DOI:
10.1039/C9RA07031A
(Paper)
RSC Adv., 2019,
9, 33890-33897
Temperature-induced structural diversity of metal–organic frameworks and their applications in selective sensing of nitrobenzene and electrocatalyzing the oxygen evolution reaction†
Received
3rd September 2019
, Accepted 14th October 2019
First published on 22nd October 2019
Abstract
Structural diversities are presented in four new Co-MOFs containing 1,5-bi(imidazolyl)anthracene and different dibenzobarrelene skeletons based on dicarboxylic acid, in which MOFs 1–3 exhibit 2D networks in a 4-connected node sql topology with the point symbol of {44·62}, while MOF 4 forms a 1D chain structure. It is clearly observed that the 2D-1D structural transformation of 2–4 has been realized by temperature modulated hydrothermal synthesis procedures from 120–160 °C, suggesting the key role of temperature for constructing MOFs. In addition, obvious π–π interactions between anthracene rings can be observed in the architectures of 1–3, which may favorably stabilize their 2D supramolecular networks. More importantly, fluorescence behaviors of 1–4 have been investigated in water among various nitro-aromatic compounds (NACs) and the results show that all samples exhibit high selectivity and fine sensitivity to nitrobenzene (NB) with fluorescence quenching, which is confirmed to be the result of electron transfer from the excited state of ligands to that of NB by density functional theory. Furthermore, MOFs 1–4 have been directly employed as electrocatalysts for the oxygen evolution reaction (OER), in which MOF 4 gives the best activity towards the OER among all as-synthesized samples with an overpotential of 398 mV at a current density of 10 mA cm−2 and a low Tafel slope of 59 mV dec−1.
Introduction
In the past decades, much attention has been paid to the exploitation of metal–organic frameworks (MOFs), a new class of porous coordination materials, owing to their advantages of tailorable porosity and tunable structures, fantastic topologies, and high surface area,1 which exhibit excellent performance in fuel gas storage, heterogeneous catalysis, drug delivery system, targeted cancer therapy, luminescent materials and so on.2 Generally, the application of MOFs largely depends on their architectural features. As a result, it is a great challenge and of significance to adopt a new synthetic method in preparing MOFs with targeted architectures and expected properties. In an earlier report, multidentate ligands, including multi-imidazole and multi-carboxylate, have been used as building blocks for constructing MOFs with fascinating structures, novel topologies and varieties of applications.2c,3 In addition, to make full utilization of the coordination sites in imidazole/carboxylate, mixed ligands of them have been employed for the construction of various functional MOFs, which achieves remarkable progress in tailoring structures and improving properties.4 However, the structural formation of MOF may suffer from several factors, such as temperature, metal centers, organic linkers, solvent, and so on.5 Among them, temperature plays key role for the formation of structural mold, and different temperature can lead to the diversity of MOFs from the same starting materials. Besides, the utilization of several aromatic rings contained rigid ligands in construction of MOFs may contribute to stable frameworks via π-stacking interactions among aromatic rings,6 which are also considered easily to predict the final topologies compared to that of the flexible ligands1b,7 Meanwhile, introduction of aromatic π-rings in organic ligands may enhance the host-guest chemical interactions between the analytes and MOFs in chemical sensing.3e,8 As far as we know, there are some examples for constructing MOFs with the same metal core involving rigid hybrid ligands of multi-imidazole and multi-carboxylate, however, the researches on the relationship between the architectures and temperature are rather rare.5a–g
Up to date, a great variety of MOFs have been applied in chemical sensing via the interactions between the target analyst and the porosity/surface area of MOFs.9 Particularly, chemical sensing based on the fluorescence changes is considered as a simple, sensitive and rapid method for recognizing ions, molecules and so on. Recently, developing MOF-based chemical sensors for detection of NACs attracts extensive attention owing to the high explosiveness and hazardousness of NACs, which are of great significance for public security and environmental protection.10 As a member of NACs, NB is widely used as solvent and intermedia in chemical synthesis, which gradually causes concerns for its environmental pollution. Though several examples of MOFs-based sensors have been reported with sensing ability towards NB,11 to our knowledge, most of them suffered from interferences caused by other NACs and thus showed low selectivity to NB among variety of NACs (Table S1†).11a–h Consequently, it is highly desirable to explore efficient MOF-based sensors that can detect NB with high selectivity and sensitivity by fluorescent response. On the other hand, developing efficient electrocatalysts for oxygen evolution reaction (OER) presents as an attractive strategy in clean energy generation and storage.12 Actually, OER is generally considered to baffle the electrocatalytic water splitting due to its kinetically sluggish, low efficiency, poorly structural stability and etc.13 As such, to utilize proper and highly efficient catalysts in electricity-driven systems for water splitting to produce oxygen is significant and challenging. For the reasons mentioned above, MOFs have emerged as a class of promising electrocatalytic catalysts owing to their advantages in porous structure, active surface area, and open metal sites, which may facilitate the enhancement of catalytic activity. So far, enormous MOF-based catalysts have been designed and developed with excellent OER activities, some of which could comparably with non-precious metal electrocatalysts, displaying great potentials for developing electrocatalytic OER materials.14 Nevertheless, developing MOF based electrocatalysts for OER is still in its infant stage, especially, a limited number of pristine MOFs have been reported for direct use in catalytic OER. Therefore, exploring low-cost but efficient pristine MOF electrocatalysts for OER is significant and challenging.
In consideration of the aforementioned as well as our continuous research for development and application of MOFs, herein, mixed organic ligands including rigid 1,5-bi(imidazolyl)anthracene and dibenzobarrelene skeletons based dicarboxylic acid were introduced to construct a series of luminescent 2D/1D Co-MOFs (1–4) for the first time (Scheme 1). Meanwhile, a facile method via varying temperature to tailor the structures of MOFs has been conducted successfully during the hydro-thermal processes. Then the luminescent behaviors of all prepared MOFs were systematically investigated, which showed obvious solvent-dependent fluorescent spectra and exhibited high selectivity and fine sensitivity towards NB among various NACs by fluorescence quenching. Moreover, the specific response to NB was further demonstrated by density functional theory (DFT) calculations. Significantly, MOFs 1–4 have been directly employed as electrocatalysts for oxygen evolution reaction (OER), in which MOF 4 showed the best activity towards OER among all as-synthesized samples, achieving overpotential of 398 mV at current density of 10 mA cm−2 and Tafel slope of 59 mV dec−1. We anticipate this work can shed light on fabricating MOFs with structural diversity via temperature regulation and applying MOFs in sensing and electrocatalytic OER materials.
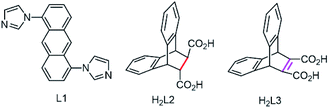 |
| | Scheme 1 The structures of bidentate imidazole ligand L1 and dibenzobarrelene based carboxylic acid ligands H2L2 and H2L3. | |
Experimental
Materials and methods
All chemicals were obtained from Aladdin (Shanghai) or Chengdu Kelong Chemical Co., Ltd. (Chengdu). H2L2 and H2L3 were synthesized according to the previous method.15 NMR spectra were recorded on a Bruker advance II 400 MHz NMR spectrometer. All FT-IR absorption spectroscopy were obtained from a Nicolet 6700 with KBr pellets. X-ray powder diffractions were collected by Rigaku Dmax/Ultima IV diffractometer. Thermogravimetric analyses (TGA) were obtained from a Netzsch STA 449 F3 thermal analyzer. Elemental analyses were acquired from a PerkinElmer 240 elemental analyzer. Nitrogen adsorption isotherms were recorded at 77 K via Autosorb-IQ gas analyzer. Brunauer–Emmett–Teller (BET) method was employed for evaluating the specific surface area and pore size distribution. Fluorescence measurements were conducted on a Cary Eclipse fluorescence spectrophotometer. Electrochemical activities of 1–4 for OER were examined using a CHI-660E electrochemical workstation equipped with a standard three electrode system.
Synthesis of 1,5-bi(imidazolyl)anthracene (L1). To a dry DMF (20 mL) solution of imidazole (5.0 g, 73.5 mmol) in a 100 mL two-necked flask were wadded NaH (3.0 g, 125.0 mmol), which were stirred at room temperature for 0.5 h, followed by addition of 1,5-dichloroanthracene (1 g, 4.1 mmol) and CuI (0.57 g, 3.0 mmol) under N2 atmosphere. The resulting mixture was heated to 140 °C for 48 hours and cooled to room temperature. After that, the reaction mixture was poured into 100 mL water and extracted with CH2Cl2 (50 mL × 3). The combined organic layers were dried over anhydrous Na2SO4 and evaporated to remove the solvent. The residue was purified by column chromatography to obtain the target product in 40% yield (0.5 g). Mp: 260–262 °C 1H NMR (400 MHz, DMSO-d6) δ 8.35 (s, 2H), 8.26–8.22 (m, 2H), 8.10 (s, 2H), 7.69 (s, 1H), 7.66–7.63 (m, 4H), 7.29 (s, 2H).13 C NMR (100 MHz, DMSO-d6) δ 138.59, 133.65, 132.16, 129.70, 129.20, 127.55, 125.59, 124.07, 122.14, 121.99. Anal. calcd (%) for C20H14N4: C, 90.29; H, 4.51; N, 20.6; found: C, 90.40; H, 4.50; N, 20.56. HRMS calcd m/z 311.1297 for [M + H]+, found m/z 311.1309 for [M + H]+.
Synthesis of {[Co2(L1)2(L2)2·(H2O)2]n·3H2O} (1). A mixture of L1 (15.5 mg, 0.05 mmol), H2L2 (14.7 mg, 0.05 mmol) and Co(NO)3·6H2O (29.1 mg, 0.1 mmol) were dissolved in a mixture solution of CH3CN (2 mL) and H2O (6 mL), which was heated to 120 °C with a heating rate of 3 °C min−1 and reacted for 96 h in a 20 mL Teflon vial. Then the reaction was cooled down to room temperature to obtain purplish red block crystals in 72% yield (based on L1 ligand). Elemental analysis (%), calcd for C76H62Co2N8O13: C, 64.53; H, 4.38; N, 7.92; found: C, 63.72; H, 4.46; N, 7.83.
Synthesis of {[Co2(L1)2(L3)2·(H2O)2]n·4H2O} (2). To a mixture of L1 (15.5 mg, 0.05 mmol) and H2L3 (29.2 mg, 0.1 mmol) in CH3CN/H2O solution (8 mL, v/v = 1/3) were added Co(NO)3·6H2O (29.1 mg, 0.1 mmol), which were heated to 120 °C at a heating rate of 3 °C min−1 in a Teflon vial and reacted for 96 hours. After cooled to room temperature, brownish blocks crystals of 2 were obtained in 48% yield (based on L1 ligand). Elemental analysis (%), calcd for C76H60Co2N8O14: C, 63.90; H, 4.20; N, 7.85; found: C, 63.87; H, 4.23; N, 7.84.
Synthesis of {[Co(L1)2(HL3)2]n·2CH3CN} (3). The prepared procedure was according to that of 2 except for altering the heating temperature to 140 °C. Brown block crystals of complex 3 were collected in 56% yield (based on L1 ligand). Elemental analysis (%), calcd for C80H56CoN10O8: C, 71.41; H, 4.17; N, 10.42; found: C, 71.47; H, 4.18; N, 10.38.
Synthesis of {[Co(L1)(HL3)2·(H2O)2]n} (4). The synthetic method was according to that of 2 only by altering the heating temperature to 160 °C. Brown block crystals of 4 were obtained in 26% yield (based on L1 ligand). Elemental analysis (%), calcd for C56H40CoN4O10: C, 68.09; H, 4.08; N, 5.67; found: 68.06; H, 4.09; N, 5.65.
Crystallographic data collection and refinement. The crystal data for 1–4 were collected on a Bruker SMART APEX-II CCD diffractometer with graphite monochromatic Mo-Kα radiation (λ = 0.71073 Å) by using the ω-scan technique at 293 °C. All structures were solved by direct methods and refined with the full-matrix least-squares procedures on F2 using the SHELXL-97 (ref. 16) and Olex-2 programs, respectively. All H atoms were generated geometrically at idealized positions at idealized positions and refined using the riding model. The CCDC deposition numbers allocated to 1–4 were 1887850, 1887851, 1887854 and 1887855, respectively. Topology analyses were carried out by using TOPOS program.17 Corresponding data for 1–4 were provided in Tables 1, S2 and Fig. S1.†
Table 1 Crystallographic data for MOFs 1–4
| R1 = ∑||Fo| − |Fc||/∑|Fo|. wR2 = ∑[w(Fo2 − Fc2)2]/∑[w(Fo2)2]1/2. |
| Compounds |
1 |
2 |
3 |
4 |
| Formula |
C76H62Co2N8O13 |
C76H60Co2N8O14 |
C80H56CoN10O8 |
C56H40CoN4O10 |
| fw |
1431.9 |
1427.18 |
1344.27 |
987.85 |
| Temp., K |
293(2) |
293.15 |
293(2) |
293(2) |
| Crystal syst |
Triclinic |
Triclinic |
Monoclinic |
Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P21/c |
P21/n |
| a (Å) |
10.353(4) |
10.336(8) |
14.1122(9) |
13.407(6) |
| b (Å) |
12.285(6) |
12.301(10) |
27.0471(18) |
12.528(4) |
| c (Å) |
14.373(5) |
14.368(11) |
9.1595(7) |
15.550(10) |
| α (deg) |
101.162(12) |
101.18(2) |
90 |
90 |
| β (deg) |
107.318(10) |
107.255(19) |
108.227(2) |
114.216(19) |
| γ (deg) |
107.004(13) |
107.055(19) |
90 |
90 |
| V (Å3) |
1588.1(11) |
1587(2) |
3320.7(4) |
2382(2) |
| Z |
1 |
1 |
2 |
2 |
| Dc (g cm−3) |
1.478 |
1.493 |
1.344 |
1.377 |
| μ (mm−1) |
0.599 |
0.601 |
0.326 |
0.427 |
| Rint |
0.0395 |
0.0633 |
0.0589 |
0.0749 |
| Reflections collected/unique |
5818/4860 |
5811/4495 |
7367/5696 |
4872/3436 |
| GOF on F2 |
1.028 |
1.063 |
1.029 |
1.041 |
| R1a, wR2b [I > 2σ(I)] |
0.0334/0.0824 |
0.0464/0.1195 |
0.0422/0.0937 |
0.0443/0.0968 |
| R1a, wR2b (all data) |
0.0439/0.0876 |
0.0668/0.1307 |
0.0644/0.1035 |
0.0773/0.1099 |
| Largest peak, hole (e Å−3) |
0.30/−0.33 |
0.81/−0.62 |
0.41/-0.30 |
0.23/−0.34 |
Results and discussion
L1 was synthesized by a modified C–N coupling under the catalyst of CuI in about 40% yield as depicted in Scheme S1.†3a H2L2 and H2L3 were facilely prepared in a high yield via Diels–Alder reaction and ester hydrolysis process according to literature.12 L1 was characterized by NMR spectra and HRMS (Fig. S1–S3†). MOFs 1–4 were characterized by FT-IR spectra (Fig. S4†).
{[Co2(L1)2(L2)2·(H2O)2]n·3H2O} (1)
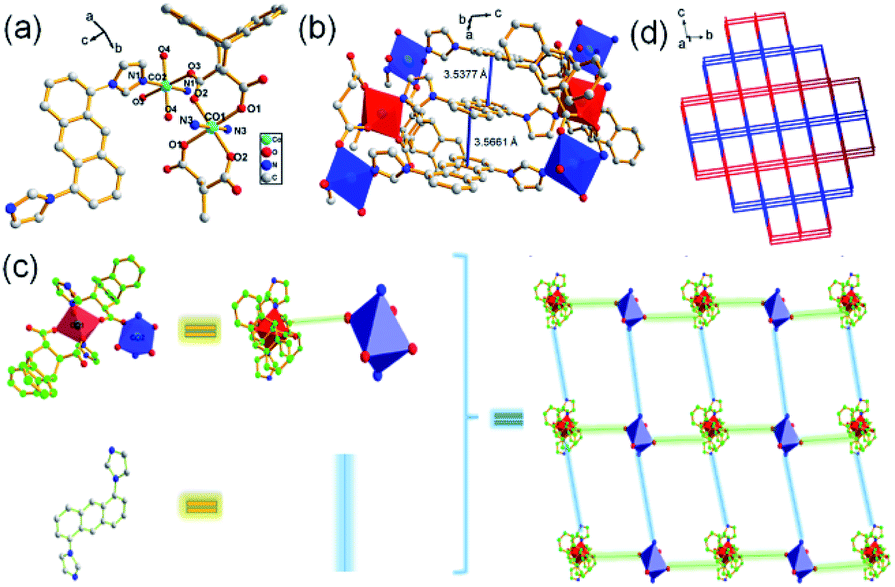
The single crystal X-ray data reveals complex 1 conforms to the triclinic crystal system, space group P. Fig. 1a shows an asymmetric unit of 1 containing two Co(II) atoms: Co1 and Co2, in which Co1 is six-coordinated by two nitrogen atoms from L1 and four oxygen atoms from two carboxylate ligands H2L2 in μ-bridged mode,4b while Co2 forms an octahedral geometry by coordinating to two oxygen atoms from two carboxylate ligands H2L2, two oxygen atoms from two water molecules, and two nitrogen atoms from two L1 molecules, respectively. Subsequently, Co1 and Co2 are connected by two oxygen atoms from a carboxylate ligand, which further coordinate to N atoms form L1 to form a 2D structure. Interestingly, π–π stacking interactions between anthracene rings can be observed with face to face distances of 3.5377–3.5661 Å, which contribute to the formation of a 2D net structure (Fig. 1b and c). The structure of compound 1 can be simplified by topology program as a 2D square grid network with the point symbol of {44·62} (Fig. 1d).
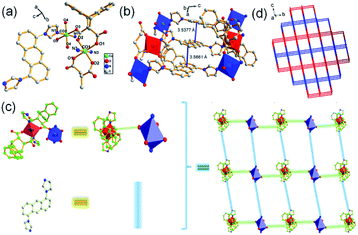 |
| | Fig. 1 (a) Asymmetric unit of 1, symmetry code: (i) = 1 − x, 1 − y, 1 − z; (ii) = 2 − x, 1 − y, 1 − z; (iii) = 2 − x, 1 − y, 2 − z; (iv) = 1 − x, 1 − y, −z; (b) π-stacking interactions in 1; (c) simplified 2D structure of 1; (d) topology structure of 1. | |
{[Co2(L1)2(L3)2·(H2O)2]n·4H2O} (2)
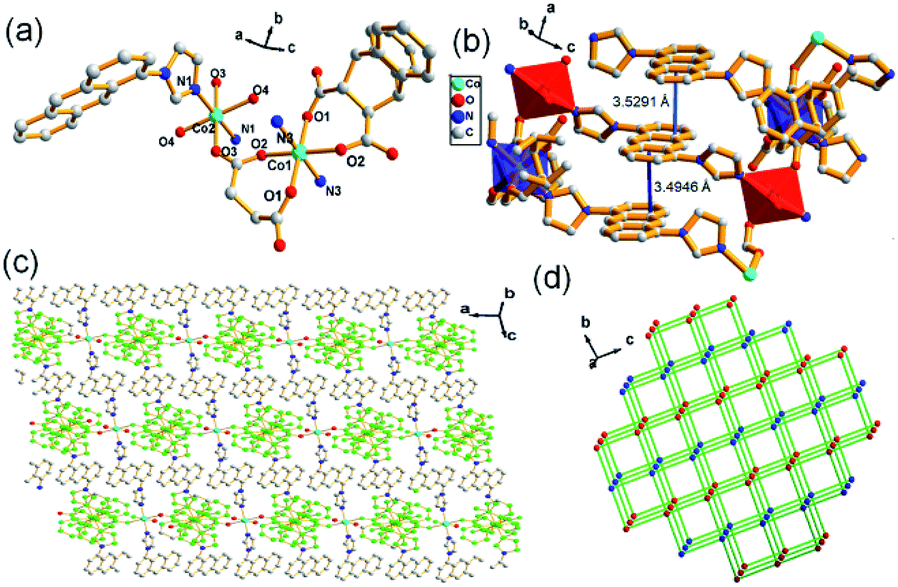
Structural analysis reveals that complex 2 belongs to a triclinic crystal system with the space group of P. Fig. 2a shows the asymmetry unit of 2 contains two six-coordinated Co(II) cations (Co1 and Co2) with octahedral geometry, respectively, in which Co1 connects with four oxygen atoms from two L3 ligands and two nitrogen atoms from ligands L1, while Co2 coordinates to two oxygen atoms from two lattice water, two oxygen atoms form two carboxylate ligands and two N atoms from L1. Co1 and Co2 are bridged by carboxylate ligand, which further links to L1 ligands respectively to form a 2D network. Meanwhile, strong face to face π–π stacking interactions between the anthracene rings can be observed with face to face distances of about 3.4946–3.5291 Å, which may be favorably for the formation of 2D structure (Fig. 2b and c). Topological analysis shows complex 2 can be simplified as a 4-connected node sql with a Schläfli symbol of {44·62} (Fig. 2d).
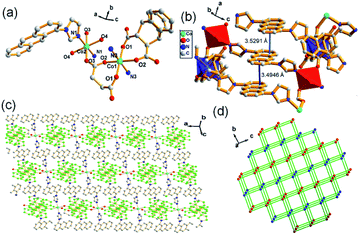 |
| | Fig. 2 (a) Asymmetric unit of 2, symmetry code: (i) = 2 − x, 1 − y, 2 − z; (ii) = 1 − x, 1 − y, 2 − z; (iii) = 1 − x, 1 − y, 1 − z; (iv) = 2 − x, 1 − y, 1 − z; (b) π-stacking interactions between the anthracene rings in 2; (c) 2D structure of 2; (d) topological analysis of 2. | |
{[Co(L1)2(HL3)2]n·2CH3CN} (3)
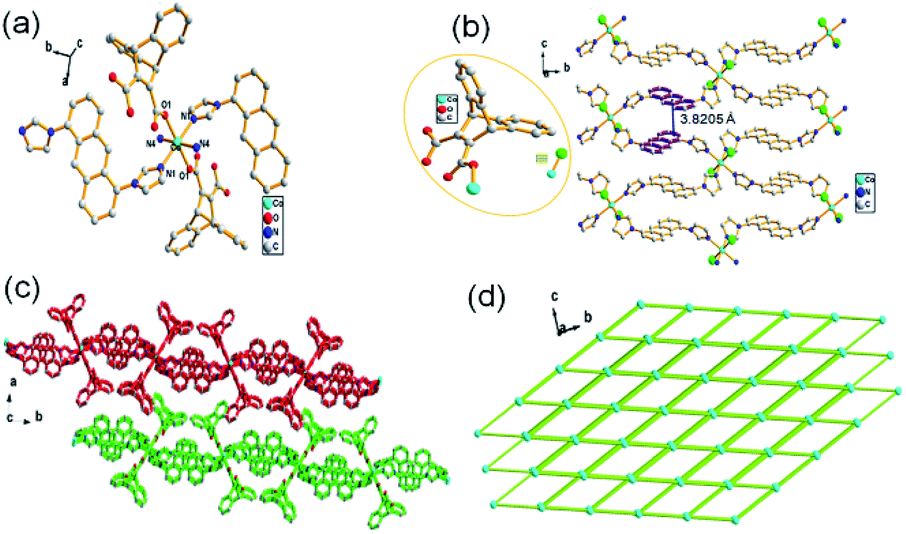
Single-crystal X-ray data shows that complex 3 crystallized in the monoclinic system with space group P21/c. The secondary building unit (SBU) is consisted by two HL3−, one Co(II) and four L1 ligands to present an octahedral geometry as depicted in Fig. 3a. The connections between Co(II) atoms and L1 further leads to the formation of a 2D network, in which extensive distorted face to face π-stacking interactions can be observed with contribution to the self-assembly of 2D supramolecular structure (face to face distances of about 3.8205 Å) (Fig. 3b and c). Topologically, MOF 3 can be depicted as a 4-connected node with the point symbol {44·62} (Fig. 3d). The coordination environment of Co(II) in SUB of MOF 3 is different to that of 2, providing the evidence for temperature-induced diversities of structure.
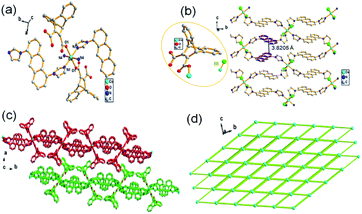 |
| | Fig. 3 (a) The SBU of 3, symmetry code: (i) = −x, 1 − y, −z; (ii) = +x, 3/2 − y, 1/2 + z; (iii) = −x, −1/2 + y, −1/2 − z; (iv) = −x, 1/2 + y, −1/2 − z; (b) infinite 2D layer of 3; (c) stack-configuration of 3; (d) topological architecture of 3. | |
{[Co(L1)(HL3)2·(H2O)2]n} (4)
Structural analysis shows that complex 4 is a monoclinic crystal system, space group P21/n. The asymmetric SBU has a six coordinated Co(II) core connecting with two O atoms from two HL3 ligands, two N atoms form two L1 and two O atoms from two lattice water molecules to form a octahedral geometry (Fig. 4a), in which the another N atom of imidazole in L1 further coordinates to Co(II) in next SBU, forming a 1D chain structure (Fig. 4b and c), further suggesting the key role of temperature in the synthesis of MOF.
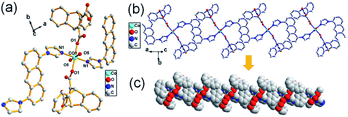 |
| | Fig. 4 (a) The SBU of 4, symmetry code: (i) = 1 − x, −y, 1 − z; (ii) = −x, −y, 1 − z; (b) 1D chain structure of 4; (c) stacking diagram of 4. | |
It is worth noting that ligands variation in synthetic processes lead to a slight pore size change from micropore in 1 to mesoporous in 2, which may due to the more rigid dibenzobarrelene derived carboxylate ligand H2L3 (bridgehead C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) compared to that of H2L2 (bridgehead C–C), while no other obvious structural changes between them can be observed. However, temperature controlled strategies employed in synthetic processes of MOFs 2–4 causes the distinct coordination environment of Co(II) (two coordination modes of dibenzobarrelene derived carboxylate ligands as depicted in Scheme S2†) as well as the transformation from 2D porous structure (2 and 3) to 1D chain structure (4) with a gradual rise in temperature. The above mentioned results imply the important roles of ligands/temperature in tailoring structure of MOFs.
C) compared to that of H2L2 (bridgehead C–C), while no other obvious structural changes between them can be observed. However, temperature controlled strategies employed in synthetic processes of MOFs 2–4 causes the distinct coordination environment of Co(II) (two coordination modes of dibenzobarrelene derived carboxylate ligands as depicted in Scheme S2†) as well as the transformation from 2D porous structure (2 and 3) to 1D chain structure (4) with a gradual rise in temperature. The above mentioned results imply the important roles of ligands/temperature in tailoring structure of MOFs.
PXRD, TGA and N2 adsorption
Fig. S5† shows the experimental PXRD patterns of MOFs 1–4 closely match that simulated patterns obtained from the single crystal data, demonstrating the phase purity of as-synthesized MOFs. As depicted in Fig. S6,† TG curves for 1–4 were recorded in the temperature range of 35–600 °C to evaluate the stability of samples. Based on the thermogravimetric analysis, complex 1 loses 2.8% (calcd 2.5%) weight from 150–180 °C associated with the loss of crystallization water, which begins to collapse beyond ca. 270 °C. Complex 2 has a weight loss of 3.3% (calcd 2.5%) from 100–228 °C, corresponding to the water molecules, and then decomposes around 270 °C. A weight loss of 6.7% (calcd 6.1%) is observed when complex 3 is heated from 35–270 °C, which should be ascribed to the escape of residual acetonitrile molecule, and a structural decomposition occurs at about 280 °C. For complex 4, it exhibits a weight loose of 3.5% (calcd 3.6%) between 100–250 °C corresponding to the lattice water, which begins to collapse at around 251 °C. The results demonstrate the good stability of these samples. In addition, the porous structures of 1–3 were evaluated by N2 adsorption at 77 K. Fig. S7† shows BET surface areas of complex 1–3 are estimated to be 13.694 m2 g−1, 10.485 m2 g−1 and 4.194 m2 g−1, respectively. Meanwhile, the pore diameters of them are calculated to be 1.754 nm, 2.000 nm, and 3.502 nm, respectively.
Sensing behaviors towards nitrobenzene
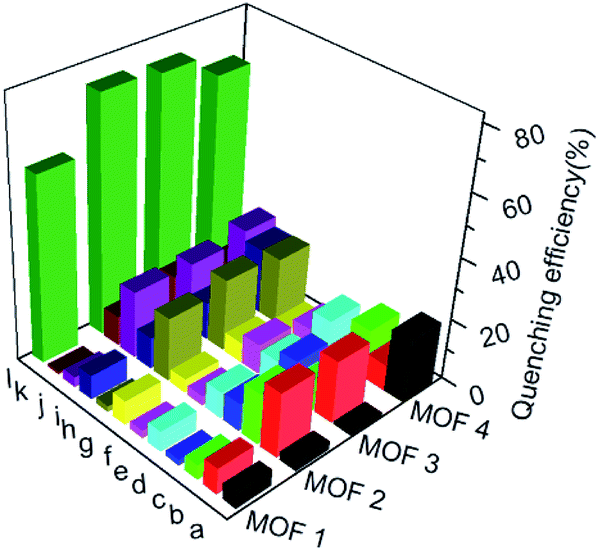
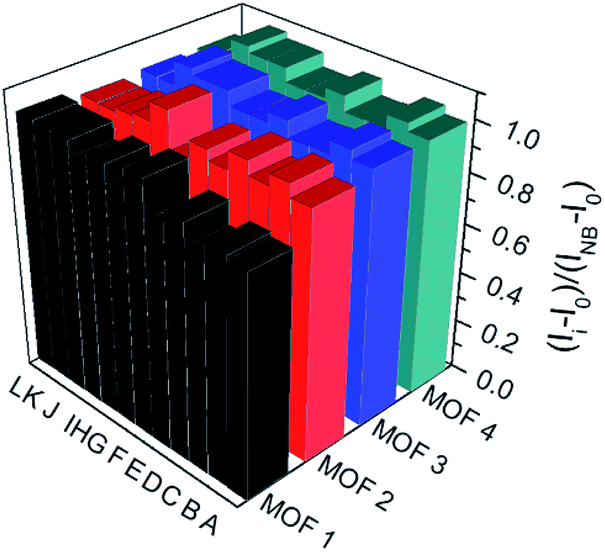
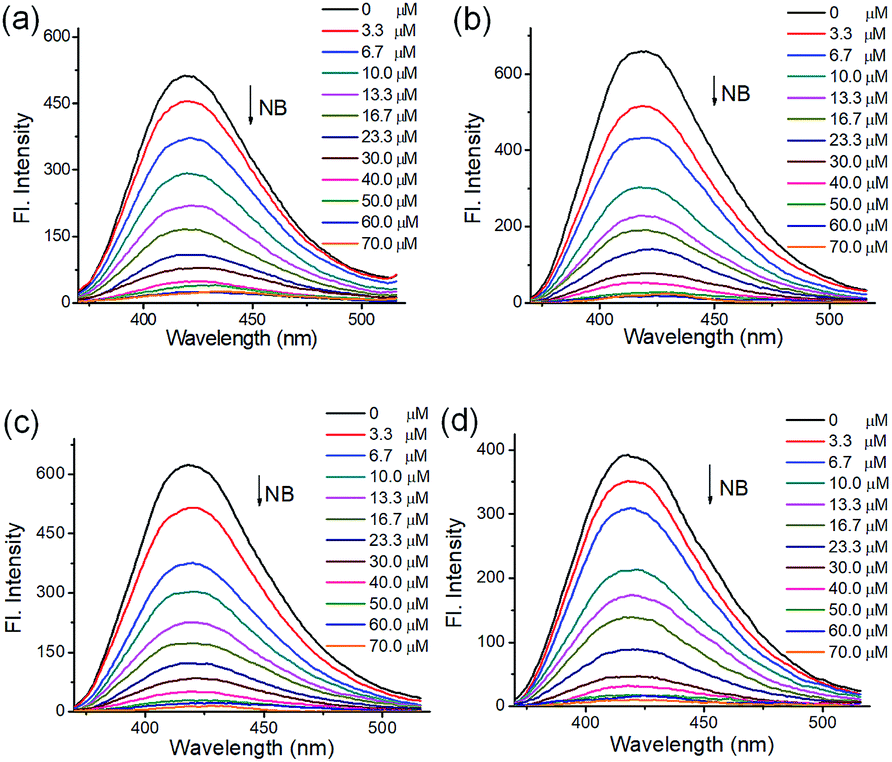
To understand the luminescent behaviors of MOFs 1–4, 0.3 mg of them were added respectively into 3 mL suitable solvents and ultrasonated for 0.5 h to obtain dispersed suspensions. Firstly, the emission profiles of 1–4 were investigated in different solvents. Fig. S8† show all samples present obvious emission peaks centered at the range of 400–430 nm, while they show almost completely quenching phenomena with nearly 100% quenching efficiency in NB, which is analogues to that of most reported MOFs.11i–m Though a slight solvation effect for the wavelength can be observed with different solvents polarity, suggesting solvent-dependence of the emission intensity, no obvious relationship between them can be found. Secondly, to further investigate the selectivity of these MOFs towards NB among various NACs, water was chosen as detection medium for its environment friendliness. Then the specific responses of MOFs towards nitrobenzene among various NACs were examined in water under 270 nm excitation by fluorescence spectra, in which the suspension solutions of 1–4 were added with equivalents of different NACs including NB, 4-hydroxynitrobenzene (4-NP), 2,4,6-trinitrophenol (TNP), 2-nitrophenol (2-NP), 1-chloro-4-nitrobenzene (4-CNB), 4-nitrotoluene (4-NTol), 2-nitro-phenylamine (2-NPA), 3-nitrophenol (3-NP), 2,4-dinitroaniline (2,4-DNA), 4-nitroaniline (4-NPA), 2,4-dinitrophenylhydrazine (2,4-NPH) and 4-nitrobenzoic acid (4-NBA). Fig. S9† and 5 show samples 1–4 exhibit obvious fluorescence bands centered at ca. 425 nm, and the addition of low concentrations (1.7 × 10−5 mol L−1) of NB causes significant decreases in fluorescence intensity at 425 nm with quenching efficiencies of 60.0%, 76.6%, 75.5% and 68.3% for MOFs 1–4, respectively, while the other NACs only elicit slight decreases in emission intensity with quenching efficiency less than 20%. The results indicate all MOFs show high selectivity towards nitrobenzene compared with that of other MOF-based sensors in Table S1,† which may be ascribed to PET process from the ligands to NB.11a In addition, the effect of other NACs for the specific responses of MOFs 1–4 towards NB were evaluated by competition experiments, in which the fluorescence profiles were obtained from the chemosensors solutions in the presence of various NACs coexisting with NB, respectively. Fig. 6 exhibits other NACs causes no obvious fluorescence changes of 1–4 compared to that of NB alone, further suggesting the high selectivity of these MOFs towards NB. To better understand the sensing behaviors of 1–4 towards NB, the fluorescence titration assays of them were recorded by treatment of increasing concentrations of NB, respectively. As can be seen from Fig. 7, low concentration of NB (3.3 μM) can trigger obvious fluorescence changes for all samples, indicating their sensitivities towards NB. Overall, the fluorescence intensity of all MOFs sharply decreases with the addition of NB below the concentrations of ca. 16.7 μM and completely quenches at high concentration of NB (ca. 70 μM) with quenching efficiencies beyond 96%. The fluorescence quenching efficiency is evaluated via Stern–Volmer formula (I0/I) = Ksv [NB] + 1, where I0 and I represent the fluorescence intensity before and after addition of an NB, respectively; ([NB] refers to the concentration of NB, and Ksv is the quenching constant). Fig. S9† shows a linear increase in the S–V plots at the low concentrations of NB, while the further increasing concentration causes divergences from the linearity, which could be probably ascribed to an analogous diffusion-controlled energy transfer process reported in literature.11l Moreover, the Ksv values of 1–4 for NB are evaluated as 4.59 × 104 M−1, 6.15 × 104 M−1, 3.28 × 104 M−1 and 4.50 × 104 M−1, respectively, which are larger than that in the previous literatures.11j,k Besides, MOFs 1–4 can be renewable and reused for four numbers of cycles via centrifugation, filtration and rinsing several times with DMF, which show insignificant quenching efficiency decreases after four repeated cycles, suggesting the stability and recyclability of MOFs 1–4 in detection of NB (Fig. S11†).
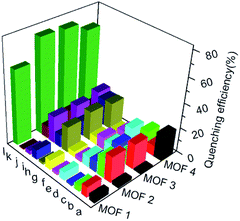 |
| | Fig. 5 Emission quenching efficiencies of 1–4 in water after the addition of various NACs (16.7 μM). Histograms (a)–(l) represent: 4-NP; TNP; 2-NP; 4-CNB; 4-NTol; 2-NPA; 3-NP; 2,4-DNA; 4-NPA; 2,4-NPH; 4-NBA and NB, respectively. | |
 |
| | Fig. 6 Emission intensity ratio changes of MOFs 1–4 towards NB (16.7 μM) and NB (16.7 μM) in presence of equivalent other NACs. Histogram (A)–(L) represent: NB + 4-NP; NB + TNP; NB + 2-NP; NB + 4-CNB; NB + 4-NTol; NB alone; NB + 2-NPA; NB + 3-NP; NB + 2, 4-DNA; NB + 4-NPA; NB + 2, 4-NPH and NB + 4-NBA, respectively. | |
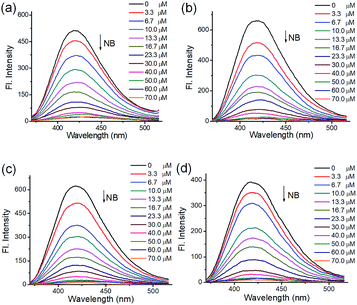 |
| | Fig. 7 Emission profiles of MOFs ((a–d) for 1–4, respectively) upon treatment of different concentrations of NB (0–70 μM) in water, λex: 270 nm. | |
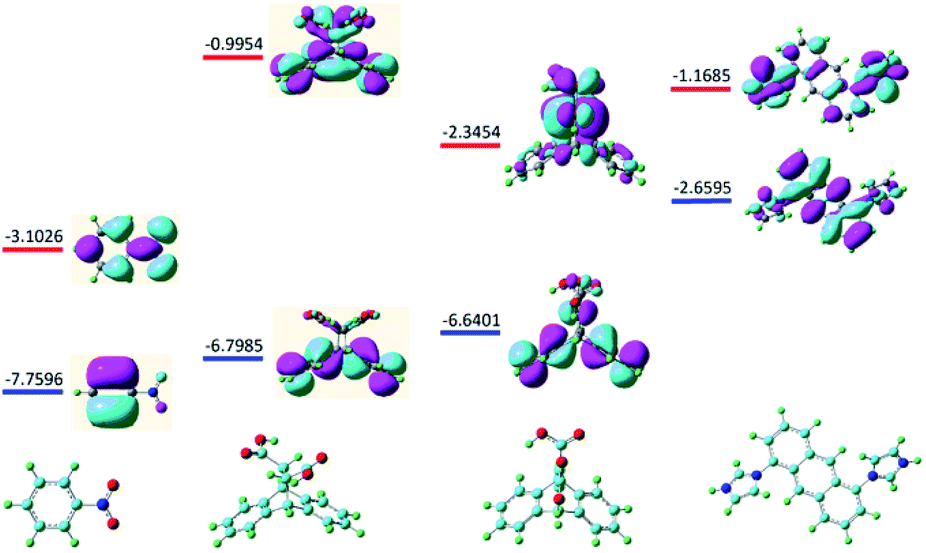
Considering either the porous frameworks of 1–3 (1.754 nm, 2.000 nm and 3.502 nm for 1–3, respectively) or 1D structure of 4 could provide suitable pores for inclusion of NB molecule (6.0 Å), the possible quenching mechanism is suggested to be the electron transfer processes from the electron donating ligands to the electron deficient NB molecules that adhered to the surface of MOFs.18 With the above idea in mind as well as to further illustrate the specific response mechanisms of MOFs 1–4 to NB, the fluorescence quenching mechanism was examined at the level of B3LYP/6-31G* according to density functional theory. Fig. 8 shows the LUMO (−3.1026 eV) of NB is lower than that of ligands H2L2, H2L3 and L1 (−0.9954 ev, −2.3454 ev and −1.1685 ev, respectively), which is favorable for electron transfer from the excited state of ligands to that of NB, resulting in luminescence quenching behaviors. As state above, all prepared MOF samples exhibit high selectivity towards NB via PET mechanism controlled fluorescence quenching process.
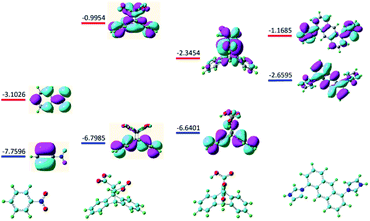 |
| | Fig. 8 Optimal structures and calculated HOMO (blue) and LUMO (red) orbits for NB and ligands H2L2, H2L3 and L1. | |
Electrocatalytic OER
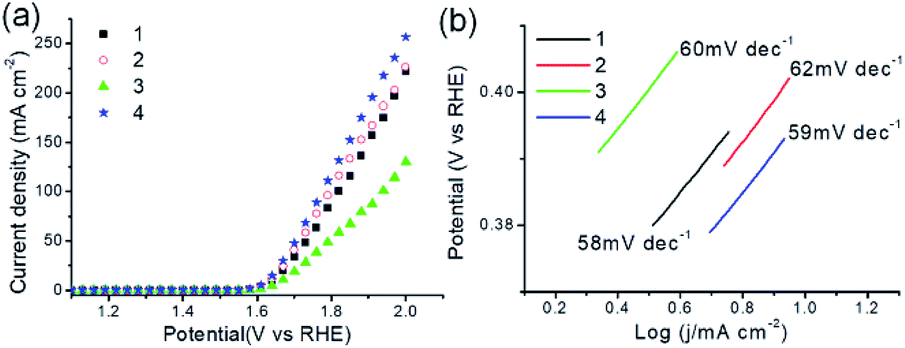
Electrocatalytic properties of MOFs 1–4 were investigated. Fig. 9a shows the polarization curves of all as-synthesized samples obtained by LSV measurements, in which 1–4 present electrocatalytic OER activities with onset overpotentials of 354, 352, 373 and 345 mV, and overpotentials of 411, 406, 438 and 398 mV for achieving current density of 10 mA cm−2 (η10), respectively. The results could compare favorably to those of OER catalysts in reported works (Table S3†).14d,e Fig. 9b shows OER kinetics of MOFs 1–4 are characterized with Tafel plots of 58, 62, 60, and 59 mV dec−1, respectively, revealing a more favorable kinetics of 4 for electrocatalytic OER. Next, electrochemical impedance spectroscopy (EIS) was conducted to evaluate the electrode activities for OER under operating conditions. As shown in Fig. S12,† all MOFs deliver more than one semicircles in the Nyquist curves. The low frequency loop may be attributed to the adsorption on the electrode surface of reaction intermediate such as Oad as reported in literature,14e and the high frequency loop corresponds to charge transfer processes. Among them, 4 holds the smallest value of charge-transfer resistance (Rct), revealing the fastest electron transfer property and high electrocatalytic activity toward oxygen production under the catalysis of 4. Furthermore, the PXRD patterns of 1–4 immersed in 1 M KOH for 2 h were acquired. As provided in Fig. S13,† the PXRD patterns of all samples remain almost unchanged, suggesting their retained crystallinity and stabilities. As described, MOFs 1–4 exhibit promising activities for electrocatalytic OER.
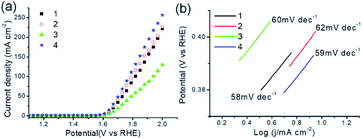 |
| | Fig. 9 (a) OER polarization curves of as-synthesized MOFs 1–4 in 1 M KOH solution with a scan rate of 5 mV s−1. (b) Tafel plots of 1–4. | |
Conclusions
In conclusion, four 2D/1D Co-MOFs have been designed and obtained with structural diversities based on 1,5-biimidazoylanthracene and different dibenzobarrelene skeletons derived dicarboxylic acid, among which 1–3 show 2D network in a 4-connected node sql with the point symbols of {44·62} and 4 presents a 1D chain structure. Notably, strategy of varying temperature to tailor the structures of MOFs have been adopted and realized in the synthesis of 2–4, suggesting the key role of temperature in constructing MOFs. In addition, MOFs 1–4 exhibit high selectivity and sensitivity towards NB among various nitro-aromatics compounds in water, which is confirmed to be a result of electron transfer from the excited state of ligands to that of NB by density functional theory. Besides, MOFs 1–4 can be directly used as electrocatalysts for OER and 4 exhibits the best activity among all as-synthesized samples with low overpotential of 398 mV to achieve current density of 10 mA cm−2 and Tafel slope of 59 mV dec−1. This work is of great importance for extensively developing structural diversity MOFs via temperature regulation and employing MOFs as efficiently sensing and electrocatalytic materials.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21671159), Meritocracy Research Funds of China West Normal University (17YC030, 17YC010), Sichuan Provincial Education Department (18ZA0470), Chengdu University of Technology Teacher Development Fund (10912-2019KYQD-07546).
Notes and references
-
(a) M. Eddaoudi, H. Li and O. M. Yaghi, J. Am. Chem. Soc., 2000, 122, 1391–1397 CrossRef CAS;
(b) X. C. Huang, Y. Y. Lin, J. P. Zhang and X. M. Chen, Angew. Chem., Int. Ed., 2006, 45, 1557–1559 CrossRef CAS PubMed;
(c) O. K. Farha, I. Eryazici, N. C. Jeong, B. G. Hauser, C. E. Wilmer, A. A. Sarjeant, R. Q. Snurr, S. T. Nguygn, A. O. Yazaydin and J. T. Hupp, J. Am. Chem. Soc., 2012, 134, 15016–15021 CrossRef CAS;
(d) J. Pang, S. Yuan, J. Qin, C. Liu, C. Lollar, M. Wu, D. Yuan, H. C. Zhou and M. Hong, J. Am. Chem. Soc., 2017, 139, 16939–16945 CrossRef CAS;
(e) X. Gu and D. Xue, CrystEngComm, 2007, 9, 471–477 RSC.
-
(a) H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed;
(b) B. Chen, N. W. Ockwig, A. R. Millward, D. S. Contreras and O. M. Yaghi, Angew. Chem., Int. Ed., 2005, 44, 4745–4749 CrossRef CAS;
(c) Z. Zhou, C. He, J. Xiu, L. Yang and C. Duan, J. Am. Chem. Soc., 2015, 137, 15066–15069 CrossRef CAS PubMed;
(d) P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, C. Férey, R. E. Morris and C. Serre, Chem. Rev., 2012, 112, 1232–1268 CrossRef CAS;
(e) M. X. Wu and Y. W. Yang, Adv. Mater., 2017, 29, 1606134 CrossRef;
(f) J. Park, Q. Jiang, D. Feng, L. Mao and H. C. Zhou, J. Am. Chem. Soc., 2016, 138, 3518–3525 CrossRef CAS;
(g) S. Khatua, S. Goswami, S. Biswas, K. Tomar, H. S. Jena and S. Konar, Chem. Mater., 2015, 27, 5349–5360 CrossRef CAS;
(h) J. X. Hou, J. P. Gao, J. Liu, X. Jing, L. J. Li and J. L. Du, Dyes Pigm., 2019, 160, 159–164 CrossRef CAS;
(i) G. Xiong, B. Yu, J. Dong, Y. Shi, B. Zhao and L. N. He, Chem. Commun., 2017, 53, 6013–6016 RSC.
-
(a) L. Luo, P. Wang, G. C. Xu, Q. Liu, K. Chen, Y. Lu, Y. Zhao and W. Y. Sun, Cryst. Growth Des., 2012, 12, 2634–2645 CrossRef CAS;
(b) H. J. Lee, P. Y. Cheng, C. Y. Chen, J. S. Shen, D. Nandi and H. M. Lee, CrystEngComm, 2011, 13, 4814–4816 RSC;
(c) Z. Wei, Z. Y. Gu, R. K. Arvapally, Y. P. Chen, R. N. McDougald Jr, J. F. Ivy, A. A. Yakovenko, D. Feng, M. A. Omary and H. C. Zhou, J. Am. Chem. Soc., 2014, 136, 8269–8276 CrossRef CAS;
(d) J. Pang, C. Liu, Y. Huang, M. Wu, F. Jiang, D. Yuan, F. Hu, K. Su, G. Liu and M. Hong, Angew. Chem., Int. Ed., 2016, 55, 7478–7482 CrossRef CAS;
(e) M. Zhang, Z. Qi, Y. Feng, B. Guo, Y. Hao, Z. Xu, L. Zhang and D. Sun, Inorg. Chem. Front., 2018, 5, 1314–1320 RSC;
(f) A. K. Crane, N. G. White and M. J. MacLachlan, CrystEngComm, 2015, 17, 4912–4918 RSC;
(g) T. Wang, K. Huang, M. Peng, X. Li, D. Han, L. Jing and D. Qin, CrystEngComm, 2019, 21, 494–501 RSC.
-
(a) W. G. Yuan, F. Xiong, H. L. Zhang, W. Tang, S. F. Zhang, Z. He, L. Jing and D. Qin, CrystEngComm, 2014, 16, 7701–7710 RSC;
(b) Y. Liang, W. G. Yuan, S. F. Zhang, Z. He, J. Xue, X. Zhang, L. Jing and D. Qin, Dalton Trans., 2016, 45, 1382–1390 RSC;
(c) C. H. Ke, G. R. Lin, B. C. Kuo and H. M. Lee, Cryst. Growth Des., 2012, 12, 3758–3765 CrossRef CAS;
(d) S. Q. Zang, M. M. Dong, Y. J. Fan, H. W. Hou and T. C. Mak, Cryst. Growth Des., 2012, 12, 1239–1246 CrossRef CAS;
(e) S. F. Zhang, F. Xiong, Z. He, Y. Liang, J. R. Xue, L. H. Jing and D. B. Qin, Polyhedron, 2015, 102, 401–409 CrossRef CAS;
(f) S. Mukherjee, D. Samanta and P. S. Mukherjee, Cryst. Growth Des., 2013, 13, 5335–5343 CrossRef CAS;
(g) L. Luo, S. Q. Liu, X. W. Xu, J. Xu and J. L. Zhao, CrystEngComm, 2018, 20, 7719–7728 RSC.
-
(a) B. Liu, L. Wei, N. N. Li, W. P. Wu, H. Miao, Y. Y. Wang and Q. Z. Shi, Cryst. Growth Des., 2014, 14, 1110–1127 CrossRef CAS;
(b) H. F. Zhou, T. He, K. F. Yue, Y. L. Liu, C. S. Zhou, N. Yan and Y. Y. Wang, Cryst. Growth Des., 2016, 16, 3961–3968 CrossRef CAS;
(c) L. Y. Du, H. Wang, G. Liu, D. Xie, F. S. Guo, L. Hou and Y. Y. Wang, Dalton Trans., 2015, 44, 1110–1119 RSC;
(d) B. Zheng, H. Dong, J. Bai, Y. Li, S. Li and M. Scheer, J. Am. Chem. Soc., 2008, 130, 7778–7779 CrossRef CAS;
(e) Y. B. Dong, Y. Y. Jiang, J. Li, J. P. Ma, F. L. Liu, B. Tang, R. Q. Huang and S. R. Batten, J. Am. Chem. Soc., 2007, 129, 4520–4521 CrossRef CAS;
(f) K. L. Gurunatha, K. Uemura and T. K. Maji, Inorg. Chem., 2008, 47, 6578–6580 CrossRef CAS;
(g) P. Cui, L. Ren, Z. Chen, H. Hu, B. Zhao, W. Shi and P. Cheng, Inorg. Chem., 2012, 51, 2303–2310 CrossRef CAS;
(h) X. Y. Duan and M. L. Wei, Cryst. Growth Des., 2017, 17, 1197–1207 CrossRef CAS;
(i) J. J. Hou, X. Q. Li, P. Gao, H. Q. Sun and X. M. Zhang, Cryst.
Growth Des., 2017, 17, 3724–3730 CrossRef CAS;
(j) Y. F. Hou, B. Liu, K. F. Yue, C. S. Zhou, Y. M. Wang, N. Yan and Y. Y. Wang, CrystEngComm, 2014, 16, 9560–9567 RSC.
-
(a) Z. He, K. Huang, F. Xiong, S. F. Zhang, J. R. Xue, Y. Liang, L. H. Jing and D. B. Qin, J. Organomet. Chem., 2015, 797, 67–75 CrossRef CAS;
(b) X. J. Li, Z. J. Yu, T. N. Guan, X. X. Li, G. C. Ma and X. F. Guo, Cryst. Growth Des., 2014, 15, 278–290 CrossRef.
-
(a) M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469–472 CrossRef CAS;
(b) N. B. Shustova, A. F. Cozzolino and M. Dincă, J. Am. Chem. Soc., 2012, 134, 19596–19599 CrossRef CAS.
- M. Zhang, L. Zhang, Z. Xiao, Q. Zhang, R. Wang, F. Dai and D. Sun, Sci. Rep., 2016, 6, 20672 CrossRef CAS.
-
(a) Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815–5840 RSC;
(b) W. P. Lustig, S. Mukherjee, N. D. Rudd, A. V. Desai, J. Li and S. K. Ghosh, Chem. Soc. Rev., 2017, 46, 3242–3285 RSC;
(c) Y. Zhang, S. Yuan, G. Day, X. Wang, X. Yang and H. C. Zhou, Coord. Chem. Rev., 2018, 354, 28–45 CrossRef CAS.
-
(a) S. S. Nagarkar, B. Joarder, A. K. Chaudhari, S. Mukherjee and S. K. Ghosh, Angew. Chem., Int. Ed., 2013, 52, 2881–2885 CrossRef CAS;
(b) C. Wang, J. Shang, Y. Lan, T. Tian, H. Wang, X. Chen, J. Y. Gu, J. Z. Liu, L. J. Wan, W. Zhu and G. Li, Adv. Funct. Mater., 2015, 25, 6009–6017 CrossRef CAS;
(c) M. Bagheri, M. Y. Masoomi, A. Morsali and A. A. Schoedel, ACS Appl. Mater. Interfaces, 2016, 8, 21472–21479 CrossRef CAS PubMed;
(d) R. B. Fu, S. M. Hu and X. T. Wu, J. Mater. Chem. A, 2017, 5, 1952–1956 RSC.
-
(a) S. Pramanik, C. Zheng, X. Zhang, T. J. Emge and J. Li, J. Am. Chem. Soc., 2011, 133, 4153–4155 CrossRef CAS;
(b) J. H. Qin, B. Ma, X. F. Liu, H. L. Lu, X. Y. Dong, S. Q. Zang and H. Hou, Dalton Trans., 2015, 44, 14594–14603 RSC;
(c) W. Xie, S. R. Zhang, D. Y. Du, J. S. Qin, S. J. Bao, J. Li, Z. M. Su, W. W. He, Q. Fu and Y. Q. Lan, Inorg. Chem., 2015, 54, 3290–3296 CrossRef CAS PubMed;
(d) L. Li, S. Zhang, L. Xu, L. Han, Z. N. Chen and J. Luo, Inorg. Chem., 2013, 52, 12323–12325 CrossRef CAS;
(e) L. Di, J. J. Zhang, S. Q. Liu, J. Ni, H. Zhou and Y. J. Sun, Cryst. Growth Des., 2016, 16, 4539–4546 CrossRef CAS;
(f) S. Zhang, L. Han, L. Li, J. Cheng, D. Yuan and J. Luo, Cryst. Growth Des., 2013, 13, 5466–5472 CrossRef CAS;
(g) X. Zhou, H. Li, H. Xiao, L. Li, Q. Zhao, T. Yang, J. Zuo and W. Huang, Dalton Trans., 2013, 42, 5718–5723 RSC;
(h) Y. N. Gong, L. Jiang and T. B. Lu, Chem. Commun., 2013, 49, 11113–11115 RSC;
(i) G. Y. Wang, L. L. Yang, Y. Li, H. Song, W. J. Ruan, Z. Chang and X. H. Bu, Dalton Trans., 2013, 42, 12865–12868 RSC;
(j) R. X. Yao, X. Cui, X. X. Jia, F. Q. Zhang and X. M. Zhang, Inorg. Chem., 2016, 55, 9270–9275 CrossRef CAS;
(k) Y. T. Yan, J. Liu, G. P. Yang, F. Zhang, Y. K. Fan, W. Y. Zhang and Y. Y. Wang, CrystEngComm, 2018, 20, 477–486 RSC;
(l) D. M. Chen, X. Z. Ma, W. Shi and P. Cheng, Cryst. Growth Des., 2015, 15, 3999–4004 CrossRef CAS;
(m) D. Liu, X. Liu, Y. Liu, Y. Yu, F. Chen and C. Wang, Dalton Trans., 2014, 43, 15237–15244 RSC.
- C. Tan and H. Zhang, Chem. Soc. Rev., 2015, 44, 2713–2731 RSC.
-
(a) S. Piccinin, A. Sartorel, G. Aquilanti, A. Goldoni, M. Bonchio and S. Fabris, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 4917–4922 CrossRef CAS;
(b) W. Zhang and K. Zhou, Small, 2017, 13, 1700806 CrossRef;
(c) S. Wang, Y. Hou, S. Lin and X. Wang, Nanoscale, 2014, 6, 9930–9934 RSC.
-
(a) J. Q. Sheng, P. Q. Liao, D. D. Zhou, C. T. He, J. X. Wu, W. X. Zhang, J. P. Zhang and X. M. Chen, J. Am. Chem. Soc., 2017, 139, 1778–1781 CrossRef;
(b) Y. T. Xu, Z. M. Ye, J. W. Ye, L. M. Cao, R. K. Huang, J. X. Wu, D. D. Zhou, X. F. Zhang, C. T. He, J. P. Zhang and X. M. Chen, Angew. Chem., Int. Ed., 2019, 131, 145–149 CrossRef;
(c) J. Duan, S. Chen and C. Zhao, Nat. Commun., 2017, 8, 15341 CrossRef CAS;
(d) F. Dai, W. Fan, J. Bi, P. Jiang, D. Liu, X. Zhang, H. Lin, C. Gong, R. Wang, L. Zhang and D. Sun, Dalton Trans., 2016, 45, 61–65 RSC;
(e) J. Jiang, L. Huang, X. Liu and L. Ai, ACS Appl. Mater. Interfaces, 2017, 9, 7193–7201 CrossRef CAS;
(f) X. Zhao, B. Pattengale, D. Fan, Z. Zou, Y. Zhao, J. Du, J. Huang and C. Xu, ACS Energy Lett., 2018, 3, 2520 CrossRef CAS;
(g) J. Li, W. Huang, M. Wang, S. Xi, J. Meng, K. Zhao, J. Jin, W. Xu, Z. Wang, X. Liu, Q. Chen, L. Xu, X. Liao, Y. Jiang, A. K. Owusu, B. Jing, C. Chen, D. Fan, L. Zhou and L. Mai, ACS Energy Lett., 2018, 4, 285–292 CrossRef;
(h) Z. Xue, Y. Li, Y. Zhang, W. Geng, B. Jia, J. Tang, S. Bao, H. Wang, Y. Fan, Z. Wei, Z. Zhang, Z. Ke, G. Li and C. Su, Adv. Energy Mater., 2018, 8, 1801564 CrossRef;
(i) F. L. Li, Q. Shao, X. Huang and J. P. Lang, Angew. Chem., Int. Ed., 2018, 57, 1888–1892 CrossRef CAS;
(j) W. Zhou, D. D. Huang, Y. P. Wu, J. Zhao, T. Wu, J. Zhang, D. S. Li, C. Su, P. Feng and X. Bu, Angew. Chem., Int. Ed., 2019, 58, 1–6 CrossRef;
(k) G. Hai, X. Jia, K. Zhang, X. Liu, Z. Wu and G. Wang, Nano Energy, 2018, 44, 345–352 CrossRef CAS;
(l) S. Zhao, Y. Wang, J. Dong, C. T. He, H. Yin, P. An, K. Zhao, X. Zhang, C. Gao, L. Zhang, J. Lv, J. Wang, J. Zhang, A. M. Khattak, N. A. Khan, Z. Wei, J. Zhang, S. Liu, H. Zhao and Z. Tang, Nat. Energy, 2016, 1, 16184 CrossRef CAS;
(m) K. Wan, J. Luo, C. Zhou, T. Zhang, J. Arbiol, X. Lu, B. W. Mao, X. Zhang and J. Fransaer, Adv. Funct. Mater., 2019, 29, 1900315 CrossRef;
(n) X. Zhang, J. Luo, K. Wan, D. Plessers, B. Sels, J. Song, L. Chen, T. Zhang, P. Tang, J. R. Morante, J. Arbiol and J. Fransaer, J. Mater. Chem. A, 2019, 7, 1616–1628 RSC.
- İ. Çapan and S. Servi, Synth. Commun., 2018, 48, 1164–1171 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- V. A. Blatov, A. P. Shevchenko and V. N. Serezhkin, J. Appl. Crystallogr., 2000, 33, 1193 CrossRef CAS.
-
(a) B. Gole, A. K. Bar and P. S. Mukherjee, Chem. Commun., 2011, 47, 12137–12139 RSC;
(b) J. S. Qin, S. J. Bao, P. Li, W. Xie, D. Y. Du, L. Zhao, Y. Q. Lan and Z. M. Su, Chem.–Asian J., 2014, 9, 749–753 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Crystal data, additional figures, TGA, PXRD, IR spectra, optical spectra or other electronic format. CCDC 1887850, 1887851, 1887854 and 1887855 for compounds 1–4. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra07031a |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Xianglin Lia,
Mengni Penga,
Linhai Jinga,
Baoyi Yub,
Zeqin Chen*ac and
Dabin Qin*a
*a,
Xianglin Lia,
Mengni Penga,
Linhai Jinga,
Baoyi Yub,
Zeqin Chen*ac and
Dabin Qin*a