
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
FeII-catalysed insertion reaction of α-diazocarbonyls into X–H bonds (X = Si, S, N, and O) in dimethyl carbonate as a suitable solvent alternative†
Nour
Tanbouza
 ,
Hoda
Keipour
and
Thierry
Ollevier
*
,
Hoda
Keipour
and
Thierry
Ollevier
*
Département de Chimie, Université Laval, 1045 Avenue de la Médecine, Québec, QC G1V 0A6, Canada
First published on 2nd October 2019
Abstract
The insertion reaction of a broad range of diazo compounds into Si–H bonds was found to be efficiently catalysed by Fe(OTf)2 in an emerging green solvent i.e. dimethyl carbonate (DMC). The α-silylated products were obtained in good to excellent yields (up to 95%). Kinetic studies showed that the extrusion of N2 to form an iron carbene intermediate is rate-limiting. The iron-catalysed insertion reaction of methyl α-phenyl-α-diazoacetate into polar X–H bonds (S–H, N–H, and O–H) was also established in DMC.
Dimethyl carbonate (DMC), the simplest among the dialkyl carbonate family (DACs), is an emerging and interesting green reagent and solvent alternative in organic synthesis due to its high biodegradability and low toxicity.1,2 It is exploited industrially for its efficiency as a solvent for numerous applications,3 however it still remains underused as a solvent in homogeneous catalysis. Even though major contributions have been achieved to promote the twelve principles of green chemistry,4 searching for efficient green solvent alternatives still remains a challenge for chemists.5 Solvent selection guides are huge influencers for rendering the choice of an appropriate solvent less tedious in terms of life cycle evaluation.6
For this study, we were successful in establishing dimethyl carbonate as an appropriate solvent for an iron-catalysed insertion reaction of acceptor/donor diazo compounds into Si–H bonds and polar X–H bonds (X = S, N, and O). Diazo compounds are the most commonly used carbene precursors. They can be de-diazonized to obtain highly reactive free carbene intermediates or metal carbene species. Then, varieties of chemical transformations can occur, such as X–H (X = C, S, N, O, and Si) insertions,7 cyclopropanation,7b ylide formation,8 and Wolff rearrangement.9,10 Notably, the transition metal-catalysed insertion reaction of diazo compounds into various X–H bonds represents a facile route towards different tailored functionalized compounds which are arduous to obtain through other routes.11
Special attention has been paid to the insertion reaction of diazo compounds into Si–H bonds, because even though silicon is vastly abundant on earth, routes towards organosilicons are not. Organosilicon compounds are increasingly mainstream within a variety of applications.12 They are pertinent in the pharmaceutical sector where the integration of silicon motifs as carbon isosteres has allowed distinctive therapeutic potential in a number of biologically active molecules.13 The transition metal-catalysed insertion reaction into Si–H bonds is a facile and atom-economic method to produce organosilicons.14 This reaction was first described by Kramer and Wright in 1963.15 Since then, pivotal breakthroughs have been made with Ru-,16 Rh-,17 Ir-,18 Ag-,19 Cu-,20 and enzyme-catalysed21 insertion reactions of diazo compounds into the Si–H bond. Iron catalysis being one of our group's major areas of research,22 we developed a highly efficient iron-catalysed insertion reaction of α-diazoesters into Si–H bonds.23 Afterwards, an enantioselective iron-catalysed version was developed by Xie and Lin.24 The use of iron salts in transition metal catalysis is gaining traction due to their low toxicity, abundance, and environmental benignity. Iron catalysts have proved on and on to be efficient and green substitutes of conventional noble metal catalysts.25
Even though iron-catalysed Si–H insertion reactions were successfully developed to render it more sustainable, they were still conducted in dichloromethane (CH2Cl2), a solvent which is commonly used in homogeneous catalysis due to its good solubility of most organics, low polarity, and non-coordinating properties. However, it is notorious for its toxicity, harmfulness to both human health and the environment, and costly disposal.26 Herein, we disclose the use of DMC as an appropriate solvent alternative to CH2Cl2 in a ferrous system for the insertion reaction of α-diazocarbonyl compounds into Si–H bonds. Kinetic studies are then employed to establish the rate determining step of an iron-catalysed insertion of α-diazoesters into Si–H bonds. This study is then further extended to cover insertion reactions of α-diazoesters into different polar X–H bonds (X = O, N, and S).
A series of FeII and FeIII salts were selected for screening in DMC with Et3SiH (Table 1).27 The following screening shows that the nature of the catalyst greatly influences the reactivity of the diazo compound. Fe(OTf)2 was capable of affording the α-silylated product 2a in a 95% yield within 6 h (entry 1).28 FeBr2 and FeCl2 led to poor conversions after 72 h (entries 2 and 3). The diazo compound 1a was unreactive towards Fe(OAc)2 and was recovered quantitatively (entry 4). Fe(BF4)2·6H2O resulted in a low 6% yield which was due to insertion with H2O and dimerization (entry 5). Interestingly, both Fe(OTf)3 and Fe(acac)3 led to the formation of the silyl enolate, which was attributed to the strong Lewis acidity of FeIII salts (entries 6 and 7).
|
|
|||
|---|---|---|---|
| Entry | MXn | Time (h) | Yield (%) |
| a 1a (0.25 mmol), catalyst (5 mol%), Et3SiH (1.25 mmol), DMC (2 ml). b Incomplete conversion. c No conversion and recovery of α-diazoester 1a. d Insertion with H2O and dimerization as major pathways. e A mixture of O-silyl enolate and dimerization product was obtained. | |||
| 1 | Fe(OTf)2 | 6 | 95 |
| 2 | FeBr2 | 72 | 4b |
| 3 | FeCl2 | 72 | 45b |
| 4 | Fe(OAc)2 | 48 | 0c |
| 5 | Fe(BF4)2·6H2O | 18 | 6d |
| 6 | Fe(OTf)3 | 48 | —e |
| 7 | Fe(acac)3 | 48 | —e |

In order to conduct an appropriate screening of solvents, and to figure out the correlation between the solvent's solvatochromic properties and how it affects the course of this reaction, a Kamlet–Taft plot of aprotic solvents was used to this advantage (Fig. 1).29 The plot includes different common organic solvents previously screened for this reaction in addition to a screening of different classical and less common green solvent alternatives (see ESI† for individual yields and reaction times). Solvents that produced very good yields of the insertion product (>90%) are coloured green ( ), whilst those leading to moderate yields (between 90% and 50%) and poor yields (<50%) are identified in yellow (
), whilst those leading to moderate yields (between 90% and 50%) and poor yields (<50%) are identified in yellow ( ) and red (
) and red ( ), respectively. As it can be seen from the distribution of solvents on the Kamlet–Taft plot, solvents with high basicity (β > 0.4) and low polarizability (π* < 0.5) resulted in moderate and low yields of the desired insertion product. It can be deduced that the reaction prefers a reaction media that is low in polarity and basicity which is delivered by all of CH2Cl2, dichloroethane (DCE, (CH2Cl)2), benzene (PhH), chlorobenzene (PhCl), bromobenzene (PhBr), and DMC. An exception is observed with toluene (PhMe) which is reactive towards metal carbenes. Another is with MeCN; this can be attributed to the low solubility of the silane. Following the solvent selection guides, DMC was chosen for this reaction. Other organic carbonates, i.e. diethyl carbonate (DEC) and propylene carbonate (PC), were not as efficient as dimethyl carbonate. Polar protic solvents, H2O and EtOH, were also tested in this reaction where no sign of the insertion product was observed, and only O–H insertion products were obtained in high yields (see ESI†).
), respectively. As it can be seen from the distribution of solvents on the Kamlet–Taft plot, solvents with high basicity (β > 0.4) and low polarizability (π* < 0.5) resulted in moderate and low yields of the desired insertion product. It can be deduced that the reaction prefers a reaction media that is low in polarity and basicity which is delivered by all of CH2Cl2, dichloroethane (DCE, (CH2Cl)2), benzene (PhH), chlorobenzene (PhCl), bromobenzene (PhBr), and DMC. An exception is observed with toluene (PhMe) which is reactive towards metal carbenes. Another is with MeCN; this can be attributed to the low solubility of the silane. Following the solvent selection guides, DMC was chosen for this reaction. Other organic carbonates, i.e. diethyl carbonate (DEC) and propylene carbonate (PC), were not as efficient as dimethyl carbonate. Polar protic solvents, H2O and EtOH, were also tested in this reaction where no sign of the insertion product was observed, and only O–H insertion products were obtained in high yields (see ESI†).
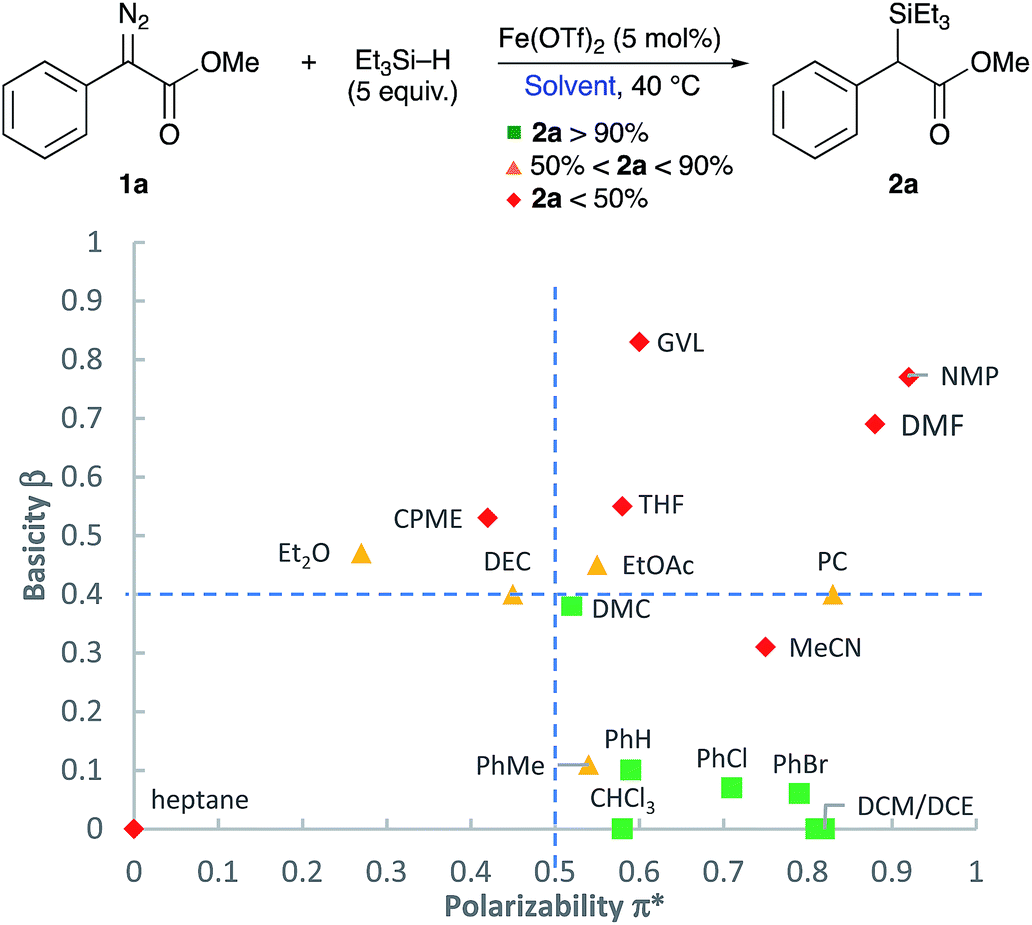 | ||
| Fig. 1 Kamlet–Taft plot of screened aprotic solvents. Reaction conditions: 1a (0.25 mmol), Fe(OTf)2 (5 mol%), Et3SiH (1.25 mmol), solvent (2 ml). | ||
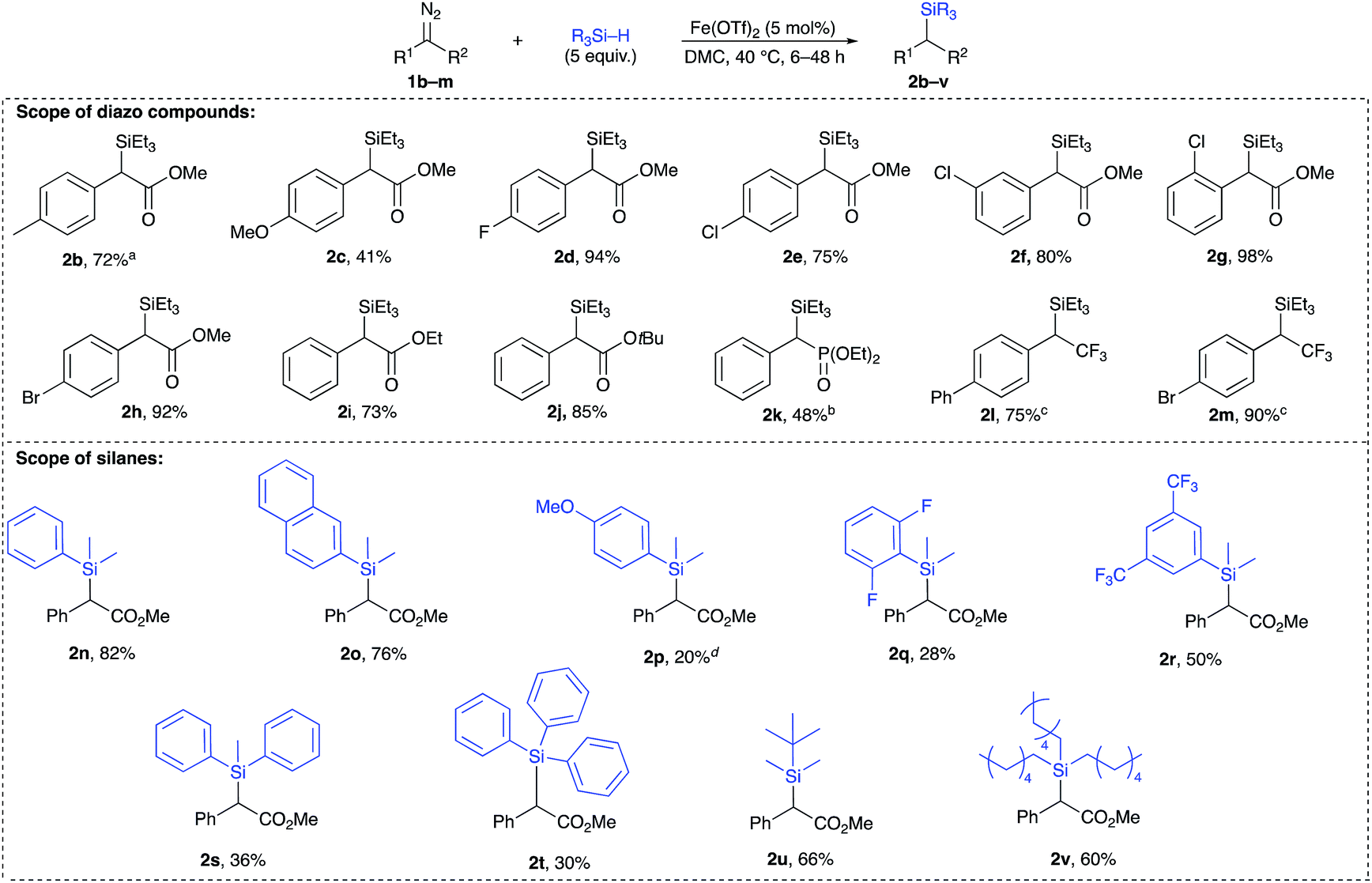
A broad range of acceptor/donor α-diazo compounds and silanes was examined to demonstrate the tolerance of this catalytic system (Table 2). Electron-donating groups in the p-position of the aryl group were tolerated as seen for cases 2b and 2c. Also, α-diazoesters with electron-withdrawing groups in the o-, p-, and m-positions of the aromatic ring (2d–h) allowed the reaction to proceed with good to excellent yields. Different ester functionalities were found to be well-tolerated (2i–k). Interestingly, the α-diazophosphonate 1l underwent complete conversion after 72 h to yield 48% of the corresponding insertion product 2l. The diazotrifluoromethyl substrates 1m and 1n efficiently underwent the insertion reaction to give rise to the α-silylated products in 75% and 90% yields, respectively. The Si–H insertion reaction of methyl α-phenyl-α-diazoacetate 1a was then conducted with different silanes. An insertion with PhMe2SiH led to an 82% yield of 2o with completion in 12 h. The reaction of the α-diazoester 1a with (2-naphthyl)Me2SiH achieved completion in 6 h with the corresponding product 2p in 76%. When tuning the aryl group of an ArMe2SiH series, it was observed that a p-OMe substituent resulted in an extended reaction time of 24 h with the corresponding silylated product in a 20% yield. An (o,o′-F)C6H3 substituted silane produced the insertion product 2r in 28%. A (m,m′-CF3)C6H3 substituted silane resulted in a moderate yield (50% of 2s) with completion in 6 h. Bulkier silanes, such as Ph2MeSiH and Ph3SiH, were tougher to insert where yields dramatically dropped to 36% and 30%, respectively, in addition to extended reaction times (up to 48 h). For alkyl silanes, the reaction of methyl α-phenyl-α-diazoacetate with (t-Bu)Me2SiH and (Hex)3SiH afforded the insertion products 2v and 2w in moderate yields (66% and 60%, respectively) previous attempts to establish the rate-determining step of an iron catalysed Si–H insertion reaction have been inconclusive.23,24 Unlike Si–H insertions with other metal catalysts, no H/D kinetic isotope effect was observed after conducting a competition experiment with a D-silane. The competition experiment between Et3Si–H and Et3Si–D was run again (Fig. 2, eqn (1)), but this time in DMC, using equimolar amounts of both silanes under the previously established optimum conditions. This experiment did not show any sign that the activation of the Si–H bond is rate-determining, where a value of 1.04 was obtained. Thus, we hypothesized that the extrusion of nitrogen to form the iron carbene is most likely the rate-determining step and that the activation of Si–H bond occurs quickly after the formation of the metal carbene.
 | ||
| Fig. 2 Kinetic studies for the determination of the rate-limiting step. | ||
So, the kinetics of this reaction were placed under scrutiny in order to validate the hypothesis of a rate-determining step governed by the formation of the iron carbene species. In a study by Wu, a nitrogen kinetic isotope effect showed that the formation of a Rh carbene is rate-limiting for a Si–H insertion (Fig. 2, eqn (2)).31 Such a study was done by using isotope ratio mass spectrometry (IRMS) for the determination of a nitrogen kinetic isotope effect at natural abundance. The ratio of 14N/15N resulting from the natural 15N-enrichment of unreacted α-diazoester 1a during the progression of the insertion reaction was measured by IRMS. This ratio can be used to calculate the kinetic isotope effect from enrichment in the heavy isotope in a substrate.32 Given the fact that the extrusion of N2 is irreversible and that it is not involved in any subsequent step, the measured nitrogen ratios are only relevant to the carbene formation step.
Here, a large and normal heavy KIE value of 1.022 ± 0.007 was obtained, which supports the hypothesis that the formation of the iron carbene intermediate is rate-determining.33 Also, the insertion reaction was run under pseudo-first order conditions under the optimum conditions with different concentrations of the α-diazoester 1a (Fig. 2, eqn (3)). The formation of 2a was monitored by GC analysis and initial rates were found to be first-order with regard to the concentration of methyl α-phenyl-α-diazoacetate 1a. A linear variation was observed between the initial rate and the concentration of 1a, with [1a] = 0.125, 0.25, and 0.5 M, resulted in initial rates of 5.55, 10.48, and 15.16 mM h−1, respectively. Such results are in accordance with those obtained from a study of an iron-catalysed insertion reaction of diazo compounds into C–H bonds.34
With these results in hand, a mechanism can be therefore drawn where the coordination of the diazo species to the iron centre enriches it thus allowing π-back bonding to extrude nitrogen gas (I, Fig. 3). The formation of the Fe carbene II is rate-determining which subsequently reacts with the activated silane in a one-step manner to yield the insertion product 2a and regenerate the iron catalyst. An alternative mechanism involving the coordination of the terminal nitrogen to the metal centre has thus been refuted.35
 | ||
| Fig. 3 Plausible mechanism for the iron-catalysed insertion reaction of methyl α-phenyl-α-diazoacetate into Si–H bonds. | ||
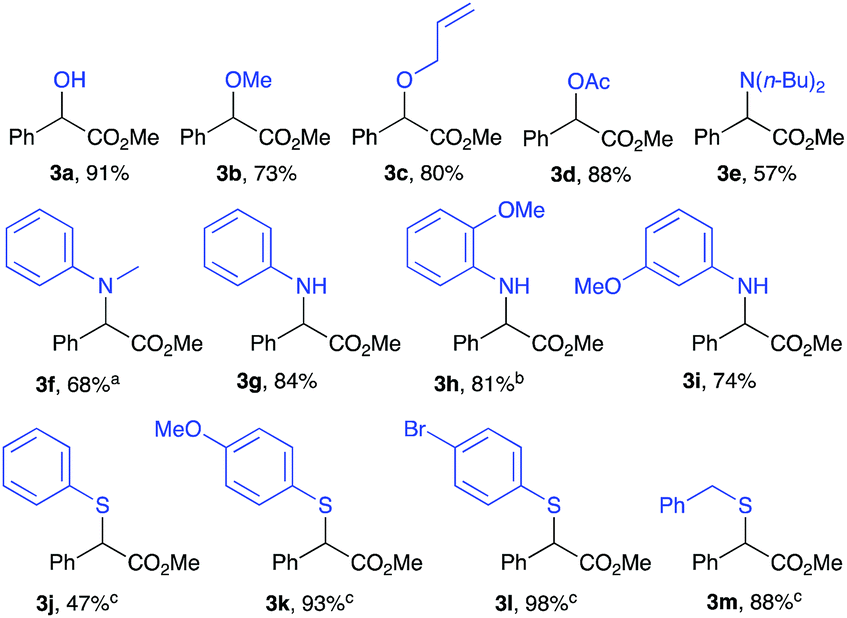
Polar X–H insertion reactions of methyl α-phenyl-α-diazoacetate 1a was then examined (Table 3). Insertions into O–H bonds were conducted in DMC where alcohol 3a and ethers 3b and 3c were obtained in very good yields with completion in 6 h.36 HOAc has been tested for O–H insertion and proceeds to give the corresponding ester 3d in an 88% yield. N–H insertions, however, were strongly dependent on the nature of the amine used. Secondary amines 3e and 3f were afforded in moderate yields of 57% and 68%, respectively, with prolonged reaction times (72 h) and even required increased temperatures (80 °C) to achieve completion. Improved yields were obtained with primary aromatic amines, where an insertion with aniline led to the corresponding product 3g in 84% yield but completion could only be reached after 72 h. Also, when using o-anisidine and m-anisidine, corresponding amines were obtained in 81% and 74%, respectively (Table 3, 3h and 3i). It is important to mention that in all of the three cases using primary amines, the reaction was selective toward a mono-insertion with no sign of a double insertion product. S–H insertions were mediated by an excess of the aryl thiol substrate and using 4 Å MS as an additive along with heating to reflux of DMC. The insertion reaction with thiophenol proceeded in a modest yield of 47% (Table 3, 3j). A higher yield was obtained when employing a p-OMe or a p-Br substituted thiophenol affording the corresponding products 3k and 3l in 93% and 98%, respectively. The reaction of 2a with benzyl thiol resulted in 3m in an 88% yield.

In conclusion, we have successfully realized an iron-catalysed insertion reaction of α-diazo compounds into X–H bonds in DMC as a suitable solvent alternative. A wide range of α-silylesters were obtained in good to excellent yields. The mechanism of the insertion reaction into Si–H bonds was studied while showing that the formation of the iron carbene intermediate is rate-limiting. This work demonstrates the efficiency of iron carbenes used in insertion reactions and also shows that chlorinated solvents can be advantageously replaced by greener alternatives in diazo chemistry. Further developments in the use of iron carbenes will be reported in due course.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery Grant RGPIN-2017-04272, the FRQNT Centre in Green Chemistry and Catalysis (CGCC) Strategic Cluster FRQNT-2020-RS4-265155-CCVC, and Université Laval. N. T. thanks the Francis & Geneviève Melançon Foundation for a graduate scholarship. H. K. thanks CGCC for a graduate scholarship. We thank Jonathan Gagnon (Laboratoire d'Océanographie, Université Laval) for IRMS measurements.Notes and references
-
(a) F. Arico and P. Tundo, Russ. Chem. Rev., 2010, 79, 479–489 CrossRef CAS
; (b) B. Schaffner, F. Schaffner, S. P. Verevkin and A. Borner, Chem. Rev., 2010, 110, 4554–4581 CrossRef CAS PubMed
-
(a)
U. Romano, F. Rivetti and N. D. Muzio, Italy Pat., 3045767, 1981
-
(a) A. Sasidharanpillai, C. H. Kim, C. H. Lee, M. T. Sebastian and H. T. Kim, ACS Sustainable Chem. Eng., 2018, 6, 6849–6855 CrossRef CAS
-
(a) P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC
- P. G. Jessop, Green Chem., 2011, 13, 1391–1398 RSC
-
(a) A. Jordan and H. F. Sneddon, Green Chem., 2019, 21, 1900–1906 RSC
-
(a) S.-F. Zhu and Q.-L. Zhou, Acc. Chem. Res., 2012, 45, 1365–1377 CrossRef CAS
-
H. M. L. Davies, in Comprehensive Asymmetric Catalysis, Supplement 1, ed. E. N. Jacobson, A. Pfaltz and H. Yamamoto, Springer, Heidelberg, Germany, 2004. pp. 83–94 Search PubMed
- W. Kirmse, Eur. J. Org. Chem., 2002, 2193–2256 CrossRef CAS
-
(a) M. P. Doyle, Chem. Rev., 1986, 86, 919–940 CrossRef CAS
-
(a) I. Fleming, A. Barbero and D. Walter, Chem. Rev., 1997, 97, 2063–2192 CrossRef CAS PubMed
-
(a) H. Yang, A. M. Swartz, H. J. Park, P. Srivastava, K. Ellis-Guardiola, D. M. Upp, G. Lee, K. Belsare, Y. Gu, C. Zhang, R. E. Moellering and J. C. Lewis, Nat. Chem., 2018, 10, 318–324 CrossRef CAS
-
(a) A. K. Franz and S. O. Wilson, J. Med. Chem., 2013, 56, 388–405 CrossRef CAS
- H. Keipour, V. Carreras and T. Ollevier, Org. Biomol. Chem., 2017, 15, 5441–5456 RSC
- K. A. W. Kramer and A. N. Wright, J. Chem. Soc., 1963, 3604–3608 RSC
- Y. Nakagawa, S. Chanthamath, I. Fujisawa, K. Shibatomi and S. Iwasa, Chem. Commun., 2017, 53, 3753–3756 RSC
-
(a) R. T. Buck, M. P. Doyle, M. J. Drysdale, L. Ferris, D. C. Forbes, D. Haigh, C. J. Moody, N. D. Pearson and Q.-L. Zhou, Tetrahedron Lett., 1996, 37, 7631–7634 CrossRef CAS
- Y. Yasutomi, H. Suematsu and T. Katsuki, J. Am. Chem. Soc., 2010, 132, 4510–4511 CrossRef CAS
-
(a) M. J. Iglesias, M. C. Nicasio, A. Caballero and P. J. Perez, Dalton Trans., 2013, 42, 1191–1195 RSC
-
(a) L. A. Dakin, P. C. Ong, J. S. Panek, R. J. Staples and P. Stavropoulos, Organometallics, 2000, 19, 2896–2908 CrossRef CAS
-
(a) S. B. Kan, R. D. Lewis, K. Chen and F. H. Arnold, Science, 2016, 354, 1048–1051 CrossRef CAS
-
(a) M. Li, V. Carreras, A. Jalba and T. Ollevier, Org. Lett., 2018, 20, 995–998 CrossRef CAS
- H. Keipour and T. Ollevier, Org. Lett., 2017, 19, 5736–5739 CrossRef CAS
- H. Gu, Z. Han, H. Xie and X. Lin, Org. Lett., 2018, 20, 6544–6549 CrossRef CAS
-
(a) I. Bauer and H.-J. Knoelker, Chem. Rev., 2015, 115, 3170–3387 CrossRef CAS
-
(a) P. M. Schlosser, A. S. Bale, C. F. Gibbons, A. Wilkins and G. S. Cooper, Environ. Health Perspect., 2015, 123, 114–119 CrossRef CAS
- 5 equiv. of the silane were required to achieve better yields. However, the remaining silane could be recovered after purification.
- In a control experiment involving Ph2MeSiH (1 equiv.) in the presence of the iron catalyst Fe(OTf)2 (5 mol%) in DMC (2 ml) at 40 °C for 24 h, the silane was found intact and completely recovered. This shows that there is no reaction occurring between the silane and DMC.
- P. G. Jessop, D. A. Jessop, D. Fu and L. Phan, Green Chem., 2012, 14, 1245–1259 RSC
- Diazo compounds 1l and 1m decompose at 40 °C.
- F. M. Wong, J. Wang, A. C. Hengge and W. Wu, Org. Lett., 2007, 9, 1663–1665 CrossRef CAS
- R. G. Duggleby and D. B. Northrop, Bioorg. Chem., 1989, 17, 177–193 CrossRef CAS
-
(a) M. Gomez-Gallego and M. A. Sierra, Chem. Rev., 2011, 111, 4857–4963 CrossRef CAS
- H. M. Mbuvi and L. K. Woo, Organometallics, 2008, 27, 637–645 CrossRef CAS
- The mechanism is similar to that obtained with a Rh catalyst. See ref. 31.
- Phenol was tested as a substrate for O–H insertion with diazo compound 1a where the crude showed complex mixtures with the corresponding ether in low yields (19%).
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and copies of 1H NMR, 13C NMR, and 19F NMR spectra. See DOI: 10.1039/c9ra07203a |
| This journal is © The Royal Society of Chemistry 2019 |