DOI:
10.1039/C9RA07139C
(Paper)
RSC Adv., 2019,
9, 33107-33116
Biomimetic heterobimetallic architecture of Ni(II) and Fe(II) for CO2 hydrogenation in aqueous media. A DFT study†
Received
5th September 2019
, Accepted 10th October 2019
First published on 16th October 2019
Abstract
In this work, density functional theory has been employed to design a heterobimetallic catalyst of Ni(II) and Fe(II) for the effective CO2 hydrogenation to HCOOH. Based on computational results, our newly designed catalyst is found to be effective for such conversion reactions with free energy as low as 14.13 kcal mol−1 for the rate determining step. Such a low value of free energy indicates that the NiFe heterobimetallic catalyst can prove to be very efficient for the above said conversion. Moreover, the effects of ligand substitutions at the active metal center and the effects due to various spin states are also explored, and can serve as a great tool for the rational design of NiFe catalyst for CO2 hydrogenation.
1 Introduction
Carbon dioxide, being a detrimental component of the atmosphere in higher concentrations, has reached freakish levels and continues to build up; this has become a cause of concern to the present day scientific community.1,2 The continued dependence on non-renewable fossils fuels for energy, high standards of living, and population explosion are exacerbating this situation even more.3,4 In this perspective, a possible way out of this problem is to convert CO2 into formic acid, which holds great promise in reversible hydrogen storage.5–8 Since, being an abundant, cheaper, and freely available chemical reservoir, recycling of CO2 forms a clean technology with zero carbon footprint for the synthesis of value added chemicals.9 Thus, hydrogenation of CO2 to HCOOH can address both the atmospheric as well as economic crisis concomitantly.7,10–12 Therefore, over the last few decades steady progress has been achieved for hydrogenation of CO2 to formic acid using homogenous metal catalysis. However, most of the effective homogenous catalysts proposed to date for such conversion utilize costly and toxic noble metals.13–25 For example, Nozaki and co-workers reported an iridium based pincer catalyst IrH3(P) (P represents 2,6-bis(diisopropylphosphinomethyl)pyridine) with TON 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500000 and TOF 150000 h−1.26 Subsequently after this, Hazari et al. also reported another Ir-based reported catalyst Ir(H3)(P) (P represents (diisopropylphosphinoethyl)amine) in which hydride from secondary coordination sphere N–H is transferred to CO2 and is very efficient catalyst with TON 348000 and TOF 18780 h−1.20 Pidko and co-workers reported a Ru-based catalyst with TOF equal to 1892000 h−1.27,28 Therefore, in quest of cheaper and nontoxic alternatives, researchers have shifted themselves towards earth abundant metal catalysts.29–35 Although significant number of earth abundant catalysts have been reported but almost all of them either require an external base or vigorous reaction conditions for the reaction to accomplish. Moreover, the catalytic activity of these reported catalysts is quite low and thus the design of efficient and highly active earth abundant metal catalysts remains elusive.
500000 and TOF 150000 h−1.26 Subsequently after this, Hazari et al. also reported another Ir-based reported catalyst Ir(H3)(P) (P represents (diisopropylphosphinoethyl)amine) in which hydride from secondary coordination sphere N–H is transferred to CO2 and is very efficient catalyst with TON 348000 and TOF 18780 h−1.20 Pidko and co-workers reported a Ru-based catalyst with TOF equal to 1892000 h−1.27,28 Therefore, in quest of cheaper and nontoxic alternatives, researchers have shifted themselves towards earth abundant metal catalysts.29–35 Although significant number of earth abundant catalysts have been reported but almost all of them either require an external base or vigorous reaction conditions for the reaction to accomplish. Moreover, the catalytic activity of these reported catalysts is quite low and thus the design of efficient and highly active earth abundant metal catalysts remains elusive.
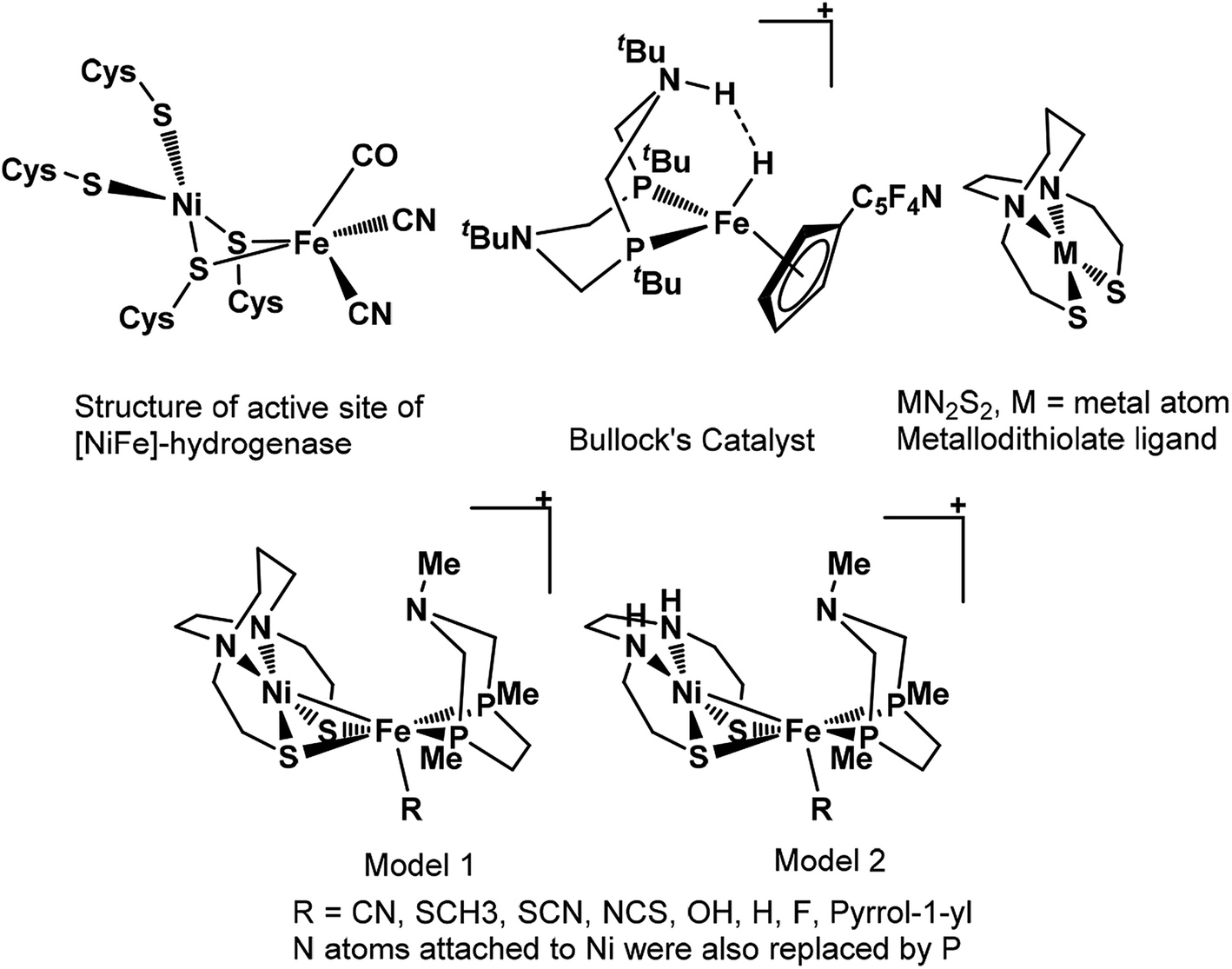
Nature's remarkable design, evolved over a long period for different reactions to accomplish, is an inspiration for rational design of catalysts by biomimicking active site of these architectures. However, such an approach of catalytic design requires in-depth understanding mechanism of action and structure of the active center of the enzyme. The remarkable structural information available for enzymes has led to design of various models for hydrogenation of CO2.36–41 The mechanistic studies of CO2 hydrogenation through DFT studies has revealed that either abstraction of hydride from the metal center or heterolytic cleavage of coordinated H2 over the metal center is usually the rate-determining step of entire catalytic cycle.36–39 The barrier height for H2 heterolytic cleavage can be significantly reduced by introducing a base in catalyst and one of such example is hydrogen oxidation catalyst reported by Bullock and co-workers containing pendant amine as base.42 Likewise, the significant role of base in heterolytic cleavage of coordinated H2 has also been reported in other studies as well.43,44 Moreover, MN2S2 metallodithiolate frameworks have been reported to be the best donors than phosphines and these ligands have been found in so many natural enzymes such as nitrile and thiocyanate hydratases.45,46 Such an architecture adopt square planar geometry and serve as bidentate ligand through two cis-configured S-donors.47
In previous study, we computationally designed biomimetic Mo and W based catalysts for hydrogenation of CO2.48 In this present work, inspired by the active site of [NiFe]-hydrogenase, Bullock's catalyst, and MN2S2 metallodithiolate framework, our aim here is to examine the catalytic activity of heterobimetallic Ni(II) and Fe(II) architecture shown in Scheme 1, for the hydrogenation of CO2 to HCOOH in aqueous media using various computational tools. We will also understand the various mechanistic aspects of CO2 hydrogenation involving various species such as carbonic acid, bicarbonate and carbonate ions formed by the reaction of CO2 with water.49 Moreover, the effects of various ligand substitutions and alteration of spin state of the metal atoms on catalytic activity will also be explored.
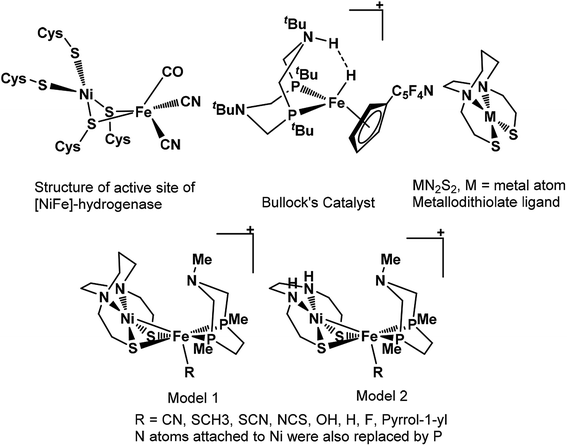 |
| | Scheme 1 The structural representation of active site of [NiFe]-hydrogenase, Bullock's catalyst, MN2S2 metallodithiolate ligand and our modelled structures used in present work. | |
2 Computational details
The geometrical calculations of all the complexes were optimized at PBE-D3 (ref. 50 and 51) level of theory in conjunction with 6-31+G(d,p)52 basis set for all atoms except for nickel and iron for which LANL2DZ53 basis set and ECP were employed. We tested the other functionals like M06-L,54 M06,55 PBE0-D3,56 and B3LYP,57,58 for computing the barrier heights for the addition of H2 over the metal center using PBE-D3 optimized geometries (refer to ESI for more details†). B3LYP and M06 procreated high results while as the best functionals for computing thermochemistry of transition metal complexes59–61 M06-L, PBE0-D3 and PBE-D3 produced almost similar results and thus PBE-D3 functional was chosen for our studies. In addition to LANL2DZ,53 the results were also gauged with Stuttgart-Dresden (SDD) basis-set, ECP10MDF including the ECP's augmented with d-polarization functions from Frenking.62 The computed results are almost same with both the basis sets (for details refer to ESI†). The solvent effects were incorporated employing integral equation formalism polarizable continuum model (IEFPCM)63 with SMD64 atomic radii corrections using water as solvent. All the calculations were performed using ultrafine numerical integration grid as defined in ‘G09’.65 The nature of all the stationary states along the reaction coordinate were confirmed by analyzing harmonic frequency data computed at the above said method. The transition states were confirmed by a single imaginary frequency along the reaction coordinate and the intermediates were without any imaginary frequency. The former were also confirmed by intrinsic reaction coordinate (IRC) calculations performed at the above level of theory. Thermal corrections within the rigid rotor and harmonic approximations were employed at 298.15 K temperature and 1 atm pressure. All the reported energies are Gibbs free energies unless mentioned otherwise. The free energy of Gaq(H+) equal to −264.0 kcal mol−1 has been taken from the literature66 and has been employed in other computational studies as well.67 Number of corrections to Gibbs free energies reported in the text were employed. First, the free energy of complexes were corrected by 1.89 kcal mol−1 considering the 1 atm to 1 M standard state conversion (added in those cases in which number of moles increases or vice versa). Second, correction equal to 2.38 kcal mol−1 for 55.4 to 1 molar standard state conversion and −6.32 kcal mol−1 correction for self-solvation were added for water molecules as well.68
All the above said calculations have been performed using Gaussian 09 software suite.65
3 Results and discussions
3.1 Mechanistic aspects of CO2 hydrogenation
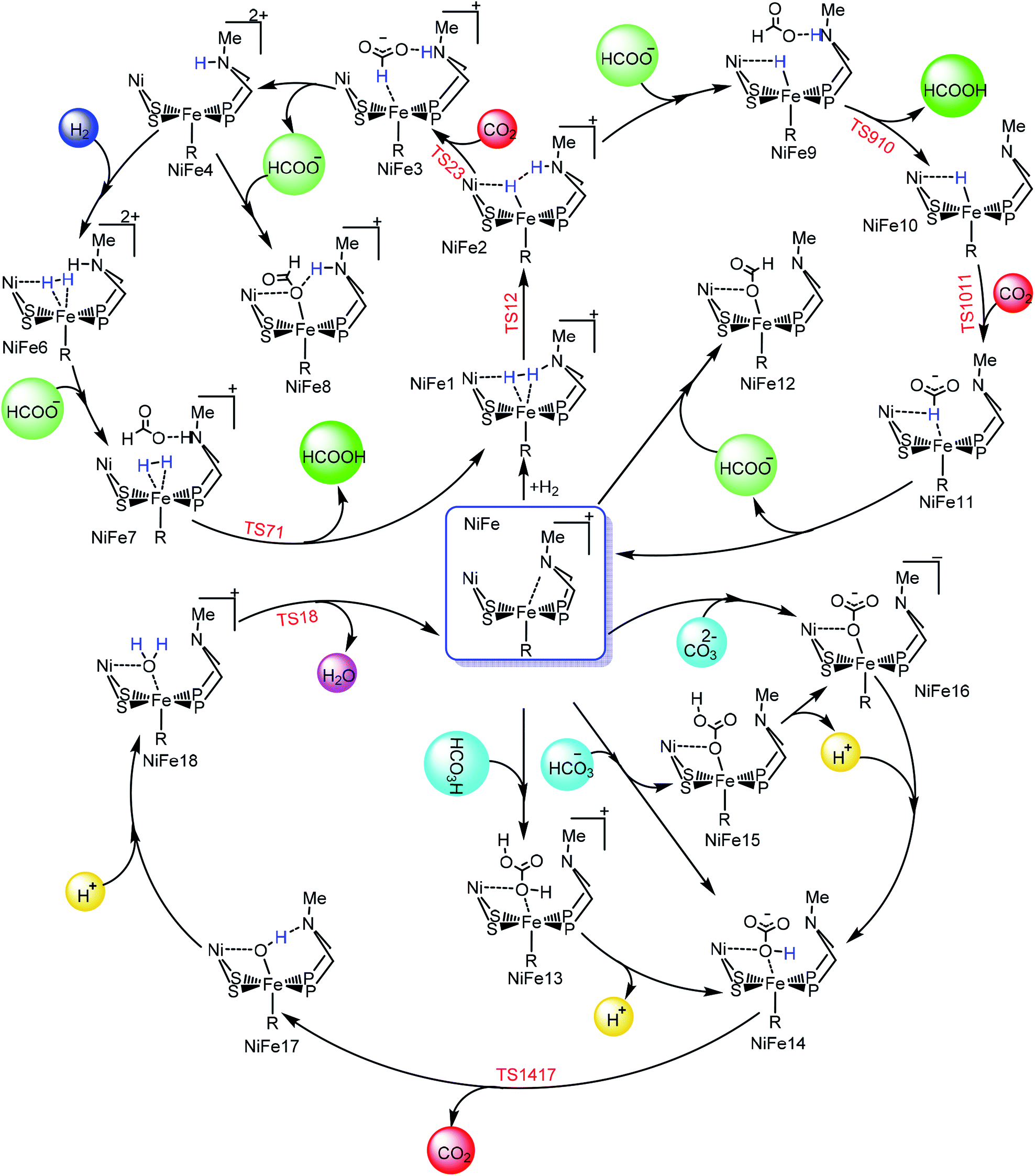
The entire mechanistic cycle of CO2 hydrogenation in aqueous media catalyzed by our NiFe-heterobimetallic catalyst has been divided into two sections, one in which HCOOH is formed and the second in which catalytic decomposition of carbonic acid, bicarbonate and carbonate ions takes place. Both these mechanistic cycles have been briefly described separately in the following two sections.
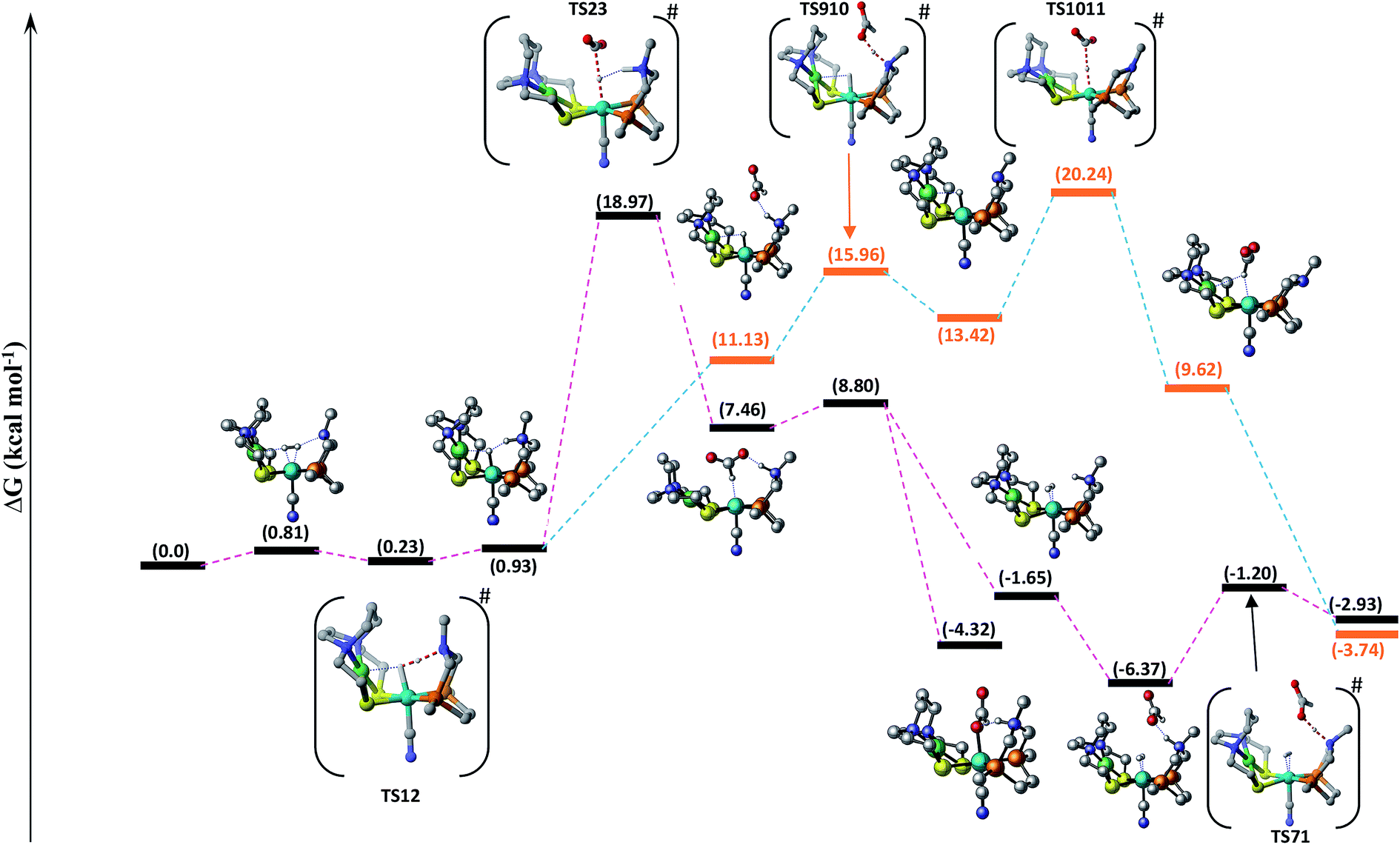
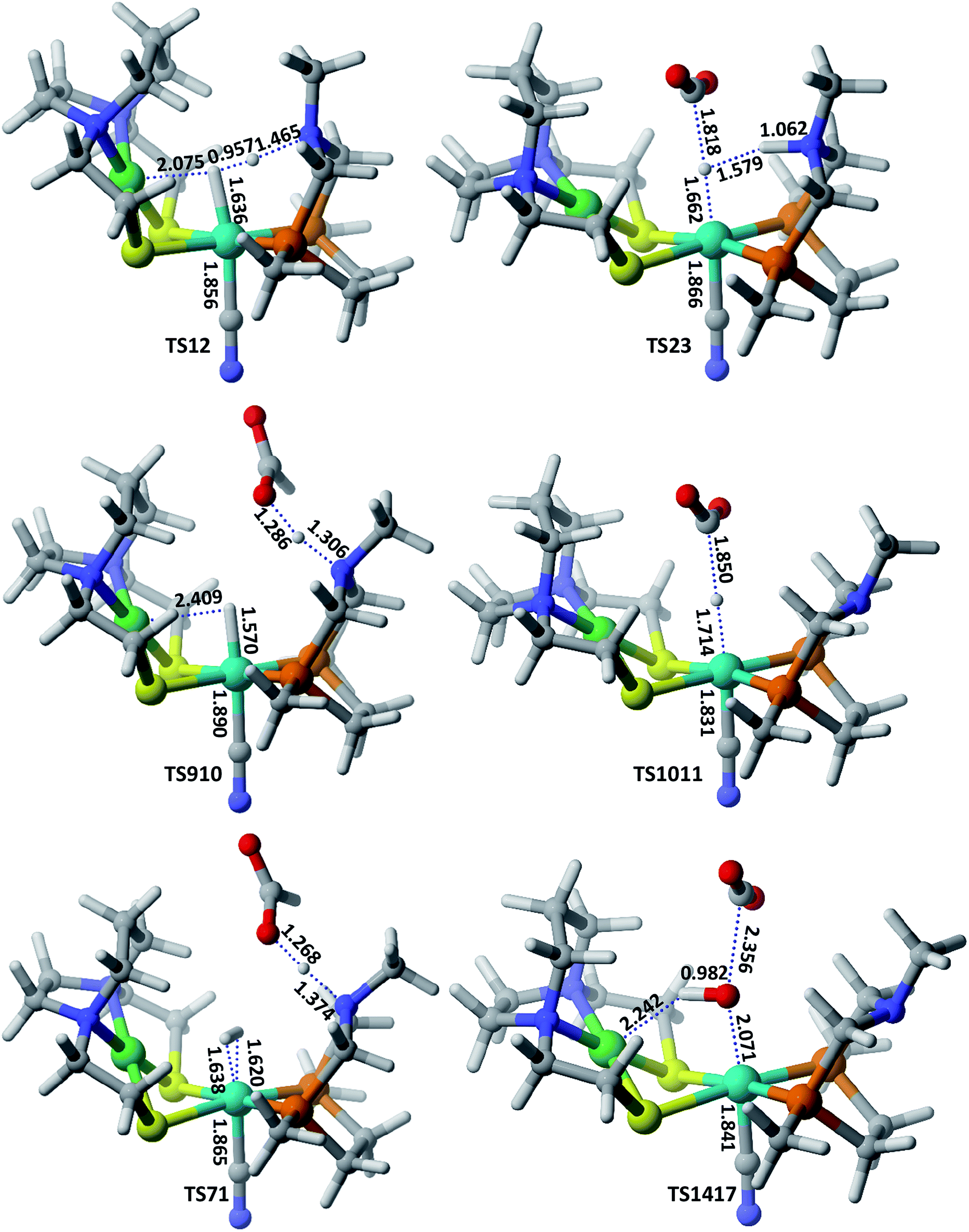
Hydrogenation of CO2 to HCOOH. Scheme 2 shows the entire catalytic cycle of CO2 hydrogenation catalyzed by our NiFe-designed catalyst and Fig. 1 shows complete energy profile of all species along the reaction coordinate for model 1 with R being CN. Fig. 2 portrays the optimized geometries of transition states with their corresponding geometrical parameters while as the complete optimized geometrical coordinates along with their computed energies have been given in ESI.† The interaction between Fe(II) and N of pendant amine is hindered by the alkyl group of metallodithiolate ligand with distance between Fe(II)⋯N equal to 3.151 Å. The vacant site over the Fe(II) is freely available for coordination and thus the catalytic cycle starts with the addition of H2 over the Fe(II)-metal center, which results in the formation of NiFe1 intermediate in an endergonic fashion with barrier height equal to 0.81 kcal mol−1. The intermediate NiFe1 undergoes heterolytic cleavage of coordinated H2 to form NiFe2 via, TS12 a barrierless transition state. The formation of intermediate NiFe2 is just 0.12 kcal mol−1 higher in energy than NiFe1 and its formation is aided by pendant amine present in the vicinity of Fe(II). The bond distance between Fe–H in NiFe2 is 1.595 Å, the N–H of pendant amine is 1.092 Å, and the Fe–Hδ−⋯δ+H–N the dihydrogen bond distance is equal to 1.376 Å. The same respective bond distances in Bullock's catalyst are 1.736 Å, 0.930 Å, and 1.558 Å.69 The Fe–H and Fe–H⋯H–N dihydrogen bond distances are slightly low while as N–H bond length being slightly on higher side when compared with the crystal structures reported by Bullock and co-workers. Our computed values of bond lengths are almost equal to those reported by Pathak and co-workers by carrying DFT studies of the same Bullock's catalyst.44 Our computed Fe–Hδ−⋯δ+H–N bond length falls short than the range 1.7–2.2 Å of metal dihydrogen bonds reported so far.70 The catalytically active metal-hydride intermediate NiFe2 thus formed can undergo two mechanistic cycles. In route first, NiFe2 interacts with CO2 molecule via metal-hydride bond and thus forms NiFe3 with barrier, TS23 equal to 18.97 kcal mol−1 as shown in Fig. 1. The transfer of hydride ion from Fe(II)-center towards CO2 via the formation of TS23 is the rate-determining step of pathway 1. In transition state, TS23 the CO2 directly interacts with metal-hydride bond instead of interacting with both Fe–Hδ− as well as δ+H–N simultaneously. Such a transition state in which both the hydridic as well protic hydrogen simultaneously interact with the incoming CO2 was not located after so many attempts. The reason for such unfavorable approach of CO2 is probably due to small cavity size of the catalyst, which hinders such an interaction. The abstraction of hydride from the Fe-center in CO2 hydrogenation catalysis as reported by some of the earlier computational studies has free energy equal to 14.9,44 23.7,36 and 18.1 kcal mol−1.37 These values of barrier energy are almost similar to our computed value 18.97 kcal mol−1. It is noteworthy to mention here that we have obtained much lower value than this after substituting CN with other ligands, which has been discussed in the next section. Yang in 2011, reported that heterolytic cleavage of coordinated H2 is energetically demanding even with using Ir, Co, and Fe PNP pincer catalysts.71 In 2016, the same researcher reported number of Co, Fe, and Mn cyclopentadienone catalysts for CO2 hydrogenation with free energies for hydride transfer step equal to 16.0, 13.9, and 14.2 kcal mol−1, respectively.72 These values are almost equal to our computed results, however, the heterolytic cleavage of coordinated H2 over the metal center is far higher (37.6, 31.4, and 29.6 kcal mol−1, respectively) than our value (0.23 kcal mol−1). The energy of coordinated H2 heterolytic cleavage step is higher even with other active metal catalysts like Rh as well.73,74 This shows the significant role played by NiFe heterobimetallic architecture as a catalyst for CO2 hydrogenation. In the next step, the NiFe3 eliminates HCOO− anion in endergonic fashion to form NiFe4 with free energy equal to 1.34 kcal mol−1 (NiFe3 → NiFe4). The NiFe4 with available coordination site has two options available, either it can undergo H2 addition over the Fe(II)-metal center to form NiFe6, or it can react with HCOO− anion to form more stable NiFe8. In either of the cases, addition of H2 or HCOO− anion over the metal center are exergonic in nature with their respective free energies equal to −10.45 and −12.22 kcal mol−1. NiFe6 thus formed interacts with HCOO− anion through the hydrogen atom of pendant amine to form NiFe7 with Gibbs free energy equal to −4.72 kcal mol−1. Finally, the loss of HCOOH from intermediate NiFe7 via TS71 regenerates NiFe1 with small energy barrier in equal to 5.17 kcal mol−1 (NiFe7 → TS71).
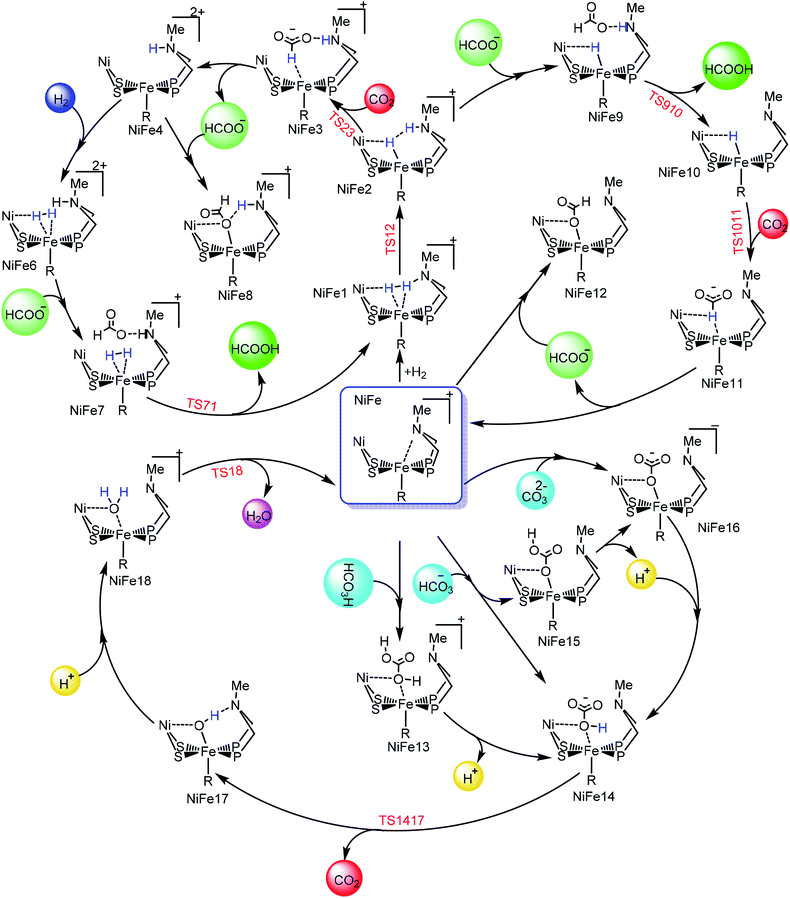 |
| | Scheme 2 The complete mechanistic cycle of CO2 hydrogenation in aqueous media catalyzed by NiFe designed catalyst. The structure of the complexes have been displayed in truncated form to have clear overview of the mechanism. | |
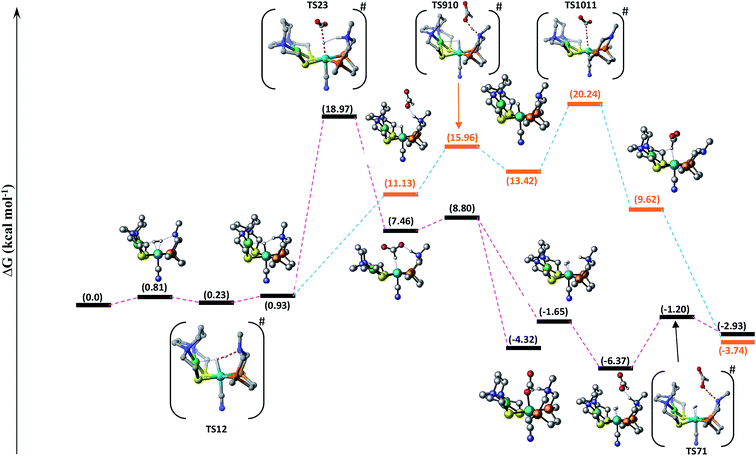 |
| | Fig. 1 Free energy profile of CO2 hydrogenation catalyzed by [NiFe] heterobimetallic catalyst along the reaction coordinate. Non-essential hydrogen atoms have been omitted for the sake of clarity. | |
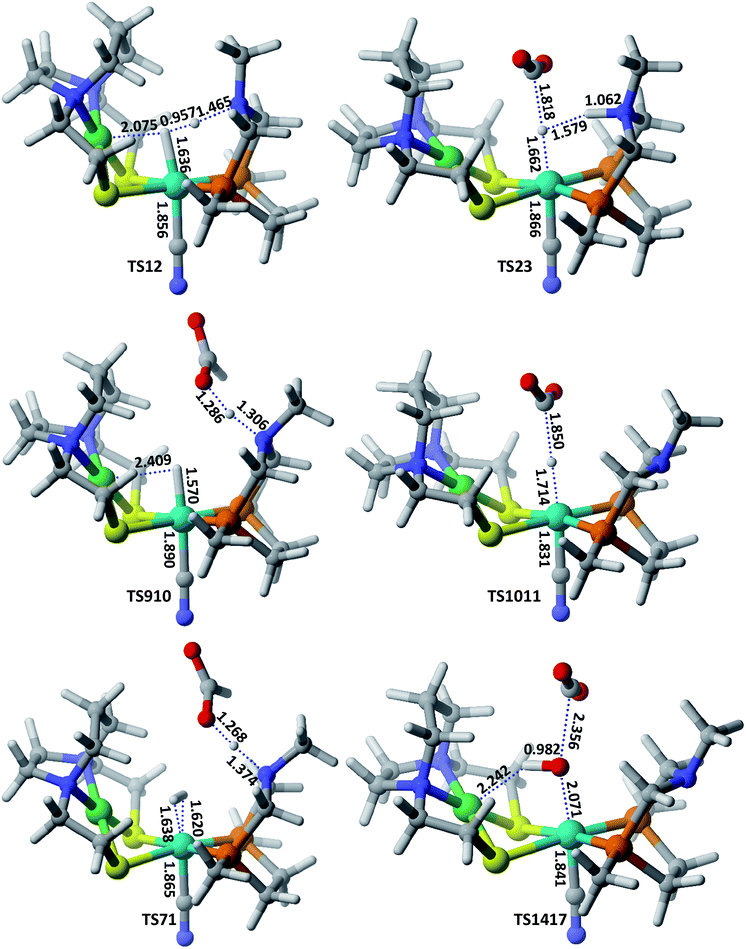 |
| | Fig. 2 Optimized geometrical parameters of some of the important transition states involved in CO2 hydrogenation catalyzed by model 2 complex. All the bond distances are given in Å. | |
In route second, instead of reacting with CO2, NiFe2 can interact with HCOO− anion to form NiFe9 with 10.2 kcal mol−1 uphill energy. In the next elementary step, the NiFe9 subsequently undergoes HCOOH elimination via TS910 transition state with an uphill barrier height equal to 4.83 kcal mol−1 (NiFe9 → TS910). Next, NiFe10 thus formed from NiFe9 interacts with CO2 through metal-hydride bond via TS1011 to form NiFe11 having an uphill barrier height equal to 6.82 kcal mol−1 (NiFe10 → TS1011). The transfer of hydride from NiFe10 to CO2, which proceeds through the transition state TS1011, is the rate-determining step of pathway 2 with total free energy of activation equal to 20.24 kcal mol−1. Finally, the loss of HCOO− anion from NiFe11 regenerates the initial catalyst.
The first route is less energy demanding compared to route second with abstraction of hydride from Fe-metal center by CO2 being the rate determining step of both the mechanistic pathways. As far as the catalysis of CO2 by model 2 is concerned, the free energy of various intermediates and transition states is almost similar to model 1. For example, free energy of H2 addition over the Fe(II)-metal center is 1.93 kcal mol−1 (NiFe1), the heterolytic cleavage of coordinated H2 is 0.59 kcal mol−1 while as the transfer of hydride from metal center to CO2 is 19.03 kcal mol−1.
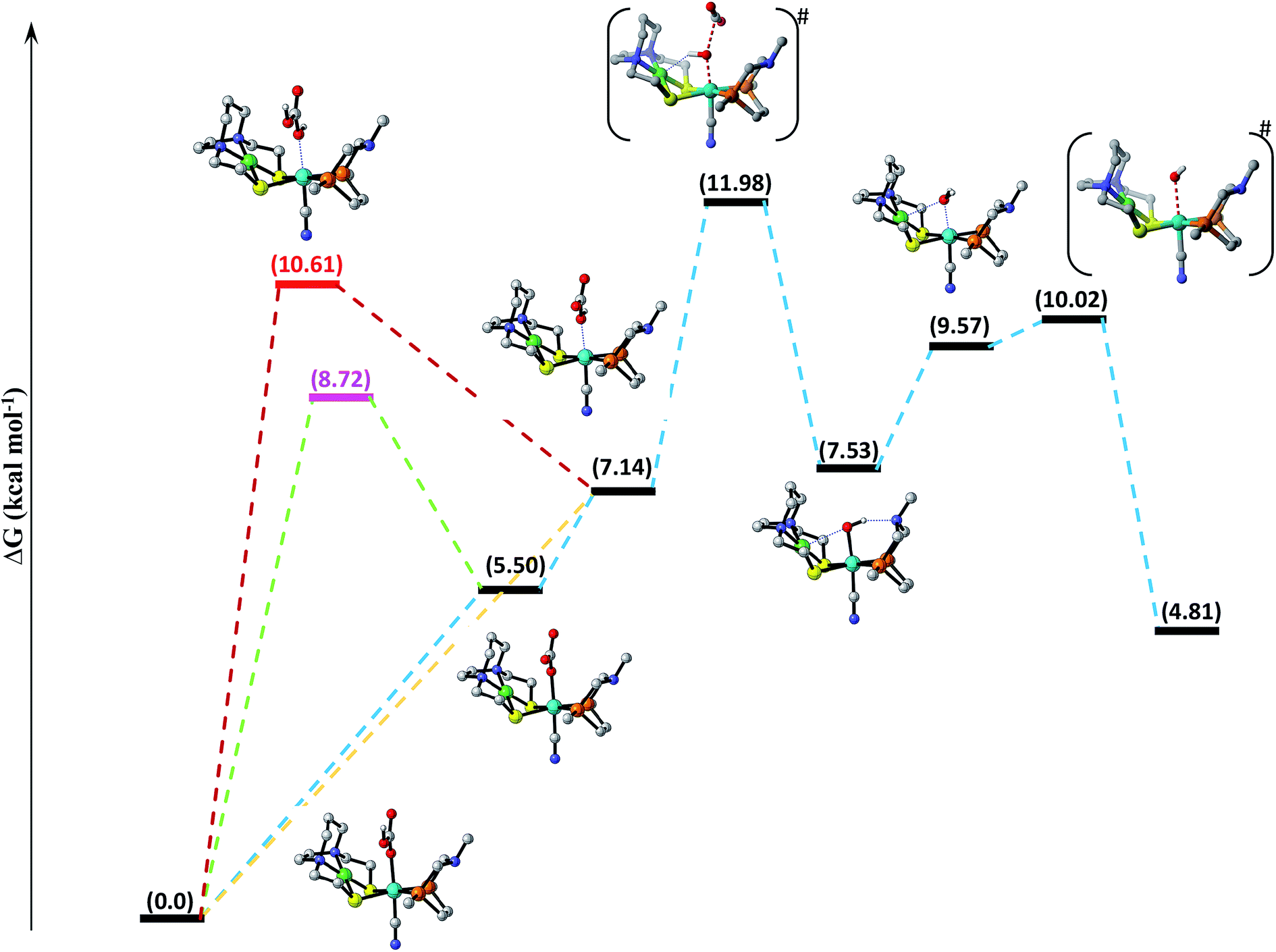
Decomposition of carbonic acid, bicarbonate and carbonate ions. Water not only increases the catalytic activity of the catalyst (ΔG0298 = −4.0 kJ mol−1) but it can also participate in the reaction by stabilizing the polar intermediates or by interacting with CO2 itself.75 In aqueous media, the CO2 combines with water to form carbonic acid, which undergoes acid/base equilibria under varying conditions of pH, temperature, and pressure to form other species like bicarbonate and carbonate ions. Thus, the interaction of above mentioned species should also be studied. According to computational assessment, H2CO3 interacts with NiFe to form NiFe13 with barrier free energy 10.61 kcal mol−1, which subsequently undergo H+ elimination in exothermic fashion with free energy equal to −3.47 kcal mol−1 to form NiFe14. The dissociation of CO2 via TS1417 from NiFe14 with total free energy of activation equal to 11.98 kcal mol−1 is the rate-determining step of the catalytic cycle. NiFe17 thus formed reacts with proton along OH coordinated over the Fe-center to form NiFe18, which finally eliminates the H2O molecule via TS18 to regenerate the initial catalyst as shown in Fig. 3. Likewise, bicarbonate ion can interact with the catalyst in two possible ways; either the ionized oxygen atom reacts with the catalyst to form NiFe15 with uphill free energy 8.72 kcal mol−1, which ultimately eliminates H+ to generate NiFe16 or it can interact directly along OH bond with Fe-metal center, which results in the formation of already mentioned intermediate NiFe14. Similarly, carbonate ion interacts with the catalyst to form NiFe16 with free energy 5.50 kcal mol−1. Out of all the species, the interaction of H2CO3 with the catalyst is energetically demanding followed by HCO3− and then finally CO32−.
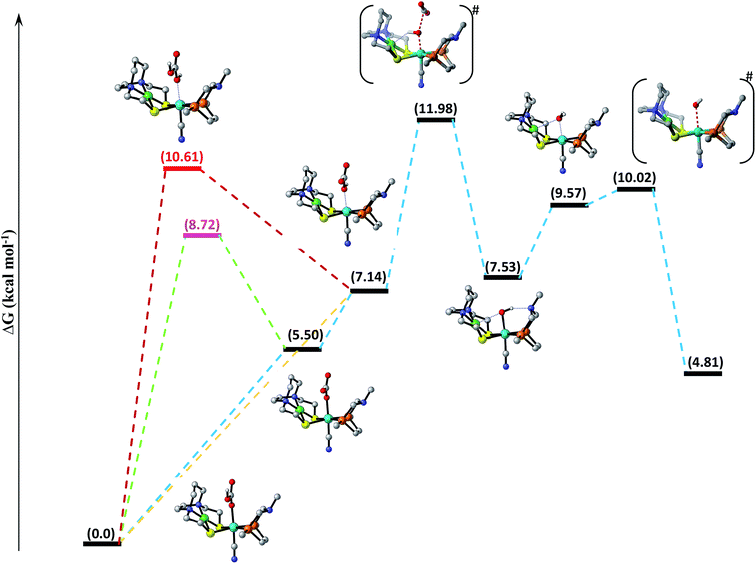 |
| | Fig. 3 Free energy profile for decomposition of carbonic acid, bicarbonate and carbonate ions catalyzed by [NiFe] heterbimetallic catalyst along the reaction coordinate. Non-essential hydrogen atoms have been omitted for the sake of clarity. | |
3.2 Effect of ligand substitution
In order to design a catalyst with higher efficiency, we examined the effects of replacing CN ligand with other substituent ligands like SCH3, SCN, NCS, OH, H, F, and pyrrol-1-yl including metallodithiolate analogous variant in which N-atoms attached with Ni(II) were replaced with P. In total nine complexes were examined and after computational assessment, it was found that the activity of [NiFe] heterobimetallic catalyst is highly sensitive to these positions. The free energy of activation for TS23 in these newly proposed complexes decreased by almost 5 kcal mol−1 with lowest value obtained for OH group equal to 14.13 kcal mol−1 as shown in Table 1. The ligands SCH3, SCN, NCS, and H decrease the free energy of activation within 1–2 kcal mol−1 range. These effects of ligand substitution can be explained based on NBO analysis, according to which the ligands with low free energy of activation increase the negative charge at the Fe(II) metal center as listed in Table 1. The ligands with strong sigma donating ability like OH and pyrrol-1-yl increase the electron density at the metal center as depicted by their high negative charges at Fe(II) (−1.673 and −1.597). The C-atom of CO2 is electrophilic in nature as predicted by its NBO charge equal to 0.987 and thus higher negative charge at metal center will favors the hydride transfer. Likewise, F destabilizes TS23 by lowering electron density at the metal center (−1.365), which increases the activation energy of hydride transfer step. Moreover, replacement of N in metallodithiolate ligand with P also decreases the free energy of activation of TS23 transition state significantly. Thus, we recommend complex with metallodiothiolate ligand containing P instead of N and strong sigma donating ligands at Fe(II) metal center for the effective hydrogenation of CO2.
Table 1 Reaction free energy barriers for the hydrogen transfer step (TS23), energy gap between Fe–H σ-bonding orbital and π* antibonding orbital of CO2 and NBO changes on Fe–H in NiFe2 substituted complexesa
| R |
ΔG#TS (kcal mol−1) |
Energy gap (eV) |
NBO Charges (e) |
| Fe |
| The atom of the ligand written first are the donor atoms with Fe(II) center. |
| SCH3 |
17.09 |
4.01 |
−1.495 |
| SCN |
17.83 |
4.11 |
−1.443 |
| NCS |
16.19 |
3.89 |
−1.546 |
| OH |
14.13 |
3.61 |
−1.673 |
| H |
17.12 |
4.02 |
−1.483 |
| F |
20.78 |
4.51 |
−1.365 |
| Pyrrol-1-yl |
14.92 |
3.72 |
−1.597 |
| CN |
18.97 |
4.27 |
−1.398 |
| N is replaced by P |
|
|
|
| P |
14.29 |
3.63 |
−1.622 |
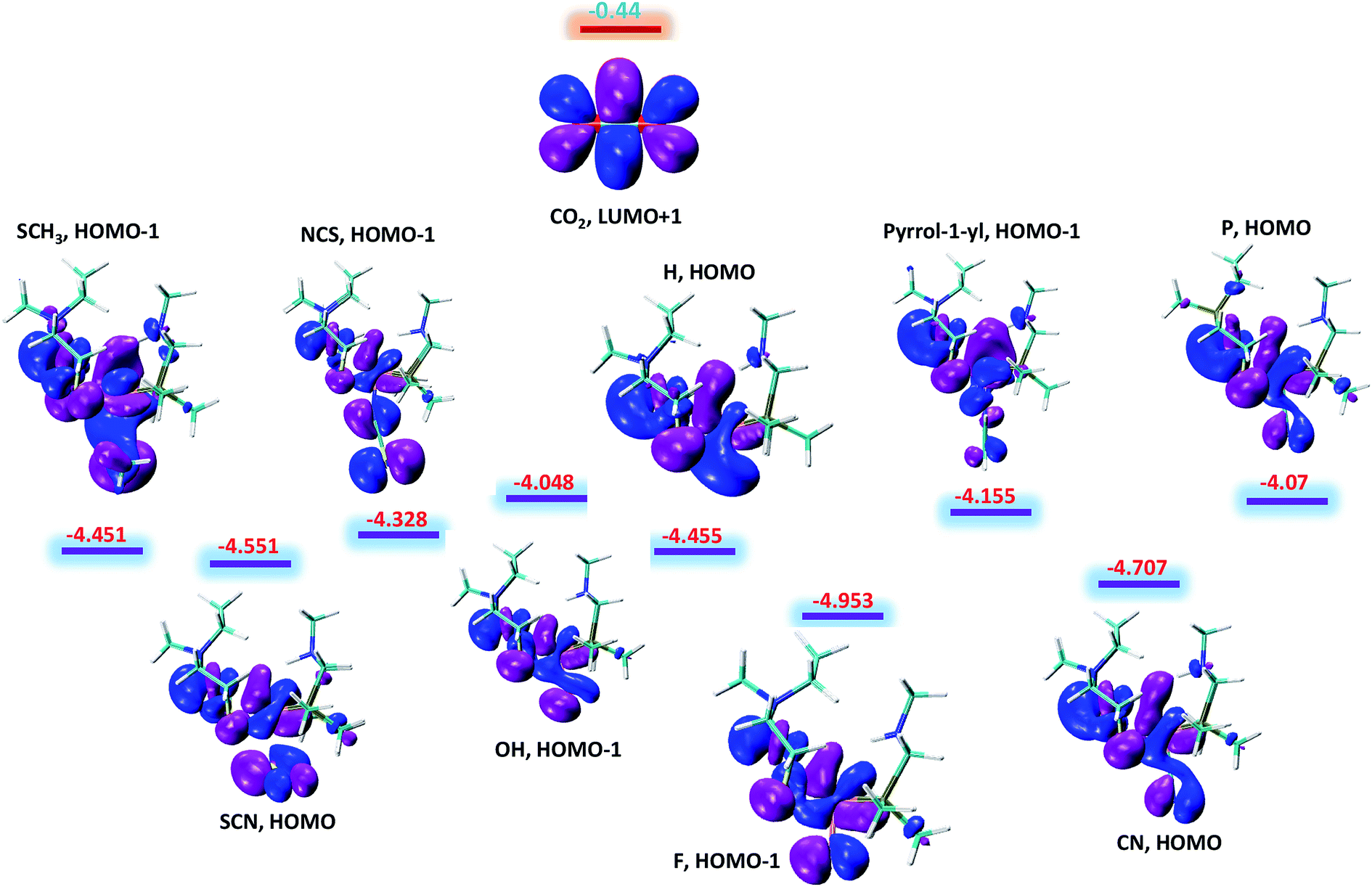
The above said effects of ligand substitution can further be explained based on energy gap between Fe–H σ-bonding orbital and the π*-antibonding orbital of CO2. According to NBO analysis Fe–H σ-bonding orbital populates the π*-antibonding orbital of CO2, which is the driving force for hydride abstraction step. The molecular orbital analysis of substituted complexes predict that HOMO and HOMO-1 represent the Fe–H σ-bonding orbital and LUMO+1 which represent π*-antibonding orbital for CO2. The LUMO+1 of CO2 is having energy equal to −0.44 eV while as the energy of other complexes falls in the range −4.048 to −4.953 eV with total difference of entire range almost equal to 1 eV as shown in Fig. 4. It is quite interesting to see that the complexes with lowest free energy of activation are having lowest energy difference between Fe–H σ-bonding orbital and π*-antibonding orbital of CO2. For example, OH and P ligands with low free energy of activation equal to 14.13 and 14.29 kcal mol−1 are having lowest energy gaps 3.61 and 3.63 eV, respectively. Similarly, F ligand with high free energy of activation 20.73 kcal mol−1 is having high-energy gap 4.51 eV as listed in Table 1. Thus, our computed free energy barriers are consistent with molecular orbital analysis.
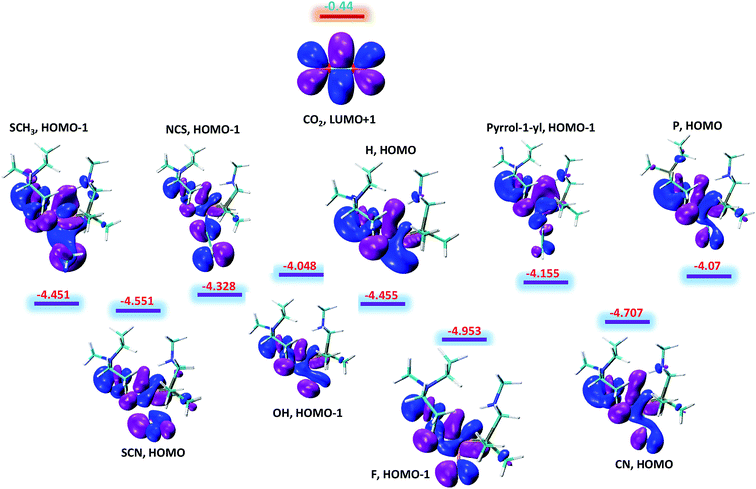 |
| | Fig. 4 The energy difference between Fe–H σ-bonding orbitals of substituted NiFe2 complexes and π*-antibonding orbital of CO2. All the given values are in eV. | |
3.3 Electronic effects
Effect of spin state. The spin state of the metal atom influences the activity of a catalyst,76 thus we studied the effects of various possible spin states of Ni(II) and Fe(II) in our newly designed catalyst. Three different complexes of spin states were considered during computational analysis, viz., Ni-singlet/Fe-triplet, Ni-triplet/Fe-singlet, and Ni-triplet/Fe-triplet. Assessment of the results demonstrate that the free energy of activation for heterolytic cleavage of H2 over the metal center (TS12) almost remains same for Ni-singlet/Fe-triplet and Ni-triplet/Fe-singlet spin state combinations (0.59 kcal mol−1). However, Ni-triplet/Fe-triplet combination in which both the Ni(II) and Fe(II) are in triplet states shows an unexpected increment in total free energy of activation for TS12 by almost 8 kcal mol−1 as listed in Table 2. Similarly, Ni-singlet/Fe-triplet and Ni-triplet/Fe-singlet combinations procreate results for hydride transfer step, TS23 equal to 24.25 kcal mol−1, which is 5.28 kcal mol−1 higher than the most stable ground state combination Ni-singlet/Fe-singlet. The Ni-triplet/Fe-triplet combination again reproduces higher results for TS23, 37.22 kcal mol−1, which is inaccessible at normal conditions. Thus, it is concluded here that CO2 hydrogenation is dominated by most stable Ni-singlet/Fe-singlet spin state combination, however, Ni-singlet/Fe-triplet and Ni-triplet/Fe-singlet combinations may also be involved sometimes.
Table 2 Free energy of transition states at different spin states. All the given values are kcal mol−1
| Complex |
Spin state |
TS12 |
TS23 |
| NiFe |
Ni, singlet: Fe, singlet |
0.22 |
18.97 |
| NiFeT |
Ni, singlet: Fe, triplet |
0.59 |
24.25 |
| NiTFe |
Ni, triplet: Fe, singlet |
0.59 |
24.25 |
| NiTFeT |
Ni, triplet: Fe, triplet |
8.95 |
37.22 |
The spin densities of some of the important transition states and intermediates located in catalytic cycle of CO2 hydrogenation has been computed using Mulliken method. The higher free energy of transition states as well as the stability of various arrangements of complexes relative to one another with increasing spin density over the metal centers can be explained based on their spin densities. As shown in Table 3, with increase in Mulliken spin density on both the metal centers the free energy of transition states (Table 2) increases, which explains the lower stability of these complexes with increasing spin density. Moreover, the higher stability of transition states with Fe being the active metal center is explained by its low spin density compared to Ni. The protonated complex, NiFe10 has much lower spin density in either of the spin combinations (NiFeT/NiTFeT) from initial value 1.831/2.383 (NiFe complex). The low spin density of Fe center and negative ESP charge over hydride ligand explains the higher stability of the arrangement NiII⋯−H–FeII over other possible combinations like NiIII–−H⋯FeII etc.
Table 3 Mulliken spin densities of some of the important intermediates and transition states located in entire catalytic cycle of our newly designed NiFe catalyst for CO2 hydrogenation
| Atom |
TS12 |
TS23 |
NiFe10 |
NiFe11 |
| NiFeT |
NiTFeT |
NiFeT |
NiTFeT |
NiFeT |
NiTFeT |
NiFeT |
NiTFeT |
| H represents the hydrogen atom attached with Fe-center. |
| Fe |
0.326 |
1.864 |
0.324 |
1.905 |
0.403 |
1.992 |
0.327 |
2.195 |
| Ni |
1.153 |
1.186 |
1.145 |
1.106 |
1.103 |
1.120 |
1.141 |
0.997 |
| Ha |
— |
— |
0.005 |
−0.007 |
0.024 |
−0.048 |
−0.001 |
−0.030 |
| S |
0.171 |
0.272 |
0.189 |
0.159 |
0.163 |
0.261 |
0.186 |
0.168 |
| N |
0.082 |
0.093 |
0.078 |
0.077 |
0.074 |
0.077 |
0.080 |
0.072 |
| P |
−0.007 |
0.110 |
−0.004 |
0.116 |
−0.012 |
0.089 |
0.000 |
0.004 |
4 Conclusions
In present work, two heterobimetallic Ni(II) and Fe(II) catalysts, model 1 and model 2 were computationally designed inspired by the active site of [NiFe] hydrogenase using density functional theory. The assessment of DFT results demonstrated that free energy of activation for both the models were almost identical. The abstraction of hydride at Fe(II)-center (TS23) is the rate-determining step of entire catalytic cycle with total free energy equal to 18.97 kcal mol−1. Such a low free energy of activation indicates that [NiFe] heterobimetallic architecture is a promising catalyst for CO2 hydrogenation. Moreover, the carbonic acid, bicarbonate and carbonate ions formed by CO2 with water are decomposed back to CO2 by the catalyst with free energy of activation for the rate-determining step equal to 11.98 kcal mol−1. In order to design a catalyst with higher activity, we examined the free energy of activation for TS23 by substituting the CN-group with SCH3, SCN, NCS, OH, H, F, and pyrrol-1-yl in our model 1 complex including the metallodithiolate analogous variant in which N-atoms attached with Ni(II) were replaced with P. After analysis of their results, the strong sigma donating ligands like OH, pyrrol-1-yl, and P analogous variant of metallodithiolate were found have lowest free energies of activation for TS23 equal to 14.13, 14.92, and 14.29 kcal mol−1. These results were further supported by the energy difference between Fe–H σ-bonding orbital of complexes and π*-antibonding orbital of CO2. In addition to this, variation of spin states of the metal centers has great influence on the catalytic activity of the catalyst. With increase in spin density of metal centers, the free energy of transition states as well as the instability of the various complexes increases. Thus, our DFT results not only provide a complete overview of mechanistic insights of CO2 hydrogenation by [NiFe] heterobimetallic architecture in aqueous media but also provide deep insights regarding effects of changing chemical environment around the metal center and spin states on the catalytic activity of the catalyst.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
This work was financially supported by University Grants Commission (UGC), Govt. of India under UGC-BSR scheme as SRF (Senior Research Fellow) vide notification number, No. F.25-1/2013-14(BSR)/5-27/2007(BSR).
References
- A. M. Appel, J. E. Bercaw and A. B. Bocarsly, et al., Chem. Rev., 2013, 113, 6621–6658 CrossRef CAS.
- H. Arakawa, M. Aresta and J. N. Armor, et al., Chem. Rev., 2001, 101, 953–996 CrossRef CAS.
- J. Bongaarts, Pop. Devel. Rev., 1992, 18, 299–319 CrossRef.
- D. Archer, J. Geophys. Res., 2005, 110, C09S05, DOI:10.1029/2004jc002625.
- M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709–1742 CrossRef CAS.
- G. A. Olah, G. K. S. Prakash and A. Goeppert, J. Am. Chem. Soc., 2011, 133, 12881–12898 CrossRef CAS.
- J. Qiao, Y. Liu, F. Hong and J. Zhang, Chem. Soc. Rev., 2014, 43, 631–675 RSC.
- J. Eppinger and K. W. Huang, ACS Energy Lett., 2017, 2, 188–195 CrossRef CAS.
- E. A. Quadrelli, G. Centi, J. L. Duplan and S. Perathoner, ChemSusChem, 2011, 4, 1194–1215 CrossRef CAS.
- M. Aresta and A. Dibenedetto, Dalton Trans., 2007, 2975–2992 RSC.
- A. K. Singh, S. Singh and A. Kumar, Catal. Sci. Technol., 2016, 6, 12–40 RSC.
- D. Mellmann, P. Sponholz, H. Junge and M. Beller, Chem. Soc. Rev., 2016, 45, 3954–3988 RSC.
- E. Fujita, J. T. Muckerman and Y. Himeda, Biochim. Biophys. Acta, 2013, 1827, 1031–1038 CrossRef CAS.
- K. W. Huang, J. H. Han, C. B. Musgrave and E. Fujita, Organometallics, 2007, 26, 508–513 CrossRef CAS.
- N. Onishi, S. Xu, Y. Manaka, Y. Suna, W. H. Wang, J. T. Muckerman, E. Fujita and Y. Himeda, Inorg. Chem., 2015, 54, 5114–5123 CrossRef CAS PubMed.
- R. Tanaka, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2009, 131, 14168–14169 CrossRef CAS.
- K. Mori, T. Taga and H. Yamashita, ACS Catal., 2017, 7, 3147–3151 CrossRef CAS.
- C. A. Huff and M. S. Sanford, ACS Catal., 2013, 3, 2412–2416 CrossRef CAS.
- J. A. Therrien, M. O. Wolf and B. O. Patrick, Inorg. Chem., 2014, 53, 12962–12972 CrossRef CAS.
- T. J. Schmeier, G. E. Dobereiner, R. H. Crabtree and N. Hazari, J. Am. Chem. Soc., 2011, 133, 9274–9277 CrossRef CAS.
- W. H. Wang, Y. Himeda, J. T. Muckerman, G. F. Manbeck and E. Fujita, Chem. Rev., 2015, 115, 12936–12973 CrossRef CAS.
- S. Sato, T. Morikawa, T. Kajino and O. Ishitani, Angew. Chem., Int. Ed., 2013, 52, 988–992 CrossRef CAS PubMed.
- R. Lalrempuia, M. Iglesias, V. Polo, P. J. S. Miguel, F. J. F. Alvarez, J. J. P. Torrente and L. A. Oro, Angew. Chem., Int. Ed., 2012, 51, 12824–12827 CrossRef CAS.
- J. F. Hull, Y. Himeda, W. H. Wang, B. Hashiguchi, R. Periana, D. J. Szalda, J. T. Muckerman and E. Fujita, Nat. Chem., 2012, 4, 383–388 CrossRef CAS PubMed.
- Y. Himeda, S. Miyazawa and T. Hirose, ChemSusChem, 2011, 4, 487–493 CrossRef CAS PubMed.
- R. Tanaka, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2009, 131, 14168–14169 CrossRef CAS.
- G. A. Filonenko, M. P. Conley, C. Coperet, M. Lutz, E. J. M. Hensen and E. A. Pidko, ACS Catal., 2013, 3, 2522–2526 CrossRef CAS.
- G. A. Filonenko, R. V. Putten, E. N. Schulpen, E. J. M. Hensen and E. A. Pidko, ChemCatChem, 2014, 6, 1526–1530 CrossRef CAS.
- A. Z. Spentzos, C. L. Barnes and W. H. Bernskoetter, Inorg. Chem., 2016, 55, 8225–8233 CrossRef CAS PubMed.
- M. S. Jeletic, M. L. Helm, E. B. Hulley, M. T. Mock, A. M. Appel and J. C. Linehan, ACS Catal., 2014, 4, 3755–3762 CrossRef CAS.
- F. Bertini, N. Gorgas, B. Stöger, M. Peruzzini, L. F. Veiros, K. Kirchner and L. Gonsalvi, ACS Catal., 2016, 6, 2889–2893 CrossRef CAS.
- S. A. Burgess, A. J. Kendall, D. R. Tyler, J. C. Linehan and A. M. Appel, ACS Catal., 2017, 7, 3089–3096 CrossRef CAS.
- J. M. Smieja, M. D. Sampson, K. A. Grice, E. E. Benson, J. D. Froehlich and C. P. Kubiak, Inorg. Chem., 2013, 52, 2484–2491 CrossRef CAS.
- R. Langer, Y. Diskin-Posner, G. Leitus, L. J. W. Shimon, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2011, 50, 9948–9952 CrossRef CAS.
- A. Dubey, L. Nencini, R. R. Fayzullin, C. Nervi and J. R. Khusnutdinova, ACS Catal., 2017, 7, 3864–3868 CrossRef CAS.
- X. Chen and X. Yang, J. Phys. Chem. Lett., 2016, 7, 1035–1041 CrossRef CAS PubMed.
- X. Yang, Chem. Commun., 2015, 51, 13098–13101 RSC.
- M. G. Mingot, J. P. Porcher, T. K. Todorova, T. Fogeron, C. M. Draznieks, Y. Li and M. Fontecave, J. Phys. Chem. B, 2015, 119, 13524–13533 CrossRef PubMed.
- X. Chen, Y. Jing and X. Yang, Chem.–Eur. J., 2016, 22, 8897–8902 CrossRef CAS.
- C. Tard and C. J. Pickett, Chem. Rev., 2009, 109, 2245–2274 CrossRef CAS.
- C. Wombwell and E. Reisner, Chem.–Eur. J., 2015, 21, 8096–8104 CrossRef CAS.
- T. Liu, D. L. DuBois and R. M. Bullock, Nat. Chem., 2013, 5, 228–233 CrossRef CAS.
- K. A. Murray, M. D. Wodrich, X. Hu and C. Corminboeuf, Chem.–Eur. J., 2015, 21, 3987–3996 CrossRef CAS.
- K. S. Rawat, A. Mahata and B. Pathak, J. Phys. Chem. C, 2016, 120, 26652–26662 CrossRef CAS.
- T. Arakawa, Y. Kawano, S. Kataoka, Y. Katayama, N. Kamiya, M. Yohda and M. Odaka, J. Mol. Biol., 2007, 366, 1497–1509 CrossRef CAS PubMed.
- S. Nagashima, M. Nakasako, N. Dohmae, M. Tsujimura, K. Takio, M. Odaka, M. Yohda, N. Kamiya and I. Endo, Nat. Struct. Mol. Biol., 1998, 5, 347–351 CrossRef CAS.
- J. A. Denny and M. Y. Darensbourg, Chem. Rev., 2015, 115, 5248–5273 CrossRef CAS.
- B. A. Shiekh, D. Kaur and S. Kumar, Phys. Chem. Chem. Phys., 2019, 21, 21370–21380 RSC.
- K. P. Zeller, P. Schuler and P. Haiss, Eur. J. Inorg. Chem., 2005, 2005, 168–172 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS.
- M. J. Frisch, J. A. Pople and J. S. Binkley, J. Chem. Phys., 1984, 80, 3265–3269 CrossRef CAS.
- EMSL Basis Set Exchange, http://bse.pnl.gov/bse/portal.
- Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2006, 125, 194101–194118 CrossRef.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- D. G. Gusev, Organometallics, 2013, 32, 4239–4243 CrossRef CAS.
- K. H. Hopmann, Organometallics, 2016, 35, 3795–3807 CrossRef CAS.
- B. B. Averkiev and D. G. Truhlar, Catal. Sci. Technol., 2011, 1, 1526–1529 RSC.
- A. Höllwarth, M. Böhme, S. Dapprich, A. W. Ehlers, A. Gobbi, V. Jonas, K. F. Köhler, R. Stegmann, A. Veldkamp and G. Frenking, Chem. Phys. Lett., 1993, 208, 237–240 CrossRef.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, J. R. Cheeseman, G. Scalmani, M. Caricato, H. P. Hratchian, X. Li, V. Barone, J. Bloino, G. Zheng, et al., Gaussian 09; Revision D.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- M. Z. Ertem, Y. Himeda, E. Fujita and J. T. Muckerman, ACS Catal., 2016, 6, 600–609 CrossRef CAS.
- M. D. Tissandier, K. A. Cowen, W. Y. Feng, E. Gundlach, M. H. Cohen, A. D. Earhart, J. V. Coe and T. R. Tuttle, J. Phys. Chem. A, 1998, 102, 7787–7794 CrossRef CAS.
- R. Fu, W. A. Goddard, M. J. Cheng and R. J. Nielsen, ACS Catal., 2017, 7, 356–364 CrossRef CAS.
- T. Liu, X. Wang, C. Hoffmann, D. L. DuBois and R. M. Bullock, Angew. Chem., Int. Ed., 2014, 53, 5300–5304 CrossRef CAS.
- R. Custelcean and J. E. Jackson, Chem. Rev., 2001, 101, 1963–1980 CrossRef CAS.
- X. Yang, ACS Catal., 2011, 1, 849–854 CrossRef CAS.
- H. Ge, X. Chen and X. Yang, Chem. Commun., 2016, 52, 12422–12425 RSC.
- C. Hou, J. Jiang, S. Zhang, G. Wang, Z. Zhang, Z. Ke and C. Zhao, ACS Catal., 2014, 4, 2990–2997 CrossRef CAS.
- B. Mondal, F. Neese and S. Ye, Inorg. Chem., 2016, 55, 5438–5444 CrossRef CAS.
- P. G. Jessop, T. Ikariya and R. Noyori, Chem. Rev., 1995, 95, 259–272 CrossRef CAS.
- G. Dong, Q. M. Phung, S. D. Hallaert, K. Pierloot and U. Ryde, Phys. Chem. Chem. Phys., 2017, 19, 10590–10601 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra07139c |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*