
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffects and formulation of silver nanoscaffolds on cytotoxicity dependent ion release kinetics towards enhanced excision wound healing patterns in Wistar albino rats†
Lakshimipriya Sethuram,
John Thomas,
Amitava Mukherjee and
Natarajan Chandrasekaran*
and
Natarajan Chandrasekaran*
Centre for Nanobiotechnology, VIT University, Vellore, Tamilnadu, India. E-mail: nchandrasekaran@vit.ac.in; Fax: +91 416 2243092; Tel: +91 416 2202624
First published on 4th November 2019
Abstract
Wound tissue regeneration and angiogenesis are dynamic processes that send physiological signals to the body. Thus, designing novel nanoscaffolds by understanding their surface modifications and toxicological response in a biological system with a potent anti-inflammatory response is a viable solution. In this respect, inspired by the surface chemistry, in the present work we focus on the chemical optimization of silver nanoscaffolds using surface cappings in order to understand their kinetic release behaviour in simulated wound fluids (SWF), to analyze their blood compatibility in human lymphocytes and erythrocytes and then embed them in a chitosan-agarose matrix (CAM) as a productive drug delivery system to evaluate in vivo excision wound tissue regeneration efficiency in Wistar rats. In this regard, polyvinyl alcohol capped silver nanocomposites (PVA-AgNPs) exhibit a dominant antibacterial efficacy with the sustained and controlled release of silver ions and percentage cell mortality and percentage hemolysis of only 10% and 16% compared with uncapped-AgNPs or silver bandaids (SBDs). Also, PVA-AgNP impregnated CAM (PVA-CAM) shows positive effects through their anti-inflammatory and angiogenic properties, with a nearly 95% healing effect within 9 days. The complete development of collagen and fibroblast constituents was also monitored in PVA-CAM by hematoxylin & eosin (H & E) and Masson trichrome (MT) staining. These results provide a clear insight into the development of a potent therapeutic formulation using CAM as a scaffold incorporated with surface functionalized PVA-AgNPs as a bioeffective and biocompatible polymer for the fabrication of efficacious silver wound dressing scaffolds in clinical practice.
1. Introduction
Skin is the protective physical barrier of the integumentary system preventing external injury and damage and which is involved in sensation, immunological surveillance, prevention of dehydration and the effective synthesis of vitamin D3.1 The skin controls body temperature by vasodilation and vasoconstriction of cutaneous blood vessels, and is formed of three main layers: the epidermis covering the outer surface; the dermis, which covers the connective tissue; and the hypodermis, which contains the hair roots and varying amounts of flat cells.2 However, the structural integrity of the skin gets disrupted by cuts, burns, scratches or surgical injuries, which can be termed wounds, and can be caused by chemicals, pressure, heat, friction, or some diseases.3,4 The condition of a wound depends on the diameter and depth of the wound caused in the epidermis and dermis layers of the skin.5,6After a skin injury/disruption, the body will eventually undergo a regeneration mechanism termed wound healing, which is a dynamic series of anatomical and biochemical cascades that involves four different phases: namely, hemostasis, inflammation, proliferation and remodeling. Various cell organelles, cytokines, enzymes, hormones and proteins are involved in these tissue repair processes.7
Briefly, at the wound site, thrombin stimulates platelet activation, which in turn produces various growth factors (chemokines, cytokines). These growth factors assist fibroblast migration and proliferation to the injured site.8 The release of histamine and serotonin forces the endothelial cells to open up their junctions for the free entry of monocytes and neutrophils (known collectively as macrophages). Lymphocytes and polymorphs are required at the injured site to kill pathogens, thereby preventing bacterial contamination.9 Later, the inflammatory phase is accompanied by the proliferative phase, in which the epithelial cells and fibroblasts form new blood vessels. During the remodeling phase, fibrous proteins, fibronectin, collagen and proteoglycans are synthesized and deposited. As the wound matures, collagen gels will be transformed into collagen fibers which will be distributed all over the wound bed,10 resulting in wound contraction, which is a dynamic process whereby a larger wound area gets closed by a secondary skin intention.11
To protect the wound from external and internal forces, a number of preventive measures, including wound dressings, the administration of painkillers and the topical application of anti-inflammatory drugs, have emerged in the market. According to recent reports, wound management products are expected to gradually increase from US$18.35 billion in 2017 to US$22.01 billion by 2022. The formulation of wound dressings is an essential and important sector in biomedical and pharmaceutical research worldwide. Modern dressings are produced on the basis of type of material (hydrocolloid, bandage, hydrogel or film).12 Hydrocolloid products are produced from colloidal suspensions combined with other media such as adhesives and elastomers.13,14 Bandages are obtained from natural materials (cellulose and cotton wool) and synthetic polymers (polyamide) which perform various functions. Gauze material dressings are produced from nonwoven and woven cotton fibers; rayon or polyester; or both.15 Hydrogels are hydrophilic materials that are found to be insoluble and which are obtained from polymethacrylates and can be applied as amorphous, solid sheet, elastic or film formats. But hydrogels are found to possess decreased mechanical strength, are difficult to handle and are found to affect patient compliance.16,17 Film dressings obtained from nylon derivatives were carried on a polyethylene frame. Most types differ in terms of adhesiveness, extensibility and vapour permeability.18,19 Even though there have been tremendous improvements in wound dressings and antibiotics in the pharmaceutical research market, wound healing bandage formulations are found to be ineffectual because of instability, high cost, low availability, low effectiveness, and cytotoxic and deleterious effects.20
Silver based biomedical products could play a most effective part in preventing bacterial infections and in healing wounds.21,22 Also, silver encapsulated nanoparticles have gained extensive attention owing to their sturdy antimicrobial activity towards a broad range of microorganisms, including fungal and bacterial species.23,24 Furthermore, silver nanoparticles (AgNPs), due to their extensive physicochemical properties, have a great aptitude to deal with multi-drug resistance against recent medicines,25,26 and have secured a new therapeutic potential using different polymer cappings of AgNPs as wound dressings for use on various types of wounds due to their safety, reliability, low toxicity, effectiveness and resistance to various microorganisms.27 It has also been demonstrated that AgNPs control the respiratory signaling pathways of cells, keeping them alive so that regeneration of skin cells might be achieved, which leads to reproliferation within the required time period and therefore reconstruction of the skin surface.28 AgNPs and silver ions have a widely known biological effect that occurs in environmental media of more complex composition. The physicochemical characteristics of AgNPs are suitable for better encapsulation, improved solubility of drugs by intravenous administration, and active and passive targeted delivery, and they allow for the controlled and sustained release of antimicrobial agents.29 Work on dissolution characteristics has been analyzed in many publications,30–35 and furthermore the rate of silver release from AgNPs can be controlled. Studies have analyzed the combination of AgNPs with various biocompatible polymers, such as polyvinyl pyrrolidone (PVP), polyvinyl alcohol (PVA), polylactic acid (PLA) and chitosan, for applications in wound healing in different types of dressings, mats and hydrogels,36–39 but there is less knowledge to be found about their behaviour in more complex media, such as in biological processes and in the environment.
The purpose of employing polymers in AgNPs is to protect against the nature of instability and to accelerate the wound healing process through an alteration in growth factors and a reduction in the matrix metalloproteinase (MMP) levels and upgraded cellular apoptosis.40,41 The role of different polymer cappings with AgNPs of smaller size and shape with an effective stability is to allow catalytic and reactive sites for quicker counteraction and healing potential during the different phases of the wound healing process.42 Polymer engulfed AgNPs have the capacity to penetrate inside the circulatory system and hence across the blood barrier medium in the human body. In that way, chitosan and agarose have attractive physicochemical properties that are essential for tissue engineering applications. Agarose is well known for controlled degradation, greater mechanical strength and its capacity to withstand cellular phenotypes.43–45 But, agarose on its own is not sufficient to mimic the environment for hepatocytes. So, the incorporation of an ECM-like polymer component could enhance the biomimetic characteristics of the scaffold matrix. In this case, chitosan has shown excellent biodegradability, biocompatibility, non-toxicity and the ability to degrade in human bodily fluids.46 Biomimetic scaffolds composed of chitosan and agarose could act as key components in balancing cell proliferation and adhesion, enabling tissue regeneration and helping in biological delivery.
In our present study, we have formulated a chemically optimized AgNP synthesis using various capping agents, such as trisodium citrate capped silver nanoparticles (TSC-AgNPs), polyvinyl pyrrolidone capped silver nanoparticles (PVP-AgNPs), and polyvinyl alcohol capped silver nanoparticles (PVA-AgNPs), and compared them with uncapped-AgNPs and conventional silver bandaids (SBD) to ensure their antibacterial efficacy against Staphylococcus aureus, to control their ion release kinetic behaviour in simulated wound fluids (SWF) and to analyze their behavioural cytotoxicity using blood compatibility studies. Further, in vivo excision wound healing potential was proven in Wistar albino rat models using a chitosan-agarose matrix (CAM) as a scaffold impregnated with AgNP formulations. Histopathological analysis of the impregnated scaffolds was carried out using hematoxylin & eosin (H & E) and Masson trichrome (MT) staining. The main aim of this work is to authenticate the role of bioeffective polymers and silver nanocomposites in forming competitive scaffolds associated with the sustained and controlled release of silver ions in biological systems as a major prospect for angiogenic response in clinical practice.
2. Experimental section
2.1. Materials used
Uncapped-AgNPs were procured from IoLiTec (Ionic Liquids Technologies GmbH, Germany). Silver nitrate (AgNO3) was procured from Sigma-Aldrich (Bangalore, India). Sodium borohydride (NaBH4), TSC, PVP, PVA, nutrient agar (NA), nutrient broth (NB), Muller Hinton broth (MHB), chitosan (degree of deacetylation ≥ 75.00%) and agarose were obtained from Himedia Laboratories (Mumbai, India). Ultrapure de-ionized water was acquired from a Cascada™ Biowater System (Pall Corporation, USA). The chemicals used in the study were of analytical reagent grade.2.2. Particle formulation
A comparative study with the procured uncapped-AgNPs, capped-AgNPs (stabilized with TSC, PVP or PVA) and SBD-suspensions (collectively termed U-C-B) was conducted and they were characterized and evaluated in in vitro and biosafety studies, their controlled release behaviour on SWF was studied and in vivo experimentation using excision wound models was coupled with histopathological evaluation on Wistar albino rats.
2.3. Particle characterizations
All characterization studies were analyzed a day after the synthesis to obtain the best stabilization. The U-C-B colloids were investigated to determine the surface plasmon resonance (SPR) peak using a UV-Visible spectrophotometer (Hitachi double beam model (U-2910), Japan) with a quartz cell size of 3 cm. The absorbance values ranged from 300 to 800 nm with 1 nm intervals at an optimum temperature, and the recorded spectra were re-plotted using Microsoft Excel. Millipore water was used as a blank and the test samples were analyzed.Analysis of nanoparticle hydrodynamic size and surface charge zeta potential measurement was carried out using a particle size analyzer (Horiba Scientific Nanoparticle analyzer model (SZ-100), Japan) at room temperature. The nanoparticle size was derived based on the measurement of time-dependent scattering of laser light undergoing Brownian motion, using a dynamic light scattering (DLS) technique. The zeta potential measurement is dependent on the velocity and direction of particles under the influence of an electric field. Triplicate measurements were taken and averaged to estimate the z-average and polydispersity index (pDI).
The morphology, particle core size and distribution were observed by transmission electron microscopy (TEM). Samples were obtained by drying a drop of U-C-B colloid on a copper grid (300 mesh) enriched with a carbon support film (Agar Scientific) at ambient temperature. The copper grid was cleaned effectively with de-ionized water and the solvent allowed to evaporate. The absolute core size was quantified through TEM images (at least 500 nanoparticles were counted) using an FEI Tecnai G2 F20 X-TWIN (accelerating voltage of 200 kV) with a W-source and a high-resolution pole piece. The AgNP suspension was dispersed on cotton fabric and oven dried at 40–50 °C for 30 minutes. The surface morphology of the cotton fabricated AgNP suspension was determined using scanning electron microscopy (SEM) (Carl Zeiss EVO 18 Research model, USA) operated at EHT 5 kV. Prepared thin fabrics were mounted on a carbon coated copper grid and allowed to dry by placing them under a mercury lamp for 5 minutes and the samples were analyzed under high-resolution magnification mounted with energy dispersive X-ray spectroscopy (EDS) (INCA x-act model: 51ADD0048, Oxford Instruments, Great Britain) in order to quantify the percentage elemental analysis in the generated U-C-B suspensions. The purified aliquots of U-C-B were evaluated using Fourier transform infrared spectroscopy (FTIR). All FTIR measurements were conducted on a PerkinElmer Spectrum One instrument, (Waltham, USA) at a resolution of 4 cm−1 in potassium bromide (KBr) pellets. The silver concentration in the AgNP stock suspensions was analyzed by ICPOES. (The names and conditions of the analytical techniques were discussed above.) The weight loss and reaction state were determined using a thermogravimetric analysis/differential thermal analysis (TGA/DTA) thermal system (DTG-60, Shimadzu, Kyoto, Japan). The electronic and chemical structures at the molecular metallic interface and the stability of the ligands in the capped AgNPs were investigated using X-ray photoelectron spectroscopy (XPS) (K-Alpha surface analysis XPS instrument, Thermo Fisher Scientific, U.K.). The ion source was operated with an acceleration voltage of 600 V at near ambient pressure.
2.4. In vitro profiles – minimum inhibitory concentration evaluation
The antimicrobial efficacy of U-C-B colloids was analyzed against Gram positive Staphylococcus aureus procured from the American Type Culture Collection (ATCC-25923). In order to optimize against MIC standard values, EUCAST clinical breakpoint tables v. 9.0 (MIC breakpoints – Gentamicin (R < 18 mm) for Staphylococcus aureus) were used as a reference for standardization. Aliquots of U-C-B of desired working concentrations were made from the stock suspension. In order to evaluate the minimum inhibitory concentration (MIC), the U-C-B colloids were diluted to appropriate concentrations and interacted with a bacterial suspension containing 106 to 107 colony forming units (CFU) in 96 well plates. Triplicate measurements were made for statistical observation.MIC was referenced as the minimal concentration of AgNP that completely inhibits the bacterial growth of a target microorganism after incubation at a temperature of 37 °C.51 Briefly, the bacterial cells were grown in NB overnight and diluted in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100 MHB (culture – set OD at 0.1) to a cell density of ∼1 × 108 CFU mL−1. Various working concentrations of U-C-B were interacted with gentamicin and MHB media as positive and negative controls, respectively, and incubation was carried out at 37 °C for 24 hours. Then, 10 μl of resazurin dye was added to all the wells. After 2 hours of incubation the MIC was evaluated for a colour change.52,53
:100 MHB (culture – set OD at 0.1) to a cell density of ∼1 × 108 CFU mL−1. Various working concentrations of U-C-B were interacted with gentamicin and MHB media as positive and negative controls, respectively, and incubation was carried out at 37 °C for 24 hours. Then, 10 μl of resazurin dye was added to all the wells. After 2 hours of incubation the MIC was evaluated for a colour change.52,53
2.5. Release studies
To determine the release kinetics of silver onto SWF, dialysis experiments for the AgNP formulations were conducted using a beaker method with a known volume of various capped AgNP suspensions (0.35 g L−1 Ag) dispersed in the freshly prepared SWF, adjusted to a neutral pH of 7.4. The dialysis method was carried out with a magnetic stirrer under slow stirring at normal ambient temperature.54,55 After appropriate time intervals (0, 1, 2, 3, 4, 5, 6, 12, 18 and 24 hours), the samples were replaced by wound fluids and the percentage of silver ions released with respect to time duration was evaluated at 400 nm. Thus, the percentage at which the release of silver becomes controlled can be evaluated for the respective AgNPs in order to understand their dissolution behaviour in the biological system within stipulated time intervals.2.6. Biosafety studies
The percentage cell viability was evaluated by a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay.
The percentage hemolysis of RBCs was calculated59,60 according to the equation:
| percentage hemolysis = ((sample absorbance − negative control absorbance)/(positive control absorbance − negative control absorbance)) × 100. |
2.7. Preparation of chitosan-agarose matrix (CAM) sponge like biocomposite
For CAM preparation, various concentrations of deacetylated chitosan powder ranging from 0.5% to 2% w/v were dissolved in 1% acetic acid solution at room temperature with the help of a magnetic stirrer. In a separate glass beaker, different concentrations of agarose ranging from 1% to 5% w/v were dissolved in ultrapure de-ionized water and heated until a clear transparent solution appeared.61,62 The prepared chitosan solution was centrifuged at 5000 rpm for 15 minutes in order to remove excess particulates. Then, the centrifuged chitosan solution was added to the dissolved agarose solution in different ratios. Once the transparent agarose solution had disintegrated with the chitosan solution, 1 N of NaOH was added under stirring mode and the polymeric solutions were cooled down to ambient temperature. Finally, PVA-AgNPs, PVP-AgNPs, TSC-AgNPs, uncapped-AgNPs and SBD-suspensions were incorporated into the formulated polymeric matrix to form PVA-AgNP impregnated CAM (PVA-CAM), PVP-AgNP impregnated CAM (PVP-CAM), TSC-AgNP impregnated CAM (TSC-CAM), uncapped-AgNP impregnated CAM (uncapped-CAM) and SBD-suspension impregnated CAM (SBD-CAM), respectively, and freeze dried for further analysis.2.8. Animal studies
Firstly, the housed animals were systematically anaesthetized with diethyl ether before the excision wound procedure.63,64 An impression was made on the dorsal region, i.e., 1.5 cm away from the vertebral column and 4.5 cm away from the ear. The predetermined area was shaved and the skin was excised to the full depth to achieve a wound area of about 400 mm2. The wound area was traced using transparent polythene graph paper and measured. Treatment was given on predetermined days, in which the prepared matrix scaffolds were used as substrates for impregnation of AgNPs.65,66 The process includes the exposure of various synthesized scaffolds (i.e., CAM alone, uncapped-CAM, capped = TSC-CAM, PVP-CAM and PVA-CAM, SBD-CAM) which were allowed to react on alternate days with the wound area in order to initiate the inflammatory phase followed by the other phases. The rate of wound contraction was observed with the help of the graph paper on the prespecified days, i.e., 0th, 3rd, 6th, 9th, 14th and 20th days, and the percentage wound contraction was calculated according to equation (eqn (1))
 | (1) |
The 0th day wound area was assigned a value of 100% to demonstrate the percentage wound healing.
2.9. Statistical analysis
The obtained results were evaluated in triplicate and expressed in terms of mean ± standard error with the help of Microsoft Excel. Statistical analysis was done using analysis of variance (ANOVA) with the Prism GraphPad v4.03 software program (San Diego, CA, USA) and the statistical difference was found to be significantly lower than p-value < 0.05.3. Results and discussion
3.1. Effects on formulation and characterization of AgNPs
The beneficial effects of silver nanomaterials in biomedical applications demand a promising strategy for the preparation of effective wound dressings embedded with bioactive polymers which could show their quality of tissue regeneration and their scarless wound healing process.68 Though the topical application of antimicrobial drugs, silver has for years been promoted in wound care,69 but the use of silver wound dressings has recently faced numerous challenges, including a perceived lack of efficiency, high cytotoxic effects, rapid and sudden release of ions, and concerns about cost-effectiveness and safety.70–72 In order to overcome the toxicological effects and to control the ionic release effects, we tried using different polymers encapsulated with silver to form polymer stabilized silver nanomaterials and compared their efficacy with uncapped-AgNPs to prove and authenticate the benefits of capping properties as a way to control ionic release into a biological system. The advantage of using polymer stabilized AgNPs is that their action against bacterial cells depends on the rate and amount of ionic release, resulting in the death of bacterial cells and in detrimental non-reversible changes in the cell wall structure, thereby inhibiting cellular growth.73In summary, the purpose of synthesizing AgNPs is to produce a relatively effective nanomaterial with a well-defined size and shape distribution, distinct morphological structure and surface functionalization properties with better efficiency.74 Upon the addition of a strong reducing agent to the metal precursor with the help of stabilizing agents, in accordance with the previously established chemical reduction method, the suspension turned from a colourless Ag+ solution to pale yellow, which confirmed the identity of colloidal AgNPs. Ultraviolet (UV) spectroscopy has been found to be an effective analytical technique for the structural determination of AgNPs. Plasmon resonance absorption spectra for the uncapped and capped (TSC, PVP, PVA) AgNPs were analytically evaluated at an average of 395–450 nm. The broadening of the peak shows that the nanoparticles were polydispersed in the medium, as shown in Table 1. The peak centered from 400 to 420 nm is mainly due to the plasmon resonance of the AgNPs. The shape and position of the plasmon resonance of a noble metal nanocluster are dependent on the shape, size, surrounding matrix and dielectric medium of the absorbed species.75,76 In addition, the variation in the SPR peak depends solely upon the activity and presence of capping agents in the surrounding surface matrix. Further, to determine the hydrodynamic particle size and stability, the DLS technique is used effectively to observe the mean particle size and aggregation behaviour of the nanoparticles in the suspension. DLS provides a mean particle size between 10 and 40 nm with an average limit of 40 nm and the pDI was no greater than 0.5 (Table 1).
| Contents | Uncapped-AgNPs | TSC-AgNPs | PVP-AgNPs | PVA-AgNPs | SBD-suspension |
|---|---|---|---|---|---|
| SPR peak | 400 | 394 | 410 | 407 | 403 |
| DLS size | 25.2 nm | 12.7 nm | 19.1 nm | 18.1 nm | 32.2 nm |
| pDI width | 0.51 | 0.46 | 0.53 | 0.589 | 0.306 |
| TEM | 19.5 ± 0.2 | 10.5 ± 0.5 | 17.8 ± 0.2 | 13.4 ± 0.4 | 20.1 ± 0.5 |
| pH value | 7.1 | 7.97 | 8 | 7.94 | 7.0 |
| Mass concentration (mg L−1) | 15.70 | 24.07 | 26.15 | 25.51 | 23.15 |
| Zeta potential (mV) | 10 | 34 | 70.3 | 90.5 | 26 |
| MIC value | 20 μg mL−1 | 10 μg mL−1 | 10 μg mL−1 | 5 μg mL−1 | 20 μg mL−1 |
The values acquired from DLS are a little larger than those from TEM due to the precipitation or accumulation of particles in the solution. A pDI value below 0.5 may be associated with high homogeneity and stability in the AgNP population. The pH value for silver colloidal suspensions was adjusted to the range of 7–8, as shown in Table 1. The successful synthesis of AgNPs is in accordance with the structural morphology, particle diameter, and dispersions of AgNPs. This was confirmed by TEM, in which the particles were found to be polydispersed, spherical in shape and distributed uniformly upon the surface, confirming the homogeneous formation of AgNPs as determined. The generated particle core size was between 8 and 20 nm, as presented in Fig. 1(1a–1e), which shows that the TEM and DLS results were similar in hydrodynamic size to the originally synthesized nanoparticle colloids.
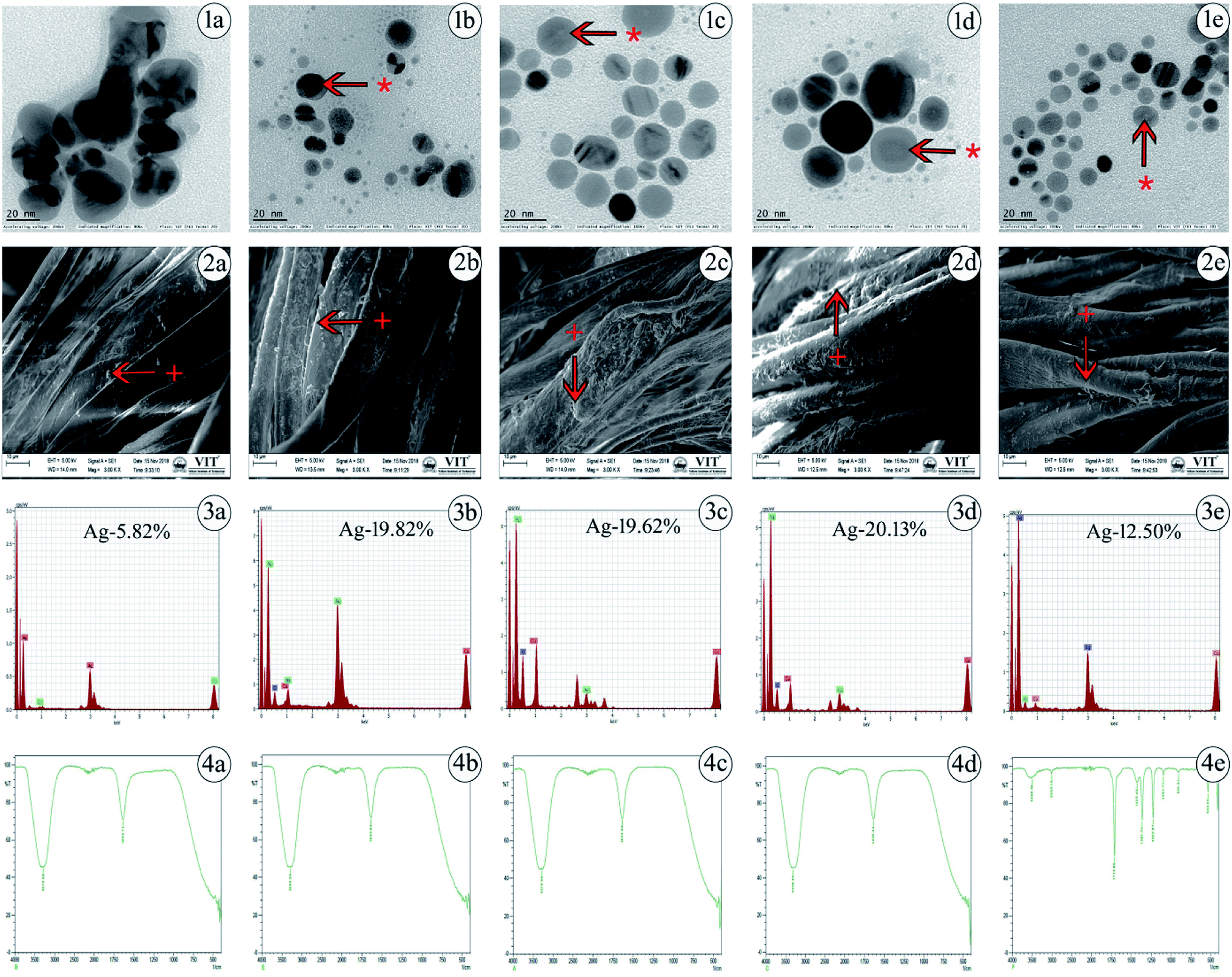 | ||
| Fig. 1 Characterization of chemically synthesized AgNPs using various capping agents. (1a–1e) Determination of particle core size using transmission electron microscopy (TEM) for the AgNPs. (2a–2e) Surface morphological features of AgNPs impregnated onto cotton fabrics using scanning electron microscopy (SEM). (3a–3e) Presence of elemental silver detected by energy dispersive X-ray spectroscopy (EDS) for the AgNPs. (4a–4e) Determination of functional groups using Fourier transform infrared (FTIR) spectroscopy for the AgNPs. Uncapped-AgNPs (1a, 2a, 3a and 4a), TSC-AgNPs (1b, 2b, 3b and 4b), PVP-AgNPs (1c, 2c, 3c and 4c), PVA-AgNPs (1d, 2d, 3d and 4d), SBD-suspension (1e, 2e, 3e and 4e). * represents the presence of capping agents on the surface of nanoparticles, + represents the presence of AgNPs and to denote where the EDS was taken. | ||
Generally, TEM is considered to be a most adaptable technique to monitor the shape, size and distribution of AgNPs. SEM images of various cappings of AgNPs at different magnifications have been depicted. SEM reveals the surface morphology of the AgNPs, which determines their topographical features for further systemic characterization. The impregnation of AgNPs in prepared cotton fabrics was monitored using SEM, which shows a prominent uniform diffusion of AgNPs in the prepared cotton fabric material, as shown in Fig. 1(2a–2e).
Further, the presence of elemental silver in the prepared cotton fabric material was analyzed using EDS, which is an analytical technique used to depict the presence of metal ions in samples with the help of metallic signals.77 Fig. 1(3a–3e) shows the EDS analytical results for the AgNPs. The strong Ag signal denotes the presence of metallic AgNPs and their crystalline nature. In this way, the percentages of silver present in uncapped-AgNPs, TSC-AgNPs, PVP-AgNPs, PVA-AgNPs and SBD-AgNPs were found to be 5.82%, 19.82%, 19.62%, 20.13% and 12.50%, respectively. This shows the quantity of silver formed and present in the state of AgNP formation. Together with these emitted signals, O and C atoms were likely to be observed from the carbon coated grid by the X-ray emission used in EDS analysis. Fig. 1(4a–4e) shows the FTIR spectra of the AgNP formulations. The spectrum of uncapped-AgNPs (Fig. 1(4a)) shows stretching vibrations of N–O peaks and N–H peaks at 1633 cm−1 and 3270 cm−1, respectively. This shows that nitrogen atoms attach to and bind upon the nanoparticle surface after the indication of nanoparticle formation. FTIR spectra of TSC-AgNPs and PVP-AgNPs show stretching vibrations of N–H peaks at 3288 cm−1 and 3276 cm−1, respectively, as shown in Fig. 1(4b and 4c). FTIR spectra of SBD-suspensions show stretching vibrations of N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, CC, O–H and N–H in peaks at 1710 cm−1, 1361 cm−1, 1425 cm−1 and 3496 cm−1, respectively. This indicates that the polymer molecules are bound onto the nanoparticle surface, as shown in Fig. 1(4e). Due to the presence of polymer molecules on the surface of the nanoparticles, the particles do not merge together. Prominent peaks of PVA-AgNPs are shown in Fig. 1(4d). The peaks are found at 3309 cm−1 and 1635 cm−1, which confirms the binding of PVA polymer on the surface of AgNPs. The binding is shown by symmetric stretching with the carboxylate anion. The peaks at 3309 cm−1 and 1635 cm−1 may be due to O–H and C–H bending. In addition, symmetric and asymmetric stretching of –CH2– was observed in PVA-AgNPs when compared with other AgNP formulations. The concentration of the silver present in AgNPs was evaluated upon oxidation with excess concentrated HNO3 (65%). After a process of microwave digestion, the concentrations of silver for all AgNPs were effectively determined by ICPOES with four-digit accuracy and the observed concentrations for all the colloidal AgNPs were found to be between 15 and 25 mg L−1 (ppm), as shown in Table 1. The zeta potential measurements were investigated for the AgNP formulations, as shown in Table 1. The value of the zeta potential for PVA-AgNPs was found to be 90.5 mV, which is highly stable and monodispersed compared to PVP-AgNPs, TSC-AgNPs and SBD suspensions, the values for which were found to be 70.3 mV, 34 mV and 26 mV, respectively. The value of the zeta potential for uncapped-AgNPs was found to be only 10 mV, which shows it to be a highly unstable suspension, which may be due to the absence of capping agents in the colloidal suspension. The stability of uncapped-AgNPs was found to be significantly lower than for the other AgNP formulations, as shown in Table 1. Although the percentage of PVA to AgNP in the colloidal suspension was only 1%, the value of the zeta potential was higher and it proved to be a more stable formulation.78 Results from the TGA curve for uncapped-AgNPs show that there is an insufficient weight loss of only 2.655% in the temperature range 100 °C to 400 °C and the DTA profile for uncapped-AgNPs also lies below 100 °C, where there is incomplete decomposition and crystallization of the sample, as shown in Fig. S1 (ESI†). Results from the TGA curve for PVA-AgNPs exhibit a predominant weight loss for the sample of nearly 64.15%, which occurs in the temperature region 200 °C to 500 °C and the DTA profile for PVA-AgNPs exhibits an exothermically intense peak between 100 °C and 200 °C, which indicates a suitable and complete crystallization and thermal decomposition of the sample,79 as shown in Fig. S2 (ESI†), compared to uncapped-AgNPs. (TGA/DTA curves for TSC-AgNPs, PVP-AgNPs and SBD suspensions – data not shown). These results show that, compared to uncapped-AgNPs, there is a systematic weight loss in PVA-AgNPs, which stabilizes when it reaches and crosses a specific temperature. Evaluation of elemental analysis using XPS for uncapped-AgNPs shows only the presence of silver (Ag3d3/2 and Ag3d5/2) and there are no other peaks connected to oxygen, carbon or nitrogen, which may be due to the absence of capping agents, as shown in Fig. S3 (ESI†). (XPS results for TSC-AgNPs, PVP-AgNPs and SBD suspensions – data not shown). But evaluation of the elemental analysis for PVA-AgNPs shows the presence of carbon (C–C peak), oxygen (O1s peak) and nitrogen (NSiO2, NSi3 and NSi2O) connected to Ag (Ag3d3/2 and Ag3d5/2), as shown in Fig. S4 (ESI†). These results for PVA-AgNPs indicate the presence of oxygen, carbon and nitrogen peaks, which may be due to the presence of capping agents on the surface of the nanoparticles. Whereas, due to the absence of capping agents on the surface of the nanoparticles, uncapped-AgNPs show no carbon or oxygen peaks connected to Ag.
O, CC, O–H and N–H in peaks at 1710 cm−1, 1361 cm−1, 1425 cm−1 and 3496 cm−1, respectively. This indicates that the polymer molecules are bound onto the nanoparticle surface, as shown in Fig. 1(4e). Due to the presence of polymer molecules on the surface of the nanoparticles, the particles do not merge together. Prominent peaks of PVA-AgNPs are shown in Fig. 1(4d). The peaks are found at 3309 cm−1 and 1635 cm−1, which confirms the binding of PVA polymer on the surface of AgNPs. The binding is shown by symmetric stretching with the carboxylate anion. The peaks at 3309 cm−1 and 1635 cm−1 may be due to O–H and C–H bending. In addition, symmetric and asymmetric stretching of –CH2– was observed in PVA-AgNPs when compared with other AgNP formulations. The concentration of the silver present in AgNPs was evaluated upon oxidation with excess concentrated HNO3 (65%). After a process of microwave digestion, the concentrations of silver for all AgNPs were effectively determined by ICPOES with four-digit accuracy and the observed concentrations for all the colloidal AgNPs were found to be between 15 and 25 mg L−1 (ppm), as shown in Table 1. The zeta potential measurements were investigated for the AgNP formulations, as shown in Table 1. The value of the zeta potential for PVA-AgNPs was found to be 90.5 mV, which is highly stable and monodispersed compared to PVP-AgNPs, TSC-AgNPs and SBD suspensions, the values for which were found to be 70.3 mV, 34 mV and 26 mV, respectively. The value of the zeta potential for uncapped-AgNPs was found to be only 10 mV, which shows it to be a highly unstable suspension, which may be due to the absence of capping agents in the colloidal suspension. The stability of uncapped-AgNPs was found to be significantly lower than for the other AgNP formulations, as shown in Table 1. Although the percentage of PVA to AgNP in the colloidal suspension was only 1%, the value of the zeta potential was higher and it proved to be a more stable formulation.78 Results from the TGA curve for uncapped-AgNPs show that there is an insufficient weight loss of only 2.655% in the temperature range 100 °C to 400 °C and the DTA profile for uncapped-AgNPs also lies below 100 °C, where there is incomplete decomposition and crystallization of the sample, as shown in Fig. S1 (ESI†). Results from the TGA curve for PVA-AgNPs exhibit a predominant weight loss for the sample of nearly 64.15%, which occurs in the temperature region 200 °C to 500 °C and the DTA profile for PVA-AgNPs exhibits an exothermically intense peak between 100 °C and 200 °C, which indicates a suitable and complete crystallization and thermal decomposition of the sample,79 as shown in Fig. S2 (ESI†), compared to uncapped-AgNPs. (TGA/DTA curves for TSC-AgNPs, PVP-AgNPs and SBD suspensions – data not shown). These results show that, compared to uncapped-AgNPs, there is a systematic weight loss in PVA-AgNPs, which stabilizes when it reaches and crosses a specific temperature. Evaluation of elemental analysis using XPS for uncapped-AgNPs shows only the presence of silver (Ag3d3/2 and Ag3d5/2) and there are no other peaks connected to oxygen, carbon or nitrogen, which may be due to the absence of capping agents, as shown in Fig. S3 (ESI†). (XPS results for TSC-AgNPs, PVP-AgNPs and SBD suspensions – data not shown). But evaluation of the elemental analysis for PVA-AgNPs shows the presence of carbon (C–C peak), oxygen (O1s peak) and nitrogen (NSiO2, NSi3 and NSi2O) connected to Ag (Ag3d3/2 and Ag3d5/2), as shown in Fig. S4 (ESI†). These results for PVA-AgNPs indicate the presence of oxygen, carbon and nitrogen peaks, which may be due to the presence of capping agents on the surface of the nanoparticles. Whereas, due to the absence of capping agents on the surface of the nanoparticles, uncapped-AgNPs show no carbon or oxygen peaks connected to Ag.
3.2. Antimicrobial efficacy
The antibacterial effect of the nanoparticles was concentration dose dependent and was marked competent against Gram-positive microorganisms.80 The MIC values of Staphylococcus aureus against AgNPs were studied by the broth dilution method, keeping 50% as the maximum MIC value. The uncapped-AgNPs, TSC-AgNPs, PVP-AgNPs, PVA-AgNPs and SBD-suspensions exhibited MIC percentage values of 20 μg mL−1, 10 μg mL−1, 10 μg mL−1, 5 μg mL−1 and 20 μg mL−1, respectively. The results after 48 hours showed high efficiency, as the MIC percentage had increased to 60%. The results indicate that Staphylococcus aureus was resistant up to 20 μg mL−1, 10 μg mL−1 and 20 μg mL−1 for uncapped-AgNPs, TSC-AgNPs and SBD-suspensions, whereas for PVP-AgNPs and PVA-AgNPs, the resistance of the Gram-positive strain was revealed as early as 5 μg mL−1. This shows that the efficiency of antibacterial activity was higher for PVP-AgNPs and PVA-AgNPs compared with uncapped-AgNPs, TSC-AgNPs or SBD-suspensions. This proves its efficiency in bacterial cell wall degradation. A few existing in vitro research studies show that chitosan/PVA dressings had excellent biodegradation and biocompatibility grades of healing against the penetration of microorganisms81 and exhibited effective antibacterial activity with minimal side effects. Also, AgNPs with four different capping agents, polyethylene glycol (PEG), ethylenediaminetetraacetic acid (EDTA), PVP and PVA, were studied and it was found that PVA-AgNPs had the smallest particle size with a high stability and also displayed the highest antimicrobial activity.82 These findings correlate well with our results and suggest that PVA capped composites would be an ideal capping agent and an effective polymer of interest for biomedical applications.3.3. Release studies
Although numerous studies have been done on the dissolution behaviour of AgNPs,30–35 the effect of the release of different cappings for nanomaterials on simulated biological fluids remains unanswered. Hence, this work could focus on ways to control the release of silver ions from synthesized AgNPs using different capping agents and correlate its findings with the toxicological effects and wound healing mechanisms. The dissolution kinetics of different cappings of AgNPs were monitored in order to observe the percentage rate of cumulative release with respect to time period. Further, to evaluate the interactions between AgNPs and SWF, we first determined the release time by providing a range between 400 and 420 nm. Then, the silver ions released from AgNPs were studied using UV spectroscopy when they interacted with SWF by monitoring the absorbance which determines the plasmon resonance band of AgNPs. As shown in Fig. 2, the rate percentage of cumulative release for uncapped-AgNPs during consecutive time periods in terms of hours (1, 2, 3, 4, 5, 6, 12, 16, 18, 20, 24, 28 and 32) was found to have a maximum release compared with SBD-suspensions, TSC-AgNPs, PVP-AgNPs and PVA-AgNPs. | ||
| Fig. 2 Silver ion release kinetics of AgNPs using different capping agents. | ||
The cumulative release observed at the end of the 24th to 32nd hours for uncapped-AgNPs was found to be nearly 98%, whereas the cumulative release for SBD-suspensions, TSC-AgNPs, PVP-AgNPs and PVA-AgNPs were found to be nearly 91%, 80%, 68% and 11%, respectively, as shown in Fig. 2. These observations imply that the maximum release of silver ions onto SWF may be due to the cytotoxic effect of the nanoparticles' behaviour on the biological environment, whereas particles with sustained and controlled release show a kinetically stable dissolution behaviour in the biological medium. The release of silver ions was preponderantly controlled by a diffusion mechanism known as Fickian diffusion. In summary, the release of silver ions can also be controlled by their capping effects and properties. In this regard, the nanoparticles embedded with PVA (i.e., PVA-AgNPs) show a sustained and controlled release of silver ions onto SWF compared with uncapped-AgNPs, SBD-suspensions, TSC-AgNPs or PVP-AgNPs. The percentage cumulative release for PVA-AgNPs was only 11% during the 32nd hour, and was found to have a minimal release of silver ions compared with the uncapped-AgNPs, TSC-AgNPs, PVP-AgNPs or SBD-suspensions. From the statistical observations made, PVA-AgNPs show a more significant difference (***) (p value ≤ 0.001) compared with uncapped-AgNPs. This may be due to the sustained and controlled release of PVA capped silver ions onto SWF with respect to consecutive time periods. Whereas, SBD-suspension shows no significant difference (ns) (p value > 0.05) compared with uncapped-AgNPs. This may be due to the sudden release of silver ions with respect to consecutive time periods. A few research studies have concluded that controlled drug release was observed from PVA capped dressings, which is required for wound care management and is in accordance with the findings proposed here.83 Blending of PVA composites enhances a convenient and useful method to control the period and rate of ion release in wound healing processes.84 PVA hydrogel stabilized with nano-Ag exhibits controlled and sustained release with a pH identical to simulated body fluids and shows effective antibacterial activity against Gram negative Escherichia coli and Gram positive Staphylococcus aureus.85 The results suggested that the release of silver ions from AgNPs may depend solely upon the antimicrobial and anti-inflammatory profiles and surface chemistry of the capping agents. Uncapped-AgNPs release a higher amount of Ag+ ions than capped ones. In capped nanoparticles, ionic release is controlled and varies according to the capping agent. From the results obtained, PVA-AgNPs can be designed as an effective capping agent/surface coating agent in wound dressing matrices for the sustained and controlled release of drugs into a biological system.
3.4. Effects on the cytotoxic analysis
The percentage cell mortality of lymphocytes can be regulated by the presence of different cappings on AgNPs in a concentration-dependent manner. In fact, a significant increase in cell mortality (%) of nearly 97% was found with a higher amount of uncapped-AgNPs, whereas at lower concentrations of uncapped-AgNPs, the percentage cell mortality was 90%. This result indicates that a decrease in the concentration of AgNPs may definitely decrease the percentage cell mortality. Another aspect is that the percentage cell mortality for uncapped-AgNPs at 20 μg mL−1 was 97% and this is significantly higher than the values for PVP-AgNPs and PVA-AgNPs, which were 42.4% and 10.43%, whereas with TSC-AgNPs and SBD-suspensions, there was no significant difference and the percentage mortalities were found to be 70.4% and 88.8%, respectively. Even at 10 μg mL−1 concentration, the percentage cell mortality for uncapped-AgNPs was 95.55%, significantly higher than the values for PVP-AgNPs and PVA-AgNPs, which were 34.14% and 9.06%, whereas with TSC-AgNPs and SBD-suspensions, there was no significant difference and the percentage mortalities were found to be nearly 63.02% and 85.1%, respectively. At 1 μg mL−1 concentration, the percentage cell mortality for uncapped-AgNPs was 90.25%, significantly higher than the values for TSC-AgNPs, PVP-AgNPs, PVA-AgNPs which were only 55.18%, 25.81%, and 5.12%, respectively, as shown in Fig. 3.
 | ||
| Fig. 3 Cell mortality rate of WBCs interacted with different AgNP formulations. | ||
Even though the percentage mortality rates for TSC-AgNPs, PVP-AgNPs and SBD-suspensions were found to be low compared with uncapped-AgNPs, PVA-AgNPs possess a very minimal cytotoxic activity (p ≤ 0.001) which may be because of the polyvinyl polymer being encapsulated with an OH group and embedded on the outer surface of the AgNP that provides the polymer with high stability and anti-angiogenic efficiency. Also, from the statistical observations made, the percentage cell mortality for PVA-AgNPs shows a more significant difference (***) (p value ≤ 0.001) than the uncapped-AgNPs. This may be due to the minimal cytotoxic effect of PVA-AgNPs that show the lowest percentage cell mortality. Although SBD fabricated with polyethylene polymers has an anti-inflammatory property, its efficiency is comparatively low compared with SBD fabricated with polyvinyl polymers. The reason for comparing different coatings of AgNPs with uncapped-AgNPs is to authenticate the role of different surface coatings in nanoparticle synthesis for enhanced stability with antibacterial efficacy and cytotoxic influence and to examine the angiogenic response in wounds.
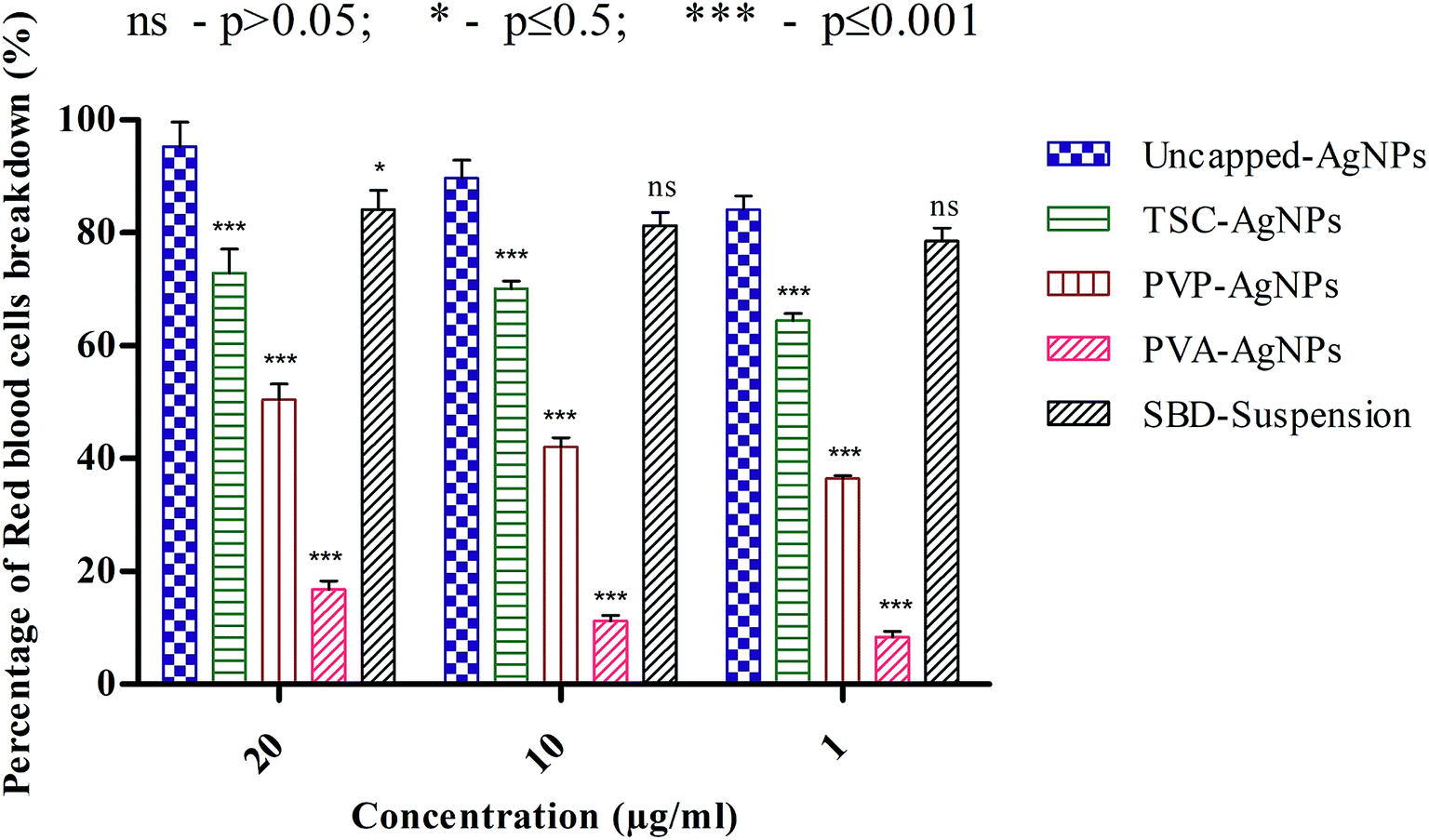 | ||
| Fig. 4 Hemolytic activity of AgNPs using different capping agents. | ||
Another point to be discussed is that the percentage RBC breakdown for uncapped-AgNPs at 20 μg mL−1 was 95.32%, which is significantly higher than the values for TSC-AgNPs, PVP-AgNPs, PVA-AgNPs and SBD-suspensions, which were 72.89%, 50.46%, 16.82% and 84.11%, respectively. Even at 10 μg mL−1, the percentage RBC breakdown for uncapped-AgNPs was 89.71%, which is higher than the values for TSC-AgNPs, PVP-AgNPs, PVA-AgNPs and SBD-suspensions, which were found to be 70.09%, 42.05%, 11.21% and 81.30%, respectively. At 1 μg mL−1, the percentage RBC breakdown for uncapped-AgNPs was found to be 84.11%, which is significantly higher than the values for TSC-AgNPs, PVP-AgNPs, PVA-AgNPs and SBD-suspensions, which were 64.48%, 36.44%, 8.41% and 78.5%, respectively, as shown in Fig. 4. Also, from the statistical observations made, the percentage RBC breakdown for PVA-AgNPs shows a more significant difference (***) (p value ≤ 0.001) than the value for uncapped-AgNPs. This may be due to the minimal cytotoxic effect of PVA-AgNPs that show the lowest percentage RBC breakdown. Whereas SBD-suspensions show no significant difference (ns) (p value > 0.05) at 10 μg mL−1 or 1 μg mL−1 concentrations compared with uncapped-AgNPs. Although the percentage RBC breakdown for TSC-AgNPs, PVP-AgNPs and SBD-suspensions was found to be low compared with uncapped-AgNPs, PVA encapsulated AgNPs show a lower response to breakage of RBCs (p ≤ 0.001), which proves they are a better antimicrobial agent for further applications in the design of films and dressings for wound healing applications.
3.5. In vivo excision wound models
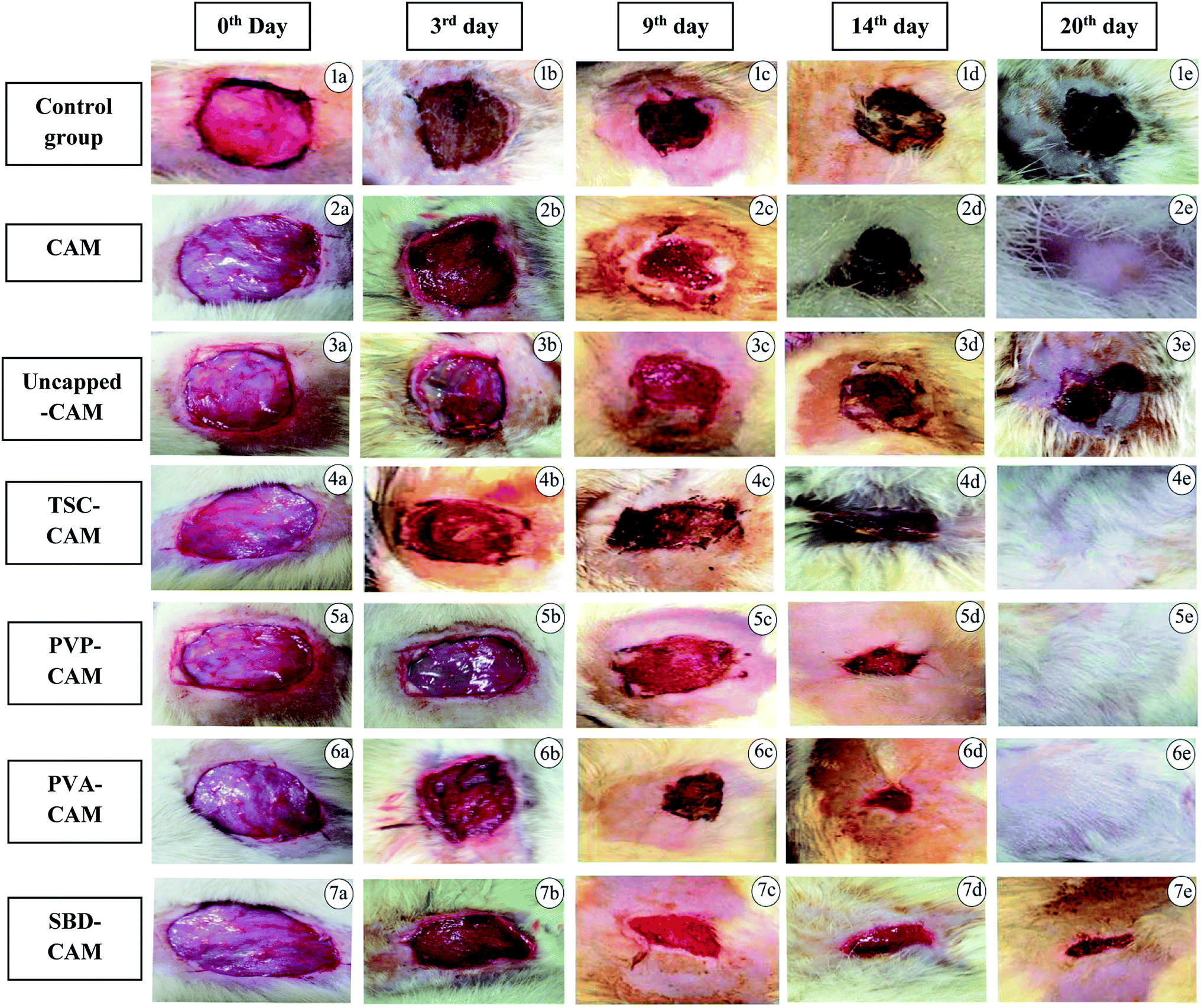 | ||
| Fig. 5 Wound healing efficiency of different CAM associated AgNP formulations during the 3rd day, 9th day, 14th day and 20th days. Healing period with respect to number of days: (1a–1e) control group, (2a–2e) CAM, (3a–3e) uncapped-CAM, (4a–4e) TSC-CAM, (5a–5e) PVP-CAM, (6a–6e) PVA-CAM, (7a–7e) SBD-CAM. | ||
From the results of Fig. 6, on the 3rd day, the percentage wound contraction for PVA-CAM, PVP-CAM, TSC-CAM, SBD-CAM, uncapped-CAM, CAM alone and the control group was found to be 69.25%, 65.5%, 34.25%, 32.75%, 20.25%, 29.5% and 18.25%, respectively. Similarly, on the 6th day, the percentage wound contraction for PVA-CAM, PVP-CAM, TSC-CAM, SBD-CAM, uncapped-CAM, CAM alone and the control group was found to be 81.75%, 78.5%, 44.25%, 37.75%, 31.5%, 35% and 28.25%, respectively. On the 9th day, the percentage wound contraction for PVA-CAM, PVP-CAM, TSC-CAM, SBD-CAM, uncapped-CAM, CAM alone and the control group was found to be 95.25%, 90%, 85.25%, 77%, 60.75%, 64% and 55.25%, respectively. On the 14th day, the percentage wound contraction for PVA-CAM, PVP-CAM, TSC-CAM, SBD-CAM, uncapped-CAM, CAM alone and the control group was found to be 99.5%, 95%, 88.5%, 84.5%, 70.75%, 72% and 68.75%, respectively. On the 20th day, the percentage wound contraction for PVA-CAM, PVP-CAM, TSC-CAM, SBD-CAM, uncapped-CAM, CAM alone and the control group was found to be nearly 100%, 100%, 100%, 98.5%, 90.25%, 99.87% and 89.75%, respectively. Although the percentage wound contraction for PVP-CAM, TSC-CAM and SBD-CAM was quite a bit higher than for uncapped-CAM and the control group, wounds treated with PVA-CAM show the maximum significant wound contraction (p ≤ 0.001) efficiency due to their biocompatibility with wound tissue. CAM is used as a base reference for impregnation with AgNPs since it contains natural polymers for better wound healing applications.
 | ||
| Fig. 6 Percentage of wound contraction of CAM associated AgNP formulations. | ||
From Fig. 6, it is proven that the percentage wound contraction for uncapped-CAM was significantly lower than the values for SBD-CAM, TSC-CAM, PVP-CAM and PVA-CAM on all the respective (3rd, 6th, 9th, 14th and 20th) days, which may be due to the absence of surface coating or capping agents in the uncapped-CAM, which are responsible for nanoparticle stability and healing potential. The results suggested that PVA-CAM has greater percentage wound contraction and shows a more significant difference (***) (p ≤ 0.001) than the control group. This may be due to the higher efficiency of the polyvinyl combined OH group for healing potential. The wound contraction efficiency for uncapped-CAM was also found to be comparatively lower (ns) (p value > 0.05) than that for PVA-CAM, which may be due to the absence of capping agents in the uncapped-CAM for wound healing applications. The degree of wound contraction was monitored using various wound models in rats.64,92–97 In some current research, antibacterial hydrogels made using a horseradish peroxidase (HPR) system were optimized for the development of wound dressings. These antibacterial hydrogels were endowed with greater tissue adhesiveness, enhanced antioxidant activity, and self-healing capacity and they possess similar mechanical properties to human skin. These wound hydrogels exhibit significant vascularization and improved wound closure with sustained and effective drug release.98 However, in spite of all this, wound dressings made with antibiotics will develop further antibiotic resistance in microbes.99 Bacterial cellulose fabricated using guided assembly based biolithography used on artificial wounds in live mouse models exhibits better fibroblast proliferation and collagen deposition with high levels of inflammation and vascularization.100 Also, collagen incorporated with gold nanoparticles used on artificial wounds showed higher neovascularization and maximum wound closure with a milder inflammatory reaction than in an untreated control.101 Composite wound dressings comprised of streptomycin and diclofenac were tested against Staphylococcus aureus, Escherichia coli and Pseudomonas aeruginosa, and showed greater antibacterial efficacy with reduced chronic wound inflammation and infection.102 Antibacterial injectable conductive cryogels were modeled using carbon nanotubes (CNT) for wound healing and hemorrhage hemostasis. These cryogels show better platelet activation and adhesion and a high blood flow uptake capacity with greater speed of absorption.103 A series of hydrogels made using chitosan-g-polyaniline and benzaldehyde functionalized polyethylene glycol presented higher antibacterial and anti-oxidant activity with better self-healing capacity. Therefore, they promote tissue thickness with greater collagen deposition and up-regulate the expression of growth factors.104 Herein, silver encapsulated PVA solutions were synthesized by a specific chemical route and characterized by analytical techniques, and this composite was found to be a potential sensor for the detection and determination of hydrogen peroxide for low-cost applications.105 These results also agree with our findings: apart from being a diagnostic tool for the detection of hydrogen peroxide, PVA-AgNPs could also be potential nanomaterials in terms of wound healing and antimicrobial efficacy with less toxicological activity. For the antimicrobial drug ibuprofen (IBP), β cyclodextrin carriers were formulated along with PVA dressings which were produced by a carbon dioxide (scCO2)-assisted process to promote rapid skin regeneration. Also, PVA-AgNPs play an important role in re-epithelization and a faster skin regeneration process, which were carried out in an excision wound healing model of Wistar albino rats.106 Formulated nanocomposite hydrogels based on PVA containing 5 and 10 wt% were synthesized by a freezing–thawing facile cyclic technique, and their characteristics show they can be used as potent dressings in wound care. But our results showed a higher amount of anti-inflammatory property with 1% of PVA synthesized by a chemical reduction method along with the silver precursor for optimum wound healing applications. Our results suggest that PVA-CAM reveals a faster recovery of healing and ends up with better proliferation of skin tissues and re-epithelization levels in the wound healing cascades.
 | ||
| Fig. 7 Histopathological observations of wounds after treatment with CAM associated AgNP formulations. Red arrows represent the development of collagen and fibroblast regeneration tissues stained using hematoxylin & eosin (H & E). Control group (1a and 2a), CAM (1b and 2b), uncapped-CAM (1c and 2c), TSC-CAM (1d and 2d), PVP-CAM (1e and 2e), PVA-CAM (1f and 2f), SBD-CAM (1g and 2g). (1 represents 9th day, 2 represents 14th day). | ||
 | ||
| Fig. 8 Histopathological observations of CAM associated AgNP treated wounds using Masson trichrome (MT) staining. Red arrows represent the maximum growth of collagen fibers and fibrin regenerative tissues. Control group (1a and 2a), CAM (1b and 2b), uncapped-CAM (1c and 2c), TSC-CAM (1d and 2d), PVP-CAM (1e and 2e), PVA-CAM (1f and 2f), SBD-CAM (1g and 2g) (1 represents 9th day, 2 represents 14th day). | ||
| Name of sample | Fibroblast | Collagen | Regeneration | Comments |
|---|---|---|---|---|
| a + Represents minimum regeneration; ++ represents moderate regeneration; +++ represents maximum regeneration. | ||||
| 9th day (control group) | — | — | — | Normal |
| 9th day (CAM) | + | + | + | Presence of collagen and fibroblast regeneration |
| 9th day (uncapped-CAM) | — | + | + | Skin tissue shows dense inflammation and minimal regeneration of collagen is present |
| 9th day (TSC-CAM) | ++ | + | + | Fibroblast regeneration |
| 9th day (PVP-CAM) | +++ | + | + | Fibroblast and collagen regeneration found to be satisfactory |
| 9th day (PVA-CAM) | +++ | ++ | ++ | Fibroblast are plentiful, but minimal inflammation is present |
| 9th day (SBD-CAM) | + | ++ | + | Skin ulceration shows dense inflammation, granulation tissue present |
| 14th day (control group) | + | + | + | Fibroblasts are present with collagen, inflammation is present |
| 14th day (CAM) | ++ | ++ | + | Presence of collagen fibers, fibrins, and erythrocytes regeneration |
| 14th day (uncapped-CAM) | + | ++ | + | Presence of minimal collagen with regeneration |
| 14th day (TSC-CAM) | ++ | + | + | Moderate regeneration |
| 14th day (PVP-CAM) | +++ | ++ | ++ | Maximum collagen and fibroblast with moderate regeneration |
| 14th day (PVA-CAM) | +++ | +++ | +++ | Equally maximum regeneration |
| 14th day (SBD-CAM) | + | ++ | ++ | Dense fibrosin and scant inflammation |
Based on the observations made, on the 9th day after the excision wound injury, there was no significant development of the fibroblast, collagen or skin regeneration in the control group. Even on the 14th day in the control group, there was only minimal development of fibroblast and collagen observed, with the presence of inflammation. Histopathological studies on treatment with PVA-CAM in the excision wound model showed a maximum amount of fibroblast presence and collagen regeneration after the 9th day of the healing process, which was found to be comparatively more effective than CAM alone, uncapped-CAM, PVP-CAM, TSC-CAM or SBD-CAM that showed a minimal amount of fibroblast and collagen regeneration, with the presence of dense inflammation. Wound treatment with uncapped-CAM after the 9th day of observation showed skin tissue with dense inflammation, an absence of fibroblast development and a very minimal amount of collagen regeneration, and was found to be comparatively less effective than CAM alone, PVA-CAM, PVP-CAM, TSC-CAM or SBD-CAM.
Similarly on the 14th day after excision wound injury, treatment with PVA-CAM showed an equal maximum regeneration of fibroblast and collagen and was significantly more effective and angiogenic compared with CAM alone, uncapped-CAM, PVP-CAM, TSC-CAM or SBD-CAM that showed a moderate amount of collagen and fibroblast regeneration, with scant inflammation during their healing process. At the same time, excision wounds treated with uncapped-CAM after the 14th day showed the presence of a minimal amount of collagen with regeneration and incomplete tissue regeneration, compared with CAM alone, PVA-CAM, PVP-CAM, TSC-CAM or SBD-CAM, as shown in the Fig. 7, 8 and Table 2. Histopathological results suggested that the angiogenic response and re-epithelization potential in PVA-CAM were effectively higher than for uncapped-CAM. The main reason was the presence of surface coating or capping agents in PVA-CAM, which leads to a better healing capacity, an effective angiogenic response and complete fibroblast and collagen regeneration.
As a whole, applications of polymers along with silver nanomaterials could be a promising strategy to study the complex wound healing process both at cellular and molecular level. Of all the optimized formulations being studied in these findings, PVA-CAM possess a predominant antimicrobial potential with an effective wound healing contraction of nearly 69% on the 3rd day after the wounding process, in contrast with the uncapped-CAM with a value of 20%. There is a consequent increase in the percentage wound contraction for PVA-CAM with a maximum amount of angiogenic response. A possible explanation is as follows: the percentage cumulative release of PVA-CAM is found to be a systematic and controlled release due to their less cytotoxic effects on blood cells, whereas the percentage cumulative release of uncapped-AgNPs is found to have a faster and sudden release profile due to the absence of a capping agent and their high toxicity to blood cells. Therefore, due to the lower toxicological effects, controlled release of silver ions and dominant antimicrobial efficacy against selective microorganism, we conclude that PVA-CAM can be considered a more effective polymer for therapeutic benefits in wound healing applications. Its beneficial use may provide an effective therapeutic route for attaining scar-less wound care in clinical practice.
4. Conclusion
These findings indicate that PVA-CAM have a predominant antibacterial activity and accelerate the healing potential of excision wound models in Wistar albino rats by enhancing angiogenesis in the wounded sections and are comparatively more efficient (among polymers) than uncapped-CAM, SBD-CAM or untreated wounds (the control). However, there are many molecular mechanisms behind the healing potential and anti-inflammatory response for the different cappings of AgNPs that are as yet unknown. The obtained histopathological results show the maximum development of collagen and fibroblast deposition when treated with PVA-CAM and these are relatively higher than for other AgNP formulations. This provides authentic evidence for the utilization of capping agents in the synthesis of AgNPs. Hence, the antimicrobial efficacy and wound contraction response observed in PVA-CAM was at a maximum rate. Also, controlled and sustained Ag+ ionic release onto a biological medium can be observed in PVA-AgNPs compared with other AgNP formulations. This PVA-AgNP formulation may be incorporated in CAM as a potential scaffold and act as a therapeutic agent for biomedical wound applications in clinical practice.Conflicts of interest
All authors declare no personal or professional conflicts of interest.Acknowledgements
We deeply acknowledge Sophisticated Analytical Instrumentation Facility (SAIF), at Indian Institute of Technology (IIT), Madras, for ICPOES analysis facility and VIT University for providing the facilities to carry out the research work.References
- W. Paul, Advances in Wound Healing Materials, Smithers Rapra, Jun 11, 2015 Search PubMed.
- A. D. Sezer and E. Cevher, Biopolymers as wound healing materials: challenges and new strategies, Biomaterials Applications for Nanomedicine, Nov 16, 2011, pp. 383–414 Search PubMed.
- J. Hutchinson, The wound programme, Centre for Medical Education, Dundee, 1992, vol. 1(2) Search PubMed.
- S. Mukherjee, S. K. Nethi and C. Patra, Particulate Technology for Delivery of Therapeutics, 2017, pp. 359–393 Search PubMed.
- S. Schreml, R. M. Szeimies, L. Prantl, S. Karrer, M. Landthaler and P. Babilas, Br. J. Dermatol., 2010, 163(2), 257–268 CrossRef CAS.
- S. Rajendran and S. C. Anand, Handbook of medical textiles, 2011, pp. 38–79 Search PubMed.
- G. Arango Duque and A. Descoteaux, Front. Immunol., 2014, 5, 491 Search PubMed.
- N. D. Evans, R. O. Oreffo, E. Healy, P. J. Thurner and Y. H. Man, J. Mech. Behav. Biomed. Mater., 2013, 28, 397–409 CrossRef.
- T. J. Koh and L. A. DiPietro, Expert Rev. Mol. Med., 2011, 13, e23 CrossRef.
- L. E. Tracy, R. A. Minasian and E. J. Caterson, Adv. Wound Care, 2016, 5(3), 119–136 CrossRef.
- K. P. Krafts, Organogenesis, 2010, 6(4), 225–33 CrossRef.
- J. S. Boateng, K. H. Matthews, H. N. Stevens and G. M. Eccleston, J. Pharm. Sci., 2008, 97(8), 2892–2923 CrossRef CAS.
- C. Dealey, Br. J. Nurs., 1993, 2(7), 358–365 CrossRef CAS.
- J. W. Millsop and A. L. Chang, Textbook of Aging Skin, 2014, pp. 1-1 Search PubMed.
- V. J. Jones, Int. Wound J., 2006, 3(2), 79–88 CrossRef PubMed.
- L. Martin, C. G. Wilson, F. Koosha, L. Tetley, A. I. Gray, S. Senel and I. F. Uchegbu, J. Controlled Release, 2002, 80(1–3), 87–100 CrossRef CAS.
- D. A. Morgan, Pharm. J., 1999, 263(7072), 820–825 Search PubMed.
- J. B. Debra and O. Cheri, Technology Catalysts International Corporation, 1998, 2, 1–85.
- S. Thomas, P. Loveless and N. P. Hay, Pharm. J., 1988, 240, 785–787 CAS.
- B. Kumar, M. Vijayakumar, R. Govindarajan and P. Pushpangadan, J. Ethnopharmacol., 2007, 114(2), 103–113 CrossRef CAS.
- R. Subbiah, M. Veerapandian and K. S. Yun, Curr. Med. Chem., 2010, 17(36), 4559–4577 CrossRef CAS.
- I. Donati, E. Marsich, A. Travan and S. Paoletti, Tissue Eng. Regener. Med., 2012, 146–181 Search PubMed.
- A. Frattini, N. Pellegri, D. Nicastro and O. De Sanctis, Mater. Chem. Phys., 2005, 94(1), 148–152 CrossRef CAS.
- T. Textor, M. M. Fouda and B. Mahltig, Appl. Surf. Sci., 2010, 256(8), 2337–2342 CrossRef CAS.
- K. Ito, A. Saito, T. Fujie, K. Nishiwaki, H. Miyazaki, M. Kinoshita, D. Saitoh, S. Ohtsubo and S. Takeoka, Acta Biomater., 2015, 24, 87–95 CrossRef CAS.
- D. Liang, Z. Lu, H. Yang, J. Gao and R. Chen, ACS Appl. Mater. Interfaces, 2016, 8(6), 3958–3968 CrossRef CAS.
- A. Ivask, K. Juganson, O. Bondarenko, M. Mortimer, V. Aruoja, K. Kasemets, I. Blinova, M. Heinlaan, V. Slaveykova and A. Kahru, Nanotoxicology, 2014, 8, 57–71 CrossRef CAS.
- C. Rigo, L. Ferroni, I. Tocco, M. Roman, I. Munivrana, C. Gardin, W. Cairns, V. Vindigni, B. Azzena, C. Barbante and B. Zavan, Int. J. Mol. Sci., 2013, 14(3), 4817–4840 CrossRef CAS.
- R. Rasouli and A. Barhoum, Advances in nanofibers for antimicrobial drug delivery, Handbook of Nanofibers, 2019, pp. 733–74 Search PubMed.
- G. A. Sotiriou and S. E. Pratsinis, Curr. Opin. Chem. Eng., 2011, 1(1), 3–10 CrossRef CAS.
- Z. M. Xiu, Q. B. Zhang, H. L. Puppala, V. L. Colvin and P. J. Alvarez, Nano Lett., 2012, 12(8), 4271–4275 CrossRef CAS PubMed.
- J. Liu and R. H. Hurt, Environ. Sci. Technol., 2010, 44(6), 2169–2175 CrossRef CAS.
- J. Liu, D. A. Sonshine, S. Shervani and R. H. Hurt, Controlled release of biologically active silver from nanosilver surfaces, ACS Nano, 2010, 4(11), 6903–6913 CrossRef CAS.
- X. Li, J. J. Lenhart and H. W. Walker, Langmuir, 2010, 26(22), 16690–16698 CrossRef CAS.
- S. Kittler, C. Greulich, J. Diendorf, M. Koller and M. Epple, Chem. Mater., 2010, 22(16), 4548–4554 CrossRef CAS.
- T. H. Nguyen, Y. H. Kim, H. Y. Song and B. T. Lee, J. Biomed. Mater. Res., Part B, 2011, 96(2), 225–233 CrossRef.
- T. H. Nguyen, K. H. Lee and B. T. Lee, Mater. Sci. Eng., C, 2010, 30(7), 944–950 CrossRef CAS.
- D. J. Ostlie, D. Juang, P. Aguayo, J. P. Pettiford-Cunningham, E. A. Erkmann, D. E. Rash, S. W. Sharp, R. J. Sharp and S. D. Peter, J. Pediatr. Surg., 2012, 47(6), 1204–1207 CrossRef.
- A. M. Abdelgawad, S. M. Hudson and O. J. Rojas, Carbohydr. Polym., 2014, 100, 166–178 CrossRef CAS PubMed.
- V. R. Netala, P. Bobbu, S. B. Ghosh and V. Tartte, Asian J. Pharm. Clin. Res., 2015, 8(3), 113–116 CAS.
- H. Jiang, K. Moon, Z. Zhang, S. Pothukuchi and C. P. Wong, J. Nanopart. Res., 2006, 8, 117–124 CrossRef CAS.
- T. Gunasekaran, T. Nigusse and M. D. Dhanaraju, J. Am. Coll. Clin. Wound Spec., 2011, 3(4), 82–96 CrossRef.
- A. Tripathi, N. Kathuria and A. Kumar, J. Biomed. Mater. Res., Part A, 2009, 90(3), 680–694 CrossRef.
- K. W. Ng, C. C. Wang, R. L. Mauck, T. A. Kelly, N. O. Chahine, K. D. Costa, G. A. Ateshian and C. T. Hung, J. Orthop. Res., 2005, 23(1), 134–141 CrossRef.
- P. D. Benya and J. D. Shaffer, Cell, 1982, 30(1), 215–224 CrossRef CAS.
- J. Hankiewicz and E. Swierczek, Lysozyme in human body fluids, Clin. Chim. Acta, 1974, 57(3), 205–209 CrossRef CAS.
- L. Mulfinger, S. D. Solomon, M. Bahadory, A. V. Jeyarajasingam, S. A. Rutkowsky and C. Boritz, J. Chem. Educ., 2007, 84(2), 322 CrossRef.
- M. Tejamaya, I. Römer, R. C. Merrifield and J. R. Lead, Environ. Sci. Technol., 2012, 46(13), 7011–7017 CrossRef CAS.
- Y. Xiong, I. Washio, J. Chen, M. Sadilek and Y. Xia, Angew. Chem., Int. Ed., 2007, 46(26), 4917–4921 CrossRef CAS.
- D. Gao, X. Zhou, Z. Gao, X. Shi, Z. Wang, Y. Wang and P. Zhang, J. Pharm. Sci., 2018, 6(3), 91–97 Search PubMed.
- E. Amato, Y. A. Diaz-Fernandez, A. Taglietti, P. Pallavicini, L. Pasotti, L. Cucca, C. Milanese, P. Grisoli, C. Dacarro, J. M. Fernandez-Hechavarria and V. Necchi, Langmuir, 2011, 27(15), 9165–9173 CrossRef CAS.
- D. Manikprabhu and K. Lingappa, J. Pharm., 2013, 6(2), 255–260 CAS.
- C. Bradford, R. Freeman and S. L. Percival, J. Am. Coll. Clin. Wound Spec., 2009, 1(4), 117–120 Search PubMed.
- K. Loza, J. Diendorf, C. Sengstock, L. Ruiz-Gonzalez, J. M. Gonzalez-Calbet, M. Vallet-Regi, M. Köller and M. Epple, J. Mater. Chem. B, 2014, 2(12), 1634–1643 RSC.
- K. A. Johnston, L. M. Stabryla, A. M. Smith, X. Y. Gan, L. M. Gilbertson and J. E. Millstone, Environ. Sci.: Nano, 2018, 5(2), 304–312 RSC.
- C. Vergallo, E. Panzarini, D. Izzo, E. Carata, S. Mariano, A. Buccolieri, A. Serra, D. Manno and L. Dini, AIP Conf. Proc., 2014, 1603, 78–85 CrossRef CAS.
- A. Zhornik, L. Baranova, I. Volotovski, S. Chizhik, E. Drozd, M. Sudas, Q. B. Ngo, H. C. Nguyen, T. H. Huynh and T. H. Dao, Adv. Nat. Sci.: Nanosci. Nanotechnol., 2015, 6(2), 025003 Search PubMed.
- L. Q. Chen, B. Kang and J. Ling, J. Nanopart. Res., 2013, 15(3), 1507 CrossRef.
- M. A. Dobrovolskaia, J. D. Clogston, B. W. Neun, J. B. Hall, A. K. Patri and S. E. McNeil, Nano Lett., 2008, 8(8), 2180–2187 CrossRef CAS.
- Y. S. Lin and C. L. Haynes, J. Am. Chem. Soc., 2010, 132(13), 4834–4842 CrossRef CAS.
- A. Tripathi and J. S. Melo, RSC Adv., 2015, 5(39), 30701–30710 RSC.
- S. P. Miguel, M. P. Ribeiro, H. Brancal, P. Coutinho and I. J. Correia, Carbohydr. Polym., 2014, 111, 366–373 CrossRef CAS.
- S. Sugumar, V. Ghosh, M. J. Nirmala, A. Mukherjee and N. Chandrasekaran, Ultrason. Sonochem., 2014, 21(3), 1044–1049 CrossRef CAS.
- S. Murthy, M. K. Gautam, S. Goel, V. Purohit, H. Sharma and R. K. Goel, BioMed Res. Int., 2013, 972028 CAS.
- F. Paladini, C. Di Franco, A. Panico, G. Scamarcio, A. Sannino and M. Pollini, Materials, 2016, 9(6), 411 CrossRef.
- B. Pannerselvam, M. K. Jothinathan, M. Rajenderan, P. Perumal, K. P. Thangavelu, H. J. Kim, V. Singh and S. K. Rangarajulu, Eur. J. Pharm. Sci., 2017, 100, 187–196 CrossRef CAS PubMed.
- R. Deepachitra, V. Ramnath and T. P. Sastry, RSC Adv., 2014, 4(107), 62717–62727 RSC.
- A. C. Burduşel, O. Gherasim, A. Grumezescu, L. Mogoantă, A. Ficai and E. Andronescu, Nanomaterials, 2018, 8(9), 681 CrossRef.
- A. B. Lansdown, Br. J. Nurs., 2004, 13, S6–S19 CrossRef.
- J. A. Michaels, B. Campbell, B. King, S. J. Palfreyman, P. Shackley and M. Stevenson, BJS Open, 2009, 96(10), 1147–1156 CrossRef CAS.
- H. Vermeulen, J. M. van Hattem, M. N. Storm-Versloot, D. T. Ubbink and S. J. Westerbos, Cochrane Database Syst. Rev., 2007,(1), CD005486 CAS.
- M. N. Storm-Versloot, C. G. Vos, D. T. Ubbink and H. Vermeulen, Cochrane Database Syst. Rev., 2010, 3, CD006478 Search PubMed.
- L. Wang, C. Hu and L. Shao, Int. J. Nanomed., 2017, 12, 1227 CrossRef CAS.
- X. F. Zhang, Z. G. Liu, W. Shen and S. Gurunathan, Int. J. Mol. Sci., 2016, 17(9), 1534 CrossRef.
- A. Moores and F. Goettmann, New J. Chem., 2006, 30(8), 1121–1132 RSC.
- G. Bauer, J. Hassmann, H. Walter, J. Haglmüller, C. Mayer and T. Schalkhammer, Nanotechnology, 2003, 14(12), 1289 CrossRef CAS.
- K. Anandalakshmi, J. Venugobal and V. Ramasamy, Appl. Nanosci., 2016, 6(3), 399–408 CrossRef CAS.
- H. K. Ardani, C. Imawan, W. Handayani, D. Djuhana, A. Harmoko and V. Fauzia, IOP Conf. Ser.: Mater. Sci. Eng., 2017, 188, 012056 Search PubMed.
- M. A. Khan, S. Kumar, M. Ahamed, S. A. Alrokayan and M. S. AlSalhi, Nanoscale Res. Lett., 2011, 6(1), 434 CrossRef.
- E. Pazos-Ortiz, J. H. Roque-Ruiz, E. A. Hinojos-Márquez, J. López-Esparza, A. Donohué-Cornejo, J. C. Cuevas-González, L. F. Espinosa-Cristóbal and S. Y. Reyes-López, J. Nanomater., 2017, 4752314 Search PubMed.
- P. I. Morgado, P. F. Lisboa, M. P. Ribeiro, S. P. Miguel, P. C. Simões, I. J. Correia and A. Aguiar-Ricardo, J. Membr. Sci., 2014, 469, 262–271 CrossRef CAS.
- B. Ajitha, Y. A. Reddy, P. S. Reddy, H. J. Jeon and C. W. Ahn, RSC Adv., 2016, 6(42), 36171–36179 RSC.
- P. I. Morgado, S. P. Miguel, I. J. Correia and A. Aguiar-Ricardo, Carbohydr. Polym., 2017, 159, 136–145 CrossRef CAS.
- M. Jannesari, J. Varshosaz, M. Morshed and M. Zamani, Int. J. Nanomed., 2011, 6, 993 CAS.
- J. Krstić, J. Spasojević, A. Radosavljević, A. Perić-Grujić, M. Đurić, Z. Kačarević-Popović and S. Popović, J. Appl. Polym. Sci., 2014, 131(11) DOI:10.1002/app.40321.
- F. M. Thiruvoth, D. P. Mohapatra, D. K. Sivakumar, R. K. Chittoria and V. Nandhagopal, Plastic. Aesthet. Res., 2015, 2, 250 CrossRef.
- P. Agrawal, S. Soni, G. Mittal and A. Bhatnagar, Int. J. Lower Extremity Wounds, 2014, 13(3), 180–190 CrossRef.
- L. I. Moura, A. M. Dias, E. Carvalho and H. C. de Sousa, Acta Biomater., 2013, 9(7), 7093–7114 CrossRef CAS.
- P. I. Morgado, A. Aguiar-Ricardo and I. J. Correia, J. Membr. Sci., 2015, 490, 139–151 CrossRef CAS.
- M. Flanagan, J. Wound Care, 2000, 9(6), 299–300 CrossRef CAS PubMed.
- S. P. Miguel, M. P. Ribeiro, H. Brancal, P. Coutinho and I. J. Correia, Carbohydr. Polym., 2014, 111, 366–373 CrossRef CAS.
- K. S. Priya, A. Gnanamani, N. Radhakrishnan and M. Babu, J. Ethnopharmacol., 2002, 83(3), 193–199 CrossRef.
- M. Shrimanker, N. Patel, H. Modi and R. Dave, Am. J. PharmTech Res., 2013, 3, 2249–3387 Search PubMed.
- M. Subalakshmi, A. Saranya, M. U. Maheswari, A. Jarina and S. Kaviman, J. Exp. Pharmacol., 2014, 4(2), 127–131 Search PubMed.
- V. I. Kumar, A. A. Khan and K. Nagarajan, Int. Bull. Drug Res., 2013, 3(5), 93–107 Search PubMed.
- M. Ahmadi and M. Adibhesami, Iran. J. Pharm. Res., 2017, 16(2), 661 CAS.
- M. A. Nasir, N. L. Mahammed, S. Roshan and M. W. Ahmed, Int. J. Res. Dev. Pharm. Life Sci., 2016, 5(2), 2080–2087 CAS.
- Y. Liang, X. Zhao, T. Hu, B. Chen, Z. Yin, P. X. Ma and B. Guo, Small, 2019, 1900046 CrossRef.
- M. A. Fischbach and C. T. Walsh, Science, 2009, 325(5944), 1089–1093 CrossRef CAS.
- S. Bottan, F. Robotti, P. Jayathissa, A. Hegglin, N. Bahamonde, J. A. Heredia-Guerrero, I. S. Bayer, A. Scarpellini, H. Merker, N. Lindenblatt and D. Poulikakos, ACS Nano, 2014, 9(1), 206–219 CrossRef PubMed.
- O. Akturk, K. Kismet, A. C. Yasti, S. Kuru, M. E. Duymus, F. Kaya, M. Caydere, S. Hucumenoglu and D. Keskin, J. Biomater. Appl., 2016, 31(2), 283–301 CrossRef CAS.
- H. V. Pawar, J. Tetteh, P. Debrah and J. S. Boateng, Int. J. Biol. Macromol., 2019, 121, 191–199 CrossRef CAS.
- X. Zhao, B. Guo, H. Wu, Y. Liang and P. X. Ma, Nat. Commun., 2018, 9(1), 2784 CrossRef.
- X. Zhao, H. Wu, B. Guo, R. Dong, Y. Qiu and P. X. Ma, Biomaterials, 2017, 122, 34–47 CrossRef CAS.
- E. Filippo, A. Serra and D. Manno, Sens. Actuators, B, 2009, 138(2), 625–630 CrossRef CAS.
- A. Jahani-Javanmardi, M. Sirousazar, Y. Shaabani and F. Kheiri, J. Biomater. Sci., Polym. Ed., 2016, 27(12), 1262–1276 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra06913e |
| This journal is © The Royal Society of Chemistry 2019 |