
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrocatalytic oxidation of water at a polyoxometalate nanoparticle modified gold electrode†
Abhinandan Mahanta a,
Koushik Barmanab and
Sk Jasimuddin*a
a,
Koushik Barmanab and
Sk Jasimuddin*a
aDepartment of Chemistry, Assam University, Silchar, Assam-788011, India. E-mail: sk.jasimuddin@aus.ac.in
bDepartment of Chemistry, The University of Utah, Salt Lake City, Utah 84112-0850, USA
First published on 26th November 2019
Abstract
Spherical polyoxometalate nanoparticles, [HPMo]NPs, were synthesized from a very well known Keggin-type polyoxometalate [H3PMo12O40] in the presence of sodium dodecyl sulphate (SDS) and polyvinyl pyrrolidine (PVP) in aqueous medium and characterized by UV-Vis spectroscopy and Transmission Electron Microscopy (TEM). The [HPMo]NPs were used to modify a gold working electrode and they were characterized by SEM, EDX, elemental mapping, cyclic voltammetry and electrochemical impedance spectroscopy and applied for the electrocatalytic oxidation of water in a phosphate buffer solution at neutral pH. The modified electrode showed excellent electrocatalytic activity towards oxidation of water at an impressively low overpotential ∼350 mV with a high current density of around 1.7 mA cm−2, good stability under exhaustive electrolysis conditions and also showed long term stability.
To fulfill the worldwide clean energy demand, development of renewable and inexpensive energy sources is the most challenging task in the present era. In this endeavor, photo and electro catalytic splitting of water to generate molecular oxygen and hydrogen is a promising technique.1 Molecular hydrogen is a clean and environmentally friendly fuel with high gravimetric energy density.2 The water splitting process consists of two steps, 2H2O → O2 + 4H+ + 4e− and 2H+ + 2e− → H2. Between them the water oxidation step is the energy controlling step in the overall water splitting process and is frequently an obstacle in the whole water splitting process due to its high overpotential and slow reaction kinetics.3 Extensive efforts have been devoted to make an efficient water oxidation catalyst for lowering the energy consumption and accelerating the kinetics of the reaction.
To date a large number of polyoxometalates (POMs) have been extensively utilized as homogeneous as well as heterogeneous water oxidation catalysts (WOC's).4–8 To enhance the catalytic activity and stability of POMs different strategies have been taken, for instances, incorporation of transition metal ions in the POM structure,9,10 immobilization of POM on appropriate surface such as carbon nanotube, graphene or TiO2 nanoparticles etc.,11–13 encapsulation of POM inside a cavity of metal organic frame work7 etc.
Catalytic activity of POMs can also be increased if POM molecules will be assembled into a defined shape and size such as in nanoparticles dimension. POM nanoparticles can be synthesised different ways such as microemulsion mediated POM nanoparticles formation14 or, self-assemble polymer or cationic micelles or substate for synthesizing nanostructured POM.15,16
In the present communication we have described the synthesis and characterization of spherical polyoxometalate nanoparticles by using the Keggin-type polyoxometalate, [H3PMo12O40] (HPMo) and sodium dodecyl sulfate (SDS) surfactant where POMs are at the surface and SDS at the core and the [HPMo]NPs were utilized for electrode modification to study the electrocatalytic water oxidation reaction at neutral pH. The POM nanoparticles modified gold electrode can efficiently oxidized water to oxygen at low overpotential (350 mV) with high current density (1.7 mA cm−2).
UV-visible spectra of [HPMo] nanoparticles (Fig. S1†) shows a broad band at 225 nm, owing to the O → Mo charge transfer transitions.17 Deferent corresponding bond stretching frequencies of [HPMo] nanoparticles were investigated by FT-IR spectroscopy using KBr pallets. Stretching frequencies at 1063, 954, 824 and 754 cm−1 were observed owing to the vibration modes νP![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, νMoO and νMo–O–Mo, respectively.17
O, νMoO and νMo–O–Mo, respectively.17
To investigate the morphology and size of the [HPMo] nanoparticles, TEM was taken. Fig. 1 shows the TEM images of [HPMo] nanoparticles indicating that the size of the nanoparticles is in the range of 20–30 nm. Each particle has a hollow space confirms that micelle directed [HPMo] nanoparticles was formed.
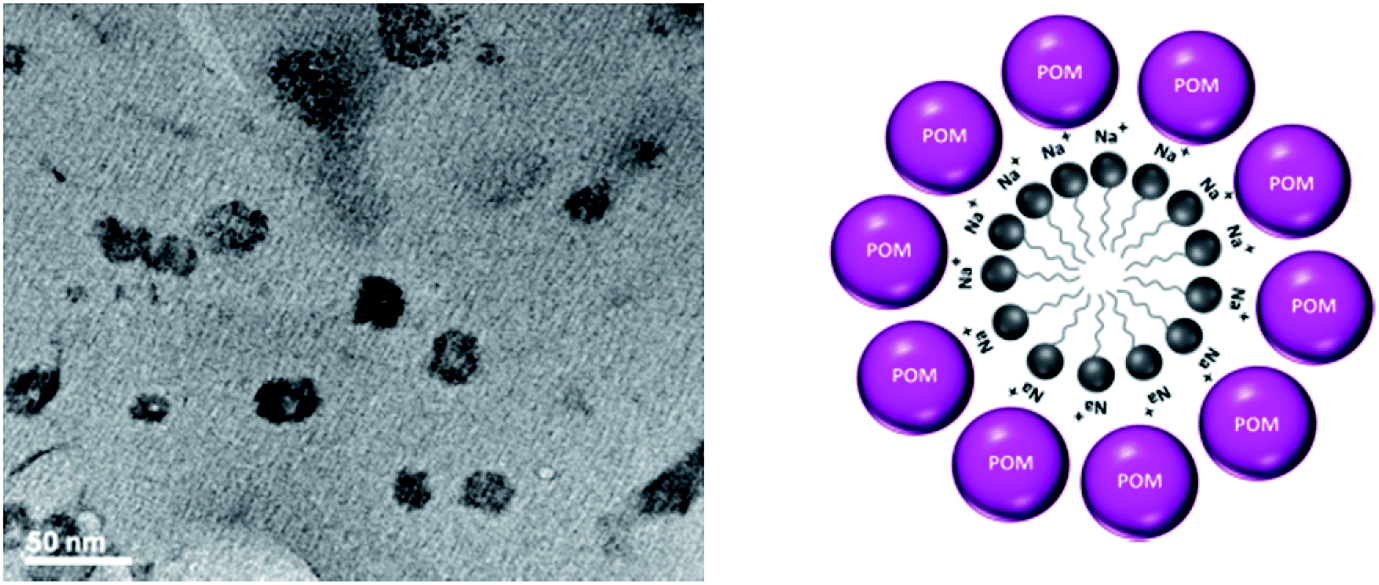 | ||
| Fig. 1 TEM image and cartoon of [HPMo] nanoparticles. | ||
The electrode modification was characterized by comparing the SEM images of the bare and [HPMo] nanoparticles modified gold electrodes. Fig. S2a† shows a smooth surface morphology for bare gold electrode whereas the modified electrode shows almost smooth surface (Fig. S2b†) due to layer formation by the [HPMo] nanoparticles. This observation indicates that the gold electrode was properly modified. The EDX spectrum of the modified gold electrode (Fig. S2c and d†) shows the presence of C, N, O, P, S, Mo and Au elements which confirms the formation of [HPMo] layer on Au electrode. Elemental mapping also supports the presence of C, N, P, O, S, Au, Mo and Na and confirms the proper modification of gold electrode by the [HPMo] nanoparticles (Fig. S3a–h†). To characterize the modified electrode electrochemically, a comparative cyclic voltammetry of the redox probe [Fe(CN)]63−/4− (0.5 mM) were carried out at bare and [HPMo]NPs modified gold electrodes in 0.1 M PBS (at pH 7.0) (Fig. S4†). The cyclic voltammograms shows a cathodic peak current (Ipc) at ∼75 μA and ∼140 μA when bare and [HPMo]NPs modified Au-electrode were used, respectively. Increase current density obtained at [HPMo] nanoparticles modified electrode indicates an enhanced electronic communication between the probe and gold electrode. This observation is also supported by the electrochemical impedance spectroscopy. Nyquist plot shows that the charge transfer resistance (Rct) is ∼0.42 × 105 Ω at [HPMo] nanoparticles modified gold electrode which is almost half than obtained at bare gold electrode (Rct = 0.80 × 105 Ω). This result also supports the higher electron transfer ability of [HPMo]NPs–Au than the bare Au electrode (Fig. S5†). Cyclic voltammograms of bare and modified gold electrodes (Fig. S6†) in 0.1 M PBS at pH 7 also supports the successful immobilization of the nanoparticles on the gold electrode surface, as the peak current associated with the [HPMo]NPs modified electrode obtained a higher value than that associated with the bare electrode. Linear response of the peak current values to the increasing scan rate as seen in the voltammograms at different scan rates (Fig. S7†) proclaims the electrochemical stability of the surface bound [HPMo]NPs/Au modified electrode.
Fig. 2 displays the linear sweep voltammogram (LSV) of 0.1 M PBS solution (pH 7.0) at bare Au, [HPMo]–Au and [HPMo]NPs–Au electrode. An anodic peak was observed at +1.58 V versus RHE with a current density of 1.7 mA cm−2 at [HPMo]NPs modified electrode. No such anodic peak was observed in this potential window (0.2 to 2.0 V versus RHE) when bare Au or [HPMo]–Au was used.
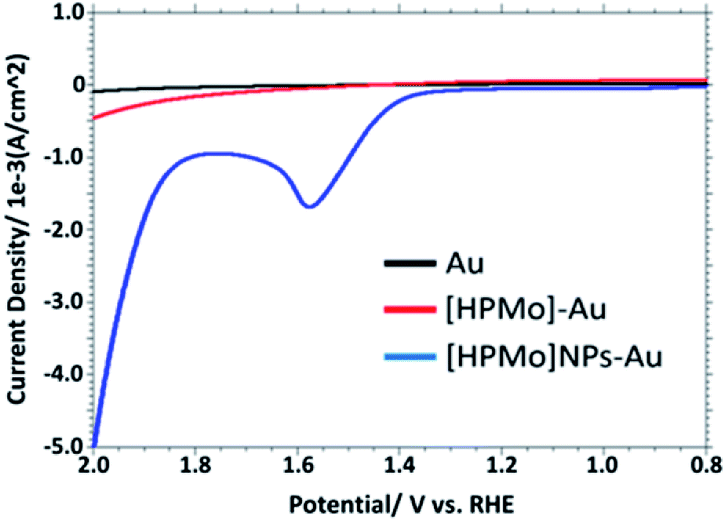 | ||
| Fig. 2 Overlaid LSV in 0.1 M PBS (pH 7.0) obtained at bare, [HPMo] and [HPMo]NPs modified gold electrode. | ||
This observation suggest that the [HPMo]NPs modified electrode can electrochemically oxidized water to oxygen, 2H2O → O2 + 4H+ + 4e−. In non-aqueous media like in CH3CN no such anodic peak was observed. When the water is added to the solution of CH3CN containing 0.1 M [Bu4N][ClO4] (pH = 7.0), the oxidative peak appeared at the same potential, +1.58 mV versus RHE (Fig. 3a). Initially, the anodic peak current density increased with increasing water concentration (up to 0.5 M) and after that the current density remained constant (Fig. 3b) as expected for surface attached species.18 The anodic peak current increased with increasing scan rate in the lower range (20–100 mV s−1), following the linear regression equation Jpa (mA cm−2) = 0.0076ν (mV s−1) + 0.943 (R2 = 0.9995) (Fig. S8†), which indicates that the electrode process is surface-controlled. However, at higher scan rate (200–700 mV s−1), the peak current varies linearly with square root of scan rate and followed the linear regression equation Jpa (mA cm−2) = 0.3556ν1/2 (mV s−1) − 2.6553 (R2 = 0.9993) which supports diffusion controlled water oxidation (Fig. S9†). Therefore, it may be concluded that the electrocatalytic oxidation of water on [HPMo]NPS–Au electrode is the mixture of adsorption and diffusion controlled process and is dependent on scan rate.19
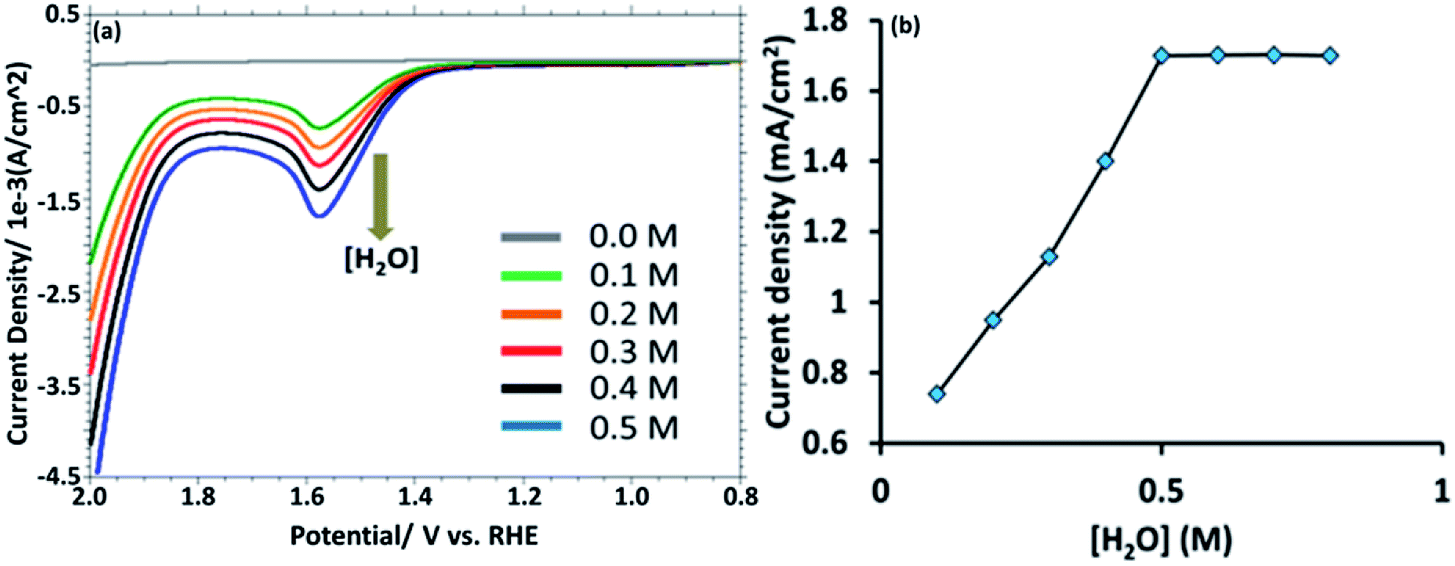 | ||
| Fig. 3 (a) LSV obtained from [HPMo]NPs–Au electrode with increasing amount of [water] (0.0 to 0.5 M). (b) A plot of anodic current density versus [H2O]. | ||
The effect of pH on the electrocatalytic oxidation of water was investigated using LSV in the pH range of 5.0–9.0 of PBS at a scan rate of 100 mV s−1 and is presented in Fig. S10.†
With increasing pH of the medium the current density for water oxidation increases. Although the water oxidation performance of [HPMo]NPs–Au electrode is better at higher pH, but the stability of the system is decreases. So, pH 7.0 was chosen as working condition for the entire water oxidation experiment. On the other hand the anodic peak potentials were shifted towards less positive potential with increasing pH following the linear regression equation Epa (V) = −0.0592pH + 1.9852 (R2 = 0.9949). The slope of 59 mV per pH unit indicates that equal number of protons and electrons are involved in the electrode reaction process.20
Controlled potential electrolysis (CPE) was performed in a stirred 0.1 M PBS solution (pH = 7.0) at +1.58 V versus RHE in a gas tight electrochemical cell using bare Au, [HPMo]–Au and [HPMo]NPs–Au electrode and overlaid CPE curves are shown in Fig. S11.† A steady-state current of 1.0 A cm−2 was obtained for up to 30 minutes at [HPMo]NPs–Au electrode, whereas a very small amount of current (0.2 A cm−2) was obtained during the electrolysis of water at bare Au and [HPMo]–Au electrodes in similar condition. During the electrolysis of water (35 minutes) around 17 μM oxygen (Fig. S12†) was produced (the amount of oxygen formed during the electrolysis was measured using fluorescent probe) with a Faraday efficiency of around 90%. Fig. 4a shows the chronoamperograms with increasing overpotential in 0.1 M PBS (pH 7.0) at [HPMo]NPs–Au electrode. The Tafel plots remained good linearity indicating a good electrical conductivity retained with the [HPMo]NPs. The Tafel slope (Fig. 4b) obtained for the [HPMo]NPs is 63 mV dec−1 indicates excellent electrocatalytic activity of the [HPMo] nanoparticles. On the other hand, the Tafel slope obtained for [HPMo] modified electrode is quite high (around 597 mV dec−1) and this high value indicates the inactivity of phosphomolybdic acid [HPMo] towards water oxidation under similar condition.21 Table 1 shows a comparative account of the previously reported polyoxometalates based nanomaterials along with present system for electrocatalytic water oxidation. It can be seen that the [HPMo]NPs–Au is quite comparable or sometimes even better water oxidation activity than the reported systems.
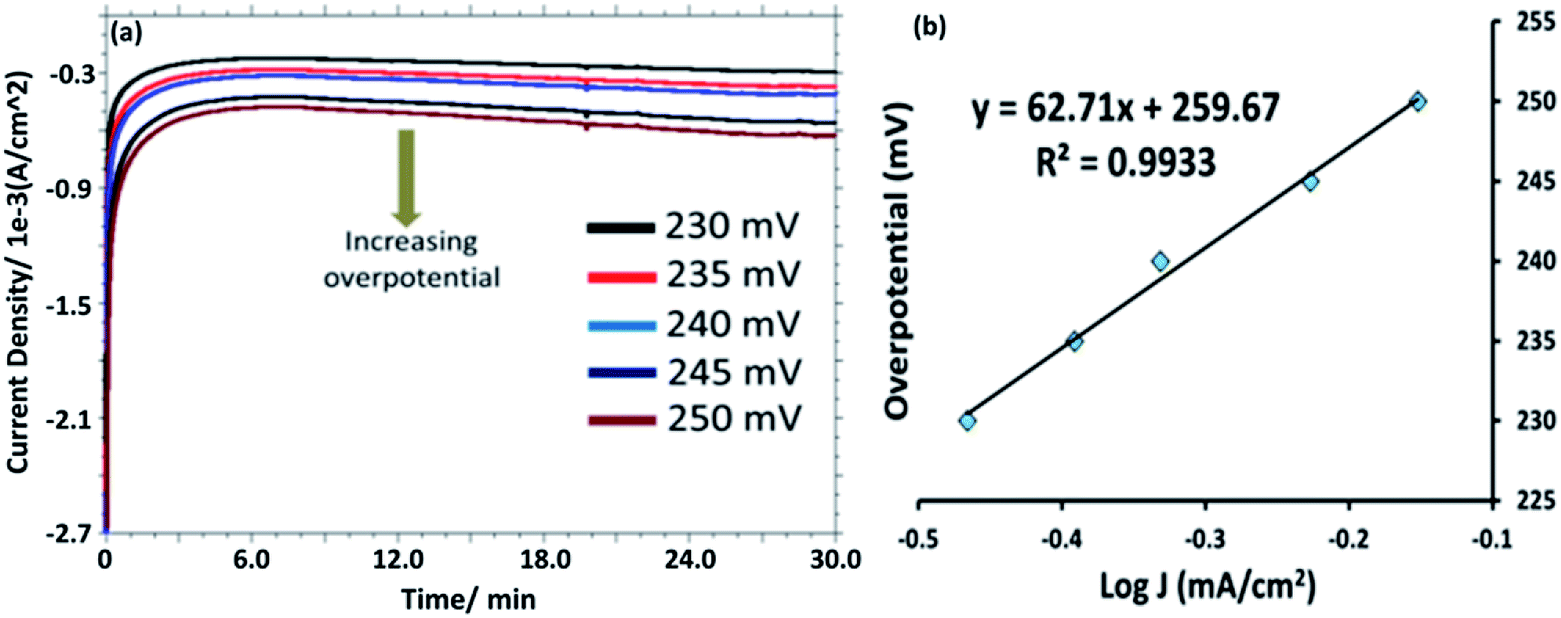 | ||
| Fig. 4 (a) Current density versus time plot as the applied potential is stepped from 230 to 250 mV. (b) Tafel plot of [HPMo]NPs–Au electrode in 0.1 M PBS (pH 7.0) at different applied potentials. The slope of the graph is around 63 mV per decade. | ||
| System | Over potential [mV] | Current density [mA cm−2] | TOF (s−1) | Tafel slope (mV dec−1) | pH | Reference |
|---|---|---|---|---|---|---|
| POM@ZIF-8 | 784.19 | 1.0 | 12.5 | 783.62 | 7.0 | 7 |
| POM@MWCNT | 310 | — | 0.01 | — | 7.0 | 11 |
| POM@graphene | 350 | — | 0.82 | — | 7.0 | 12 |
| [Rubpy]5[Ru4POM] | 490 | — | 0.35 | — | 7.0 | 22 |
| [Co9]POM | 695 | 1.0 | — | 65 | 7.0 | 23 |
| Cu6Co7 POM/CC | 500 | 10.0 | — | 147 | 7.0 | 24 |
| POM Ru4(SiW10)2 | 300 | 0.15 | 0.26 | 222 | 7.0 | 25 |
| NiCo2O3@OMC | 281 | — | — | 96.80 | 14.0 | 26 |
| IrO2@Ir | 255 | 10.0 | 0.026 | 45 | 14.0 | 27 |
| IrOx (OH)y film | 240 | — | 6.0 | 42 | 7.0 | 28 |
| [HPMo]NPs | 350 | 1.7 | 3.6 | 63 | 7.0 | Present work |
The stability of the [HPMo]NPs modified Au electrode was examined by measuring chronopotentiometry (Fig. S13†) at a fixed current density 1.5 mA cm−2 for 30 minutes. The oxidation potential (∼1.5 V) remained stable for the entire period of experiment revealing that the modified electrode had good stability. The stability of the system was also confirmed by using controlled potential electrolysis of water (pH 7.0) at a fixed potential 1.58 V versus RHE for 400 minutes (around 7 h) (Fig. S14†).CPE shows a rapid decline of current density (J) up to 20 seconds and thereafter the current remains stable for the entire period of electrolysis.
This result again supports the high stability of [HPMo]NPs–Au electrode. The long-time stability of the modified electrode was checked by taking LSV in 0.1 M PBS (pH = 7.0) after ten days interval up to 30 days (Fig. S15†). The LSV response was retained up to 30 days of measurement taken and confirmed the long term stability of [HPMo]NPs–Au electrode.18 The modified electrode was kept by covering a rubber cap when not in use.
The mechanistic pathway of electrocatalytic water oxidation by [HPMo]NPs is beyond the scope of our study. In the present article we have shown that [HPMo]–Au electrode is unable to oxidized water and this may be due to the highest oxidation state of molybdenum(VI)-oxo species in Keggin H3PMo12O40 and the (VI/V) reduction potentials are well below the reversible water oxidation.29 On the other hand [HPMo]@SDS nanoparticles modified Au electrode can efficiently oxidized water in neutral pH (Scheme 1). It may be presumed that the water molecule is adsorbed over the larger surface of the micelle stabilized [HPMo] nanoparticles through hydrogen bonding interaction which facilitates electron coupled proton transfer process and cleavage of O–H bonds in water molecule. Recent reports of photocatalytic water oxidation by dual hydrogen bonding structure on surface fluorinated TiO2 (ref. 30) and Keggin based POM@ZIF-8 catalysed7 oxidation of water supports such logical consideration.
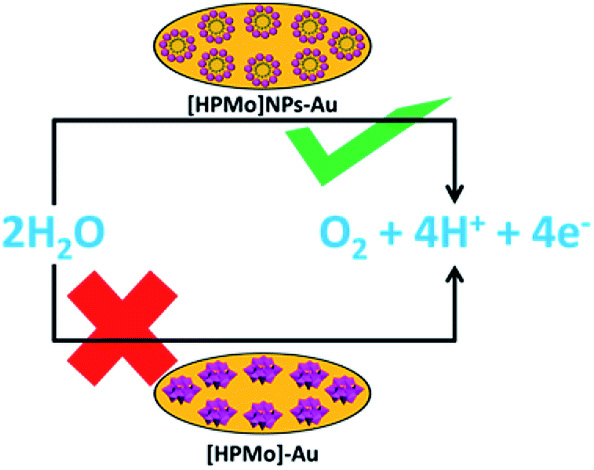 | ||
| Scheme 1 Activation of [HPMo] for electrocatalytic water oxidation by micelle directed nanoparticles formation. | ||
In summary, sodium dodecyl sulfate surfactant directed [H3PMo12O40] nanoparticles was synthesized and used to modify gold electrode surface for the electrocatalytic oxidation of water at neutral pH. The modified electrode showed excellent electrocatalytic activity towards water oxidation at an overpotential of 350 mV with a current density of 1.7 mA cm−2 and are quite better than many polyoxometalates modified electrode systems. The unique electrode system was stable during 30 minutes of electrolysis and also has long term stability (30 days). We believed such micelle directed materials can be applied for future renewable energy technology.
Conflicts of interest
There are no conflicts to declare.References
- S. D. Tilley, Adv. Eng. Mater., 2019, 9, 1802877 Search PubMed.
- S. Singh, S. Jain, P. S. Venkateswaran, A. K. Tiwari, M. R. Nouni, J. K. Pandey and S. Goel, Renewable Sustainable Energy Rev., 2015, 51, 623–633 CrossRef CAS.
- J. D. Blakemore, R. H. Crabtree and G. W. Brudvig, Chem. Rev., 2015, 115, 12974–13005 CrossRef CAS PubMed.
- M. B. Ahicart, J. S. Lopez, J. J. Carbo, J. M. poblet and J. R. G. Mascaros, Nat. Chem., 2018, 10, 24–30 CrossRef PubMed.
- L. Yu, J. Lin, M. Zheng, M. Chen and Y. Ding, Chem. Commun., 2018, 54, 354–357 RSC.
- J. J. Stracke and R. G. Finke, J. Am. Chem. Soc., 2011, 133, 14872–14875 CrossRef CAS PubMed.
- S. Mukhopadhyay, J. Debgupta, C. Singh, A. Kar and S. K. Das, Angew. Chem., Int. Ed., 2018, 57, 1918–1923 CrossRef CAS PubMed.
- M. Vazylyev, D. S. Rozner, A. Haimov, G. Maayan and R. Neumann, Top. Catal., 2005, 34, 93–99 CrossRef CAS.
- J. W. Vickers, H. Lv, J. M. Sumliner, G. Zhu, Z. Luo, D. G. Musaev, Y. V. Geletii and C. L. Hill, J. Am. Chem. Soc., 2013, 135, 14110–14118 CrossRef CAS PubMed.
- F. Song, Y. Ding, B. Ma, C. Wang, Q. Wang, X. Du, S. Fu and J. Song, Energy Environ. Sci., 2013, 6, 1170–1184 RSC.
- F. M. Toma, A. Sartorel, M. Iurlo, M. Carraro, P. Parisse, C. Maccato, S. Rapino, B. R. Gonzalez, H. Amenitsch, T. Da. Ros, L. Casalis, A. Goldoni, M. Marcaccio, G. Scorrano, G. Scoles, F. Paolucci, M. Prato and M. Bonchio, Nat. Chem., 2010, 2, 826–831 CrossRef CAS PubMed.
- S. X. Guo, Y. Liu, C. Y. Lee, A. M. Bond, J. Zhang, Y. V. Geletii and C. L. Hill, Energy Environ. Sci., 2013, 6, 2654–2663 RSC.
- S. M. Lauinger, J. M. Sumliner, Q. Yin, Z. Xu, G. Liang, E. N. Glass, T. Lian and C. L. Hill, Chem. Mater., 2015, 27, 5886–5891 CrossRef CAS.
- L. Y. Geun, Y. S. Ki, K. S. Man and O. S. Geun, Microporous Mesoporous Mater., 2005, 86, 134–144 CrossRef CAS; M. P. Pileni, Catal. Today, 2000, 58, 151–166 CrossRef.
- A. Imhop and J. D. Pine, Nature, 1997, 389, 948–951 CrossRef.
- D. Zhao, J. Feng, Q. Huo, N. Melosh, G. H. Fredrickson, B. F. Chmelka and G. D. Stucky, Science, 1998, 279, 548–552 CrossRef CAS PubMed.
- S. Li, E. Wang, C. Tian, B. Mao, Y. Song, C. Wang and L. Xu, Mater. Res. Bull., 2008, 43, 2880–2886 CrossRef CAS.
- E. Mirzakulova, R. Khatmullin, J. Walpita, T. Corrigan, N. M. V. Barbosa, S. Vyas, S. Oottikkal, S. F. Manzer, C. M. Hadad and K. D. Glusac, Nat. Chem., 2012, 4, 794–801 CrossRef CAS PubMed.
- L. Fotouhi, M. Fatollahzadeh and M. M. Heravi, Int. J. Electrochem. Sci., 2012, 7, 3919–3928 CAS.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods, Fundamentals and Applications, Wiley, New York, 2001 Search PubMed.
- T. Hayashi, N. Bonnet-Mercier, A. Yamaguchi, K. Suetsugu and R. Nakamura, R. Soc. Open Sci., 2019, 6, 190122–190136 CrossRef PubMed.
- S. X. Guo, C. Y. Lee, J. Zhang, A. M. Bond, Y. V. Geletii and C. L. Hill, Inorg. Chem., 2014, 53, 7561–7570 CrossRef CAS PubMed.
- S. G. Ferron, L. Vigara, J. S. Lopez and J. R. G. Mascaros, Inorg. Chem., 2012, 51, 11707–11715 CrossRef PubMed.
- M. Wang, W. Zhong, S. Zhang, R. Liu, J. Xing and G. Zhang, J. Mater. Chem. A, 2018, 6, 9915–9921 RSC.
- M. Quintana, A. M. López, S. Rapino, F. M. Toma, M. Iurlo, M. Carraro, A. Sartorel, C. Maccato, X. Ke, C. Bittencourt, T. Da Ros, G. Van Tendeloo, M. Marcaccio, F. Paolucci, M. Prato and M. Bonchio, ACS Nano, 2013, 7, 811–817 CrossRef CAS PubMed.
- Y. Zhang, X. Wang, F. Luo, Y. Tan, L. Zeng, B. Fang and A. Liu, Appl. Catal., B, 2019, 256, 117852–117861 CrossRef CAS.
- W. Zhong, Z. Lin, S. Feng, D. Wang, S. Shen, Q. Zhang, L. Gu, Z. Wang and B. Fang, Nanoscale, 2019, 11, 4407–4413 RSC.
- D. Chandra, D. Takama, T. Masaki, T. Sato, N. Abe, T. Togashi, M. Kurihara, K. Saito, T. Yui and M. Yagi, ACS Catal., 2016, 6, 3946–3954 CrossRef CAS.
- L. Yang, W. Liu, Z. Zhang, X. Du, L. Dong and Y. Deng, J. Power Sources, 2019, 420, 99–107 CrossRef CAS.
- H. Sheng, H. Zhang, W. Song, H. Ji, W. Ma, C. Chen and J. Zhao, Angew. Chem., Int. Ed., 2015, 54, 5905–5909 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, synthetic process of [HPMo]NPs, electrode modification, figures etc. See DOI: 10.1039/c9ra07450c |
| This journal is © The Royal Society of Chemistry 2019 |