
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Suzuki coupling-based synthesis of VATPase inhibitor archazolid natural product derived fragments†
Cooper T.
Vincent
a,
Evan T.
Long
a,
Holly C.
Jones
a,
Jeffrey C.
Young
b,
P. Clint
Spiegel
a and
Gregory W.
O'Neil
 *a
*a
aDepartment of Chemistry, Western Washington University, Bellingham, WA, USA 98229. E-mail: oneilg@wwu.edu
bDepartment of Biology, Western Washington University, Bellingham, WA, USA 98229
First published on 10th October 2019
Abstract
An archazolid natural product fragment that displays dose-dependent inhibition of the vacuolar-type ATPase (VATPase) has been synthesized by a high-yielding Suzuki coupling of two complex subunits. Similarly, a further simplified fragment was prepared and evaluated for VATPase inhibitory activity. This compound did inhibit the VATPase, as evidenced by growth inhibition of etiolated Arabidopsis seedlings, however at approximately 10× lower potency than the more complex fragment. Cyclooxygenase (COX) enzyme inhibition was not observed for either fragment.
Introduction
Recently our group reported the synthesis of an archazolid natural product fragment (1) that, after removal of the TBS protecting groups, displayed dose-dependent inhibition of the vacuolar-type ATPase (VATPase).1 The VATPase has continued to emerge as a promising therapeutic target associated with several severe forms of cancer, as evidenced by a number of recent studies involving the archazolids.2–10 Key to our synthesis of 1 was a high-yielding Stille coupling between iodide 2 and stannane 3 to complete the conjugated Z,Z,E-triene (Scheme 1).11 This particular triene is unique to the archazolids and happens to link two important pharmacophoric elements, the C7- and C15 hydroxyl groups.12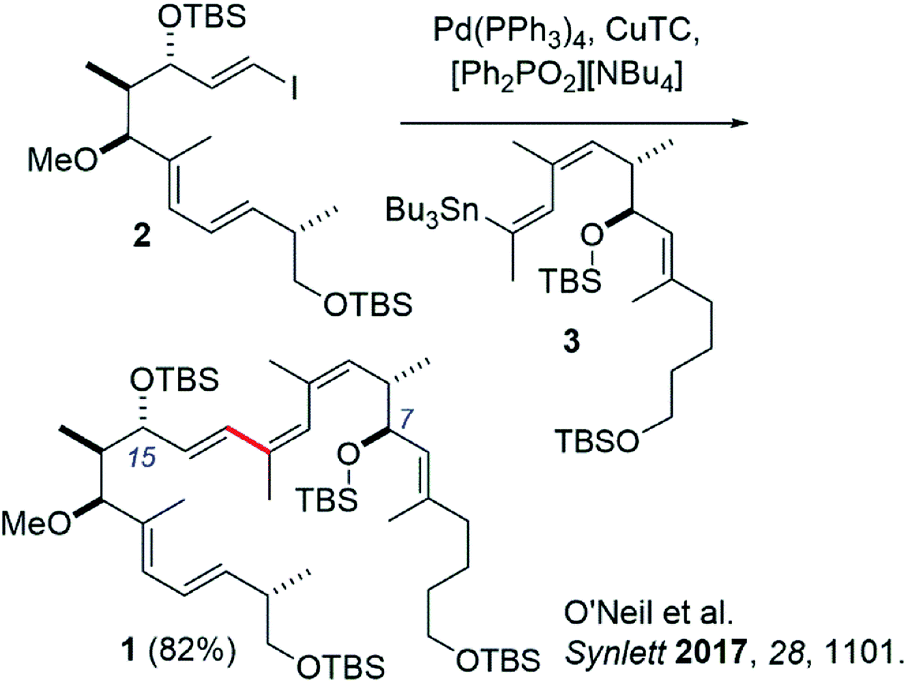 | ||
| Scheme 1 Previous synthesis of archazolid-based VATPase inhibitor compound 1 by Stille coupling. | ||
While promising, others have commented on the drawbacks of Stille couplings due to the toxicity associated with organotin compounds.13,14 In line with these recommendations, we set out to investigate a tin-free synthesis of our VATPase inhibitor lead compound. Herein we report a similarly effective Suzuki coupling strategy to synthesize compound 1, in which tin has been replaced by a boronic acid derivative. We argue that this new approach represents to date the most direct, convergent, and a greener method for synthesizing the important triene region of this exciting class of compounds.
Results and discussion
Tin featured not only in our Stille coupling, but also in our synthesis of vinyl iodide 2,1 which was prepared by palladium catalyzed hydrostannylation followed by iododestannylation (Scheme 2). To circumvent the use of tin in this instance, we opted instead to carry the TMS-protected alkynyl phosphonate 4, prepared in a single step from known Weinreb amide 5,15 into a Horner–Wadsworth–Emmons (HWE) olefination16 with aldehyde 6.17 This reaction delivered ketone 7 in 72% yield and >10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 E:Z selectivity by NMR analysis. The ketone in 7 was diastereoselectively reduced with NaBH4 and the resulting hydroxyl converted to the corresponding methyl ether by deprotonation with LiHMDS and alkylation with methyl iodide. Removal of the TMS group (K2CO3/MeOH) revealed the terminal alkyne in 8 and set the stage for vinyl iodide installation by treatment with Schwartz's reagent and trapping with iodine.18 In this way, vinyl iodide coupling partner 2 was completed in comparable yields and one fewer step from compound 4 than had previously been described (i.e. 6-steps and 36% overall yield vs. 7-steps and 25% yield) while also avoiding the use of tin.
:1 E:Z selectivity by NMR analysis. The ketone in 7 was diastereoselectively reduced with NaBH4 and the resulting hydroxyl converted to the corresponding methyl ether by deprotonation with LiHMDS and alkylation with methyl iodide. Removal of the TMS group (K2CO3/MeOH) revealed the terminal alkyne in 8 and set the stage for vinyl iodide installation by treatment with Schwartz's reagent and trapping with iodine.18 In this way, vinyl iodide coupling partner 2 was completed in comparable yields and one fewer step from compound 4 than had previously been described (i.e. 6-steps and 36% overall yield vs. 7-steps and 25% yield) while also avoiding the use of tin.
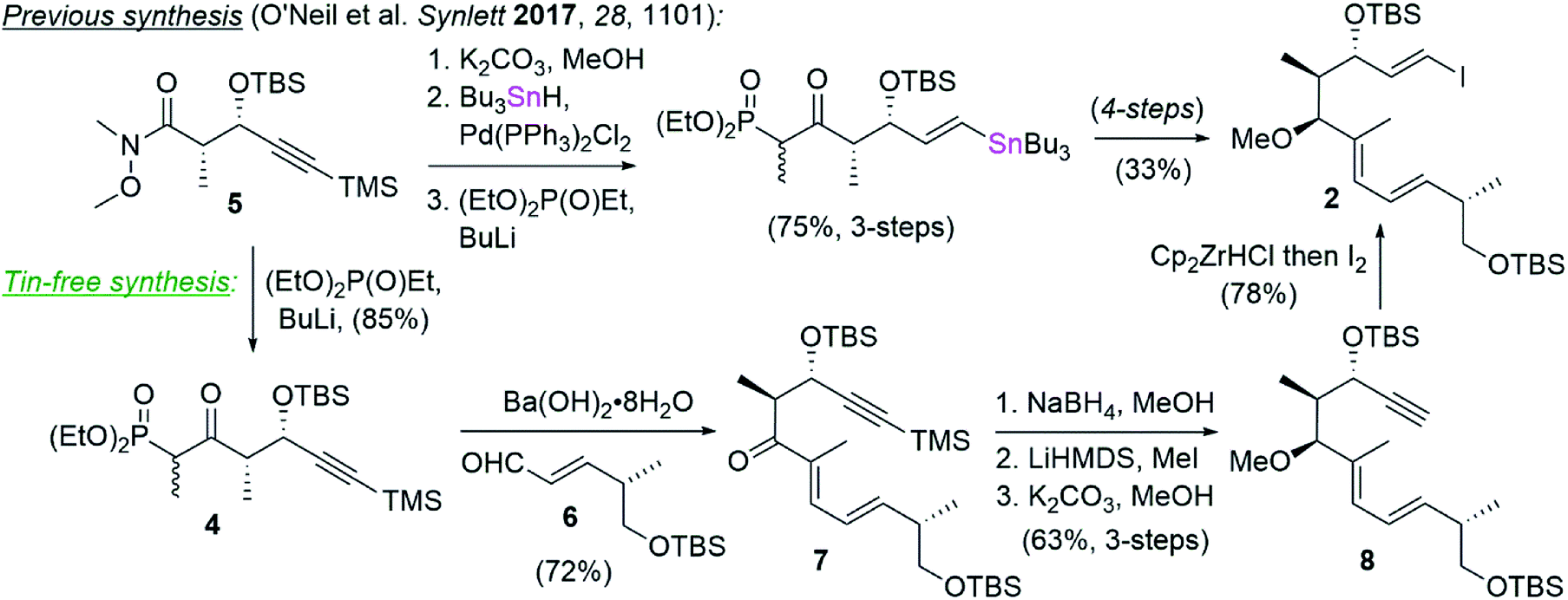 | ||
| Scheme 2 An alternative tin-free synthesis of vinyl iodide 2. | ||
Adapting our vinyl stannane synthesis to incorporate a vinyl boronic ester was possible from advanced vinyl iodide 9 by trapping with B(iPrO)3 rather than Bu3SnCl after lithium–halogen exchange (Scheme 3).19 Treatment with pinacol then allowed for isolation of vinyl pinacol boronate 10 in 93% yield. In an attempt to reduce the step count of our vinyl boronic ester synthesis, we became interested in evaluating a potential boron-Wittig reaction. This chemistry was pioneered by Endo and coworkers20 and more recently has been expanded upon by the Morken group.21,22 To that end, intermediate 11 was acylated with phosphonate acid 1223 using DCC, giving ester 13 in 72% yield. Removal of the PMB protecting group and oxidation of the resulting primary hydroxyl then allowed for an intramolecular HWE reaction. It was found that Ba(OH)2 gave the best overall yields of lactone 14, with some other reagents (e.g. KHMDS, DBU/LiCl) leading primarily to elimination. Reduction of 14 with DIBAL-H produced the corresponding lactol, that we thought could be converted directly to the desired E-vinyl boronic ester 15 by treatment with the lithium anion of a geminal bis(boronate). However, using Morken's procedure21 for the conversion of aldehydes to tri-substituted vinyl pinacol boronates (LiTMP, −78 °C) using 1,1-bis(pinacolboronato)ethane, no reaction was observed.
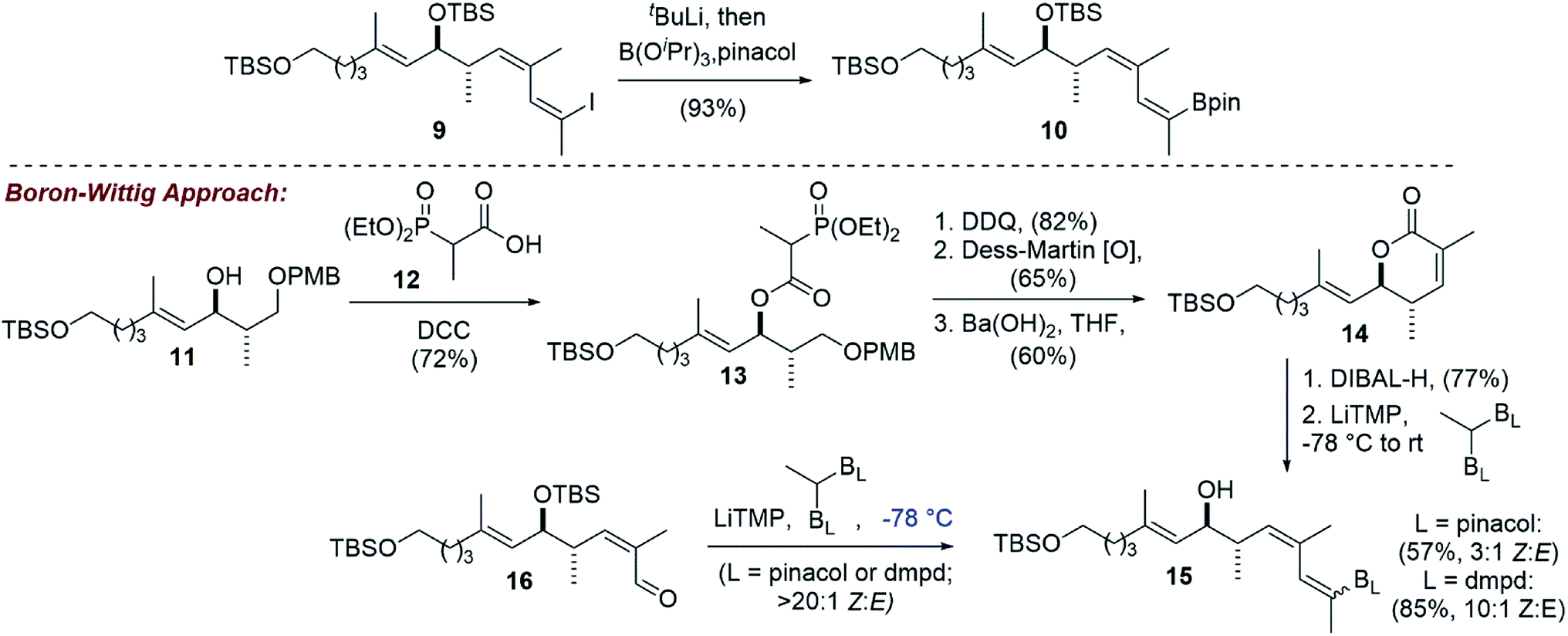 | ||
| Scheme 3 Vinyl boronic ester synthesis by lithium–halogen exchange and an alternative boron-Wittig approach that gave primarily the undesired Z-vinyl boronate. | ||
We reasoned that the lactol was less reactive than an aldehyde in these reactions, and therefore repeated the experiment allowing the reaction to slowly warm to room temperature. Under these conditions, the vinyl boronate ester product 15 was indeed obtained, albeit as a 3:1 mixture of geometrical isomers by NMR analysis. Using 2D-NMR techniques (e.g. COSY and NOESY), it was determined that the major product from this reaction was the undesired Z-vinyl boronate. Morken's group had reported that in some cases switching to the dimethylpentanediolato (dpmd) diboronate led to a reversal of stereoselectivity when compared to the pinacol diboronate.21 Unfortunately, the reaction with 1,1-bis(dimethylpentanediolato)ethane was even more selective for the undesired Z-vinyl boronic ester (10:1 Z:E). When using aldehyde 16, reactions that proceeded at the colder temperatures (−78 °C) prescribed by Endo and Morken,20,21 both diboronates gave exclusively the undesired Z-vinyl boronic ester products. We can perhaps rationalize the stereochemical outcome of these reactions by considering a steric clash between the R group from the substrate and one of the boronic esters after addition of the lithiated diboronate and upon rotation into a conformation with the O- and B-groups syn-coplanar that is required for elimination (Fig. 1).20,22
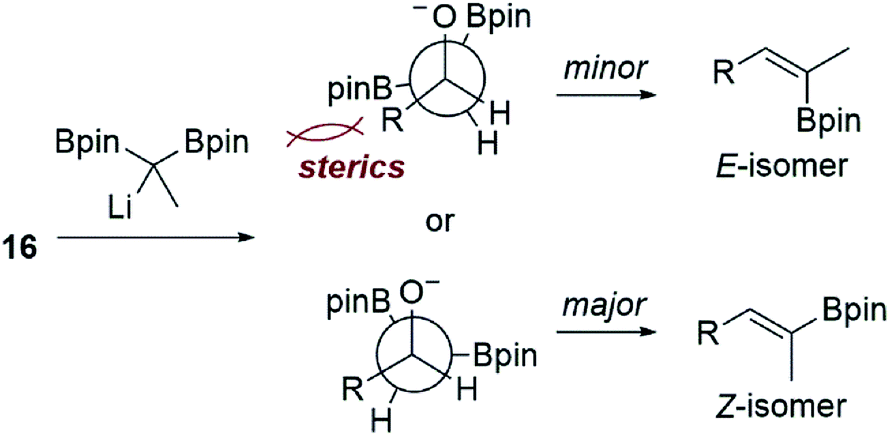 | ||
| Fig. 1 Possible stereochemical rationale for the E:Z selectivity observed in our boron-Wittig reactions with the lactol derived from 13 or alkdehyde 15 shown in Scheme 3. | ||
Nonetheless, the E/Z isomers from the boron-Wittig reactions were separable by chromatography on silica. Combined with our lithium–halogen exchange approach, sufficient quantities of the desired Z,E-boronic esters 10 and 15 were obtained to then investigate a Suzuki coupling with vinyl iodide 2.1 Gratifyingly, it was found that the use of 10 mol% Pd(dppf)Cl2 and Ba(OH)2 as base24 in DMF at 50 °C for 6 h gave the coupled product 1 in comparable yield to what was previously obtained from a Stille coupling1 (83% for the Suzuki vs. 82% from Stille coupling, Scheme 4).
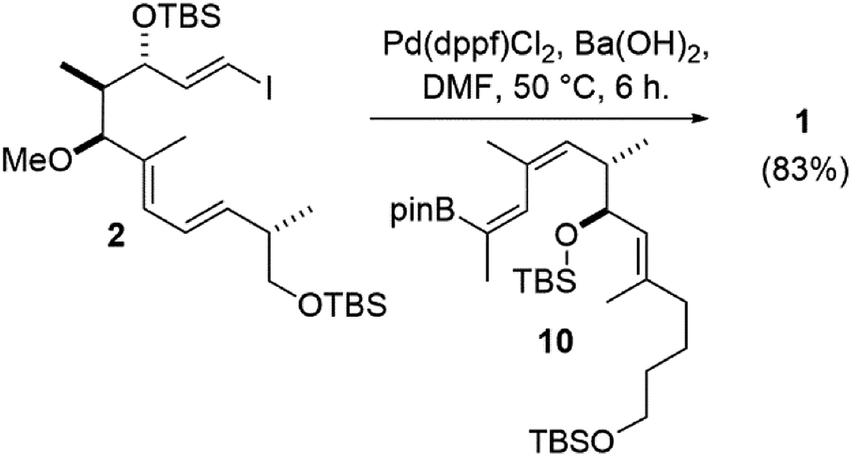 | ||
| Scheme 4 Synthesis of the archazolid conjugated triene by Suzuki coupling. | ||
We also took this opportunity to prepare a further simplified archazolid fragment 17 (Scheme 5). The requisite vinyl iodide 18 was prepared from alkynol 19, available in by enantioselective Noyori reduction25 of the corresponding alkynone.26 Red-Al reduction of the alkyne in 19 to the trans-alkene, TES protection of the alcohol, and iododesilylation of the TMS group gave 18 in 37% yield for the three steps. The Suzuki coupling with boronic ester 10 again proceeded efficiently, in this case using Pd(dppf)Cl2 and Cs2CO3 as base in DMF at room temperature,27 giving the coupled product 17 in 80% yield.
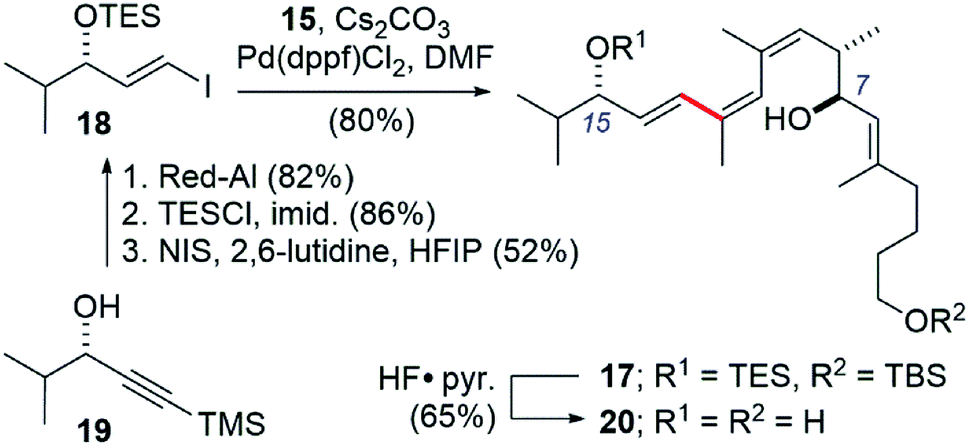 | ||
| Scheme 5 Synthesis of a simplified archazolid fragment 20 containing the pharmacophoric C7- and C15-hydroxyls linked by the Z,Z,E-triene. | ||
Given that 17 contained an intact C7–C15 region of the archazolids, we suspected this compound might exhibit VATPase inhibitory activity and opted to assay the desilylated compound 20 using our Arabidopsis-based method.28,29 As shown in Fig. 2, while 20 did exhibit dose dependent growth inhibition of etiolated Arabidopsis as an indicator for impaired VATPase function, its activity was significantly lower (∼10× less potent) than compound 1. This data suggests that while the C7–C15 segment of the archazolids is important for VATPase binding, additional functionality present in compound 1 also contributes to its effectiveness as a VATPase inhibitor.
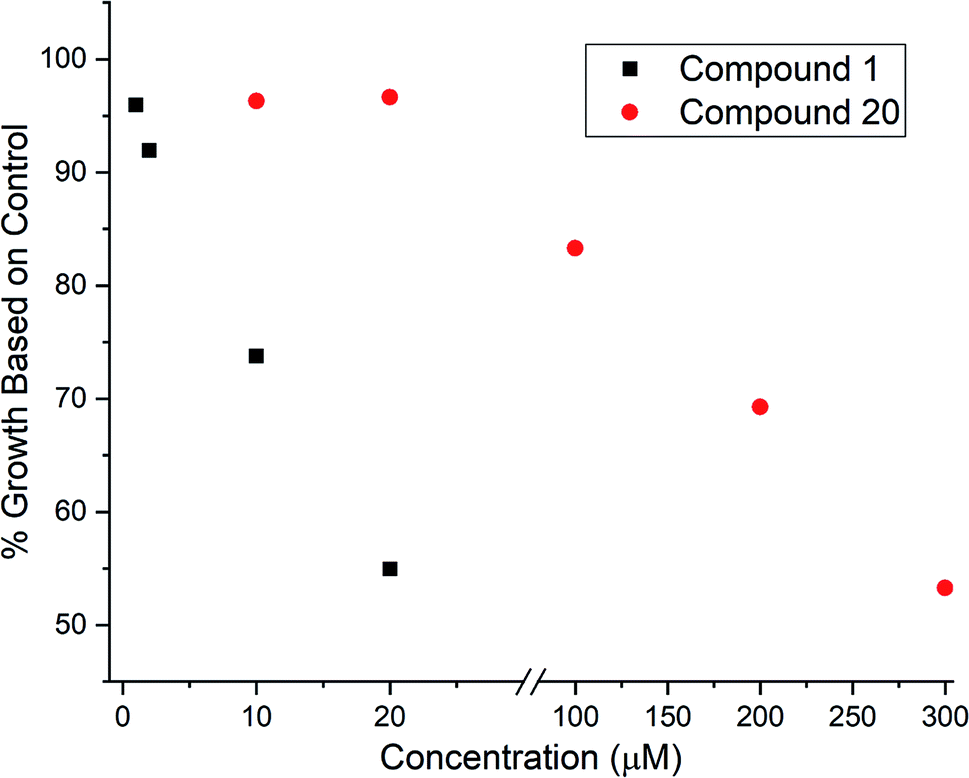 | ||
| Fig. 2 VATPase assay results for compound 1 (after TBS removal) and compound 20 where % growth relative to control represents the average hypocotyl length for 40–60 Arabidopsis seedlings at the concentrations indicated relative to the average hypocotyl length of Arabidopsis seedlings grown in the absence of any inhibitors. | ||
Compound 20 is similar the predicted cyclooxygenase (COX) enzyme inhibitor ArcA-130 (Fig. 3). When tested using a commercial COX screening assay for inhibition of COX-1 and COX-2,31 no measurable inhibition was observed at concentrations up to 200 μM. This was also true for compound 1, which we previously postulated could be due to its large size and/or lack of a carboxylic acid terminus.1 The latter might also apply to compound 20, however neither rationale would explain the modest reported COX inhibitory activity of archazolid A30 which is both sterically large and lacks a carboxylic acid.
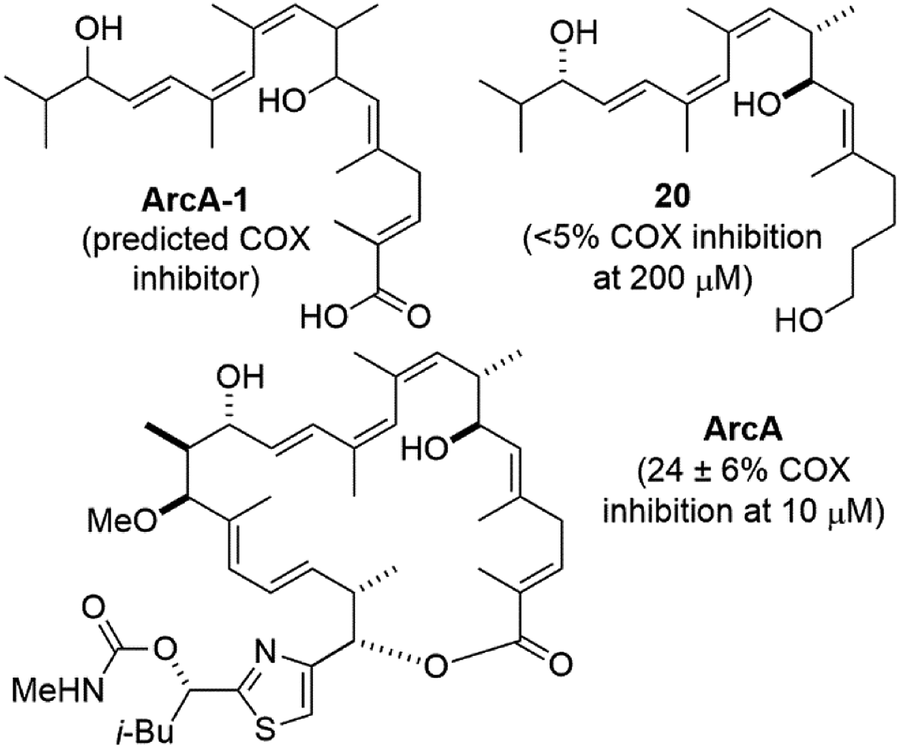 | ||
| Fig. 3 Structures and cyclooxygenase (COX) inhibitory activity of proposed COX inhibitor ArcA-1,30 archazolid A (ArcA), and compound 20. | ||
Conclusions
We have devised and executed a high-yielding Suzuki coupling-based synthesis of the challenging yet biologically important conjugated triene region of the archazolid natural products. The vinyl iodide synthesis reported here represents an improvement over our previous approach in terms of step count, yield, and avoidance of hazardous tin compounds. Two different vinyl boronic ester syntheses were investigated. Ultimately it was found that while a boron-Wittig reaction did give some of the desired compound, the majority of product obtained was the undesired geometrical isomer. The isomers were, however, separable and combined with an alternative iodo-Wittig followed by lithium–halogen exchange-based synthesis, sufficient amounts of the correct boronic ester isomer were obtained to then conduct Suzuki couplings. In this way, an archazolid natural product fragment 1 that had previously exhibited dose dependent VATPase inhibition along with a further simplified fragment 20 were synthesized. Compound 20 also showed dose dependent VATPase inhibition in our Arabidopsis VATPase assay, however with approximately 10× less potency than compound 1. The assay results support the importance of the C7–C15 region of the archazolids for VATPase inhibitory activity, but suggest additional functionality is needed for efficient VATPase inhibition. Compound 20 was also assayed for COX inhibitory activity based on its similarity to a predicted COX inhibitor. However, like compound 1, in our assay no measurable COX inhibition was observed for compound 20. Further studies are underway to better understand the interactions of compounds 1 and 20 with VATPase and COX enzymes toward the design of more efficient inhibitors with therapeutic potential.Experimental section
General information
All reactions were carried out under N2 in flame-dried glassware. IR: Nicolet iS10 spectrometer, wavenumbers (![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) ) in cm−1. The solvents used were dried by passing the solvent through a column of activated alumina under nitrogen immediately prior to use. All reagents were purchased and used as received unless otherwise mentioned. All TLC analysis used 0.25 mm silica layer fluorescence UV254 plates. Flash chromatography: SilaCycle silica gel P60 (230–400 mesh). NMR: spectra were recorded on a Varian Mercury 300, or Inova 500 spectrometer in the solvents indicated; chemical shifts (δ) are given in ppm, coupling constants (J) in Hz. The solvent signals were used as references (C6D6: δC ≡ 128.0 ppm; residual C6H6 in C6D6: δH ≡ 7.16 ppm; CDCl3: δC ≡ 77.0 ppm; residual CHCl3 in CDCl3: δH ≡ 7.26 ppm).
15 (1.0 g, 2.8 mmol) was then added and the reaction was stirred for 30 min before quenching with aq. NH4Cl (50 mL) and extracting with EtOAc (2 × 50 mL). The combined organic extracts were dried over MgSO4, filtered, and concentrated in vacuo. Purification by flash column chromatography on silica (1:1 hexanes:ethyl acetate) afforded phosponate 4 (1.1 g, 85%) as an oil.
) in cm−1. The solvents used were dried by passing the solvent through a column of activated alumina under nitrogen immediately prior to use. All reagents were purchased and used as received unless otherwise mentioned. All TLC analysis used 0.25 mm silica layer fluorescence UV254 plates. Flash chromatography: SilaCycle silica gel P60 (230–400 mesh). NMR: spectra were recorded on a Varian Mercury 300, or Inova 500 spectrometer in the solvents indicated; chemical shifts (δ) are given in ppm, coupling constants (J) in Hz. The solvent signals were used as references (C6D6: δC ≡ 128.0 ppm; residual C6H6 in C6D6: δH ≡ 7.16 ppm; CDCl3: δC ≡ 77.0 ppm; residual CHCl3 in CDCl3: δH ≡ 7.26 ppm).
15 (1.0 g, 2.8 mmol) was then added and the reaction was stirred for 30 min before quenching with aq. NH4Cl (50 mL) and extracting with EtOAc (2 × 50 mL). The combined organic extracts were dried over MgSO4, filtered, and concentrated in vacuo. Purification by flash column chromatography on silica (1:1 hexanes:ethyl acetate) afforded phosponate 4 (1.1 g, 85%) as an oil.
Spectral data for the major diastereomer. IR (ATR): 3036, 29
078, 1710, 1635, 1460, 1364, 1191, 1077, 910, 778 cm−1. 1H NMR (500 MHz, CDCl3): δ 4.24 (d, J = 8.1 Hz, 1H), 4.13 (q, J = 6.4 Hz, 2H), 4.11 (q, J = 6.4 Hz, 2H), 3.70 (dq, JHP = 26.0 Hz, JHH = 6.7 Hz, 1H), 3.30 (m, 1H), 1.33 (d, J = 7.0 Hz, 3H), 1.32 (t, J = 7.0 Hz, 3H), 1.31 (t, J = 7.0 Hz, 3H), 1.14 (d, J = 6.6 Hz, 3H), 0.89 (s, 9H), 0.14 (s, 3H), 0.13 (s, 9H), 1.11 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 207.1, 105.0, 91.2, 65.5, 62.5 (t, JCP = 6.4 Hz, 1C), 54.2, 48.4, 47.5, 25.70, 25.67, 18.2, 16.4 (t, JCP = 5.4 Hz, 1C), 13.2, 10.6 (JCP = 6.2 Hz, 1C), −0.3, −0.4, −4.5, −5.1. HRMS-TOF (ESI+) calcd for C21H43O5PSi2Na+ (M + Na): 485.2284. Found 485.2279.
17 (0.66 g, 2.9 mmol) was then added and the reaction was stirred for 8 h before quenching with aq. NaHCO3 (30 mL) and extracting with MTBE (2 × 30 mL). The combined organic extracts were dried over MgSO4, filtered, and concentrated in vacuo. Purification by flash column chromatography on silica (20:1 to 10:1 hexanes:ethyl acetate) afforded 7 (0.93 g, 72%) as an oil.
[α]20D = −2.8 (c 1.0, CH2Cl2). IR (ATR) 3064, 2903, 1735, 1640, 1350, 1194, 1067, 910, 730 cm−1. 1H NMR (500 MHz, CDCl3): δ 7.07 (d, J = 10.9 Hz, 1H), 6.48 (ddd, J = 15.0, 10.9, 1.1 Hz, 1H), 6.08 (dd, J = 15.2, 7.5 Hz, 1H), 4.38 (d, J = 10.5 Hz, 1H), 3.60–3.47 (m, 3H), 2.50 (p, J = 6.6 Hz, 1H), 1.88 (s, 3H), 1.15 (d, J = 7.0 Hz, 3H), 1.06 (d, J = 7.0 Hz, 3H), 0.91 (s, 9H), 0.89 (s, 9H), 0.14 (s, 3H), 0.11 (s, 3H), 0.06 (s, 9H), 0.05 (s, 3H), 0.04 (s, 3H). 13C NMR (125 MHz, CDCl3): δ13C NMR (126 MHz, CDCl3) δ 203.5, 146.2, 139.3, 134.7, 126.3, 106.1, 90.1, 67.4, 65.7, 46.8, 40.3, 25.9, 25.8, 18.3, 16.3, 14.6, 11.8, −0.4, −4.5, −5.1, −5.3. HRMS-TOF (ESI+) calcd for C29H56O3Si3Na+ (M + Na): 559.3435. Found 559.3428.
:1 to 10:1 hexanes:ethyl acetate) afforded the alcohol (0.93 g, 72%) as an oil.
[α]20D = −4.6 (c 1.0, CH2Cl2). IR (ATR) 3201, 2958, 2930, 2859, 1447, 1371, 1308, 1221, 1143, 1085, 1024, 755, 699 cm−1. 1H NMR (500 MHz, C6D6): δ 6.37 (ddd, J = 15.2, 10.8, 1.2 Hz, 1H), 6.13 (dd, J = 11.0, 1.5 Hz, 1H), 5.58 (dd, J = 15.2, 7.5 Hz, 1H), 5.03 (d, J = 2.5 Hz, 1H), 4.14 (dd, J = 9.0, 2.8 Hz, 1H), 3.49 (dd, J = 9.6, 5.9 Hz, 1H), 3.38 (dd, J = 9.6, 6.8 Hz, 1H), 2.41 (hept, J = 6.7 Hz, 1H), 2.12 (d, J = 2.9 Hz, O–H), 2.00 (dqd, J = 9.3, 6.9, 2.5 Hz, 1H), 1.64 (s, 3H), 1.08 (d, J = 7.0 Hz, 3H), 1.04 (d, J = 6.8 Hz, 3H), 1.02 (s, 9H), 0.97 (s, 9H) 0.29 (s, 3H), 0.19 (s, 3H), 0.17 (s, 9H), 0.05 (s, 6H). 13C NMR (125 MHz, C6D6): δ 137.3, 137.0, 126.1, 107.6, 90.0, 79.6, 68.2, 65.0, 43.0, 40.2, 31.9, 26.1, 26.0, 23.0, 18.52, 18.45, 16.9, 14.3, 11.6, 11.5, −0.1, −4.2, −5.0, −5.20, −5.24. HRMS-TOF (ESI+) calcd for C29H58O3Si3Na+ (M + Na): 561.3591. Found 561.3591.
:1 to 10:1 hexanes:ethyl acetate) afforded 8 (0.11 g, 63% 3-steps) as an oil.
[α]20D = −3.4 (c 1.0, CH2Cl2). IR (ATR) 2958, 2930, 2859, 1447, 1371, 1308, 1221, 1143, 1085, 1024, 755, 699 cm−1. 1H NMR (500 MHz, C6D6): δ 6.39 (dd, J = 15.2, 10.8 Hz, 1H), 6.09 (d, J = 10.0 Hz, 1H), 5.55 (dd, J = 15.2, 7.6 Hz, 1H), 5.22 (t, J = 2.0 Hz, 1H), 3.53 (d, J = 10.0 Hz, 1H), 3.46 (dd, J = 9.7, 6.1 Hz, 1H), 3.37 (dd, J = 9.7, 6.8 Hz, 1H), 3.09 (s, 3H), 2.40 (p, J = 6.9 Hz, 1H), 2.03 (d, J = 2.1 Hz, 1H), 2.02 (m, 1H), 1.59 (d, J = 1.3 Hz, 3H), 1.12 (d, J = 6.9 Hz, 3H), 1.04 (s, 9H), 1.02 (d, J = 6.7 Hz, 3H), 0.97 (s, 9H), 0.29 (s, 3H), 0.18 (s, 3H), 0.04 (s, 6H). 13C NMR (126 MHz, C6D6) δ 137.3, 133.6, 130.3, 125.5, 87.6, 85.7, 72.3, 67.8, 61.8, 55.2, 42.7, 39.9, 25.7, 18.2, 18.1, 16.4, 10.3, 10.0, −4.6, −5.5, −5.57, −5.59. HRMS-TOF (ESI+) calcd for C27H52O3Si2Na+ (M + Na): 503.3353. Found 503.3354.
:1 hexanes:ethyl acetate) afforded 2 (0.1 g, 78%) as an oil.
[α]20D = −7.8 (c 1.0, CH2Cl2). IR (ATR) 2955, 2928, 2865, 1450, 1370, 1310, 1235, 1121, 1065, 978, 884, 755, 699 cm−1. 1H NMR (500 MHz, CDCl3): δ 6.55 (dd, J = 14.4, 6.0 Hz, 1H), 6.29 (ddd, J = 15.2, 10.8, 1.2 Hz, 1H), 6.19 (dd, J = 14.4, 1.3 Hz, 1H), 5.89 (dd, J = 10.7, 1.5 Hz, 1H), 5.60 (dd, J = 15.3, 7.4 Hz, 1H), 4.62 (d, J = 6.0 Hz, 1H), 3.51 (dd, J = 9.7, 6.4 Hz, 1H), 3.43 (dd, J = 9.7, 6.9 Hz, 1H), 3.31 (d, J = 9.9 Hz, 1H), 3.11 (s, 3H), 2.40 (p, J = 6.7 Hz, 1H), 1.62 (m, 1H), 1.59 (s, 3H), 1.03 (d, J = 6.8 Hz, 3H), 0.92 (s, 9H), 0.89 (s, 9H), 0.63 (d, J = 7.0 Hz, 3H), 0.06 (s, 3H), 0.04 (s, 3H), 0.03 (s, 3H), 0.02 (s, 3H). 13C NMR (126 MHz, CDCl3): δ 149.2, 137.3, 133.4, 130.3, 125.4, 87.9, 75.5, 73.2, 67.9, 55.5, 41.2, 39.7, 29.7, 18.4, 18.2, 16.5, 10.5, 8.9, −4.1, −5.29, −5.32. HRMS-TOF (ESI+) calcd for C27H53IO3Si2Na+ (M + Na): 631.2476. Found 631.2476.
:1 hexanes:ethyl acetate) afforded 10 (45 mg, 93%) as an oil.
[α]20D = +3.2 (c 1.0, CH2Cl2). IR (ATR) 3056, 2973, 1640, 1565, 1350, 1194, 1067, 910, 730 cm−1. 1H NMR (500 MHz, CDCl3): δ 6.70 (s, 1H), 5.10 (dd, J = 9.1, 1.2 Hz, 1H), 5.05 (d, J = 9.8 Hz, 1H), 4.16 (dd, J = 9.0, 5.5 Hz, 1H), 3.60 (t, J = 6.2 Hz, 2H), 2.65 (ddd, J = 9.7, 6.9, 5.6 Hz, 1H), 1.97 (s, 2H), 1.88 (d, J = 1.6 Hz, 3H), 1.85 (d, J = 1.4 Hz, 3H), 1.57 (d, J = 1.4 Hz, 3H), 1.53–1.40 (m, 4H), 1.28 (s, 9H), 0.89 (s, 12H), 0.87 (d, J = 7.0 Hz, 3H), 0.85 (s, 9H), 0.04 (s, 6H), −0.01 (s, 3H), −0.04 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 138.1, 135.3, 133.4, 132.7, 127.2, 83.3, 72.9, 63.0, 39.4, 39.4, 32.5, 26.0, 25.90, 25.86, 24.8, 24.7, 24.0, 23.9, 23.0, 18.4, 18.2, 16.7, 16.0, −4.3, −4.8, −5.3. HRMS-TOF (ESI+) calcd for C34H67BO4Si2Na+ (M + Na): 629.4569. Found 629.4572.
:1 to 1:1 to 1:2 hexanes:ethyl acetate) afforded ester 13 (604 mg, 82%) as an oil.
Spectral data for the mixture of diastereomers. IR (ATR) 2931, 2856, 1730, 1612, 1586, 1513, 1460, 1386, 1301, 1246, 1172, 1093, 1023, 962, 904, 834, 818, 774, 734, 661 cm−1. 1H NMR (500 MHz, CDCl3): δ 7.25–7.22 (m, 4H), 6.86 (d, J = 8.7 Hz, 4H), 5.61 (dd, J = 9.7, 6.5 Hz, 1H), 5.57 (dd, J = 9.7, 6.7 Hz, 1H), 5.09 (d, J = 9.7 Hz, 2H), 4.41 (d, J = 10.2 Hz, 2H), 4.37 (d, J = 10.2 Hz, 2H), 4.16–4.07 (m, 8H), 3.80 (s, 6H), 3.59 (t, J = 5.9 Hz, 4H), 3.42 (dd, J = 9.3, 5.8 Hz, 1H), 3.35 (dd, J = 9.3, 6.1 Hz, 1H), 3.30 (dd, J = 9.3, 6.3 Hz, 1H), 3.31 (dd, J = 9.3, 6.1 Hz, 1H), 2.95 (dq, J = 23.3, 7.4 Hz, 2H), 2.16 (m, 12H), 2.11 (m, 2H), 2.02 (m, 4H), 1.72 (s, 6H), 1.47–1.36 (m, 8H), 1.33–1.27 (m, 6H), 0.93 (d, J = 7.0 Hz, 3H), 0.92 (d, J = 7.0 Hz, 3H), 0.88 (s, 18H), 0.03 (s, 12H). 13C NMR (125 MHz, CDCl3): δ 159.1, 142.3, 130.6, 129.2, 120.4, 113.72, 113.70, 73.7, 72.7, 71.7, 62.9, 62.5, 55.3, 40.0, 39.5, 37.8, 32.41, 32.35, 26.0, 24.0, 18.4, 16.8, 12.7, 11.8, −5.3. HRMS-TOF (ESI+) calcd for C32H57O8PSiNa+ (M + Na): 651.3458. Found 651.3458.
:H2O mixture (20 mL:0.2 mL), DDQ (180 mg, 0.795 mmol) was added portion wise and stirred at room temperature for 1 h. The resulting solution was quenched with aqueous sodium bicarbonate (50 mL) and extracted with DCM (2 × 50 mL). The combined organic extracts were dried over MgSO4, filtered and concentrated in vacuo. Purification by flash column chromatography on silica (1:2 to 0:1 hexanes:ethyl acetate) afforded the corresponding alcohol (168 mg, 83%) as an oil.
Spectral data for the mixture of diastereomers. IR (ATR) 3406, 2928, 2856, 1471, 1455, 1384, 1360, 1253, 1100, 1081, 1030, 1002, 977, 935, 834, 773, 737, 661 cm−1. 1H NMR (500 MHz, CDCl3): δ 5.43 (dd, J = 8.7, 8.7 Hz, 1H), 5.39 (dd, J = 9.4, 9.4 Hz, 1H), 5.11 (d, J = 9.2 Hz, 1H), 5.06 (d, J = 9.8 Hz, 1H), 4.17–4.09 (m, 8H), 3.75 (dd, J = 11.5, 3.3 Hz, 1H), 3.64 (dd, J = 11.5, 3.8 Hz, 1H), 3.58 (t, J = 6.1 Hz, 4H), 3.50 (dd, J = 11.0, 4.9 Hz, 1H), 3.48 (dd, J = 11.5, 4.5 Hz, 1H), 2.99 (dq, J = 23.0, 7.5 Hz, 2H), 2.00 (m, 12H), 1.83 (m, 2H), 1.73 (d, J = 1.2 Hz, 3H), 1.71 (d, J = 1.2 Hz, 3H), 1.70 (m, 2H), 1.65 (m, 2H), 1.48–1.41 (m, 4H), 1.39 (d, J = 7.0 Hz, 3H), 1.35 (d, J = 7.0 Hz, 3H), 1.34–1.28 (m, 4H), 0.91 (d, J = 7.0 Hz, 3H), 0.89 (d, J = 7.0 Hz, 3H), 0.87 (s, 18H), 0.02 (s, 12H). 13C NMR (125 MHz, CDCl3): δ 169.3, 141.9, 122.3, 75.6, 75.1, 64.2, 62.9, 40.3, 39.4, 32.3, 26.0, 23.9, 18.3, 16.8, 16.4, 13.8, 11.7, −5.3. HRMS-TOF (ESI+) calcd for C24H49O7PSiNa+ (M + Na): 531.2883. Found 531.2883.
Ba(OH)2·8H2O (340 mg, 1.1 mmol) was heated at 120 °C for 1.5 hours under vacuum and then allowed to cool to room temperature. THF (7 mL) was added to the flask and the solution was stirred for 10 minutes before adding the aldehyde (181 mg, 0.36 mmol) and the resulting mixture was stirred at room temperature for 15 h. The reaction was quenched with aq. NaHCO3 (20 mL) and extracted with MTBE (2 × 20 mL). The combined organic extracts were dried over MgSO4, filtered and concentrated in vacuo. Purification by flash column chromatography on silica (20:1 to 10:1 to 4:1 hexanes:ethyl acetate) afforded lactone 14 (72.2 mg, 57%) as an oil.
[α]20D = −2.8 (c 1.0, CH2Cl2). IR (ATR) 2952, 2928, 2856, 1716, 1460, 1359, 1253, 1226, 1199, 1154, 1134, 1099, 1005, 981, 834, 810, 773, 731, 661. 1H NMR (500 MHz, CDCl3): δ 6.34 (s, 1H), 5.22 (d, J = 9.0 Hz, 1H), 4.68 (dd, J = 10.5, 9.0 Hz, 1H), 3.57 (m, 2H), 2.45 (ddtt, J = 14.6, 7.2, 4.8, 2.4 Hz, 1H), 2.03 (t, J = 5.4 Hz, 2H), 1.87 (dd, J = 2.4, 1.5 Hz, 3H), 1.67 (d, J = 1.5 Hz, 3H), 1.50–1.41 (m, 3H), 0.98 (d, J = 7.3 Hz, 3H), 0.85 (d, J = 6.0 Hz, 1H), 0.85 (s, 9H), 0.00 (s, 6H). 13C NMR (125 MHz, CDCl3): δ 165.96, 145.52, 143.82, 127.37, 121.50, 80.36, 62.86, 39.24, 34.59, 32.30, 25.95, 23.78, 18.31, 16.88, 15.94, −5.30. HRMS-TOF (ESI+) calcd for C20H36O3SiH+ (M + H): 353.2512. Found 353.2516.
1H NMR (500 MHz, CDCl3): δ 5.48 (s, 1H), 5.16 (m, 2H), 4.24 (t, J = 9.3 Hz, 1H), 3.61 (t, J = 6.0 Hz, 3H), 2.89 (s, 1H), 2.09 (m, 1H), 2.06 (m, 2H), 1.73 (m, 3H), 1.71 (d, J = 1.5 Hz, 3H), 1.50 (m, 3H), 0.89 (s, 9H), 0.86 (d, J = 7.1 Hz, 3H), 0.04 (s, 6H). 13C NMR (125 MHz, CDCl3): δ 141.98, 132.03, 129.75, 12.65, 91.94, 69.63, 63.01, 39.38, 35.14, 32.41, 25.97, 23.96, 18.93, 18.35, 16.95, 16.38, −5.27.
To a solution of tetramethylpiperidine (137 mg, 0.97 mmol) in THF (0.5 mL) at −78 °C was added n-BuLi (0.388 mL, 2.5 M). The solution was allowed to warm to 0 °C before adding 1,1-bis(pinacolboronato)ethane (187 mg, 0.65 mmol). After 10 minutes, the lactol (114.6 mg, 0.32 mmol) was added and the solution was allowed to slowly warm to room temperature and stirred for 15 h. The solution was diluted with diethyl ether (10 mL), filtered through a silica plug, and concentrated in vacuo. Purification by flash column chromatography on silica (20:1 to 10:1 to 4:1 hexanes:ethyl acetate) afforded the vinyl boronate 15 (90 mg, 57%) as a 3:1 mixture of Z:E isomers.
Spectral data for Z-15. IR (ATR) 2977, 2933, 2879, 1460, 1305, 1268, 1215, 1142, 1105, 1016, 967, 846, 775, 736, 669. 1H NMR (500 MHz, CDCl3): δ 6.74 (s, 1H), 5.17–5.09 (m, 2H), 4.04 (dd, J = 8.9, 7.5 Hz, 1H), 3.61 (t, J = 5.9 Hz, 2H), 2.34 (m, 1H), 2.03 (m, 2H), 1.84 (s, 3H), 1.71 (s, 3H), 1.65 (s, 3H) 1.53–1.43 (m, 4H), 1.29 (s, 12H), 1.22 (d, J = 3.8 Hz, 3H), 0.89 (s, 9H), 0.04 (s, 6H). 13C NMR (125 MHz, CDCl3): δ 142.5, 139.7, 136.5, 130.3, 125.7, 83.1, 72.2, 63.1, 39.5, 32.5, 29.7, 26.0, 24.8, 24.0, 23.7, 18.4, 16.7, 16.2, 15.4, 10.4, −5.3. HRMS-TOF (ESI+) calcd for C28H53BO4SiNa+ (M + Na): 515.3704. Found 515.3706.
Spectral data for E-15. 1H NMR (500 MHz, CDCl3): δ 6.68 (s, 1H), 5.13 (d, 9.24 Hz, 1H) 5.07 (d, J = 9.24 Hz, 1H), 4.01 (t, J = 8.46 Hz, 1H), 3.60 (m, 2H), 2.50 (m, 1H), 2.04 (d, J = 5.90 Hz, 2H), 1.88 (dd, J = 5.34, 1.34 Hz, 4H) 1.66 (s, 3H), 1.48 (m, 1H) 1.24 (d, J = 3.31 Hz, 3H), 1.21 (d, 4.15 Hz, 12H), 1.04 (d, J = 7.31 Hz, 2H), 0.89 (s, 9H), 0.84 (d, J = 6.73 Hz, 2H), 0.72 (q, J = 7.29 Hz, 1H) 0.04 (s, 6H). 13C NMR (125 MHz, CDCl3): δ 140.12, 139.32, 136.49, 130.68, 126.10, 82.85, 72.41, 63.01, 40.19, 39.50, 32.45, 25.94, 24.81, 24.51, 24.08, 23.94, 23.23, 18.31, 16.80, 9.03, −5.30.
:1 to 10:1 hexanes:ethyl acetate) afforded the coupled product 1 (77 mg, 80%) as an oil.
[α]20D = −5.2 (c 0.5, CH2Cl2). IR (ATR) 3062, 2983, 1736, 1614, 1415, 1274, 1267, 1129, 1078, 930 cm−1. 1H NMR (500 MHz, C6D6): δ 6.80 (d, J = 16.0 Hz, 1H), 6.44 (ddd, J = 15.3, 10.7, 0.9 Hz, 1H), 6.17 (d, J = 10.4 Hz, 1H), 6.02 (s, 1H), 5.90 (dd, J = 16.0, 7.2 Hz, 1H), 5.58 (dd, J = 15.4, 8.0 Hz, 1H), 5.34 (dd, J = 8.4, 0.8 Hz, 1H), 5.29 (ddd, J = 9.8, 2.0, 1.0 Hz, 1H), 5.07 (d, J = 7.4 Hz, 1H), 4.29 (dd, J = 9.0, 6.0 Hz, 1H), 3.63 (d, J = 10.0 Hz, 1H), 3.58–3.53 (m, 2H), 3.47 (dd, J = 9.7, 6.2 Hz, 1H), 3.38 (dd, J = 9.7, 6.8 Hz, 1H), 3.18 (s, 3H), 2.71 (m, 1H), 2.42 (m, 1H), 1.96 (m, 2H), 1.91 (s, 3H), 1.86 (m, 1H), 1.89 (d, J = 1.0 Hz, 3H), 1.71 (d, J = 1.0 Hz, 3H), 1.59 (s, 3H), 1.50 (m, 2H), 1.39 (m, 2H), 1.08 (s, 6H), 1.03 (s, 6H), 1.02 (s, 3H), 1.01 (s, 6H), 1.00 (s, 3H), 0.99 (d, J = 7.0 Hz, 3H), 0.98 (s, 3H), 0.96 (d, J = 7.0 Hz, 3H), 0.95 (s, 6H), 0.94 (d, J = 7.0 Hz, 3H), 0.93 (s, 3H), 0.19 (s, 3H), 0.17 (s, 3H), 0.13 (s, 6H), 0.08 (s, 6H), 0.05 (s, 6H). 13C NMR (125 MHz, C6D6): δ13C NMR (126 MHz, C6D6) δ 137.8, 136.0, 134.9, 134.7, 133.4, 133.3, 133.0, 132.5, 130.9, 130.3, 126.3, 125.4, 73.8, 72.9, 68.5, 63.4, 63.4, 56.0, 43.7, 41.5, 40.6, 40.1, 33.1, 31.4, 30.3, 28.5, 26.7, 26.6, 26.5, 25.4, 24.8, 24.8, 24.0, 21.0, 19.1, 18.9, 18.8, 17.4, 17.2, 17.1, 16.2, 14.3, 11.2, 9.8, 8.7, −3.0, −3.6, −4.2, −4.3, −4.4, −4.7, −4.8, −4.9. HRMS (ESI+): calcd for C55H108O5Si4Na+ (M + Na)+: 983.7172. Found 983.7179.
26 (1.0313 g, 6.05 mmol) in diethyl ether (15 mL) at 0 °C and the mixture was stirred to room temperature for 15 hours. The reaction was quenched with ethyl acetate (50 mL) and stirred vigorously with aq. Rochelle's salt (50 mL, 2.0 M) for 1 hour. The layers were separated and the aqueous phase was extracted with ethyl acetate (2 × 50 mL). The combined organic fractions were dried over MgSO4, filtered through cotton, and concentrated under vacuum. Purification by flash chromatography on silica (20:1 to 10:1 hexanes:ethyl acetate) gave the trans-alkene product (0.859 g, 4.98 mmol, 82.4%) as an oil.
[α]20D = +20.2 (c 1.0, CH2Cl2). IR (ATR): 3355, 3057, 3025, 2956, 2923, 1721, 1621, 1601, 1582, 1493, 1452, 1247, 1025, 989, 866, 835, 762, 742, 694. 1H NMR (500 MHz, CDCl3): δ 6.06 (dd, J = 9.5, 5.6 Hz, 1H), 5.88 (dd, J = 9.4, 1.3 Hz, 1H), 3.88 (s, 1H), 1.76 (o, J = 6.9 Hz, 1H), 0.94 (d, J = 7.1 Hz, 3H), 0.92 (d, J = 7.2 Hz, 3H), 0.10 (s, 9H). 13C NMR (500 MHz, CDCl3): δ 146.98, 130.48, 79.61, 33.55, 18.35, 17.64, −1.26. HRMS (ESI+): calcd for C9H20OSiNa+ (M + Na)+: 195.1181. Found 195.1180.
:1 hexanes:ethyl acetate) gave the TES-protected alcohol (0.1453 g, 0.501 mmol, 86.3%) as an oil.
[α]20D = +18.0 (c 1.0, CH2Cl2). IR (ATR): 2954, 2911, 2876, 1620, 1459, 1414, 1259, 1059, 1004, 992, 872, 834, 721, 691. 1H NMR (500 MHz, CDCl3): δ 5.97 (dd, J = 18.7, 6.5 Hz, 1H), 5.75 (dd, J = 18.8, 1.1 Hz, 1H), 3.77 (ddd, J = 6.4, 1.0 Hz, 1H), 1.66 (o, J = 6.5 Hz, 1H), 0.96 (t, J = 8.0 Hz, 9H), 0.90 (d, J = 6.8, 3H), 0.85 (d, J = 6.8 Hz, 3H), 0.59 (dq, J = 7.7, 2.5 Hz, 6H), 0.08 (s, 9H). 13C NMR (500 MHz, CDCl3): δ 147.91, 129.98, 81.17, 34.35, 18.39, 18.18, 6.88, 5.00, −1.32. HRMS (ESI+): calcd for C15H34OSi2Na+ (M + Na)+: 309.2046. Found 309.2045.
:1 hexanes:ethyl acetate) gave 18 (0.0885 g, 0.260 mmol, 52.0%) as an oil.
[α]20D = +8.2 (c 0.5, CH2Cl2). IR (ATR): 2955, 2910, 2875, 1607, 1459, 1414, 1384, 1364, 1238, 1182, 1162, 1118, 1103, 1071, 1004, 948, 828, 723. 1H NMR (500 MHz, CDCl3): δ 6.53 (dd, J = 14.4, 6.8 Hz, 1H), 6.20 (dd, J = 14.4, 1.1 Hz, 1H), 3.83 (ddd, J = 6.7, 5.7, 1.1 Hz), 1.69 (do, J = 6.8, 1.1 Hz, 1H), 0.97 (t, J = 8.0 Hz, 9H), 0.91 (d, J = 6.8, 3H), 0.87 (d, J = 6.8, 3H), 0.60 (q, J = 8.2 Hz, 6H). 13C NMR (500 MHz, CDCl3): 147.81, 80.18, 76.26, 34.34, 18.01, 17.82, 6.82, 4.90. HRMS (ESI+): calcd for C12H25IOSiNa+ (M + Na)+: 363.0617. Found 363.0612.
:1 to 4:1 hexanes:ethyl acetate) afforded 17 (93 mg, 80%) as an oil. The product was unable to be purified away from a side product and this mixture was taken direction into the next reaction.
[α]20D = −3.2 (c 0.5, CH2Cl2). IR (ATR) 2955, 2930, 2875, 1460, 1352, 1344, 1254, 1143, 1103, 1004, 969, 835, 774, 741, 724, 666. 1H NMR (500 MHz, CDCl3): δ 6.37 (d, J = 15.7 Hz, 1H), 5.84 (s, 1H), 5.64 (dd, J = 15.7, 7.9 Hz, 1H), 5.16 (d, 9.8 Hz, 1H), 5.10 (d, J = 8.8 Hz, 1H), 4.02 (t, J = 8.8 Hz, 1H), 3.82 (t, J = 7.0 Hz, 1H), 3.60 (t, J = 5.9 Hz, 2H), 2.34 (m, 1H), 2.02 (m, 2H), 1.86 (s, 3H), 1.84 (s, 3H), 1.67 (m, 1H), 1.66 (s, 3H), 1.53–1.43 (m, 4H), 1.24 (d, J = 3.8 Hz, 3H), 0.93 (t, J = 7.9 Hz, 9H), 0.89 (s, 9H), 0.84 (d, J = 5.8 Hz, 3H), 0.82 (d, J = 5.8 Hz, 3H), 0.56 (q, J = 7.9 Hz, 6H) 0.04 (s, 6H). 13C NMR (125 MHz, CDCl3): δ 139.8, 135.1, 132.4, 130.9, 129.0, 128.7, 125.7, 82.9, 72.3, 63.0, 40.5, 39.5, 35.1, 32.5, 26.0, 24.8, 24.6, 20.4, 18.4, 16.7, 16.4, 9.1, 6.9, 5.0, −5.3. HRMS (ESI+): calcd for C34H66O3Si2Na+ (M + Na)+: 601.4448. Found 601.4446.
:1 to 1:2 hexanes:ethyl acetate) afforded 20 (30.1 mg, 65%) as an oil.
[α]20D = −2.8 (c 0.5, CH2Cl2). IR (ATR) 3338, 2957, 2929, 2870, 1667, 1638, 1585, 1447, 1378, 1366, 1328, 1257, 1203, 1175, 1136, 1106, 1065, 1003, 973, 935, 858, 817, 769, 736, 697, 667. 1H NMR (500 MHz, C6D6): δ 6.84 (d, J = 15.8, 1H), 5.78 (dd, J = 15.8, 6.6 Hz, 1H), 5.73 (s, 1H), 5.27–5.24 (m, 2H), 4.12 (t, J = 8.0 Hz, 1H), 3.90 (t, J = 6.2 Hz, 1H), 3.40 (m, 2H), 2.60 (m, 1H), 1.89 (m, 2H), 1.81 (s, 3H), 1.78 (s, 3H), 1.57 (s, 3H), 1.40–1.34 (m, 5H), 1.03 (d, J = 6.2 Hz, 3H) 0.95 (dd, J = 11.6, 7.1 Hz, 6H). 13C NMR (125 MHz, C6D6): δ 138.5, 133.6, 132.8, 132.0, 130.5, 129.1, 77.7, 72.5, 62.1, 40.4, 39.1, 34.1, 32.1, 24.4, 23.7, 19.9, 18.5, 18.1, 16.8, 16.4. HRMS (ESI+): calcd for C22H38O3Na+ (M + Na)+: 373.2719. Found 373.2711.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Camille & Henry Dreyfus Foundation is gratefully acknowledged.Notes and references
- G. W. O'Neil, A. M. Craig, J. R. Williams, J. C. Young and P. C. Spiegel, Synlett, 2017, 28, 1101 CrossRef.
- K. von Schwarzenberg, T. Lajtos, L. Simon, R. Mueller, G. Vereb and A. M. Vollmar, Mol. Oncol., 2014, 8, 9 CrossRef CAS.
- R. Hamm, M. Zeino, S. Frewert and T. Efferth, Toxicol. Appl. Pharmacol., 2014, 281, 78 CrossRef CAS.
- S. Zhang, L. S. Schneider, B. Vick, M. Grunert, I. Jeremias, D. Menche, R. Müller, A. M. Vollmar and J. Liebl, OncoTargets Ther., 2015, 6, 43508 Search PubMed.
- L. S. Schneider, K. von Schwarzenberg, T. Lehr, M. Ulrich, R. Kubisch-Dohmen, J. Liebl, D. Trauner, D. Menche and A. M. Vollmar, Cancer Res., 2015, 75, 2863 CrossRef CAS PubMed.
- O. Scherer, H. Steinmetz, C. Kaether, C. Weinigel, D. Barz, H. Kleinert, D. Menche, R. Müller, C. Pergola and O. Werz, Biochem. Pharmacol., 2014, 91, 490 CrossRef CAS.
- L. S. Schneider, M. Ulrich, T. Lehr, D. Menche, R. Müller and K. von Schwarzenberg, Mol. Oncol., 2016, 10, 1054 CrossRef CAS.
- L. Thomas, Z. Rao, J. Gerstmeier, M. Raasch, C. Weinigel, S. Rummler, D. Menche, R. Müller, C. Pergola, A. Mosig and O. Werz, Biochem. Pharmacol., 2017, 130, 71 CrossRef CAS.
- K. Bartel, M. Winzi, M. Ulrich, A. Koeberle, D. Menche, O. Werz, R. Müller, J. Guck, A. M. Vollmar and K. von Schwarzenberg, OncoTargets Ther., 2016, 8, 9576 Search PubMed.
- B. Luong, R. Schwenk, J. Bräutigam, R. Müller, D. Menche, I. Bischoff and R. Fürst, PLoS One, 2018, 13, e0203053 CrossRef PubMed.
- For alternative approaches to synthesizing the archazolid conjugated triene see: (a) D. Menche, J. Hassfeld, J. Li and S. Rudolph, J. Am. Chem. Soc., 2007, 129, 6100 CrossRef CAS; (b) P. A. Roethle, T. C. Ingrid and D. Trauner, J. Am. Chem. Soc., 2007, 129, 8960 CrossRef CAS; (c) Z. Huang and E.-i. Negishi, J. Am. Chem. Soc., 2007, 129, 14788 CrossRef CAS; (d) S. M. Swick, S. L. Schaefer and G. W. O'Neil, Tetrahedron Lett., 2015, 56, 4039 CrossRef CAS PubMed; (e) S. Scheeff and D. Menche, Org. Lett., 2019, 21, 271 CrossRef CAS.
- Glycosylation at either the C7- or C15-hydroxyls (archazolids C and E respectively) significantly reduces their V-ATPase inhibitory activity, see: N. Horstmann, S. Essig, S. Bockelmann, H. Wieczorek, M. Huss, F. Sasse and D. Menche, J. Nat. Prod., 2011, 74, 1100 CrossRef CAS.
- M. E. Mowery and P. DeShong, J. Org. Chem., 1999, 64, 1684 CrossRef CAS.
- G. C. Fortman and S. P. Nolan, Chem. Soc. Rev., 2011, 40, 5151 RSC.
- E. A. Crane, T. P. Zabawa, R. L. Farmer and K. A. Scheidt, Angew. Chem., Int. Ed., 2011, 50, 9112 CrossRef CAS.
- S. E. Kelly, in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon, Oxford, 1991, vol. 1, pp. 729–817 Search PubMed.
- A. K. Mandal, J. S. Schneekloth, K. Kuramochi and C. M. Crews, Org. Lett., 2006, 8, 427 CrossRef CAS.
- R. E. Boer, J. A. Giménez-Bastida, O. Boutaud, J. Somnath, C. Schneider and G. A. Sulikowski, Org. Lett., 2018, 20, 4020 CrossRef CAS.
- N.-W. Tseng and M. Lautens, J. Org. Chem., 2009, 74, 2521 CrossRef CAS.
- K. Endo, M. Hirokami and T. Shibata, J. Org. Chem., 2010, 75, 3469 CrossRef CAS PubMed.
- J. R. Coombs, L. Zhang and J. P. Morken, Org. Lett., 2015, 17, 1708 CrossRef CAS.
- S. Namirembe, C. Gao, R. P. Wexler and J. P. Morken, Org. Lett., 2019, 21, 4392 CrossRef CAS.
- J. Gorges and U. Kazmaier, Org. Lett., 2018, 20, 2033 CrossRef CAS.
- D. Menche, H. Irschik, F. Sasse, A. Raja and M. Altendorfer, Org. Biomol. Chem., 2013, 11, 2116 RSC.
- R. Noyori and T. Ohkuma, Angew. Chem., Int. Ed., 2001, 40, 40 CrossRef CAS.
- B. M. Trost and D. Laurent, Chem. Sci., 2016, 7, 4985 RSC.
- For a recent review see: A. T. K. Koshvandi and M. M. Heravi, Appl. Organomet. Chem., 2018, 32, e4210 CrossRef.
- A. B. Tran, G. Melly, R. Doucette, B. Ashcraft, L. Sebren, J. Young and G. W. O'Neil, Org. Biomol. Chem., 2011, 9, 7671 RSC.
- L. B. Smart, F. Vojdani, M. Maeshima and T. A. Wilkins, Plant Physiol., 1998, 116, 1539 CrossRef CAS.
- D. Reker, A. M. Perna, T. Rodrigues, P. Schneider, M. Reutlinger, B. Mönch, A. Koeberle, C. Lamers, M. Gabler, H. Steinmetz, R. Müller, M. Schubert-Zsilavecz, O. Werz and G. Schneider, Nat. Chem., 2014, 6, 1072 CrossRef CAS.
- A COX activity assay kit was purchased from Cayman Chemical, https://www.caymanchem.com/product/760151, accessed 8/5/2019). See ESI† for details.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of NMR spectra for compounds 1, 2, 4, 7, 8, 10, 13–15, and 17–20. VATPase and COX assay procedures and results. See DOI: 10.1039/c9ra07050h |
| This journal is © The Royal Society of Chemistry 2019 |