2012, 4, 973–984 CrossRef CAS PubMed.
- X. Chen, T. Pradhan, F. Wang, J. S. Kim and J. Yoon, Chem. Rev., 2012, 112, 1910–1956 CrossRef CAS PubMed.
- X. Li, X. Gao, W. Shi and H. Ma, Chem. Rev., 2014, 114, 590–659 CrossRef CAS PubMed.
- M. Abo, Y. Urano, K. Hanaoka, T. Terai, T. Komatsu and T. Nagano, J. Am. Chem. Soc., 2011, 133, 10629–10637 CrossRef CAS PubMed.
- B. C. Dickinson, V. S. Lin and C. J. Chang, Nat. Protoc., 2013, 8, 1249–1259 CrossRef PubMed.
- C. Yik-Sham Chung, G. A. Timblin, K. Saijo and C. J. Chang, J. Am. Chem. Soc., 2018, 140, 6109–6121 CrossRef CAS PubMed.
- W. Zhang, W. Liu, P. Li, F. Huang, H. Wang and B. Tang, Anal. Chem., 2015, 87, 9825–9828 CrossRef CAS PubMed.
- Y. Zhou, W. Pei, X. Zhang, W. Chen, J. Wu, C. Yao, L. Huang, H. Zhang, W. Huang, J. S. Chye Loo and Q. Zhang, Biomaterials, 2015, 54, 34–43 CrossRef CAS PubMed.
- T. B. Ren, W. Xu, W. Zhang, X. X. Zhang, Z. Y. Wang, Z. Xiang, L. Yuan and X. B. Zhang, J. Am. Chem. Soc., 2018, 140, 7716–7722 CrossRef CAS PubMed.
- X. Jiao, Y. Xiao, Y. Li, M. Liang, X. Xie, X. Wang and B. Tang, Anal. Chem., 2018, 90, 7510–7516 CrossRef CAS PubMed.
- X. Wu, X. X. Chen, B. N. Song, Y. J. Huang, W. J. Ouyang, Z. Li, T. D. James and Y. B. Jiang, Chem. Commun., 2014, 50, 13987–13989 RSC.
- S. Y. Xu, X. Sun, H. Ge, R. L. Arrowsmith, J. S. Fossey, S. I. Pascu, Y. B. Jiang and T. D. James, Org. Biomol. Chem., 2015, 13, 4143–4148 RSC.
- M. Reverte, A. Vaissiere, P. Boisguerin, J.-J. Vasseur and M. Smietana, ACS Sens., 2016, 1, 970–974 CrossRef CAS.
- X. Sun, Q. Xu, G. Kim, S. E. Flower, J. P. Lowe, J. Yoon, J. S. Fossey, X. Qian, S. D. Bull and T. D. J. C. S. James, Chem. Sci., 2014, 5, 3368–3373 RSC.
- J. Zhang, Y. Li and W. Guo, Anal. Methods, 2015, 7, 4885–4888 RSC.
- J. Zhou, Y. Li, J. Shen, Q. Li, R. Wang, Y. Xu and X. Qian, RSC Adv., 2014, 4, 51589–51592 RSC.
- W. Chen, Z. Li, W. Shi and H. Ma, Chem. Commun., 2012, 48, 2809–2811 RSC.
- J. Kim, J. Park, H. Lee, Y. Choi and Y. Kim, Chem. Commun., 2014, 50, 9353–9356 RSC.
- G. Chen, Q. Fu, F. Yu, R. Ren, Y. Liu, Z. Cao, G. Li, X. Zhao, L. Chen, H. Wang and J. You, Anal. Chem., 2017, 89, 8509–8516 CrossRef CAS PubMed.
- W. Wang, O. Rusin, X. Xu, K. K. Kim, J. O. Escobedo, S. O. Fakayode, K. A. Fletcher, M. Lowry, C. M. Schowalter, C. M. Lawrence, F. R. Fronczek, I. M. Warner and R. M. Strongin, J. Am. Chem. Soc., 2005, 127, 15949–15958 CrossRef CAS PubMed.
- Z. X. Han, X. B. Zhang, Z. Li, Y. J. Gong, X. Y. Wu, Z. Jin, C. M. He, L. X. Jian, J. Zhang, G. L. Shen and R. Q. Yu, Anal. Chem., 2010, 82, 3108–3113 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra07517h |
| This journal is © The Royal Society of Chemistry 2019 |