
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and characterization of rigid [2.2]paracyclophane–porphyrin conjugates as scaffolds for fixed-distance bimetallic complexes†
Daniel M.
Knoll‡
 a,
Thomas B.
Wiesner‡
a,
Stefan M.
Marschner
a,
Zahid
Hassan
a,
Patrick
Weis
b,
Manfred
Kappes
be,
Martin
Nieger
c and
Stefan
Bräse
*ad
a,
Thomas B.
Wiesner‡
a,
Stefan M.
Marschner
a,
Zahid
Hassan
a,
Patrick
Weis
b,
Manfred
Kappes
be,
Martin
Nieger
c and
Stefan
Bräse
*ad
aInstitute of Organic Chemistry, Karlsruhe Institute of Technology (KIT), Fritz-Haber-Weg 6, 76131 Karlsruhe, Germany. E-mail: braese@kit.edu
bInstitute of Physical Chemistry, Karlsruhe Institute of Technology (KIT), Fritz-Haber-Weg 2, 76131 Karlsruhe, Germany
cDepartment of Chemistry, University of Helsinki, P. O. Box 55 (A. I. Virtasen Aukio 1), 00014 Helsinki, Finland
dInstitute of Toxicology and Genetics, Karlsruhe Institute of Technology (KIT), Herman-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
eInstitute of Nanotechnology, Karlsruhe Institute of Technology, Herman-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
First published on 26th September 2019
Abstract
This work presents a new approach to prepare mono- and disubstituted linear rigid bimetallic [2.2]paracyclophane–porphyrin conjugates via palladium-mediated Stille cross-coupling reaction. The metalated porphyrin moiety can be varied allowing convenient access to modular metal–metal fixed-distance Cu/Zn complexes.
Catalysis is a key concept in chemistry that allows conducting reactions efficiently, using milder conditions.1 In homogeneous catalysis, metal-catalysed reactions traditionally comprise one catalyst with one transition metal centre that activates one substrate. This paradigm has been pursued for several decades. More recently, alternate catalytic approaches are being explored. They promise to allow access to difficult or otherwise unachievable reactions by for example activating two or more substrates (synergistic catalysis)2 or using two metals to activate one substrate (bimetallic catalysis).3 To understand the interplay between metal centres when introducing two or more catalytic sites in one molecule, fundamental research on cooperativity is needed. This work proposes a molecular platform for the investigation of fixed-distance metal–metal interactions relying on metalated-porphyrins and [2.2]paracyclophane as building blocks.
Because of their rigid and planar geometry, wide spectral range, absorption and emission properties, strong aromaticity and rich metal coordination chemistry, porphyrins have been extensively investigated for their vast applicability ranging from photodynamic therapy (PDT),4 mimicking enzymes,5 metal sensing,6 to organic photovoltaics.7,8 To gain access to tailor-made porphyrin derivatives, which are necessary for specific applications, rational design and targeted functionalization have been applied systematically.9–14 The porphyrin core, a tetradentate ligand, can coordinate to a large range of transition metal ions and form stable metal complexes which constitute useful synthetic intermediates for diverse applications.15,16 These complexes are facile in preparation and can change their photophysical properties if the metal is influenced by e.g., redox state variation, thus making porphyrins suitable scaffolds for the study of metal–metal interactions.17
[2.2]Paracyclophane (PCP) was chosen as backbone due to its rigidity and the possibility to precisely control the distance of the metal centres through exploiting its regioselective substitution/functionalization pattern (Fig. 1). PCP consists of two co-facially stacked, strongly interacting benzene rings with an average ring-to-ring distance of 3.09 Å. Furthermore the unique electronic structure of the PCP backbone allows through bond as well as through space π–π interactions of the substituents because of the close proximity of the co-facially stacked benzene rings and resulting π-system overlapping.18
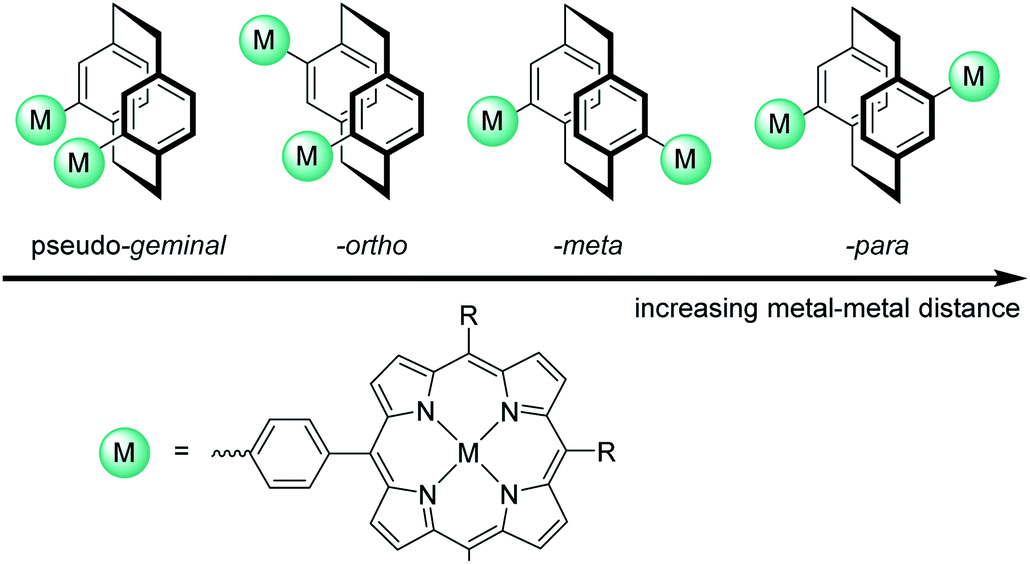 | ||
| Fig. 1 The metal–metal distance can be controlled by using different substitution patterns on the [2.2]paracyclophane. | ||
Metalated porphyrins linked to [2.2]paracyclophanes prepared by condensation of PCP–carbaldehyde and pyrrole have been described in literature.19–21 A theoretical study to understand the importance of through-space interactions and the rather strong effects of a PCP moiety in zinc–porphyrin molecular wires and their comparison to oligo(p-phenylenevinylene) for photovoltaic applications have been investigated.22,23 Furthermore, Malinski et al. reported the formation of a directly linked conjugate from a mixed condensation in low yields requiring tedious workup.21 Martin et al. formed a vinylic bridge between PCP and porphyrin via a Horner–Wadsworth–Emmons reaction in about 40% yield.14 However, a linear linker would allow for a more careful planning of the molecular geometry while exploiting the high rigidity of [2.2]paracyclophane.24 The facile variation of the metal coordinated to the porphyrin moiety and the possibility of tuning the distance between the metal centres using the different [2.2]paracyclophane regioisomers make this ensemble a suitable scaffold for assessing modular fixed-distance bimetallic complexes.
Since the previously reported optimization of reaction conditions to achieve this did not lead to satisfying yields,21 a different approach was pursued in this study.
The Stille cross-coupling reaction protocol was shown to be applicable to both, porphyrins and [2.2]paracyclophanes.25,26 The reaction parameters including palladium source, temperature, and solvent were carefully optimized.
The use of free-base porphyrin 2a and (rac)-4-stannyl[2.2]paracyclophane 1, which was synthesized according to a literature known procedure starting from (rac)-4-bromo[2.2]paracyclophane,26,27 resulted in the formation of the copper-complex 3a in 20% yield (Scheme 1). The copper(II)-ions are assumed to originate from impurities of CuBr2 in the used CuBr or from an undesired oxidation side reaction. The absence of the free-base product indicates that the reaction only proceeds with metalated porphyrins and the free-base porphyrin remains inert under these conditions. Therefore, protection of the porphyrin core with Cu(II)-ions (2b) or Zn(II)-ions (2c) was performed,25 leading to an increase of the yield of the respective resulting coupling products up to 48 and 52% (Scheme 1). The molecular structure of compound 3a was unambiguously confirmed by X-ray single crystal analysis (Fig. 2) and further characterized using collision induced dissociation (CID) experiments in ESI mass spectrometry (see ESI†).
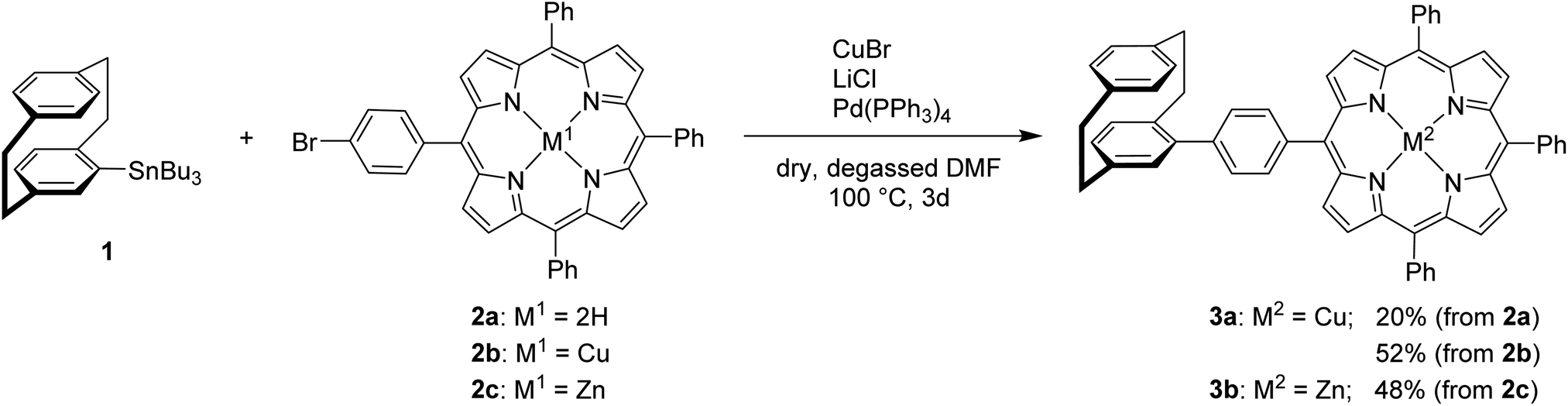 | ||
| Scheme 1 Synthesis of [2.2]paracyclophane–porphyrin conjugates linked by a phenyl unit via Stille cross-coupling protocol. | ||
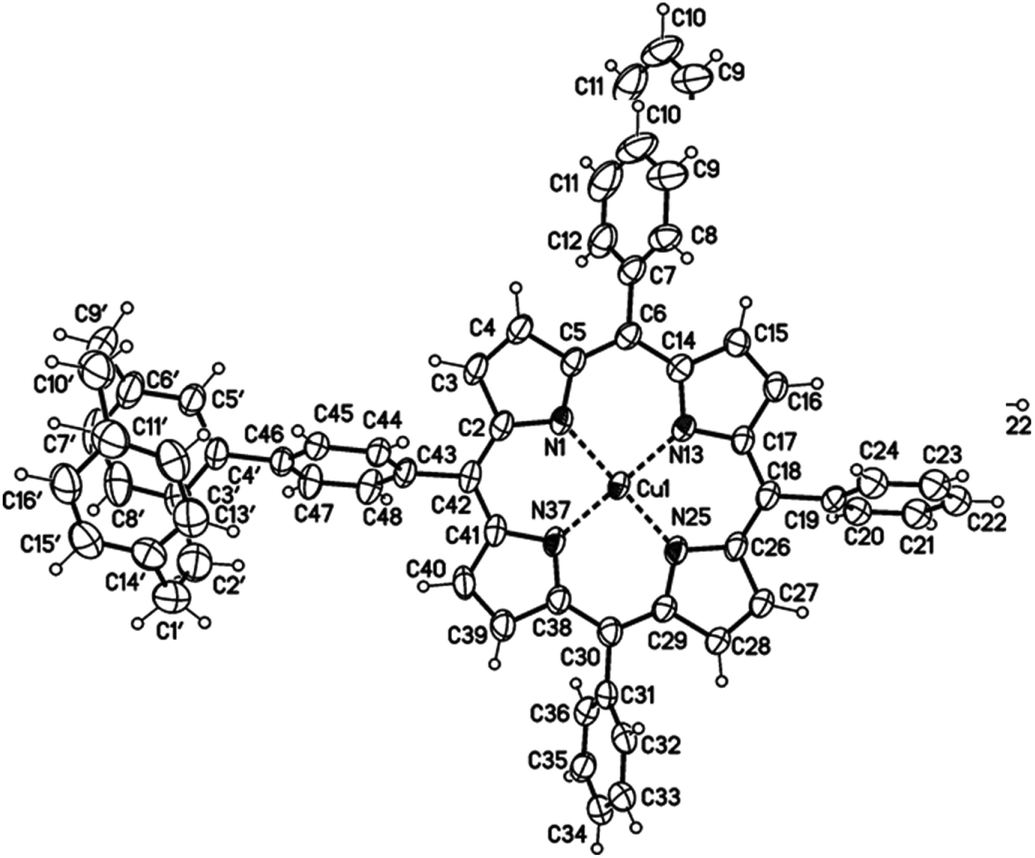 | ||
| Fig. 2 Molecular structure of the paracyclophane–porphyrin conjugate 3a (minor disordered part omitted for clarity, displacement parameters are drawn at 50% probability level; details see ESI†). | ||
In a further approach the functionalities of the two coupling reactants as described in Scheme 1 were mutually exchanged to investigate the comparative reactivity and improve yields, by reacting (rac)-4-bromo[2.2]paracyclophane with the corresponding zinc containing stannyl-porphyrin 6 as nucleophilic reacting partner. The reaction gave 3b with a similarly high yield of 41%.
After successful demonstration of mono-functionalization, disubstitution using the bifunctionalized [2.2]paracyclophane derivatives 4 and 5 was attempted (Scheme 2). Purification of the distannyl [2.2]paracyclophane 4 proved to be challenging and the crude reaction product was used in the synthesis of the pseudo-para disubstituted derivative 7. The impurities that remained are most likely the reason for the significant drop in yield in the reaction with brominated porphyrin 2c. The desired product was obtained in a yield of 2% and serves as a first proof of concept. The functionalities of the reactants were exchanged again, in order to use compounds which are easier to handle. When the porphyrin stannane 6 and the pseudo-para dibromo[2.2]paracyclophane 5, which was synthesized according to a literature known procedure,28 were used, the yield increased up to 30%. The bisporphyrin 7 was characterized via mass spectrometry. Methanol and formic acid needed to be added to the solution in DCM before the compound could be detected with the nano-ESI method in positive mode, because of its low polarity and difficult ionization.
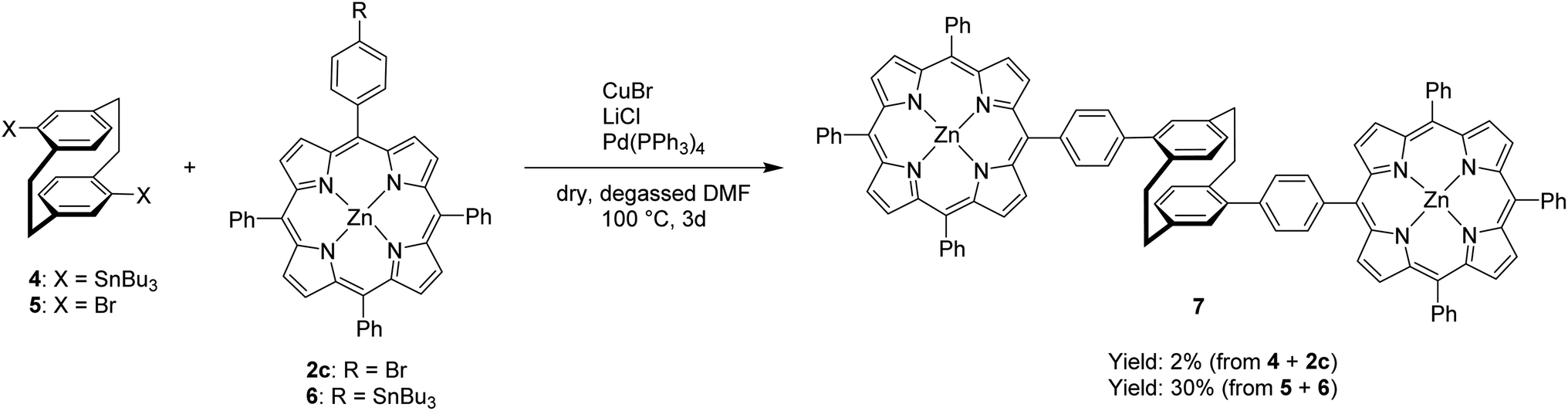 | ||
| Scheme 2 Synthesis of (rac)-4,16-di-(4-((10,15,20-triphenylporphyrin)zinc(II))phenyl) [2.2]paracyclophane(7) | ||
This new class of modular compounds might serve a molecular platform for the investigations of fixed-distance metal–metal interactions, held together by a rigid [2.2]paracyclophane. To access enantiomerically pure mono- and disubstituted PCP derivatives, various procedures of chiral resolution using optimum chiral auxiliaries, for instance, derivatives of L-amino acids, (+)-naproxen, (S)-(−)-camphanoyl chloride, (S)-(+)-10-camphorsulfonic acid, (−)-menthol and others have been reported as promising methodology towards a wide range of precursors in an enantiopure form.29–33 Synthesis of homo- and heterobimetallic pseudo-ortho, -meta and -geminal derivatives of this class and screening of different metals for catalytic activities is currently underway and we believe that may serve to highlight the immense potential for the study of intermetallic cooperative effects in complexes.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Deutsche Forschungsgemeinschaft (DFG) for the support of this work through the collaborative research center SFB/TRR 88“3MET” (Cooperative Effects in homo- und heterometallic complexes), projects B2 and C6.Notes and references
- K. P. C. Vollhardt, and N. E. Schore, Organic Chemistry; Palgrave Version: Structure and Function, Macmillan International Higher Education, 2014 Search PubMed.
- A. E. Allen and D. W. MacMillan, Chem. Sci., 2012, 3, 633–658 RSC.
- D. R. Pye and N. P. Mankad, Chem. Sci., 2017, 8, 1705–1718 RSC.
- J. P. Celli, B. Q. Spring, I. Rizvi, C. L. Evans, K. S. Samkoe, S. Verma, B. W. Pogue and T. Hasan, Chem. Rev., 2010, 110, 2795–2838 CrossRef CAS.
- S. L. Rebelo, M. Linhares, M. M. Simões, A. M. Silva, M. G. P. Neves, J. A. Cavaleiro and C. Freire, J. Catal., 2014, 315, 33–40 CrossRef CAS.
- D. Vlascici, E. Fagadar-Cosma, E. Pica, V. Cosma, O. Bizerea, G. Mihailescu and L. Olenic, Sensors, 2008, 8, 4995–5004 CrossRef CAS.
- M. R. Wasielewski, Acc. Chem. Res., 2009, 42, 1910–1921 CrossRef CAS.
- D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2009, 42, 1890–1898 CrossRef CAS.
- M. O. Senge and X. Feng, J. Chem. Soc., Perkin Trans. 1, 2000, 3615–3621 RSC.
- M. O. Senge, Chem. Commun., 2011, 47, 1943–1960 RSC.
- P. D. Rao, S. Dhanalekshmi, B. J. Littler and J. S. Lindsey, J. Org. Chem., 2000, 65, 7323–7344 CrossRef CAS.
- S. Plunkett and M. O. Senge, ECS Trans., 2011, 35, 147–157 CAS.
- S. Hiroto, Y. Miyake and H. Shinokubo, Chem. Rev., 2016, 117, 2910–3043 CrossRef.
- J. S. Lindsey, Acc. Chem. Res., 2009, 43, 300–311 CrossRef.
- J. L. Sessler, Z. Gross and H. Furuta, Chem. Rev., 2017, 117, 2201–2202 CrossRef CAS.
- M. Kielmann and M. O. Senge, Angew. Chem., Int. Ed., 2019, 58, 418–441 CrossRef CAS.
- J. P. Collman, C. M. Elliott, T. R. Halbert and B. S. Tovrog, Proc. Natl. Acad. Sci., 1977, 74, 18 CrossRef CAS.
- M. Wielopolski, A. Molina-Ontoria, C. Schubert, J. T. Margraf, E. Krokos, J. Kirschner, A. Gouloumis, T. Clark, D. M. Guldi and N. Martín, J. Am. Chem. Soc., 2013, 135, 10372–10381 CrossRef CAS.
- L. Czuchajowski and M. Lozynski, J. Heterocycl. Chem., 1988, 25, 349–350 CrossRef CAS.
- S. Banfi, A. Manfredi, F. Montanari, G. Pozzi and S. Quici, J. Mol. Catal. A: Chem., 1996, 113, 77–86 CrossRef CAS.
- L. Czuchajowski, J. Bennett, S. Goszczynski, D. Wheeler, A. Wisor and T. Malinski, J. Am. Chem. Soc., 1989, 111, 607–616 CrossRef CAS.
- Y. Kobayashi, T. Katayama, T. Yamane, K. Setoura, S. Ito, H. Miyasaka and J. Abe, J. Am. Chem. Soc., 2016, 138, 5930–5938 CrossRef CAS.
- Y. Orimoto, K. Kato and Y. Aoki, J. Phys. Chem. C, 2017, 121, 17703–17711 CrossRef CAS.
- Z. Hassan, E. Spuling, D. M. Knoll, J. Lahann and S. Bräse, Chem. Soc. Rev., 2018, 47, 6947–6963 RSC; Z. Hassan, E. Spuling, D. M. Knoll and S. Bräse, Angew. Chem., Int. Ed., 2019 DOI:10.1002/anie.201904863.
- N. N. Sergeeva, A. Scala, M. A. Bakar, G. O'Riordan, J. O'Brien, G. Grassi and M. O. Senge, J. Org. Chem., 2009, 74, 7140–7147 CrossRef CAS.
- C. Braun, E. Spuling, N. B. Heine, M. Cakici, M. Nieger and S. Bräse, Adv. Synth. Catal., 2016, 358, 1664–1670 CrossRef CAS.
- D. J. Cram and A. C. Day, J. Org. Chem., 1966, 31, 1227–1232 CrossRef CAS.
- H. J. Reich and D. J. Cram, J. Am. Chem. Soc., 1969, 91, 3534–3543 CrossRef CAS.
- G. J. Rowlands and R. J. Seacome, Beilstein J. Org. Chem., 2009, 5, 9 Search PubMed.
- R. Parmar, M. P. Coles, P. B. Hitchcock and G. J. Rowlands, Synthesis, 2010, 4177–4187 CAS.
- R. Zhuravsky, Z. Starikova, E. Vorontsov and V. Rozenberg, Tetrahedron: Asymmetry, 2008, 19, 216–222 CrossRef CAS.
- Y. Morisaki, R. Hifumi, L. Lin, K. Inoshita and Y. Chujo, Chem. Lett., 2012, 41, 990–992 CrossRef CAS.
- Y. Morisaki, M. Gon, T. Sasamori, N. Tokitoh and Y. Chujo, J. Am. Chem. Soc., 2014, 136, 3350–3353 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Cif-files for 3a. CCDC 1877701 (3a). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra07055a |
| ‡ These two authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |