DOI:
10.1039/C9RA07087G
(Paper)
RSC Adv., 2019,
9, 37747-37758
Molecular design and properties of bridged energetic pyridines derivatives†
Received
4th September 2019
, Accepted 7th November 2019
First published on 19th November 2019
Abstract
A series of bridged pyridine-based energetic derivatives were designed and their geometrical structures, electronic structures, heats of formation, detonation properties, thermal stabilities, thermodynamic properties and electrostatic potential were fully investigated using density functional theory. The results show that the steric hindrance effect is a decisive factor for structural stability, and the formation of intramolecular or intermolecular hydrogen bonds doesn't provide advantages to stabilize molecular structure, which was demonstrated by insertion of 3,4,5-trinitro-1H-pyrazole, 3,4-dinitro-1H-pyrazol-5-amine, 3,5-dinitro-1H-pyrazol-4-amine and 3-nitro-1H-1,2,4-triazol-5-amine. The azide group and azo bridge play an important role in improving the heats of formation of energetic pyridine-based materials. All designed molecules were found to have values of density ranging from 1.70 g cm−3 (E6, F6) to 2.11 g cm−3 (D3), values of detonation velocity ranging from 7.1 km s−1 (F1) to 9.77 km s−1 (D8), and values of detonation pressure ranging from 21.5 GPa (F1) to 46.0 GPa (D8). When a p-π conjugation formed between the nitrogen atom and pyridine ring, the bond between nitrogen and hydrogen atoms may be broken as the trigger bond.
1 Introduction
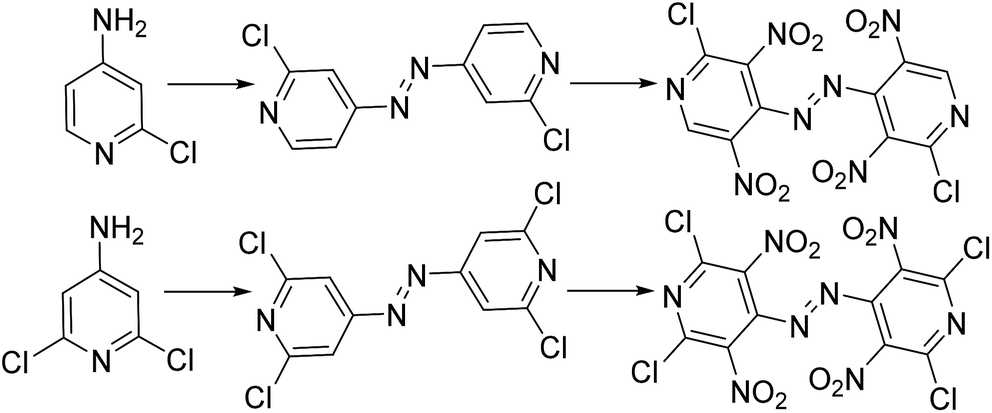
Compared with carbocyclic aromatic energetic compounds, nitrogen heterocyclic nitro compounds have higher density and better detonation performance. However, the difficulty of synthesizing some nitro heteroaromatic systems may attributed to their electron deficiency, making electrophilic aromatic substitution problematic.1 By the addition of electron donation substituents to the heteroaromatic ring, especially the amino group, nitration may proceed relatively smoothly. Additionally, high-nitrogen materials, which have large numbers of N–N and C–N bonds and few or no nitro groups, typically gain energy from their very high positive heats of formation rather than from the conventional organic energetic materials oxidizing the carbon in the backbone to carbon dioxide. Meanwhile, due to their lower carbon and hydrogen content, some properties of high-nitrogen such as density and oxygen balance have distinct advantages over conventional energetic materials.2 As a result of these properties, scientists have designed and synthesized energetic azo materials with useful detonation properties. Licht and Ritter3,4 reported the synthesis of 2,4,6-trinitropyridine (TNPy), 2,4,6-trinitropydine-1-oxide (TNPyO) and 2,6-diamino-3,5-dinitropyridine-1-oxide (ANPyO) in the twentieth century, illustrating that the density and thermal stability increase with an alternating array of amino- and nitro-groups. Zhao5 reported two promising heat-resistant energetic materials based on the pyridine and tetrazole ring were synthesized with a high decomposition temperature in excess of 300 °C. Recently, a kind of azo-bridged pyridine precursor was synthesized for the first time to develop new energetic pyridine-based compounds.6 Thus, this lead to the attempt to design and calculate bridged energetic pyridines derivatives (Scheme 1).
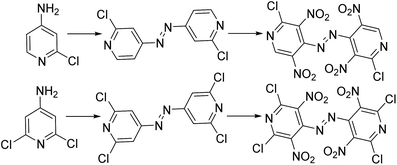 |
| | Scheme 1 Template reactions of energetic pyridine molecules. | |
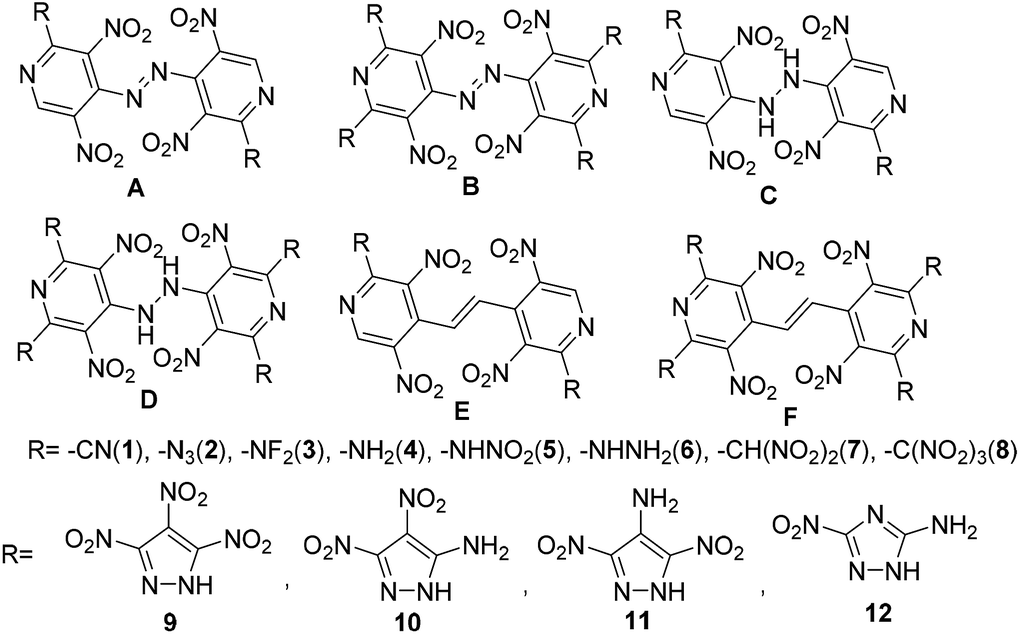
In this study, a systematic research on electronic structures, heats of formation (HOF), detonation properties, thermal stabilities and thermodynamic properties of bridged energetic pyridines derivatives (such as –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–, –NH–NH– and –NN–) with various energetic groups (such as –CN, –N3, –NF2, –NH2, –NHNO2, –NHNH2, –CH(NO2)2, –C(NO2)3)7 or common energetic materials containing active hydrogen atoms (such as 3,4,5-trinitro-1H-pyrazole, 3,4-dinitro-1H-pyrazol-5-amine, 3,5-dinitro-1H-pyrazol-4-amine, and 3-nitro-1H-1,2,4-triazol-5-amine) were investigated (Scheme 2, series A–F). It is certain that all results will provide useful information for a better understanding of the physical and chemical properties of energetic pyridines.
C–, –NH–NH– and –NN–) with various energetic groups (such as –CN, –N3, –NF2, –NH2, –NHNO2, –NHNH2, –CH(NO2)2, –C(NO2)3)7 or common energetic materials containing active hydrogen atoms (such as 3,4,5-trinitro-1H-pyrazole, 3,4-dinitro-1H-pyrazol-5-amine, 3,5-dinitro-1H-pyrazol-4-amine, and 3-nitro-1H-1,2,4-triazol-5-amine) were investigated (Scheme 2, series A–F). It is certain that all results will provide useful information for a better understanding of the physical and chemical properties of energetic pyridines.
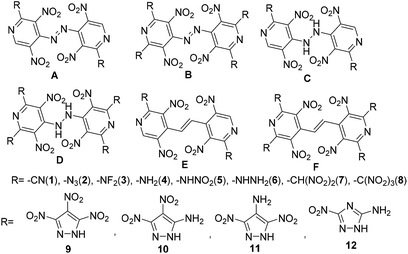 |
| | Scheme 2 Designed energetic molecules based on bridged pyridines. | |
2 Results and discussion
2.1 Electronic structures
The Highest Occupied Molecular Orbital (HOMO) and the Lowest Unoccupied Molecular Orbital (LUMO), which is called Frontier Molecular Orbital Theory (FMO Theory), has crucial effects on reaction mechanisms. Particularly, their energy gap can provide many useful information on optical polarizability, kinetic stability and chemical reactivity.8 The frontier molecular orbital energies and their energy gaps (ΔELUMO–HOMO) for designed compounds were listed in Table 1. For homologous bridged pyridine derivatives, it is found that HOMO energy levels increase evidently while –NH2 and –NHNH2 groups are introduced to the pyridine ring, so as LUMO energy levels. Oppositely, HOMO energy levels decrease when –CN, –CH(NO2)2, –C(NO2)3 or 3,4,5-trinitro-1H-pyrazole are attached, so as LUMO energy levels. Furthermore, HOMO and LUMO energy levels present no regularity when different bridges (such as –CHCH–, –NH–NH– and –NN–) are incorporated.
Table 1 Calculated HOMO and LUMO energies (eV) and energy gaps (ΔELUMO–HOMO) of designed compounds
| |
A1 |
A2 |
A3 |
A4 |
A5 |
A6 |
A7 |
A8 |
A9 |
A10 |
A11 |
A12 |
| HOMO |
−8.67 |
−7.90 |
−8.28 |
−7.32 |
−8.34 |
−7.30 |
−8.75 |
−8.80 |
−8.91 |
−7.73 |
−7.45 |
−7.78 |
| LUMO |
−5.08 |
−4.41 |
−4.49 |
−4.04 |
−4.84 |
−4.09 |
−5.19 |
−5.22 |
−5.37 |
−4.88 |
−4.78 |
−5.23 |
| ΔEHOMO–LUMO |
3.59 |
3.49 |
3.79 |
3.28 |
3.50 |
3.21 |
3.56 |
3.58 |
3.54 |
2.85 |
2.67 |
2.55 |
| |
B1 |
B2 |
B3 |
B4 |
B5 |
B6 |
B7 |
B8 |
B9 |
B10 |
B11 |
B12 |
| HOMO |
−9.27 |
−7.59 |
−8.78 |
−6.80 |
−8.39 |
−6.45 |
−9.30 |
−9.39 |
−9.25 |
−7.88 |
−7.50 |
−7.73 |
| LUMO |
−5.66 |
−4.44 |
−5.49 |
−3.54 |
−4.95 |
−3.54 |
−5.72 |
−5.80 |
−5.73 |
−5.54 |
−4.87 |
−5.67 |
| ΔEHOMO–LUMO |
3.61 |
3.15 |
3.29 |
3.26 |
3.44 |
2.91 |
3.58 |
3.59 |
3.52 |
2.34 |
2.63 |
2.06 |
| |
C1 |
C2 |
C3 |
C4 |
C5 |
C6 |
C7 |
C8 |
C9 |
C10 |
C11 |
C12 |
| HOMO |
−8.48 |
−7.78 |
−8.26 |
−7.15 |
−8.07 |
−6.83 |
−8.50 |
−8.66 |
−8.85 |
−7.64 |
−7.36 |
−7.86 |
| LUMO |
−4.23 |
−3.48 |
−3.96 |
−3.18 |
−4.10 |
−3.17 |
−4.42 |
−4.59 |
−4.51 |
−4.06 |
−3.99 |
−4.41 |
| ΔEHOMO–LUMO |
4.25 |
4.30 |
4.30 |
3.97 |
3.97 |
3.66 |
4.08 |
4.07 |
4.34 |
3.58 |
3.37 |
3.45 |
| |
D1 |
D2 |
D3 |
D4 |
D5 |
D6 |
D7 |
D8 |
D9 |
D10 |
D11 |
D12 |
| HOMO |
−8.97 |
−7.50 |
−8.69 |
−6.77 |
−8.15 |
−6.82 |
−9.13 |
−9.21 |
−9.18 |
−7.84 |
−7.27 |
−7.63 |
| LUMO |
−4.81 |
−3.40 |
−4.24 |
−2.82 |
−4.23 |
−2.95 |
−5.02 |
−5.24 |
−5.28 |
−4.59 |
−4.28 |
−4.46 |
| ΔEHOMO–LUMO |
4.16 |
4.10 |
4.45 |
3.95 |
3.92 |
3.87 |
4.11 |
3.97 |
3.90 |
3.25 |
2.99 |
3.17 |
| |
E1 |
E2 |
E3 |
E4 |
E5 |
E6 |
E7 |
E8 |
E9 |
E10 |
E11 |
E12 |
| HOMO |
−8.71 |
−7.89 |
−8.73 |
−7.15 |
−8.23 |
−7.01 |
−8.71 |
−8.77 |
−8.95 |
−7.66 |
−7.40 |
−7.51 |
| LUMO |
−4.42 |
−3.78 |
−4.33 |
−3.25 |
−3.88 |
−3.03 |
−4.23 |
−4.40 |
−4.59 |
−4.15 |
−4.09 |
−3.84 |
| ΔEHOMO–LUMO |
4.29 |
4.11 |
4.40 |
3.90 |
4.35 |
3.98 |
4.48 |
4.37 |
4.36 |
3.51 |
3.31 |
3.67 |
| |
F1 |
F2 |
F3 |
F4 |
F5 |
F6 |
F7 |
F8 |
F9 |
F10 |
F11 |
F12 |
| HOMO |
−9.22 |
−7.60 |
−8.92 |
−6.73 |
−8.22 |
−6.77 |
−9.30 |
−9.35 |
−9.20 |
−7.88 |
−7.46 |
−7.57 |
| LUMO |
−4.77 |
−3.63 |
−4.31 |
−2.78 |
−4.02 |
−2.91 |
−4.91 |
−5.12 |
−5.18 |
−4.50 |
−4.23 |
−4.55 |
| ΔEHOMO–LUMO |
4.45 |
3.97 |
4.61 |
3.94 |
4.20 |
3.86 |
4.39 |
4.23 |
4.02 |
3.38 |
3.23 |
3.02 |
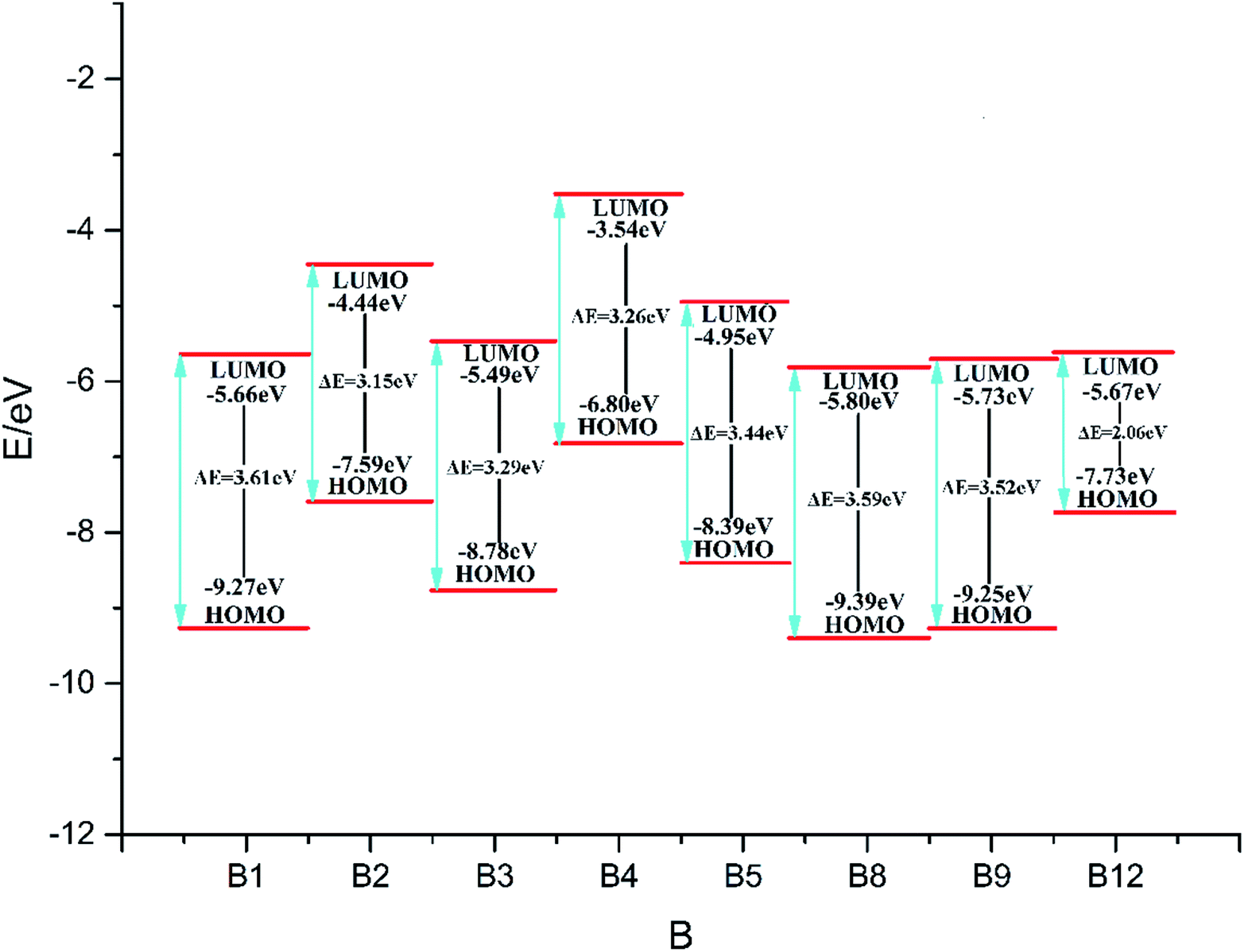
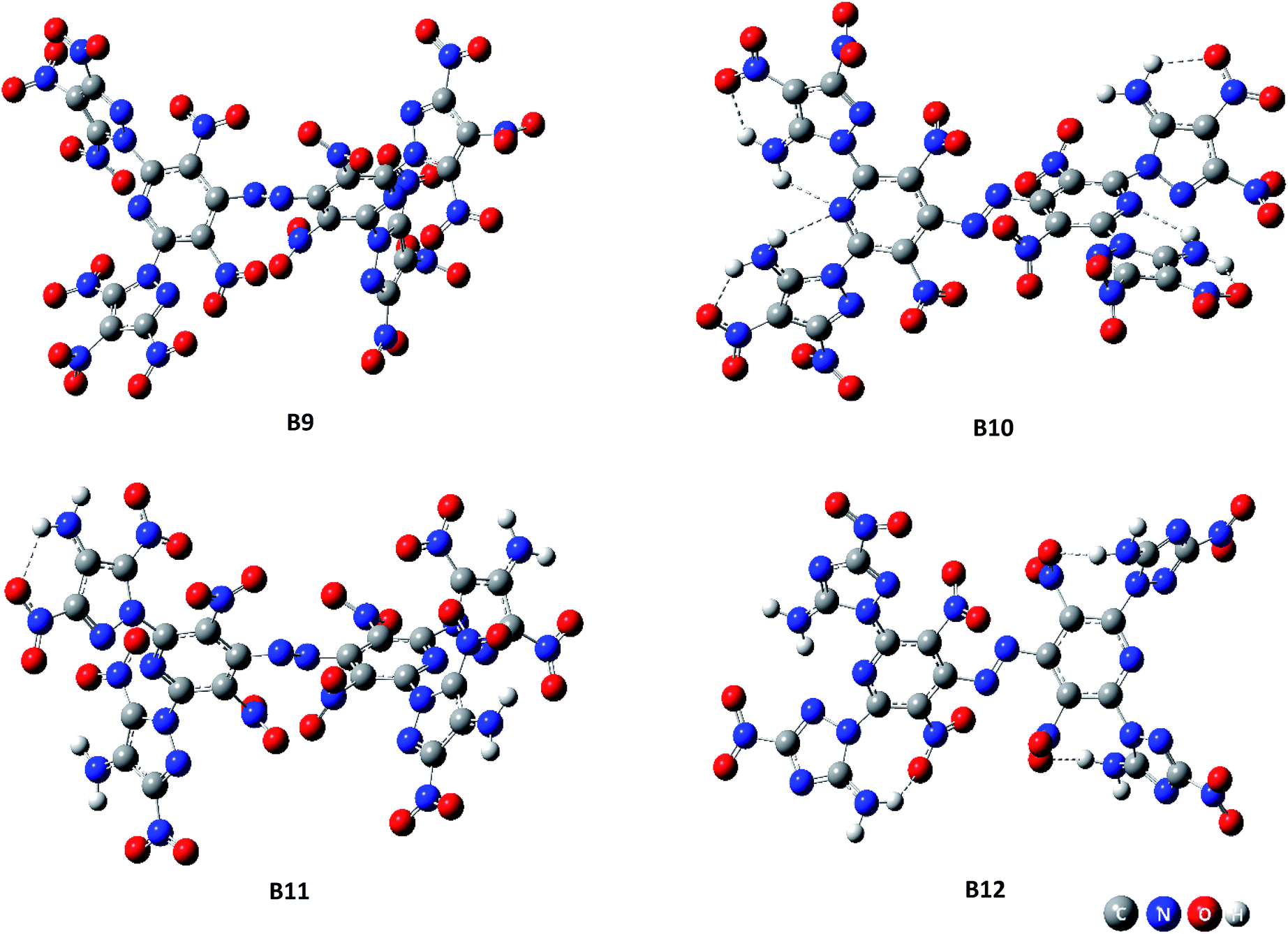
Fig. 1 (the HOMO–LUMO pictures are in ESI Table S1†) displays the variation trends of highest occupied orbital, lowest unoccupied molecular orbital energy levels and energy gaps of representative designed compounds B1, B2, B3, B4, B5, B8, B9, B12. It reveals that ΔELUMO–HOMO decreases evidently while 3,4-dinitro-1H-pyrazol-5-amine (10), 3,5-dinitro-1H-pyrazol-4-amine (11), and 3-nitro-1H-1,2,4-triazol-5-amine (12) are incorporated for series A–F, whereas the addition of other groups or energetic compounds makes similar contributions to ΔELUMO–HOMO. Surprisingly, ΔELUMO–HOMO is demonstrated to be much higher when 3,4,5-trinitro-1H-pyrazole (9) was incorporated than that of other three energetic compounds (10–12), convincing that steric hindrance effect is a decisive factor for structural stability caused by a combination of amino and nitro groups (Fig. 2, see ESI Table S2†). In general, ΔELUMO–HOMO increases gradually as different bridges incorporated in order of –NN–, –NH–NH–, –CHCH–. In view of all designed compounds, compound F3 has the highest ΔELUMO–HOMO of 4.61 eV while compounds B12 has the smallest ΔELUMO–HOMO of 2.06 eV, showing that F3 is the most inactive and B12 is the most active under external stimulus.
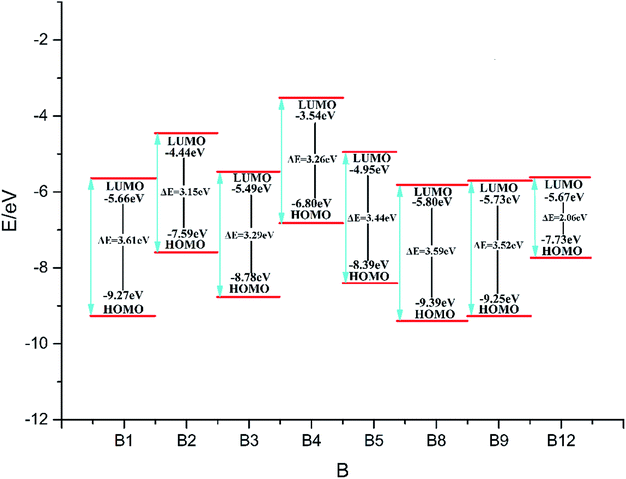 |
| | Fig. 1 The variation trends of ΔELUMO–HOMO of representative designed compounds. | |
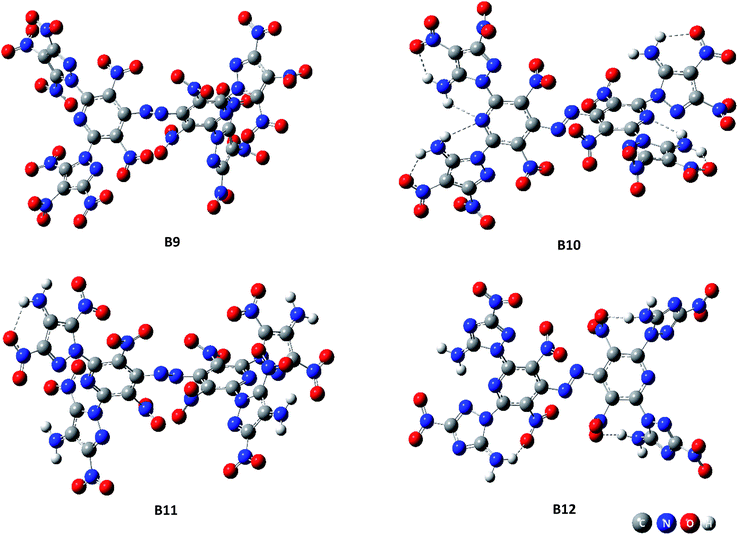 |
| | Fig. 2 Spatial structures of representative compounds B9–B12. | |
2.2 Heat of formation
Heat of formation (HOF), calculated by atomization reaction or isodesmic reaction, is an important parameter in predicting detonation properties of energetic materials. Compounds CH3CH(NO2)2 and CH3C(NO2)3 were calculated via isodesmic reactions (Scheme 3) at G4 level (Table 2). Total energies, zero-point energies, and thermal corrections for the reference compounds using the isodesmic reactions (Scheme S1, see ESI†) are listed in Table 3.
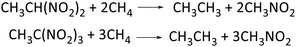 |
| | Scheme 3 Isodesmic reactions for polynitromethane. | |
Table 2 HOF calculated for polynitromethane at G4 level
| Compd |
E0a (a.u.) |
ZPEa (kJ mol−1) |
HTa (kJ mol−1) |
ΔHf,gas (kJ mol−1) |
| Calculated at the G4 level. Obtained from http://webbook.nist.gov. Obtained by isodesmic reaction. |
| CH4 |
−40.506540 |
117.5 |
10.0 |
−74.6b |
| CH3CH3 |
−79.808058 |
195.7 |
11.6 |
−84.0b |
| CH3NO2 |
−244.957724 |
130.7 |
14.0 |
−81.0b |
| CH3CH(NO2)2 |
−488.711186 |
212.5 |
22.9 |
−105.1c |
| CH3C(NO2)3 |
−693.145352 |
216.1 |
29.3 |
−73.9c |
Table 3 Calculated total energies (E0), zero-point energies (ZPE), thermal corrections (HT) and heats of formation (HOFs) of the reference compounds
| Compd |
E0a (a.u.) |
ZPEb (kJ mol−1) |
HTb (kJ mol−1) |
ΔHf,gas (kJ mol−1) |
| Calculated at the B3LYP/def2-TZVPP level. Calculated at the B3LYP/6-311G(d,p) level, and the scaling factor is 0.9888 for ZPE and 1.0062 for HT.14 Obtained from http://webbook.nist.gov. Calculated at G4 level. Obtained by isodesmic reaction. |
| CH4 |
−40.541022 |
115.8 |
10.1 |
−74.6c |
| NH3 |
−56.589815 |
89.1 |
10.1 |
−45.9c |
| CH3NO2 |
−245.127760 |
128.9 |
11.7 |
−81.0c |
| CH3CN |
−132.816078 |
117.4 |
12.0 |
74.0c |
| CH3N3 |
−204.186410 |
130.3 |
14.3 |
293.4 (ref. 9) |
| CH3NF2 |
−294.351870 |
121.4 |
13.7 |
−98.0d |
| CH3NH2 |
−95.908614 |
165.7 |
11.5 |
−23.5c |
| CH3NHNO2 |
−300.491413 |
174.6 |
16.1 |
−73.2 (ref. 10) |
| CH3NHNH2 |
−151.253386 |
211.3 |
13.5 |
94.5c |
| CH3CH(NO2)2 |
−489.043812 |
210.0 |
23.0 |
−105.1e |
| CH3C(NO2)3 |
−693.610990 |
213.7 |
29.4 |
−73.9e |
| CH3NNCH3 |
−189.363781 |
218.2 |
16.1 |
146.0d |
| CH3NHNHCH3 |
−190.578383 |
282.9 |
17.1 |
90.0 (ref. 11) |
| CH3CHCHCH3 |
−157.306017 |
278.9 |
17.0 |
−10.8c |
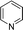 |
−248.398244 |
229.8 |
13.8 |
140.6 (ref. 12) |
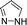 |
−226.304546 |
184.6 |
12.4 |
177.4 (ref. 12) |
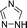 |
−242.358564 |
155.0 |
11.9 |
192.7 (ref. 13) |
Table 4 presents the ΔHf,gas, A, v, σtot2, ΔHsub and ΔHf,solid of bridged pyridine derivatives. It is seen that all compounds have positive ΔHf,gas range from to 142.8 (F4) to 1876.9 kJ mol−1 (B2). It reveals that energetic materials attached with –N3 group often have the highest ΔHf,solid, while –NH2, –CH(NO2)2, –NHNO2 groups are not conducive to improvement of ΔHf,solid. For each series F, the influence of different energetic groups on ΔHf,solid can be described as the following order: –N3 > –CN > 3,4,5-trinitro-1H-pyrazole > 3-nitro-1H-1,2,4-triazol-5-amine > 3,5-dinitro-1H-pyrazol-4-amine > 3,4-dinitro-1H-pyrazol-5-amine > –NHNH2 > –NF2 > –C(NO2)3 > –NHNO2 > –CH(NO2)2 > –NH2. Meanwhile, azo bridged pyridine derivatives usually have better HOFs than hydrazine or vinyl bridged compounds. This phenomenon may be caused by π–π conjugated system and large amount of nitrogen content. Besides, these high positive HOFs make great contribution to increasing detonation properties such as detonation velocities and detonation pressures.
Table 4 Calculated molecular properties and heats of formation of the designed compounds
| Compd |
ΔHf,gas (kJ mol−1) |
A (Å2) |
ν |
σtot2 (kcal mol−1)2 |
ΔHsub (kJ mol−1) |
ΔHf,solid (kJ mol−1) |
Compd |
ΔHf,gas (kJ mol−1) |
A (Å2) |
ν |
σtot2 (kcal mol−1)2 |
ΔHsub (kJ mol−1) |
ΔHf,solid (kJ mol−1) |
| Calculated at the B3LYP/def2-TZVPP level. Calculated at the B3LYP/6-311G(d,p) level, and the scaling factor is 0.9888 for ZPE and 1.0062 for HT.14 |
| A1 |
929.7 |
361.4 |
0.1629 |
168.3 |
194.5 |
735.2 |
D1 |
1102.5 |
382.9 |
0.1418 |
268.6 |
218.9 |
883.7 |
| A2 |
1209.7 |
380.5 |
0.2410 |
109.4 |
209.6 |
1000.1 |
D2 |
1671.6 |
420.8 |
0.2479 |
114.2 |
246.9 |
1424.6 |
| A3 |
602.9 |
353.6 |
0.1600 |
141.1 |
184.9 |
418.1 |
D3 |
467.3 |
387.8 |
0.0520 |
230.6 |
204.4 |
263.0 |
| A4 |
471.3 |
336.8 |
0.2254 |
167.6 |
181.6 |
289.7 |
D4 |
145.6 |
344.2 |
0.2484 |
233.9 |
197.4 |
−51.8 |
| A5 |
560.6 |
395.8 |
0.1514 |
139.3 |
219.2 |
341.5 |
D5 |
339.4 |
455.4 |
0.1536 |
186.9 |
281.1 |
58.3 |
| A6 |
726.8 |
366.9 |
0.2431 |
129.2 |
201.5 |
525.3 |
D6 |
653.6 |
402.8 |
0.2463 |
190.3 |
241.0 |
412.6 |
| A7 |
558.4 |
450.2 |
0.0975 |
197.1 |
269.1 |
289.3 |
D7 |
360.2 |
558.5 |
0.0774 |
310.9 |
394.8 |
−34.6 |
| A8 |
687.8 |
477.7 |
0.0678 |
170.4 |
290.8 |
397.0 |
D8 |
628.3 |
612.6 |
0.0363 |
230.8 |
451.6 |
176.7 |
| A9 |
1178.8 |
586.4 |
0.1231 |
185.8 |
429.6 |
749.2 |
D9 |
1616.9 |
836.3 |
0.0712 |
259.5 |
823.3 |
793.6 |
| A10 |
971.9 |
553.6 |
0.1878 |
212.4 |
398.4 |
573.5 |
D10 |
1216.5 |
762.0 |
0.1238 |
309.5 |
703.8 |
512.7 |
| A11 |
984.5 |
554.5 |
0.2110 |
136.2 |
392.9 |
591.6 |
D11 |
1217.2 |
765.3 |
0.2059 |
173.1 |
708.0 |
509.2 |
| A12 |
1065.4 |
502.7 |
0.2232 |
275.0 |
348.9 |
716.5 |
D12 |
1300.3 |
662.0 |
0.1805 |
276.2 |
550.8 |
749.5 |
| B1 |
1296.9 |
407.9 |
0.1216 |
228.3 |
234.7 |
1062.2 |
E1 |
721.7 |
362.8 |
0.1804 |
166.8 |
197.3 |
524.4 |
| B2 |
1876.9 |
443.0 |
0.2402 |
120.6 |
268.8 |
1608.1 |
E2 |
1069.3 |
377.9 |
0.2073 |
123.5 |
206.9 |
862.4 |
| B3 |
640.6 |
391.9 |
0.1404 |
199.4 |
220.5 |
420.0 |
E3 |
403.0 |
373.6 |
0.0909 |
149.6 |
193.8 |
209.2 |
| B4 |
357.9 |
355.9 |
0.2413 |
238.0 |
206.2 |
151.6 |
E4 |
276.0 |
337.8 |
0.2483 |
163.3 |
183.9 |
92.2 |
| B5 |
536.8 |
472.8 |
0.1331 |
169.2 |
295.0 |
241.8 |
E5 |
348.5 |
389.7 |
0.1862 |
139.7 |
217.3 |
131.2 |
| B6 |
891.6 |
417.0 |
0.2386 |
240.8 |
259.0 |
632.5 |
E6 |
546.4 |
363.6 |
0.2475 |
215.8 |
210.5 |
335.8 |
| B7 |
552.3 |
581.1 |
0.0696 |
260.6 |
419.1 |
133.2 |
E7 |
347.6 |
442.0 |
0.1104 |
190.9 |
262.4 |
85.2 |
| B8 |
806.7 |
630.5 |
0.0344 |
149.4 |
472.2 |
334.5 |
E8 |
467.4 |
473.0 |
0.0801 |
188.2 |
289.2 |
178.2 |
| B9 |
1814.7 |
846.4 |
0.0712 |
230.2 |
840.7 |
974.0 |
E9 |
972.7 |
587.0 |
0.1287 |
185.3 |
431.1 |
541.6 |
| B10 |
1409.3 |
787.7 |
0.1153 |
294.7 |
745.8 |
663.5 |
E10 |
761.8 |
551.8 |
0.2108 |
199.5 |
397.3 |
364.5 |
| B11 |
1405.2 |
785.0 |
0.1988 |
145.8 |
738.1 |
667.1 |
E11 |
779.7 |
555.4 |
0.2176 |
132.8 |
394.1 |
385.5 |
| B12 |
1516.2 |
682.2 |
0.1962 |
287.5 |
584.1 |
932.1 |
E12 |
804.2 |
492.1 |
0.2362 |
212.1 |
331.8 |
472.4 |
| C1 |
721.6 |
338.5 |
0.1841 |
168.0 |
178.8 |
542.8 |
F1 |
1091.1 |
408.8 |
0.1271 |
247.8 |
237.9 |
853.2 |
| C2 |
999.7 |
359.1 |
0.2373 |
107.7 |
191.4 |
808.4 |
F2 |
1695.3 |
440.8 |
0.2272 |
116.2 |
265.0 |
1430.3 |
| C3 |
397.5 |
343.4 |
0.1063 |
154.4 |
172.2 |
225.3 |
F3 |
424.6 |
415.5 |
0.0629 |
186.3 |
228.9 |
195.7 |
| C4 |
259.5 |
324.5 |
0.2353 |
169.9 |
173.7 |
85.8 |
F4 |
142.8 |
352.2 |
0.2493 |
251.6 |
205.7 |
−62.8 |
| C5 |
355.4 |
382.8 |
0.1629 |
163.8 |
211.8 |
143.6 |
F5 |
322.2 |
462.1 |
0.1698 |
167.2 |
287.7 |
34.5 |
| C6 |
514.5 |
354.0 |
0.2483 |
149.5 |
194.5 |
320.0 |
F6 |
652.5 |
411.1 |
0.2499 |
199.3 |
249.9 |
402.6 |
| C7 |
356.8 |
434.7 |
0.1005 |
209.7 |
255.2 |
101.6 |
F7 |
354.2 |
564.8 |
0.0749 |
288.0 |
400.8 |
−46.6 |
| C8 |
475.9 |
422.2 |
0.0799 |
155.9 |
235.9 |
240.0 |
F8 |
581.1 |
624.6 |
0.0348 |
187.7 |
465.9 |
115.2 |
| C9 |
974.1 |
570.3 |
0.1266 |
219.4 |
412.1 |
562.0 |
F9 |
1601.7 |
852.6 |
0.0711 |
249.0 |
853.6 |
748.1 |
| C10 |
754.5 |
531.4 |
0.2053 |
190.4 |
371.0 |
383.5 |
F10 |
1206.4 |
794.9 |
0.1198 |
297.1 |
759.5 |
446.9 |
| C11 |
783.0 |
538.4 |
0.2094 |
162.9 |
376.5 |
406.5 |
F11 |
1195.9 |
789.0 |
0.2009 |
142.8 |
744.9 |
451.0 |
| C12 |
852.1 |
469.9 |
0.2191 |
274.5 |
312.6 |
539.5 |
F12 |
1278.2 |
676.5 |
0.2042 |
227.0 |
570.7 |
707.5 |
2.3 Detonation property
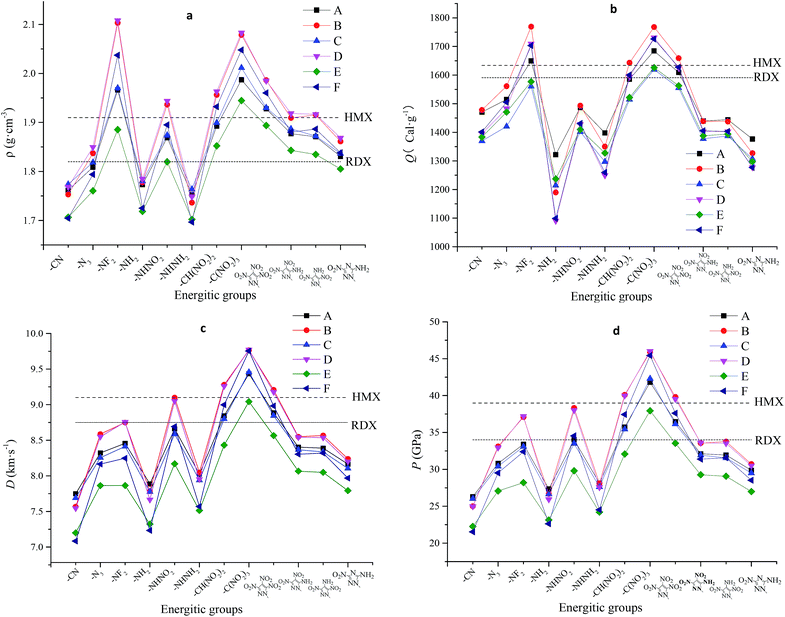
Detonation properties that related to oxygen balance (OB), density (ρ), heat of detonation (Q), detonation velocity (D) and detonation pressure (P) are summarized in Table 5. For a comparison, detonation properties of two well-known explosives 1,3,5-trinitro-1,3,5-triazinane (RDX) and 1,3,5,7-tetranitro-1,3,5,7-tetrazocane (HMX) were also presented. It is seen that most of the designed compounds have negative OB, and energetic materials attached with –C(NO2)3 group always have OB of nearly zero, proving that polynitro compounds contribute to good values of OB. Meanwhile, energetic materials attached with –CN, –NH2, or –NHNH2 group always have dissatisfied OB, proving that cyano and amino compounds are not conducive to good oxygen balance. Bridged pyridine derivatives with different substituents were also found to have different ρ, Q, D and P values: values of ρ range from 1.70 (E6, F6) to 2.11 g cm−3 (D3); values of Q range from 1090.5 (D4) to 1769.2 cal g−1 (B3). Compound F1 has the smallest value of detonation velocity (7.1 km s−1) and detonation pressure (21.5 GPa), while Compound D8 has the highest value of detonation velocity (9.77 km s−1) and detonation pressure (46.0 GPa), which is also better than that of RDX and HMX.
Table 5 Predicted densities (ρ), heats of detonation (Q), detonation velocities (D)
| Compd |
Q (cal g−1) |
D (km s−1) |
P (GPa) |
ρ (g cm−3) |
OB (%) |
Compd |
Q (cal g−1) |
D (km s−1) |
P (GPa) |
ρ (g cm−3) |
OB (%) |
| A1 |
1470.6 |
7.75 |
26.3 |
1.76 |
−65.7 |
D2 |
1483.0 |
8.55 |
33.0 |
1.85 |
−39.2 |
| A2 |
1515.0 |
8.32 |
30.8 |
1.81 |
−46.6 |
D3 |
1709.2 |
8.76 |
37.2 |
2.11 |
−36.5 |
| A3 |
1649.6 |
8.46 |
33.4 |
1.97 |
−44.6 |
D4 |
1090.5 |
7.67 |
25.9 |
1.78 |
−63.8 |
| A4 |
1321.9 |
7.89 |
27.3 |
1.77 |
−60.9 |
D5 |
1428.1 |
9.05 |
38.0 |
1.94 |
−18.5 |
| A5 |
1486.5 |
8.66 |
34.0 |
1.87 |
−33.0 |
D6 |
1249.7 |
7.95 |
27.6 |
1.75 |
−62.5 |
| A6 |
1397.7 |
8.03 |
28.1 |
1.75 |
−60.3 |
D7 |
1593.9 |
9.26 |
40.0 |
1.96 |
−14.3 |
| A7 |
1585.6 |
8.84 |
35.7 |
1.89 |
−28.0 |
D8 |
1730.9 |
9.77 |
46.0 |
2.08 |
5.0 |
| A8 |
1684.6 |
9.44 |
41.8 |
1.99 |
−12.1 |
D9 |
1624.9 |
9.18 |
39.5 |
1.99 |
−17.8 |
| A9 |
1608.9 |
8.89 |
36.5 |
1.93 |
−27.1 |
D10 |
1402.3 |
8.54 |
33.6 |
1.92 |
−38.1 |
| A10 |
1439.7 |
8.40 |
32.1 |
1.88 |
−43.0 |
D11 |
1402.4 |
8.54 |
33.5 |
1.92 |
−38.1 |
| A11 |
1444.0 |
8.39 |
31.9 |
1.87 |
−43.0 |
D12 |
1277.3 |
8.20 |
30.5 |
1.87 |
−45.7 |
| A12 |
1376.6 |
8.16 |
29.9 |
1.83 |
−49.2 |
E1 |
1383.2 |
7.20 |
22.3 |
1.71 |
−85.4 |
| B1 |
1478.0 |
7.57 |
25.0 |
1.75 |
−68.9 |
E2 |
1470.5 |
7.86 |
27.1 |
1.76 |
−64.8 |
| B2 |
1561.2 |
8.59 |
33.1 |
1.84 |
−36.3 |
E3 |
1577.0 |
7.87 |
28.2 |
1.89 |
−62.0 |
| B3 |
1769.2 |
8.75 |
37.1 |
2.10 |
−33.8 |
E4 |
1237.1 |
7.32 |
23.1 |
1.72 |
−81.6 |
| B4 |
1189.8 |
7.78 |
26.7 |
1.78 |
−60.3 |
E5 |
1409.8 |
8.17 |
29.8 |
1.82 |
−49.8 |
| B5 |
1493.1 |
9.10 |
38.3 |
1.94 |
−15.9 |
E6 |
1327.6 |
7.51 |
24.2 |
1.70 |
−79.6 |
| B6 |
1350.1 |
8.05 |
28.1 |
1.74 |
−59.5 |
E7 |
1521.7 |
8.43 |
32.1 |
1.85 |
−42.1 |
| B7 |
1643.1 |
9.28 |
40.1 |
1.96 |
−12.3 |
E8 |
1626.2 |
9.04 |
37.9 |
1.94 |
−24.2 |
| B8 |
1767.7 |
9.77 |
45.9 |
2.08 |
6.7 |
E9 |
1562.7 |
8.57 |
33.5 |
1.89 |
−37.7 |
| B9 |
1659.0 |
9.21 |
39.8 |
1.99 |
−16.4 |
E10 |
1387.8 |
8.07 |
29.3 |
1.84 |
−54.5 |
| B10 |
1438.7 |
8.55 |
33.6 |
1.91 |
−36.6 |
E11 |
1393.9 |
8.05 |
29.1 |
1.83 |
−54.5 |
| B11 |
1437.7 |
8.57 |
33.8 |
1.92 |
−36.6 |
E12 |
1297.3 |
7.79 |
27.0 |
1.81 |
−62.3 |
| B12 |
1327.0 |
8.24 |
30.7 |
1.86 |
−44.0 |
F1 |
1401.2 |
7.08 |
21.5 |
1.70 |
−86.5 |
| C1 |
1369.9 |
7.69 |
26.0 |
1.77 |
−69.2 |
F2 |
1505.1 |
8.17 |
29.5 |
1.79 |
−51.7 |
| C2 |
1420.3 |
8.26 |
30.4 |
1.82 |
−50.0 |
F3 |
1703.3 |
8.25 |
32.4 |
2.04 |
−48.0 |
| C3 |
1560.6 |
8.41 |
33.1 |
1.97 |
−47.8 |
F4 |
1099.2 |
7.23 |
22.6 |
1.73 |
−79.6 |
| C4 |
1214.7 |
7.78 |
26.6 |
1.78 |
−64.6 |
F5 |
1430.7 |
8.69 |
34.5 |
1.90 |
−29.2 |
| C5 |
1401.6 |
8.59 |
33.5 |
1.87 |
−36.2 |
F6 |
1259.6 |
7.57 |
24.5 |
1.70 |
−76.3 |
| C6 |
1297.3 |
7.94 |
27.6 |
1.76 |
−63.8 |
F7 |
1600.3 |
9.00 |
37.4 |
1.93 |
−22.6 |
| C7 |
1514.9 |
8.80 |
35.4 |
1.90 |
−30.6 |
F8 |
1726.3 |
9.75 |
45.4 |
2.05 |
−1.7 |
| C8 |
1619.5 |
9.46 |
42.3 |
2.01 |
−14.5 |
F9 |
1627.4 |
8.98 |
37.6 |
1.96 |
−23.3 |
| C9 |
1555.1 |
8.84 |
36.1 |
1.93 |
−29.2 |
F10 |
1405.3 |
8.30 |
31.4 |
1.88 |
−44.3 |
| C10 |
1377.5 |
8.37 |
31.9 |
1.89 |
−45.2 |
F11 |
1402.9 |
8.32 |
31.6 |
1.89 |
−44.3 |
| C11 |
1387.1 |
8.34 |
31.5 |
1.87 |
−45.2 |
F12 |
1277.1 |
7.97 |
28.5 |
1.84 |
−53.3 |
| C12 |
1307.4 |
8.10 |
29.5 |
1.84 |
−51.6 |
RDX15 |
1590.7 |
8.75 |
34.0 |
1.82 |
−21.6 |
| D1 |
1395.1 |
7.54 |
25.0 |
1.77 |
−72.1 |
HMX15 |
1633.9 |
9.10 |
39.0 |
1.91 |
−21.6 |
Fig. 3 displays variation trends of ρ, Q, D and P of designed compounds, in which Fig. 3a shows the influence of different energetic groups on density. It can be seen that compounds substituted by –NF2 group have the highest density while –CN, –NH2 or –NHNH2 substituted ones have smaller density. Compounds with different bridged groups have similar values in detonation velocity, detonation pressure and density, indicating that –CHCH–, –NH–NH– and –NN– bridged groups have little effect in improving the values of ρ, Q, D and P. Fig. 3b shows the influence of different energetic groups on Q. It can be seen that compounds substituted by –NF2 and –C(NO2)3 group have the highest heat of detonation while –NH2, –NHNH2 and 3-nitro-1H-1,2,4-triazol-5-amine substituted ones have smaller heat of detonation. It is clearly seen that different bridged groups have different effects on Q values in the order of –NN–, –CHCH–, –CHCH–.
 |
| | Fig. 3 (a–d) Variation trends of ρ, Q, D and P of designed compounds. | |
In view of Fig. 3c and d, it is seen that the influence of different energetic groups on values of D and P were approximately the same throughout all series. The general influence order of different energetic groups on values of D and P can be described as follows: –C(NO2)3 > –CH(NO2)2 ≈ 3,4,5-trinitro-1H-pyrazole > –NHNO2 > –NF2 ≈ –N3 ≈ 3,4-dinitro-1H-pyrazol-5-amine ≈ 3,5-dinitro-1H-pyrazol-4-amine > –NHNH2 > –NH2 > –CN while the influence of different bridges on values of D and P can be described in the order of –NN– ≈ –NH–NH– > –CHCH–. It can be concluded that polynitro groups are the most effective groups in improving D and P values while it is on the opposite side for insertion of –CN, –NH2 and –NHNH2.
2.4 Thermal stability
The bond dissociation energies (BDEs) are important in understanding thermal stability and enhancing controllability of kinetic energy release. In general, the smaller the energy is needed for breaking a bond, the weaker the bond is, and thus, this chemical bond may be acted as the trigger bond. The possible trigger bond with the weakest BDE is selected as the breaking bond based on bond order analyses. The bond order and BDEs (specific data are shown in ESI Table S3†) of all designed compounds were summarized and listed in Table 6. In most cases, the possible trigger bond was likely C–NO2 in 3- or 5-position of the pyridine ring (such as A1, A2, A9). In particular, when trinitromethyl or dinitromethyl was incorporated to the pyridine ring, C–N bond in aliphatic carbon chain was weaker than that of aromatics (such as A7, A8). When N–NO2 or N–NH2 occurred, they both appeared to be the weakest bond in one curtain designed molecule. The nitrogen atom and fluorine atom both have strong ability to attract electron, making N–F bond broken more easily. It is shocking that N–H bond were known as the trigger bond in compound A4 and B4, for example, which were contradictory with the concept of increasing thermal stability by the use of an alternating array of amino and nitro-groups. The results show that a p-π conjugation effect formed between the nitrogen atom and pyridine ring, revealing that the nitrogen atom has the weaker ability to attract the hydrogen atom. The ultimate cause of this situation seems to have been the steric hindrance effect between four nitro groups directly attached to the pyridine ring. Besides, incorporating the bridge –NN– (series A and B) or –CHCH– (series E and F) leads to the formation of π–π conjugation between double bonds and the pyridine ring, so as to stabilize the bridged atoms.
Table 6 Bond dissociation energies (BDE, kJ mol−1) for the weakest bonds
| Compd |
C–N |
N–N |
N–H |
N–F |
h50 |
Compd |
C–N |
N–N |
N–H |
N–F |
h50 |
| BO |
BDE |
BO |
BDE |
BO |
BDE |
BO |
BDE |
BO |
BDE |
BO |
BDE |
BO |
BDE |
BO |
BDE |
| A1 |
0.676 |
252.8 |
|
|
|
|
|
|
35.1 |
D1 |
|
|
|
|
0.624 |
390.6 |
|
|
29.4 |
| A2 |
0.67 |
262.5 |
|
|
|
|
|
|
54.3 |
D2 |
|
|
|
|
0.687 |
384.4 |
|
|
56 |
| A3 |
|
|
|
|
|
|
0.137 |
230.6 |
34.5 |
D3 |
|
|
|
|
|
|
0.14 |
225.6 |
7.7 |
| A4 |
|
|
|
|
0.677 |
444.5 |
|
|
50.3 |
D4 |
|
|
|
|
0.605 |
383.8 |
|
|
55.7 |
| A5 |
|
|
0.57 |
180.1 |
|
|
|
|
32.4 |
D5 |
|
|
0.531 |
191.1 |
|
|
|
|
32.7 |
| A6 |
|
|
0.61 |
315.8 |
|
|
|
|
54.8 |
D6 |
|
|
|
|
0.603 |
384.3 |
|
|
55.3 |
| A7 |
0.539 |
183.4 |
|
|
|
|
|
|
19 |
D7 |
0.526 |
182.3 |
|
|
|
|
|
|
13.4 |
| A8 |
0.457 |
174.4 |
|
|
|
|
|
|
11.9 |
D8 |
0.448 |
163.9 |
|
|
|
|
|
|
3.9 |
| A9 |
0.674 |
269 |
|
|
|
|
|
|
25.3 |
D9 |
|
|
|
|
0.637 |
375.4 |
|
|
12.2 |
| A10 |
|
|
0.614 |
279.1 |
|
|
|
|
40.9 |
D10 |
|
|
0.612 |
278 |
|
|
|
|
24.8 |
| A11 |
0.671 |
256.6 |
|
|
|
|
|
|
46.9 |
D11 |
|
|
|
|
0.624 |
375.4 |
|
|
45.5 |
| A12 |
|
|
0.655 |
297.9 |
|
|
|
|
49.3 |
D12 |
|
|
0.629 |
285.8 |
|
|
|
|
38.8 |
| B1 |
0.687 |
252.2 |
|
|
|
|
|
|
24.7 |
E1 |
0.667 |
279.2 |
|
|
|
|
|
|
39.3 |
| B2 |
0.677 |
269.3 |
|
|
|
|
|
|
54.1 |
E2 |
0.673 |
261.6 |
|
|
|
|
|
|
46 |
| B3 |
|
|
|
|
|
|
0.139 |
229 |
29.4 |
E3 |
|
|
|
|
|
|
0.142 |
228.7 |
17.7 |
| B4 |
|
|
|
|
0.667 |
449.8 |
|
|
53.9 |
E4 |
|
|
|
|
0.671 |
443.2 |
|
|
55.9 |
| B5 |
|
|
0.546 |
183.5 |
|
|
|
|
27.8 |
E5 |
|
|
0.568 |
183.3 |
|
|
|
|
40.8 |
| B6 |
|
|
0.619 |
318.9 |
|
|
|
|
53.2 |
E6 |
|
|
0.626 |
302.2 |
|
|
|
|
55.6 |
| B7 |
0.526 |
181.8 |
|
|
|
|
|
|
11.8 |
E7 |
0.524 |
186.6 |
|
|
|
|
|
|
22.2 |
| B8 |
0.453 |
173.6 |
|
|
|
|
|
|
3.9 |
E8 |
0.463 |
163.8 |
|
|
|
|
|
|
14.8 |
| B9 |
0.668 |
239.4 |
|
|
|
|
|
|
12.4 |
E9 |
0.665 |
278.8 |
|
|
|
|
|
|
26.6 |
| B10 |
|
|
0.585 |
266.1 |
|
|
|
|
22.8 |
E10 |
|
|
0.618 |
281 |
|
|
|
|
46.6 |
| B11 |
|
|
|
|
0.673 |
442.6 |
|
|
43.9 |
E11 |
0.67 |
263 |
|
|
|
|
|
|
48.5 |
| B12 |
|
|
0.619 |
281.6 |
|
|
|
|
42.6 |
E12 |
|
|
0.649 |
295.2 |
|
|
|
|
52.7 |
| C1 |
|
|
|
|
0.632 |
402.9 |
|
|
40.2 |
F1 |
0.681 |
260 |
|
|
|
|
|
|
25.9 |
| C2 |
|
|
|
|
0.628 |
405.4 |
|
|
53.4 |
F2 |
0.675 |
271.2 |
|
|
|
|
|
|
50.9 |
| C3 |
|
|
|
|
0.626 |
380.1 |
|
|
21.4 |
F3 |
|
|
|
|
|
|
0.138 |
227.6 |
10.6 |
| C4 |
|
|
|
|
0.616 |
382.3 |
|
|
52.7 |
F4 |
|
|
|
|
0.669 |
462.8 |
|
|
56 |
| C5 |
|
|
0.556 |
182.8 |
|
|
|
|
35.1 |
F5 |
|
|
0.559 |
187.5 |
|
|
|
|
36.7 |
| C6 |
|
|
0.609 |
324.4 |
|
|
|
|
56 |
F6 |
|
|
0.62 |
323.6 |
|
|
|
|
56.3 |
| C7 |
0.53 |
186.1 |
|
|
|
|
|
|
19.6 |
F7 |
0.525 |
185.2 |
|
|
|
|
|
|
13 |
| C8 |
0.463 |
169.8 |
|
|
|
|
|
|
14.9 |
F8 |
0.455 |
175.7 |
|
|
|
|
|
|
3.8 |
| C9 |
|
|
|
|
0.626 |
380.5 |
|
|
25.9 |
F9 |
0.674 |
275.3 |
|
|
|
|
|
|
12.3 |
| C10 |
|
|
|
|
0.624 |
394.6 |
|
|
45.3 |
F10 |
|
|
0.591 |
268.5 |
|
|
|
|
23.9 |
| C11 |
|
|
|
|
0.629 |
379.6 |
|
|
46.4 |
F11 |
|
|
|
|
0.672 |
446.7 |
|
|
44.4 |
| C12 |
|
|
|
|
0.625 |
394.9 |
|
|
48.3 |
F12 |
|
|
0.629 |
286.1 |
|
|
|
|
44.8 |
2.5 Thermal dynamic properties
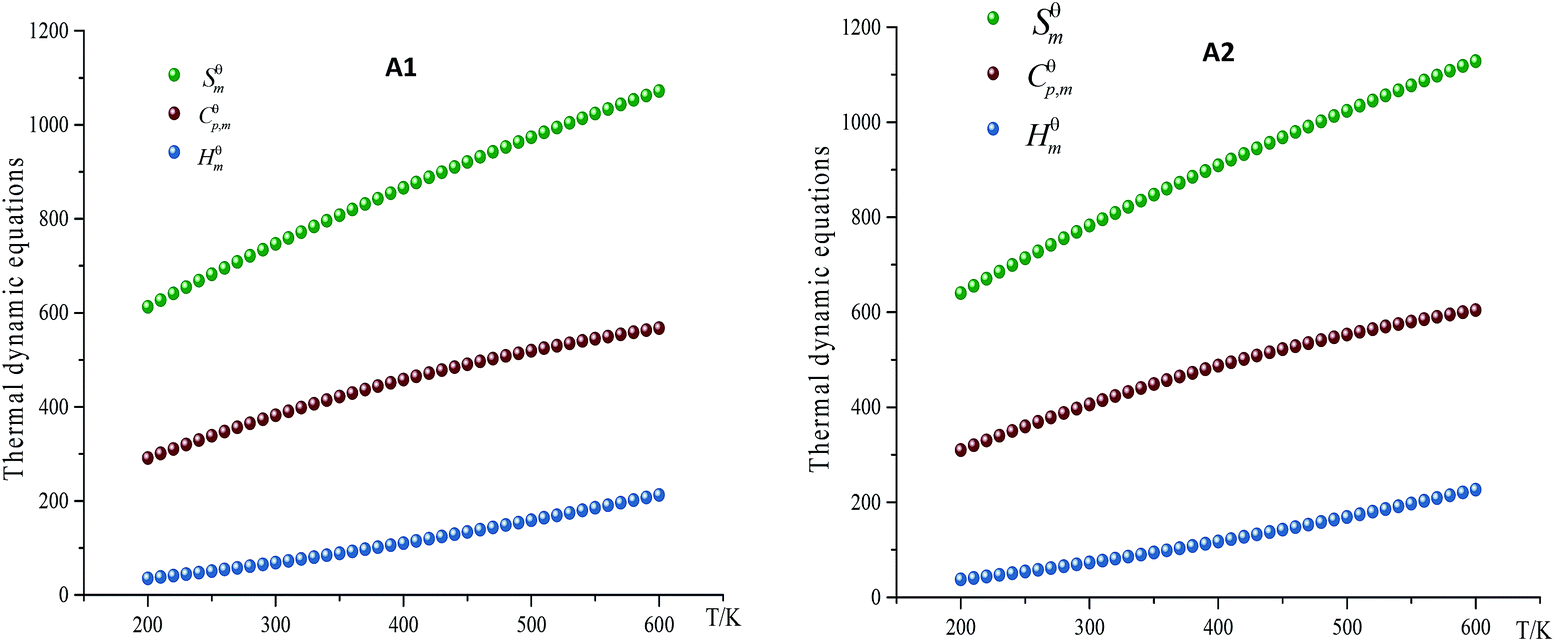
As the main contents of thermal dynamic parameters, standard molar heat capacity (Cθp,m), standard molar entropy (Sθm) and standard molar enthalpy (Hθm) can provide useful information in state equation, macroscopic properties and chemical reactions of energetic materials.16 The variation trends of Cθp,m, Sθm and Hθm of designed compounds at different temperatures (every 10 K from 200 K to 600 K, specific data are shown in ESI Table S4†) were investigated. The related equation for these parameters at different temperatures can be written in the following form:
X = a + bT + dis![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cT2 (X = Cθp,m, Sθm and Hθm) cT2 (X = Cθp,m, Sθm and Hθm) |
where a, b and c were constant and summarized in Table 7. Take compound A1 and A2 for example, as can be seen from Fig. 4, Cθp,m, Sθm and Hθm of all designed compounds improved as the temperature increasing. However, there exists differences in the growth rates of these parameters. The case is that the growth rates Cθp,m, Sθm decreased evidently as the temperature increasing while growth rate of Hθm increased. The reason is that the translations and rotations of chemical bonds were the main influencing factors at a low while vibrational movement occurred and intensified at a high temperature. At certain temperature, it is also found that the values of these thermal dynamic parameters increase as the volume of energetic groups increasing. This phenomenon may be caused by the strong space steric effects of energetic groups. For example, the Cθp,m, Sθm and Hθm of compound A7 and A8 are 382.2 J mol−1 K−1, 746.8 J mol−1 K−1, 68.8 kJ mol−1 and 406.2 J mol−1 K−1, 782.6 J mol−1 K−1, 73.2 kJ mol−1, respectively (300k). Obviously, Cθp,m, Sθm and Hθm of compound A7 are lower than those of compound A8.
Table 7 Calculated Cθp,m, Sθm, and Hθm of designed compounds
| Compd |
Hθm |
Sθm |
Cθp,m |
R2 |
| a |
b |
C × 10−4 |
a |
b |
C × 10−4 |
a |
b |
C × 10−4 |
| A1 |
−13.67 |
0.17 |
3.43 |
316.24 |
1.61 |
−5.84 |
66.83 |
1.27 |
−7.25 |
0.9999 |
| A2 |
−14.27 |
0.18 |
3.66 |
325.37 |
1.71 |
−6.17 |
70.84 |
1.35 |
−7.64 |
0.9999 |
| A3 |
−15.32 |
0.19 |
3.77 |
319.51 |
1.75 |
−6.29 |
62.71 |
1.43 |
−8.48 |
0.9999 |
| A4 |
−12.84 |
0.15 |
3.72 |
294.40 |
1.53 |
−4.97 |
35.94 |
1.36 |
−7.71 |
0.9999 |
| A5 |
−13.78 |
0.18 |
4.20 |
351.56 |
1.82 |
−6.19 |
60.51 |
1.52 |
−8.48 |
0.9999 |
| A6 |
−13.07 |
0.17 |
4.02 |
317.23 |
1.70 |
−5.69 |
62.74 |
1.39 |
−7.30 |
0.9999 |
| A7 |
−16.28 |
0.22 |
4.81 |
377.39 |
2.15 |
−7.54 |
88.02 |
1.71 |
−9.30 |
0.9999 |
| A8 |
−23.61 |
0.28 |
5.21 |
353.52 |
2.53 |
−9.44 |
114.15 |
1.95 |
−11.30 |
0.9999 |
| A9 |
−22.53 |
0.30 |
6.18 |
457.74 |
2.84 |
−10.20 |
123.00 |
2.22 |
−12.20 |
0.9999 |
| A10 |
−23.04 |
0.27 |
6.17 |
384.24 |
2.69 |
−9.18 |
88.28 |
2.25 |
−12.70 |
0.9999 |
| A11 |
−23.25 |
0.27 |
6.18 |
357.26 |
2.72 |
−9.29 |
66.62 |
2.28 |
−12.70 |
0.9999 |
| A12 |
−18.78 |
0.23 |
5.54 |
376.74 |
2.34 |
−7.76 |
70.06 |
2.00 |
−11.10 |
0.9999 |
| B1 |
−18.17 |
0.22 |
3.50 |
334.52 |
1.89 |
−7.53 |
100.10 |
1.36 |
−8.23 |
0.9999 |
| B2 |
−19.47 |
0.24 |
3.98 |
351.63 |
2.08 |
−8.16 |
106.47 |
1.53 |
−9.09 |
0.9999 |
| B3 |
−16.65 |
0.17 |
4.09 |
344.90 |
2.17 |
−8.48 |
97.69 |
1.67 |
−10.40 |
0.9999 |
| B4 |
−16.65 |
0.17 |
4.09 |
280.65 |
1.73 |
−5.74 |
37.36 |
1.56 |
−9.16 |
0.9999 |
| B5 |
−18.31 |
0.24 |
5.04 |
402.39 |
2.30 |
−8.20 |
86.93 |
1.87 |
−10.70 |
0.9999 |
| B6 |
−17.37 |
0.21 |
4.71 |
321.91 |
2.07 |
−7.16 |
86.48 |
1.63 |
−8.59 |
0.9999 |
| B7 |
−23.43 |
0.32 |
6.28 |
448.67 |
2.98 |
−10.90 |
141.26 |
2.25 |
−12.40 |
0.9999 |
| B8 |
−37.47 |
0.44 |
7.07 |
410.61 |
3.74 |
−14.80 |
197.03 |
2.71 |
−16.20 |
0.9999 |
| B9 |
−36.12 |
0.48 |
9.01 |
592.24 |
4.36 |
−16.30 |
211.88 |
3.27 |
−18.30 |
0.9999 |
| B10 |
−36.04 |
0.42 |
8.99 |
469.33 |
4.05 |
−14.20 |
146.79 |
3.31 |
−18.80 |
0.9999 |
| B11 |
−37.54 |
0.42 |
9.01 |
447.09 |
4.05 |
−14.20 |
143.92 |
3.33 |
−19.00 |
0.9999 |
| B12 |
−28.17 |
0.33 |
7.77 |
442.25 |
3.32 |
−11.20 |
98.66 |
2.84 |
−16.10 |
0.9999 |
| C1 |
−14.12 |
0.17 |
3.67 |
301.20 |
1.62 |
−5.59 |
51.87 |
1.36 |
−7.81 |
0.9999 |
| C2 |
−14.62 |
0.18 |
3.90 |
317.79 |
1.72 |
−5.95 |
57.52 |
1.44 |
−8.16 |
0.9999 |
| C3 |
−15.40 |
0.18 |
3.99 |
318.38 |
1.76 |
−6.07 |
49.68 |
1.52 |
−8.99 |
0.9999 |
| C4 |
−12.85 |
0.14 |
3.99 |
273.86 |
1.52 |
−4.54 |
16.55 |
1.46 |
−8.25 |
0.9999 |
| C5 |
−14.04 |
0.18 |
4.44 |
335.05 |
1.82 |
−5.89 |
44.90 |
1.61 |
−9.01 |
0.9999 |
| C6 |
−13.06 |
0.15 |
4.32 |
291.35 |
1.68 |
−5.14 |
38.75 |
1.50 |
−7.93 |
0.9999 |
| C7 |
−16.84 |
0.22 |
5.06 |
359.46 |
2.16 |
−7.29 |
72.97 |
1.80 |
−9.87 |
0.9999 |
| C8 |
−24.15 |
0.27 |
5.48 |
318.83 |
2.53 |
−9.09 |
95.53 |
2.05 |
−11.90 |
0.9999 |
| C9 |
−22.69 |
0.30 |
6.41 |
454.62 |
2.86 |
−10.00 |
110.83 |
2.31 |
−12.80 |
0.9999 |
| C10 |
−23.31 |
0.27 |
6.42 |
371.62 |
2.69 |
−8.91 |
73.25 |
2.34 |
−13.20 |
0.9999 |
| C11 |
−23.37 |
0.27 |
6.39 |
390.57 |
2.71 |
−9.07 |
78.71 |
2.33 |
−13.10 |
0.9999 |
| C12 |
−18.71 |
0.22 |
5.81 |
353.64 |
2.33 |
−7.34 |
51.40 |
2.09 |
−11.60 |
0.9999 |
| D1 |
−18.79 |
0.22 |
3.75 |
320.11 |
1.90 |
−7.25 |
83.47 |
1.47 |
−8.87 |
0.9999 |
| D2 |
−19.61 |
0.23 |
4.23 |
338.98 |
2.09 |
−7.87 |
91.89 |
1.62 |
−9.57 |
0.9999 |
| D3 |
−21.61 |
0.24 |
4.40 |
328.94 |
2.18 |
−8.19 |
80.65 |
1.77 |
−11.10 |
0.9999 |
| D4 |
−16.07 |
0.16 |
4.40 |
262.33 |
1.70 |
−5.15 |
12.66 |
1.67 |
−9.75 |
0.9999 |
| D5 |
−18.11 |
0.23 |
5.33 |
388.11 |
2.30 |
−7.77 |
66.44 |
1.97 |
−11.30 |
0.9999 |
| D6 |
−16.07 |
0.20 |
5.02 |
315.42 |
2.04 |
−6.56 |
64.90 |
1.72 |
−8.96 |
0.9999 |
| D7 |
−24.23 |
0.31 |
6.53 |
424.35 |
2.98 |
−10.60 |
124.57 |
2.35 |
−13.00 |
0.9999 |
| D8 |
−37.69 |
0.43 |
7.31 |
404.55 |
3.75 |
−14.50 |
181.19 |
2.81 |
−16.80 |
0.9999 |
| D9 |
−36.03 |
0.47 |
9.25 |
587.67 |
4.37 |
−16.00 |
197.90 |
3.35 |
−18.70 |
0.9999 |
| D10 |
−36.95 |
0.42 |
9.18 |
449.95 |
4.08 |
−14.20 |
140.18 |
3.38 |
−19.20 |
0.9999 |
| D11 |
−37.67 |
0.42 |
9.24 |
443.85 |
4.07 |
−14.00 |
130.66 |
3.42 |
−19.60 |
0.9999 |
| D12 |
−29.21 |
0.33 |
7.98 |
417.04 |
3.35 |
−11.10 |
88.59 |
2.93 |
−16.60 |
0.9999 |
| E1 |
−12.46 |
0.17 |
3.65 |
329.32 |
1.61 |
−5.60 |
59.19 |
1.32 |
−7.32 |
0.9999 |
| E2 |
−13.74 |
0.18 |
3.90 |
330.14 |
1.72 |
−5.92 |
61.45 |
1.41 |
−7.84 |
0.9999 |
| E3 |
−13.05 |
0.18 |
3.96 |
354.61 |
1.76 |
−6.12 |
63.25 |
1.45 |
−8.15 |
0.9999 |
| E4 |
−11.93 |
0.14 |
3.96 |
296.80 |
1.53 |
−4.66 |
25.02 |
1.43 |
−7.89 |
0.9999 |
| E5 |
−13.25 |
0.18 |
4.43 |
348.30 |
1.82 |
−5.91 |
50.65 |
1.58 |
−8.65 |
0.9999 |
| E6 |
−12.68 |
0.16 |
4.26 |
312.81 |
1.71 |
−5.44 |
50.83 |
1.47 |
−7.68 |
0.9999 |
| E7 |
−16.12 |
0.22 |
5.05 |
368.84 |
2.16 |
−7.29 |
78.45 |
1.77 |
−9.50 |
0.9999 |
| E8 |
−23.01 |
0.27 |
5.44 |
355.09 |
2.54 |
−9.20 |
106.03 |
2.00 |
−11.40 |
0.9999 |
| E9 |
−21.50 |
0.29 |
6.41 |
468.24 |
2.85 |
−9.93 |
114.80 |
2.27 |
−12.30 |
0.9999 |
| E10 |
−21.82 |
0.27 |
6.40 |
408.30 |
2.69 |
−8.91 |
79.88 |
2.30 |
−12.70 |
0.9999 |
| E11 |
−22.17 |
0.27 |
6.40 |
397.63 |
2.70 |
−8.96 |
81.77 |
2.30 |
−12.70 |
0.9999 |
| E12 |
−18.63 |
0.22 |
5.77 |
374.58 |
2.34 |
−7.51 |
59.98 |
2.06 |
−11.30 |
0.9999 |
| F1 |
−17.15 |
0.22 |
3.72 |
337.71 |
1.89 |
−7.28 |
93.04 |
1.41 |
−8.26 |
0.9999 |
| F2 |
−18.75 |
0.23 |
4.22 |
357.10 |
2.09 |
−7.89 |
96.91 |
1.59 |
−9.25 |
0.9999 |
| F3 |
−19.10 |
0.24 |
4.39 |
372.18 |
2.17 |
−8.16 |
90.36 |
1.71 |
−10.30 |
0.9999 |
| F4 |
−15.95 |
0.16 |
4.34 |
279.41 |
1.73 |
−5.40 |
24.56 |
1.63 |
−9.41 |
0.9999 |
| F5 |
−17.51 |
0.24 |
5.28 |
408.60 |
2.31 |
−7.90 |
75.88 |
1.93 |
−10.90 |
0.9999 |
| F6 |
−16.23 |
0.20 |
4.96 |
320.00 |
2.06 |
−6.80 |
75.92 |
1.69 |
−8.65 |
0.9999 |
| F7 |
−23.28 |
0.31 |
6.53 |
445.24 |
2.98 |
−10.60 |
129.40 |
2.32 |
−12.70 |
0.9999 |
| F8 |
−36.86 |
0.43 |
7.30 |
411.23 |
3.74 |
−14.50 |
187.82 |
2.77 |
−16.30 |
0.9999 |
| F9 |
−35.17 |
0.47 |
9.22 |
597.80 |
4.37 |
−16.00 |
207.33 |
3.30 |
−18.20 |
0.9999 |
| F10 |
−34.88 |
0.42 |
9.16 |
498.70 |
4.07 |
−14.20 |
148.64 |
3.33 |
−18.60 |
0.9999 |
| F11 |
−36.47 |
0.42 |
9.23 |
462.29 |
4.06 |
−14.00 |
136.87 |
3.38 |
−19.10 |
0.9999 |
| F12 |
−27.23 |
0.33 |
8.00 |
444.36 |
3.32 |
−10.90 |
89.91 |
2.89 |
−16.10 |
0.9999 |
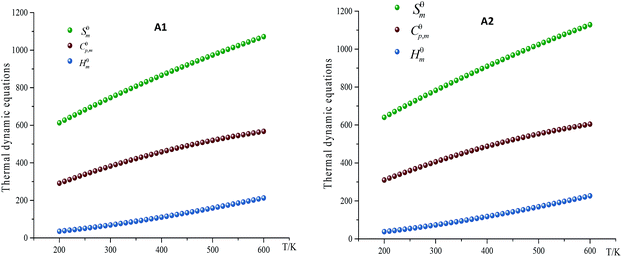 |
| | Fig. 4 Relationships between the thermodynamic equations and temperature for compound A1 and A2. | |
The scaling factor is 0.9888 for ZPE, 1.0062 for variation of enthalpy from 0 K to T and 1.0104 for entropy.14
2.6 Electrostatic potential
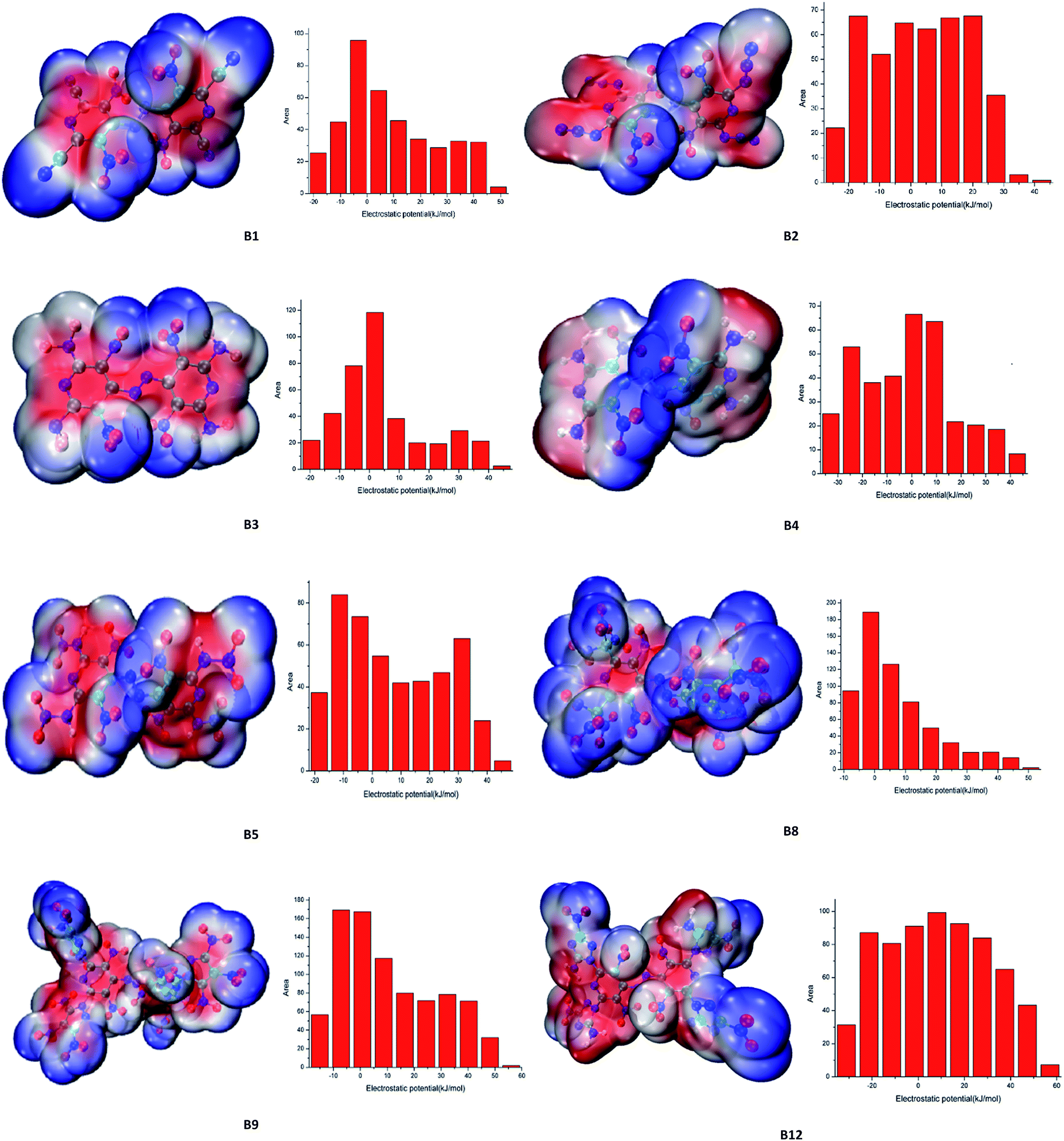
Electrostatic potential (ESP), which provides meaningful insight into charge distributions and intermolecular interaction on the molecular surface of designed compounds were fully investigated. The ESP mapped surfaces and the surface areas of designed compounds were plotted in Fig. 5, where significant surface local minima and maxima of ESP are represented in blue and red, respectively. From Fig. 5, it is found that the positive potential (red areas) mainly concentrates on the parent skeleton while the negative potential (blue areas) distributed mostly on the nitro groups and difluoramino groups, especially on the fluorine atom, which mainly due to its higher electronegativity. The positive areas of compounds B1, B2, B3, B4, B5, B8, B9 and B12 were calculated as 248 Å2 (ratio 61%), 251 Å2 (ratio 57%), 218 Å2 (ratio 56%), 180 Å2 (ratio 51%), 272 Å2 (ratio 58%), 398 Å2 (ratio 63%), 549 Å2 (ratio 65%), 426 Å2 (ratio 62%) respectively. As we can see that the positive regions are larger but also stronger in magnitude than the negative ones, which is consistent with Murray's research.17 Generally speaking, for organic molecules the negative regions cover a smaller portion of the total surface area but are significantly stronger (in terms of average magnitudes) than the positive ones. However, in the case of energetic molecules, the positive regions are not only larger but also stronger than the negative ones. From Table 6, the h50 values of compounds B1, B2, B3, B4, B5, B8, B9 and B12 were calculated as 24.7 J, 54.1 J, 29.4 J, 53.9 J, 27.8 J, 3.9 J, 12.4 J, 42.6 J respectively. It is interesting to find that the surface electrostatic potentials of the less sensitive molecules do not differ dramatically from those of the others. The delocalization of the π electronic charge that is such an important factor in stabilizing aromatic systems is counteracted in the nitroaromatics by the strongly electron-withdrawing effect of NO2 groups, which is reflected in the surface electrostatic potentials being positive rather than negative above the rings and in high degrees of internal charge separation.18 As a result, positive areas of designed compounds were higher than negative areas which may stabilize the molecular structure.
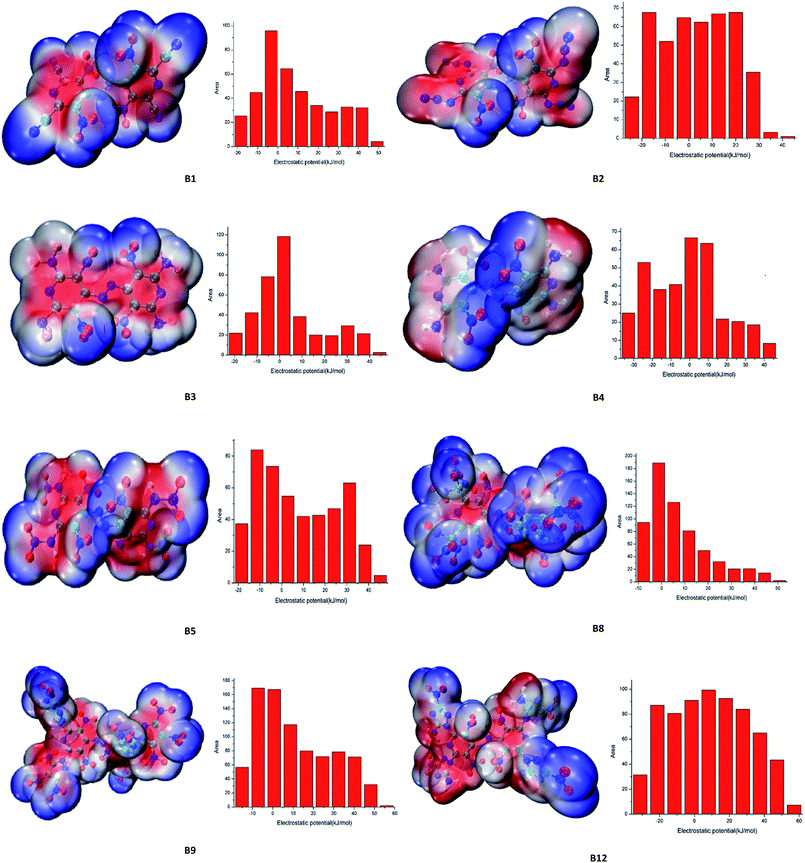 |
| | Fig. 5 Electrostatic potential of representative designed molecules. | |
3 Conclusions
In this work, a series of bridged pyridine energetic derivatives were designed and investigated by density functional theory method at DFT-D3(BJ)/B3LYP/6-311G(d,p) level. The results show that steric hindrance effect is a decisive factor for structural stability, and formation of intramolecular or intermolecular hydrogen bonds doesn't have advantages to stabilize molecular structure. The azide group and azo bridge can improve heats of formation of energetic pyridine-based materials. Compound D8 has the highest value of detonation velocity (9.77 km s−1) and detonation pressure (46.0 GPa), which is better than that of RDX and HMX, and can be selected as a potential candidate of promising high energy density materials. Analysis of the bond dissociation energies suggests that When a p-π conjugation formed between the nitrogen atom and pyridine ring, the bond between nitrogen and hydrogen atoms may be broken as the trigger bond.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (No. 11702129). We are so grateful to the High Performance Computing Center of Nanjing Tech University for doing the numerical calculations in this paper on its x-Flex enterprise blade cluster system.
Notes and references
- P. F. Pagoria, G. S. Lee, A. R. Mitchell and R. D. Schmidt, Thermochim. Acta, 2002, 384, 187–204 CrossRef CAS.
- M. A. Hiskey, N. Goldman and J. R. Stine, J. Energ. Mater., 1998, 16, 119–127 CrossRef CAS.
- H. H. Licht and H. Ritter, Propellants, Explos., Pyrotech., 1988, 13(1), 25–29 CrossRef CAS.
- H. Ritter and H. H. Licht, J. Heterocycl. Chem., 1995, 32, 585–590 CrossRef CAS.
- K. Zhao, Z. L. Liu and C. M. Ma, Chin. J. Energy Mater., 2015, 23, 1099–1102 CAS.
- C.-M. Ma, Z. L. Liu and Q. Z. Yao, Chin. J. Energy Mater., 2015, 23, 1176–1180 CAS.
- Y. Chen, H. X. Hao, S. Y. Xu and W. F. Zheng, J. Ordnance Equip. Eng., 2017, 38(6), 125–134 Search PubMed.
- A. Stobiecka, M. Sikora, R. Bonikowski and J. Kula, J. Mol. Struct., 2016, 1107, 82–90 CrossRef CAS.
- K. E. Gutowski, R. D. Rogers and D. A. Dixon, J. Phys. Chem. B, 2007, 111, 4788–4800 CrossRef CAS PubMed.
- E. A. Miroshnichenko, T. S. Kon’kova and Y. N. Matyushin, Dokl. Phys. Chem., 2003, 392, 253–255 CrossRef CAS.
- Á. Furka, Struct. Chem., 2009, 20, 605–616 CrossRef.
- W. N. Hubbard, F. R. Frow and G. Waddington, J. Phys. Chem., 1961, 65, 1326–1328 CrossRef CAS.
- P. Jiménez, M. V. Roux and C. Turrión, J. Chem. Thermodyn., 1989, 21, 759–764 CrossRef.
- J. P. Merrick, D. Moran and L. Radom, J. Phys. Chem. A, 2007, 111, 11683–11700 CrossRef CAS PubMed.
- M. B. Talawar, R. Sivabalan, T. Mukundan, H. Muthurajan, A. K. Sikder, B. R. Gandhe and A. S. Rao, J. Hazard. Mater., 2009, 161, 589–607 CrossRef CAS PubMed.
- J. Zhang and H. Xiao, J. Chem. Phys., 2002, 116, 10674–10683 CrossRef CAS.
- J. S. Murray, P. Lane and P. Politzer, Mol. Phys., 1998, 93, 187–194 CrossRef CAS.
- J. S. Murray, P. Lane and P. Politzer, Mol. Phys., 1995, 85, 1–8 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Computational methods, DFT/B3LYP/6-311G(d,p) optimized geometries of B9, B10, B11 and B12; the specific data of bond order for all designed compounds. See DOI: 10.1039/c9ra07087g |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Jinpeng Wang,
Lina Hao,
Congming Ma
,
Jinpeng Wang,
Lina Hao,
Congming Ma