
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAuNP and ssDNA capped mesoporous silica nanoparticles for laser controlled drug release†
Lu Zhou a,
Guojie Liua,
Yang Wanga,
Jianling Liua,
Yajie Zhangab and
Yong Ma*a
a,
Guojie Liua,
Yang Wanga,
Jianling Liua,
Yajie Zhangab and
Yong Ma*a
aDepartment of Chemistry, School of Fundamental Sciences, China Medical University, Shenyang 110122, China. E-mail: yma10@cmu.edu.cn
bDepartment of Gastroenterology, Shengjing Hospital of China Medical University, Shenyang 110004, China
First published on 29th October 2019
Abstract
In order to improve drug efficacy, and reduce drug toxicity and side effects, a novel drug controlled release system was developed based on mesoporous silica nanoparticles (MSNs) with gold nanoparticles (AuNPs) acting as pore caps and short single-stranded DNA (ssDNA) oligomers as the linker. The synthesised composites were characterized by Fourier transform infrared spectroscopy (FTIR), powder X-ray diffraction (XRD), thermogravimetric analysis (TGA), zeta potential measurement, transmission electron microscopy (TEM) and UV-vis spectroscopy. The anticancer drug doxorubicin (Dox) was applied as a model drug to investigate the 808 nm near infrared (NIR) laser-controlled drug release behavior at different pH by fluorescence measurements. The investigation results demonstrate that this nanocarrier could achieve drug controlled release by external near-infrared (NIR) laser stimulation, which is expected to be applied in cancer therapy.
1. Introduction
With the rapid progress of modern medicine, the research and application of drug controlled release systems has attracted increasing attention. Mesoporous silica nanoparticles (MSNs) have been developed as excellent drug nanocarriers and delivery vehicles due to their ordered mesoporous structures, uniform pore size distributions, high specific surface areas, high pore volumes, easy surface functionalization and good biocompatibility.1 These unique characteristics allow MSNs to load a great quantity of drugs in the pores, and can improve efficacy, and reduce toxicity and side effects. MCM-41 type MSNs were first introduced as a drug delivery system (DDS) in 2001.2 With the gradually increasing research, MSNs have been proved to be able to load pharmaceutical drugs such as ibuprofen, doxorubicin, camptothecin and cisplatin.3However, pore-uncapped MSNs will inevitably release drugs prematurely during the process of drug delivery. Contrarily, pore-capped MSNs are excellent carriers that can avoid early release. Diverse materials can serve as pore-caps in controlled DDS, such as polymers, quantum dots (QDs), metal nanoparticles and various biomacromolecules.4 Among these materials, gold nanoparticles (AuNPs) are known as less cytotoxic compared with other inorganic nanomaterials in clinical applications.5 AuNPs have special absorption from ultraviolet (UV) to near-infrared (NIR) range, and the absorption can be precisely controlled by changing particle size and shape. AuNPs also have good photothermal conversion efficiency. These unique optical and thermal properties, which is attributed to the intense collective plasmonic resonance characteristic,6 make it attractive in photothermal therapy (PTT),7 photodynamic therapy (PDT), photothermal imaging,5 radiotherapy8 and other medicine fields. Using AuNPs to cap MSNs, i.e. constructing a smart DDS with two or more nanoparticles, will make the characteristics of different nanoparticles complement each other and greatly improve the superior performance of the release system.
A pore-cap should serve like a switch, which can help avoid premature leakage in the delivery process and have a specific response when reach the target, such as pH,9 redox potential, temperature, biomolecules, light, or a combination of these responses.10 For example, in 2009, Vivero-Escoto11 et al. first designed and synthesized an organically derivatized AuNPs capped MSNs system by electrostatic interaction and proved that the system could be controlled by low-power UV light (365 nm) irradiation. Aznar12 et al. reported a hybrid system of nanoscopic MCM-41-based materials interacted with functionalized AuNPs that can be controlled by both pH and NIR laser (1064 nm) radiation. Yang13 et al. prepared a glutathione (GSH)-responsive MSN–Au DDS and explored the AuNP-induced chemo-photothermal synergistic therapy. Comprehensively consider the capping materials and stimulus strategies, combining MSNs with AuNPs and NIR laser can provide a possibility to realize synergistic therapy to benefit cancer treatment. Unlike ultraviolet light and visible light which can damage protein and DNA, NIR laser (ideal region 650–900 nm) can penetrate deep tissues without destroying biological tissues.14,15 MSN–AuNPs system can kill cancer cells directly by local high temperature caused by photothermal effect of AuNPs, and the drug release can be controlled remotely for non-invasive treatment without producing harmful intermediate metabolites and minimal damage to normal tissues. Deoxyribonucleic acid (DNA) can not only serve as the carrier of genetic information, but also a flexible functional polymer.16,17 Wen18 et al. designed a UV light (365 nm)-responsive hairpin-loop structure DNA nanomachine and investigated the photoinduced controlled release property of the MSN–DNA–AuNPs system in aqueous solution. However, DNA as the linker used in NIR laser-response system has not been reported.
Herein, we employed short single-stranded DNA (ssDNA) oligomers as the linker to combine AuNPs with MSNs and used an 808 nm NIR laser to control the loaded molecules release. The anticancer drug doxorubicin hydrochloride (Dox) was loaded in the mesopores as a drug model to study the functional close-open protocols. NIR laser stimuli allows MSN–AuNPs to integrate physical therapy and chemotherapy due to the excellent photothermal properties of AuNPs. Meanwhile, DNA possesses many functions as a polymer material,19 and the join of DNA will broaden the composite functions and applied fields of this model.
2. Results and discussion
The preparation process of AuNPs serving as caps and connecting with MCM-41 by short ssDNA oligomers is depicted in Scheme 1. The diameter of AuNP purchased is 3 nm (Table S1†) which approaches to the pore diameter of MCM-41 purchased (Table S2†), so AuNPs can be used as the caps to block the pores and prevent premature drug release. Firstly, amine groups modified MCM-41 (MCM-41-NH2) was prepared by a post-grafting method using 3-aminopropyltriethoxy-silane (APTES) as the alkyl coupling reagent. Next, the ssDNA (sequence: 5′-COOH-AAA AAA AAA ACG-C6SH-3′) anchored on the pore outlets (MCM-41–ssDNA) through amidation reaction. Then Dox were loaded into the pore of the nanocarrier (MCM-41–ssDNA/Dox) by stirring for 24 h. After that, the AuNPs self-assembled onto the pore outlets through ssDNA (MCM-41–ssDNA–AuNP) by Au–S bonds formed between the thiol-group and AuNPs20 as well as the binding between AuNP surface and polyA block in the sequence.21 | ||
| Scheme 1 Preparation of MCM-41–ssDNA–AuNP. | ||
The surface functionalizations of MCM-41 were identified by Fourier transform infrared spectroscopy (FTIR), powder X-ray diffraction (XRD), thermogravimetric analysis (TGA) and zeta potentials. The FTIR spectrum of MCM-41-NH2 is shown in Fig. 1(a). The spectrum of original MCM-41 is also included for comparison. The typical Si–O–Si absorption bands around 1079, 803 and 465 cm−1 associated with the formation of silica framework are present in both samples. The weak band around 962 cm−1 associated with Si-groups is present in the pure MCM-41,22 but disappears after modified with APTES. It suggests that the –NH2 have been anchored on the surface of MCM-41 through the interaction with the surface silanol groups (Scheme S1†). Apart from the disappearance of the IR signals of the silanol group, some other new vibration signals appear as –NH2 groups grafted onto MCM-41. C–N stretching vibration is normally observed at around 1000–1200 cm−1. The band at 1488 cm−1 can be attributed to the deformation of –NH2.23 The band at 697 cm−1 can be assigned to N–H wagging vibration. All new peaks illustrate that the –NH2 groups have been successfully incorporated on the surface of MCM-41 via covalent bonds.
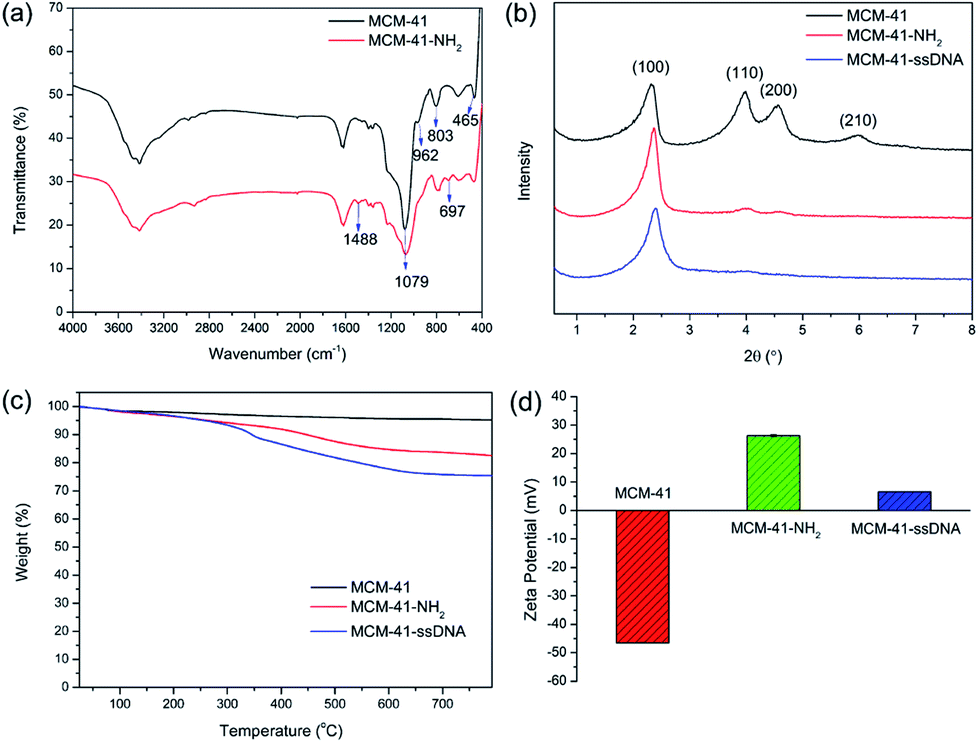 | ||
| Fig. 1 (a) The FTIR spectra of MCM-41 and MCM-41-NH2; (b) XRD of MCM-41, MCM-41-NH2 and MCM-41–ssDNA; (c) TGA curves of MCM-41, MCM-41-NH2 and MCM-41–ssDNA; (d) zeta potentials of MCM-41, MCM-41-NH2 and MCM-41–ssDNA. The data were measured in pure water at 25 °C. | ||
XRD patterns of MCM-41, MCM-41-NH2 and MCM-41–ssDNA are shown in Fig. 1(b). The XRD of MCM-41 shows four small-angle reflections at 2θ = 2.3, 4.0, 4.5 and 6.0°, which are typical features of a hexagonal unit cell, corresponding to (100), (110), (200) and (210) planes. This illustrates the raw material MCM-41 has a highly ordered hexagonal structure. After surface modification with both amine groups and ssDNA, the order degree and regularity will inevitably decrease. Thus the peak intensities for (110), (200) and (210) planes reduce greatly or disappeared comparing with the original MCM-41. The XRD diffraction patterns provide a further evidence for the successful modification of the amine groups and ssDNA on MCM-41. Nevertheless, the value and intensity of the (100) peak in this pattern strongly indicate that the further functionalizations with –NH2 and ssDNA did not damage the MCM-41 framework.12 Furthermore, for MCM-41–ssDNA, an overall fraction of ssDNA on the surface of silica nanoparticles is estimated to be 8.4% by TGA as shown in Fig. 1(c). The corresponding zeta potentials of MCM-41, MCM-41-NH2 and MCM-41–ssDNA are (−46.5 ± 0.17), (26.3 ± 0.41) and (6.57 ± 0.06) mV as shown in Fig. 1(d) and S1.† Due to –NH2 has a positive charge, negatively charged MCM-41 turn to positively charged after modified with amine groups. And the potential becomes much low after further modified with ssDNA owing to a large decrease of –NH2.
After Dox were loaded into MCM-41–ssDNA particles, the pores capped by AuNPs was confirmed by UV-visible spectroscopy and transmission electron microscopy (TEM). The UV-vis spectra of Dox, AuNPs, MCM-41–ssDNA–AuNP aqueous solution and the reaction supernatant for preparing MCM-41–ssDNA–AuNP were shown in Fig. 2. Typically, 3 nm AuNPs have distinctively absorption peak at 518 nm. After AuNPs capped MCM-41–ssDNA/Dox, the absorption spectrum of MCM-41–ssDNA–AuNP tells there is plasmonic coupling of AuNPs, indicating the successful binding of AuNPs. Meanwhile, there was no absorption signal of Dox at 480 nm in the supernatant, proving there were few leakages of Dox during the capping process.
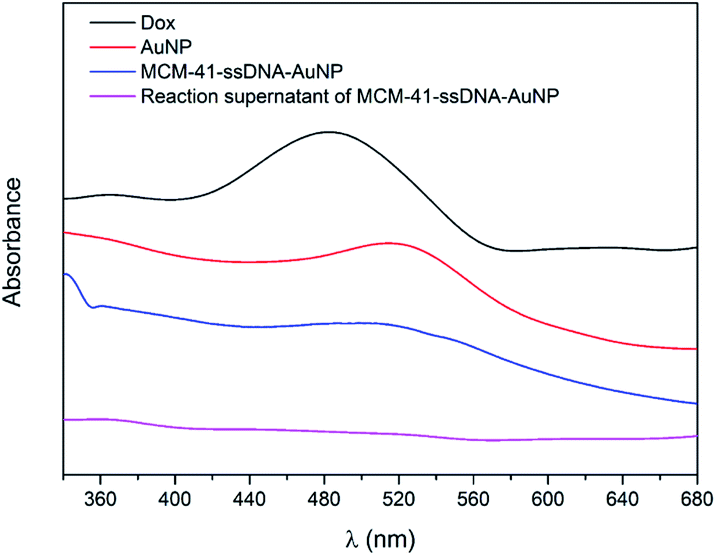 | ||
| Fig. 2 UV-vis spectra of Dox, AuNPs, MCM-41–ssDNA–AuNP aqueous solution and the reaction supernatant of MCM-41–ssDNA–AuNP. | ||
The morphology of MCM-41–ssDNA–AuNP was also observed from the TEM. An array of well ordered mesopores of the MCM-41 matrix can be seen from Fig. 3(a). For the MCM-41–ssDNA–AuNP, as shown in Fig. 3(b) and S2(a),† the black spots on the outside edges of the mesopores further confirm AuNPs have been successfully immobilized on the pores.
 | ||
| Fig. 3 TEM (a) of MCM-41; (b) of MCM-41–ssDNA–AuNP; (c) after MCM-41–ssDNA–AuNP was irradiated by 808 nm laser. | ||
Dox was chosen as the drug molecule model, which could show the release process by fluorescence measurements. To prevent possible effects of ions on Dox release, MCM-41–ssDNA–AuNP were dispersed in ultrapure water to evaluate the laser power effect at first. As shown in Fig. 4(a), when there was no laser irradiation but only stir, Dox fluorescence signals were negligible, because the AuNPs blocked the pores and prevented drug release. In contrast, after the MCM-41–ssDNA–AuNP suspension was irradiated at 808 nm under 2.0, 4.0, 6.0, 8.0 W cm−2 power in 30 min intervals, drugs released from the carrier because AuNPs separated from the pores after laser irradiation as conformed by TEM shown in Fig. 3(c) and S2(b),† and the pores were open to make Dox diffuse from the MSNs. This is attributed to the photothermal effect of AuNPs triggered by the laser which heats up the silica surface and presumably changes interaction of linker DNA with surface, allowing the AuNPs to dissociate into the surrounding medium. Moreover, the amount of drug release increased as power increased, which indicated the power dependency of the release effect. Because it could only remove part of AuNPs at low power while remove more even full at higher power. The cumulative release of Dox reached equilibrium around 5 h later. But if there is no pore-cap as shown in Fig. S3,† Dox released quickly and massively from MCM-41 no matter whether there is laser irradiation, demonstrating it cannot achieve sustained release and controlled release without AuNPs. While MCM-41–ssDNA–AuNP system can control the release of loaded molecules as needed by the photothermal effect of AuNPs. It can trigger the release of all or part of the loaded molecules precisely by simply changing the intensity of laser. In addition, during the irradiation process, the temperature of the samples increased correspondingly. As shown in Fig. 4(b), when 1 mL suspension containing 2 mg MCM-41–ssDNA–AuNP was irradiated under 2.0, 4.0, 6.0, 8.0 W cm−2, its temperature could reach ca. 28.0, 32.2, 37.3 and 41.1 °C from room temperature within 10 min. This local high temperature is beneficial to directly kill the tumor cells.13
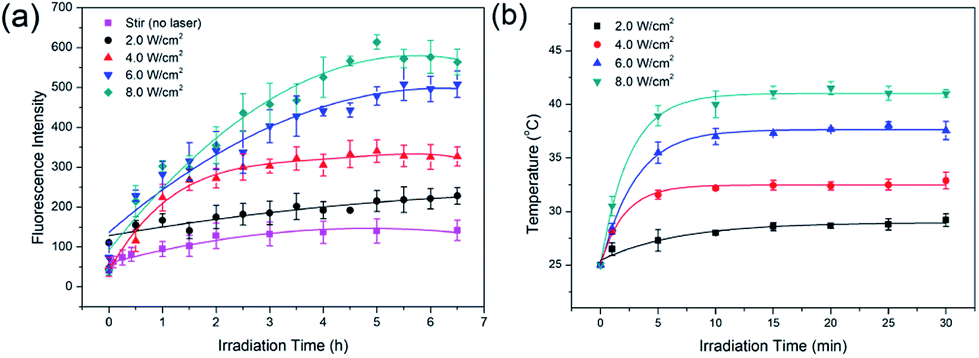 | ||
| Fig. 4 (a) Fluorescence intensity curves of Dox release under different laser power irradiations; (b) the temperature variations under different laser power irradiations generated by 1 mL suspension which contained 2 mg MCM-41–ssDNA–AuNP. | ||
Based on the results of laser power effect, 4.0 W cm−2 and 8.0 W cm−2 were chosen to study the release of Dox under different pH conditions. The MCM-41–ssDNA–AuNP were dispersed in three release media: pH 7.4 PBS (blood circulation and normal tissues), pH 6.8 PBS (tumor extracellular) and pH 5.0 PBS (endosomes and lysosomes). As shown in Fig. 5(a), the release amount of Dox is few at any pH in the absence of laser. From Fig. 5(b), Dox release was not significantly different in variant pH media under 4.0 W cm−2. This result shows Dox release is not affected by ambient pH when controlled by low-power irradiation. From Fig. 5(c), there are obvious similarities and differences in Dox release at different pH under 8.0 W cm−2. The release of Dox in pH 7.4 PBS was almost the same as that in ultrapure water; while in acidic environment with pH 6.8 and 5.0, the release amount increased and the release rate was relatively faster in pH 5.0 PBS which could be attributed to the increasingly acidic conditions weakened the electrostatic interaction between Dox and the negatively charged MCM-41.13,24,25 The Dox release curves in the same pH media under different power laser irradiations are shown in Fig. S2.† It shows the amount of released Dox at 8.0 W cm−2 is always more than that of 4.0 W cm−2 in corresponding circumstance, indicating that no matter how the circumstances change, the amount of released Dox always depends on the power of the NIR laser.
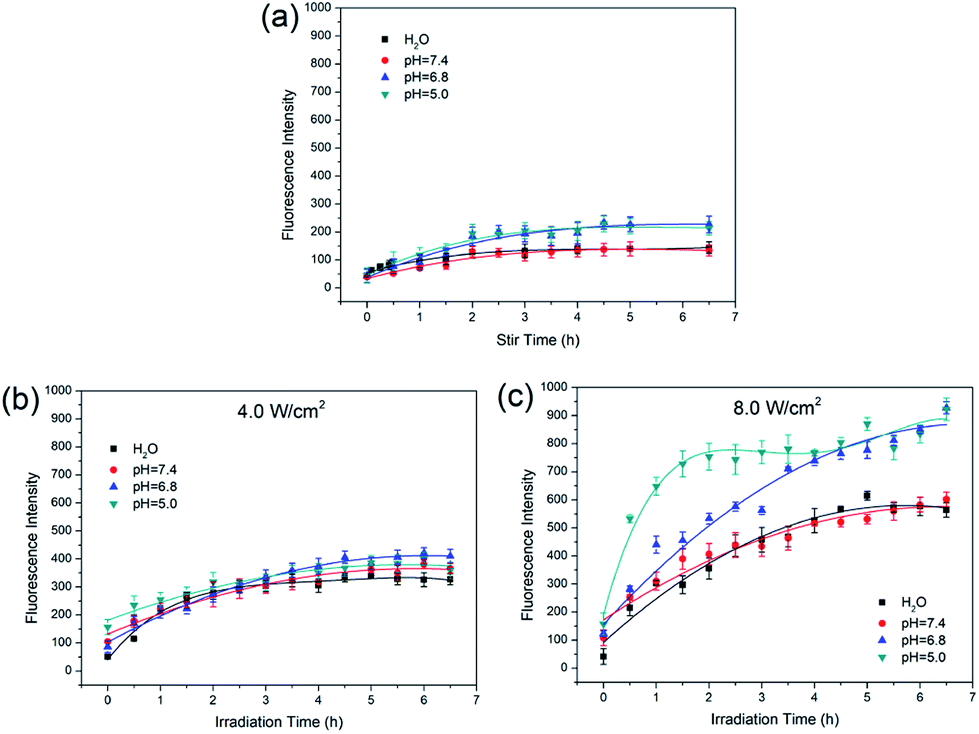 | ||
| Fig. 5 Fluorescence intensity curves of (a) Dox release at different pH when only stir without laser irradiation; (b) 4.0 W cm−2 NIR laser induced Dox release at different pH; (c) 8.0 W cm−2 NIR laser induced Dox release at different pH. | ||
3. Experimental
3.1. Materials
Commercial gold nanoparticles (AuNPs) with average diameter of 3 nm were purchased from Nanopartz Inc. Commercial mesoporous silica particles MCM-41 with pore diameters of 3–5 nm were purchased from Nanjing XFNANO Materials Tech Co., Ltd. (Nanjing, China). Modified short ssDNA oligomers were purchased from Sangon Biotech Co., Ltd. (Shanghai, China). 3-Aminopropyltriethoxy-silane (APTES, 99%), 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC, 99%) and N-hydroxysuccinimide (NHS, 98%) were purchased from Sigma-Aldrich. Doxorubicin hydrochloride (Dox, 98%) were purchased from Aladdin (Shanghai, China). Phosphate buffer saline (PBS, pH 7.4) were purchased from Huibai Biotech Co., Ltd. (Shenyang, China). All prepared buffers were prepared with ultrapure water (>18 MΩ cm) produced from a ELGA PURELAB Classic water system (ELGA, UK).3.2. Measurements
Fourier transform infrared spectrophotometric (FTIR) spectra were measured on a Spectrum One spectrometer (PerkinElmer, USA) using the KBr pellet technique. Power X-ray diffraction (XRD) measurements were performed on a X'Pert PRO diffractometer (PANalytical, Holland). Thermogravimetric analysis (TGA) was performed on a TGA/SDTA85le instrument (METTLER TOLEDO, Switzerland) with a heating rate of 10 °C min−1 under a nitrogen flow. Zeta potentials were determined by a Nano-zs90 laser particle size and zeta potential analyzer (Malvern, UK). UV-vis spectra were measured on a UV-160A spectrophotometer (Shimadzu, Japan). The morphology and the mesoporous structure of the nanoparticles were confirmed on JEM-2100 TEM (JEOL, Japan) and G20 TEM (FEI, USA). The fluorescence spectra were carried out with a F2700 fluorescence spectrophotometer (Hitachi, Japan). The Laser used an 808 nm NIR laser equipment (Changchun New Industries electronics Tech Co., Ltd, China). The temperature of suspension was monitored by a thermocouple.3.3. Methods
4. Conclusions
A novel drug nanocarrier based on AuNP-capped mesoporous silica was developed via a connection of ssDNA. FTIR, XRD, TGA and zeta potentials results confirm MCM-41 successfully modified with ssDNA. The TEM photographs and UV-visible spectra further suggest the formation of MCM-41–ssDNA–AuNP. Dox was applied as a model drug to explore the NIR laser-controlled drug release behaviors in different pH conditions at different power irradiations. The fluorescence emission spectra indicate that there are few leakages after MSNs capped with AuNPs. When irradiated with 808 nm NIR laser, AuNPs removed to control Dox release from the particles. The in vitro study confirms that MCM-41–ssDNA–AuNP can realize drug controlled release by NIR laser stimulation. One can control the release as needed and precisely trigger the release by changing the power intensity of laser. Moreover, MCM-41–ssDNA–AuNP release Dox faster and more at pH 5.0 than that at pH 7.4 under high power laser irradiation, which has great significance in cancer therapy without affecting normal tissues.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 21372262) and Basic Research Program of Higher Education of Liaoning Province of China (Grant No. LQNK201742).References
- P. Yang, S. Gai and J. Lin, Chem. Soc. Rev., 2012, 41, 3679–3698 RSC.
- M. Vallet-Regi, A. Rámila, R. P. del Real and J. Pérez-Pariente, Chem. Mater., 2001, 13, 308–311 CrossRef CAS.
- F. Tang, L. Li and D. Chen, Adv. Mater., 2012, 24, 1504–1534 CrossRef CAS.
- J. Wen, K. Yang, F. Liu, H. Li, Y. Xu and S. Sun, Chem. Soc. Rev., 2017, 46, 6024–6045 RSC.
- C. J. Murphy, A. M. Gole, J. W. Stone, P. N. Sisco, A. M. Alkilany, E. C. Goldsmith and S. C. Baxter, Acc. Chem. Res., 2008, 41, 1721–1730 CrossRef CAS PubMed.
- P. K. Jain, X. Huang, I. H. El-Sayed and M. A. El-Sayed, Acc. Chem. Res., 2008, 41, 1578–1586 CrossRef CAS.
- J. Chen, C. Ning, Z. Zhou, P. Yu, Y. Zhu, G. Tan and C. Mao, Prog. Mater. Sci., 2019, 99, 1–26 CrossRef CAS.
- S. Her, D. A. Jaffray and C. Allen, Adv. Drug Delivery Rev., 2017, 109, 84–101 CrossRef CAS.
- L. Chen, J. Di, C. Cao, Y. Zhao, Y. Ma, J. Luo, Y. Wen, W. Song, Y. Song and L. Jiang, Chem. Commun., 2011, 47, 2850–2852 RSC.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS.
- J. L. Vivero-Escoto, I. I. Slowing, C.-W. Wu and V. S.-Y. Lin, J. Am. Chem. Soc., 2009, 131, 3462–3463 CrossRef CAS PubMed.
- E. Aznar, M. D. Marcos, R. Martínez-Máñez, F. Sancenón, J. Soto, P. Amorós and C. Guillem, J. Am. Chem. Soc., 2009, 131, 6833–6843 CrossRef CAS.
- Y. Yang, Y. Lin, D. Di, X. Zhang, D. Wang, Q. Zhao and S. Wang, J. Colloid Interface Sci., 2017, 508, 323–331 CrossRef CAS.
- R. Weissleder, Nat. Biotechnol., 2001, 19, 316–317 CrossRef CAS.
- A. Vogel and V. Venugopalan, Chem. Rev., 2003, 103, 577–644 CrossRef CAS PubMed.
- O. Okay, J. Polym. Sci., Part B: Polym. Phys., 2011, 49, 551–556 CrossRef CAS.
- M. Vybornyi, Y. Vyborna and R. Haner, Chem. Soc. Rev., 2019, 48, 4347–4360 RSC.
- Y. Wen, L. Xu, W. Wang, D. Wang, H. Du and X. Zhang, Nanoscale, 2012, 4, 4473–4476 RSC.
- A. Samanta and I. L. Medintz, Nanoscale, 2016, 8, 9037–9095 RSC.
- U. Abraham, Chem. Rev., 1996, 96, 1533–1554 CrossRef.
- H. Pei, F. Li, Y. Wan, M. Wei, H. Liu, Y. Su, N. Chen, Q. Huang and C. Fan, J. Am. Chem. Soc., 2012, 134, 11876–11879 CrossRef CAS.
- H. Wang, X. Gao, Y. Wang, J. Wang, X. Niu and X. Deng, Ceram. Int., 2012, 38, 6931–6935 CrossRef CAS.
- X. Wang, V. Schwartz, J. C. Clark, X. Ma, S. H. Overbury, X. Xu and C. Song, J. Phys. Chem. C, 2009, 113, 7260–7268 CrossRef CAS.
- Q. Zhao, J. Liu, W. Zhu, C. Sun, D. Di, Y. Zhang, P. Wang, Z. Wang and S. Wang, Acta Biomater., 2015, 23, 147–156 CrossRef CAS.
- H. Tang, J. Guo, Y. Sun, B. Chang, Q. Ren and W. Yang, Int. J. Pharm., 2011, 421, 388–396 CrossRef CAS.
- L. Chen, Y. Wen, B. Su, J. Di, Y. Song and L. Jiang, J. Mater. Chem., 2011, 21, 13811–13816 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: More details of experimental are provided in the supporting information. See DOI: 10.1039/c9ra07404j |
| This journal is © The Royal Society of Chemistry 2019 |