
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSeed-mediated synthesis of Au@PtCu nanostars with rich twin defects as efficient and stable electrocatalysts for methanol oxidation reaction†
Ting Bianad,
Biao Suna,
Sai Luo c,
Long Huanga,
Shan Sub,
Chunfeng Mengd,
Shichuan Suae,
Aihua Yuan*b and
Hui Zhang*c
c,
Long Huanga,
Shan Sub,
Chunfeng Mengd,
Shichuan Suae,
Aihua Yuan*b and
Hui Zhang*c
aSchool of Energy and Power Engineering, Jiangsu University of Science and Technology, Zhenjiang 212003, People's Republic of China
bSchool of Environmental and Chemical Engineering, Jiangsu University of Science and Technology, Zhenjiang 212003, People's Republic of China. E-mail: aihua.yuan@just.edu.cn
cState Key Laboratory of Silicon Materials and School of Materials Science and Engineering, Zhejiang University, Hangzhou 310027, People's Republic of China. E-mail: msezhanghui@zju.edu.cn
dSchool of Materials Science and Engineering, Jiangsu University of Science and Technology, Zhenjiang 212003, People's Republic of China
eZhenjiang Key Laboratory of Marine Power Equipment Performance, Zhenjiang 212003, People's Republic of China
First published on 4th November 2019
Abstract
Pt-based nanocrystals with a twinned structure are highly desirable to achieve high performances in both catalytic activity and durability for methanol oxidation reaction (MOR). However, it still remains great challenge for producing such twinned nanocrystals due to the high internal strain energy of Pt. Here we present a seed-mediated approach to generate Au@PtCu nanostars with a five-fold twinned structure using Au decahedra as seeds. The composition of Pt/Cu in the nanocrystals was tuned by varying the molar ratio of Pt to Cu salt precursors with the amount of Au seeds being the same. Through composition optimization, Au@Pt1.2Cu nanostars achieved the highest specific (1.06 mA cm−2) and mass (0.18 mA mgPt−1) activities for MOR, which were about 5.9 and 1.6 times higher than those of commercial Pt/C, respectively. After accelerated stability test (ADT) for 1500 cycles, such nanostars remained ∼95% of specific activity compared to a loss of ∼28% for commercial Pt/C, indicting their superior durability for MOR. We believed that this enhancement may arise from the unique twinned structure and possible synergetic effect between Pt and Cu components.
1. Introduction
Renewable and clean energy has recently attracted much attention owing to the increasing demand for lowering our dependence on fossil fuels.1–3 Direct methanol fuel cells (DMFC) are considered as promising green energy conversion devices due to their high efficiency, high energy density and convenient storage of liquid fuel.4,5 In spite of many attempts in the last decade to design and fabricate non-Pt catalysts for methanol oxidation reaction (MOR) at the anode, Pt still remains the choice of preferred catalyst for DMFC.6 However, the high cost, limited abundance and severe poisoning effect from CO have been the key obstacles of Pt for the extensive use in DMFC.7 To alleviate these problems, it is highly urgent to develop new types of Pt-based nanocrystals with intriguing catalytic performance and high utilization efficiency of Pt atoms. One of the appealing approaches to improve the catalytic activity toward MOR is incorporating non-noble metal into Pt nanocrystals to construct bimetallic alloys. Some pioneer works have demonstrated that the improved activity of bimetallic electrocatalysts can be ascribed to electronic effect and/or bi-functional mechanism arising from the interaction between each component in the alloy.8–10 Among various Pt-based electrocatalysts, PtCu nanocrystals have been regarded as a promising candidate due to their relatively low cost, excellent catalytic activity, and super anti-CO poisoning capability.11,12Apart from chemical composition, morphology also has a strong influence on the electrocatalytic activity caused by the difference in the arrangement of surface atoms (geometric effect).13 Accordingly, significant progress has been made in the synthesis of PtCu alloyed nanocrystals with well-defined shapes, such as cube,14 octahedron,15 nanocage,16 nanowire17 and nanosheet.18 In addition, Pt-based alloys with rich defects are expected to induce positive effects on electrocatalytic reactions relative to their perfect counterparts due to the modification of surface reactivity and electronic structure.19,20 It is generally known that there are three different types of defects, including point defect, linear defect, and planar defect. As a kind of typical surface defects, twin structure has attracted much attention.21 For example, Sun and co-workers have recently demonstrated that the specific activity of the icosahedral Pt3Cu catalysts for MOR was 1.31 times higher than that of the octahedral Pt3Cu catalysts, even though with the same exposed {111} facets and composition.22 Similarly, the electrocatalytic activity of the five-fold-twinned PtCu nanoframes for MOR were highly evaluated.23 However, in such designs, the twinned structure is easily corroded under harsh operating conditions, which prevents the further application of twin defects in optimizing catalytic performance.
Core–shell structures are a class of nanostructured materials that have received increased attention owing to their fantastic properties. On the one hand, the electronic coupling between core and shell is advantageous for achieving high catalytic performances.24,25 On the other hand, by sophisticated selecting of the seeds with well-defined shapes, seeded growth method has emerged as a powerful tool for shape-controlled synthesis of core–shell nanocrystals.26,27 As demonstrated in our previous work, small size Au particle tends to assume a multiply twinned structure (e.g., <10 nm), which is an appropriate core material for establishing Pt-based catalysts with twin configurations.28 Moreover, it has been found that the addition of Au effectively increases the dissolution potential of Pt layer as a result of preventing Pt-based catalysts from degradation.29,30 Sun and co-workers demonstrated an oil-phase method for the synthesis of Au/CuPt core–shell nanoparticles, showing enhanced catalytic activity and duality for MOR.31 However, it still remains a challenge to precise control of the growth of PtCu alloys on twinned Au cores to form multimetallic core–shell nanocrystals with well-defined shapes and rich defects on surface.
Herein, we report a seed-mediated approach to synthesize Au@PtCu nanostars with rich twin defects as a new kind of high-performance electrocatalysts. Impressively, the Au@PtCu nanostars exhibited better electrocatalytic properties in terms of activity and stability toward MOR relative to commercial Pt/C, Au@PtCu decahedra, Au@Pt nanoparticles (NPs), PtCu alloyed nanobranches under acid conditions due to the unique twin structure and synergetic effect between Pt and Cu.
2. Experimental
2.1 Chemicals and materials
Chloroauric acid tetrahydrate (HAuCl4·4H2O, 99.9%), hexadecyltrimethyl ammonium bromide (CTAB, 99%), ammonium bromide (NH4Br, AR), ethanol (C2H5OH, AR), Trichloromethane (CHCl3, 99%), and cyclohexane (C6H12, AR) were purchased from Sinopharm Chemical Reagent Co., Ltd. Oleylamine (OAm, 80–90%), chloroplatinic acid hexahydrate (H2PtCl6·6H2O, 99.9%), cupric nitrate trihydrate (Cu(NO3)2·3H2O, 99.99%), tert-butylamine (C4H11N, 99%) and perchloric acid (HClO4, 99.999%) were purchased from Aladdin. Methanol (CH3OH, 99.9%) and Nafion (5 wt%) were purchased from Sigma-Aldrich. All the chemicals and materials were all used as received.2.2 Synthesis of Au decahedral seeds
In a typical synthesis, 7 mL of OAm was preheated at 180 °C for 10 min in air under magnetic stirring. 3 mL of an OAm solution containing 12.4 mg of HAuCl4 was injected quickly, which was then kept at 180 °C for additional 3 h. The product was collected by centrifugation, washed with ethanol and hexane for three times, and then re-dispersed in 10 mL of OAm for further use.2.3 Synthesis of Au@PtCu nanostars
In a standard preparation, 2 mL of Au seeds and 4 mL of OAm solution containing 300 mg CTAB were added to a 25 mL three-necked flask. Subsequently, 4 mL of OAm solution containing H2PtCl6 (0.02 mmol) and Cu(NO3)2 (0.06 mmol) were injected. After that, the reaction was heated at 240 °C in nitrogen under magnetic stirring for 3 h. The solution was centrifuged and washed three times with ethanol and hexane to remove CTAB and OAm for further applications. We also systematically investigated the effects of various parameters, including reaction time, composition, amounts of CTAB, as well as temperature, on the morphology of resultant products.2.4 Synthesis of Au@Pt3 NPs and PtCu alloy nanobranches
The Au@Pt3 NPs and PtCu alloy nanobranches were generated using the aforementioned standard procedure without adding Cu precursors or Au seeds, respectively.2.5 Preparation of carbon-supported catalysts
In a standard preparation, carbon black particles were dispersed in chloroform and sonicated for 30 min. The as prepared Au@Pt1.2Cu nanostars, Au@Pt3.2Cu decahedra, Au@Pt3 NPs, and Pt1.2Cu nanobranches were added to this dispersion at the mass ratio (nanocrystals-to-carbon-black) of 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :80. This mixture was further sonicated for 10 min and stirred overnight. After that, the products were collected by centrifugation and re-dispersed in tert-butylamine at a concentration of 0.5 mg-nanocrystals mL−1. The mixture was kept under magnetic stirring for 3 days and then centrifuged and washed three times with ethanol.
:80. This mixture was further sonicated for 10 min and stirred overnight. After that, the products were collected by centrifugation and re-dispersed in tert-butylamine at a concentration of 0.5 mg-nanocrystals mL−1. The mixture was kept under magnetic stirring for 3 days and then centrifuged and washed three times with ethanol.
2.6 Morphological, structural, and elemental characterizations
Morphology and elemental distributions of the samples were analyzed using transmission electron microscope (TEM, Hitachi HT-7700, 100 kV), high-resolution TEM (HRTEM, Tecnai G2 F20, 200 kV), and high-angle annular dark-field scanning TEM equipped with a super energy dispersive X-ray (EDX) spectroscope (HAADF-STEM, Titan G2 80-200, 200 kV). The atomic ratio of Au, Pt and Cu in the samples was determined by using inductively coupled plasma atomic emission spectrometry (ICP-AES, IRIS Intrepid II XSP, TJA Co., USA). X-ray powder diffraction (XRD) patterns were recorded by Shimadzu XRD-6000 diffractometer (Cu-Kα radiation). X-Ray Photoelectron Spectrometer (XPS) was performed on ESCALAB 250Xi (Thermo UK). Thermogravimetric analysis (TGA) was carried out on a thermogravimetric analyzer (Pyris Diamond TG-DTA) from room temperature to 800 °C at a heating rate of 10 °C min−1.2.7 Electrochemical measurements
The electrochemical measurements were performed at a rotating disk electrode (RDE, Pine Research Instrumentation, United States) connected to an electrochemical workstation (CHI 760E). A conventional three-electrode system was adopted with a glassy carbon electrode (GCE, 0.5 mm in diameter) as the working electrode, a platinum wire as the auxiliary electrode, and a silver chloride electrode (Ag/AgCl) as the reference electrode, respectively. 5 mg of the as-prepared Au@Pt1.2Cu/C, Au@Pt3.2Cu/C, Au@Pt3/C, Pt1.2Cu/C, and commercial Pt/C catalysts were dispersed in 1 mL of a mixed solvent containing water, isopropanol and Nafion (5%) at the volumetric ratio of 1:1:0.025. Thereafter, 10 μL of the catalyst ink was added onto the GCE to make the modified working electrode. Cyclic voltammograms (CVs) were recorded in a N2-saturated 0.1 M HClO4 and/or 0.1 M HClO4 + 1 M CH3OH solution with a scan rate of 50 mV s−1. The accelerated stability test (ADT) was carried out at a scan rate of 100 mV s−1 for 1500 cycles in N2-saturated 0.1 M HClO4 + 1 M CH3OH solution. The chronoamperometry curves were obtained at 0.6 V for 10000 s.
3. Results and discussion
3.1 Characterization of the prepared Au@PtCu nanostars
The five-fold twinned Au@PtCu core–shell nanostars were prepared by simultaneous reduction of H2PtCl6 and Cu(NO3)2 in oleylamine (OAm) containing cetyltrimethylammonium bromide (CTAB) with Au decahedra as the seeds. Representative transmission electron microscopy (TEM) image (Fig. S1a†) shows that the Au seeds were highly uniform with an average size of ca. 8 nm. From high-resolution TEM (HRTEM) image (Fig. S1b†), apparent five-fold twins were clearly observed from the center of a nanocrystal, which was generally recognized as decahedral nuclei. Fig. 1 shows the morphological, structural, compositional characterizations of Au@PtCu nanostars that prepared using the standard procedure with the molar ratio of Pt and Cu salt precursors being 1:3. From TEM image (Fig. 1a), most of the nanocrystals were star-shaped with an approximate size of 26.7 ± 2.5 nm. The size was defined as the diameter of the circumscribed circle projected by the pentagram, as shown in Fig. S2.† In addition, HRTEM image clearly indicates five-fold twins existed in the Au@PtCu nanostars with rich defects (Fig. 1b). The marked fringes with a lattice spacing of 2.25 Å correspond well to {111} plane of face-centered cubic (fcc) PtCu, confirming the epitaxy of highly crystalline PtCu layers on Au seeds. Energy dispersive X-ray (EDX) mapping images of an individual Au@PtCu nanostar show that the core region is essentially made by Au (red color), while the shell is dominated by Pt (blue color) and Cu (green color), demonstrating a core–shell structure (Fig. 1c). The core–shell structure was further confirmed by line-scan analysis of an Au@PtCu nanostar through the arrow direction (Fig. 1d). Moreover, inductively coupled plasma atomic emission spectrometry (ICP-AES) shows that the atomic ratio of Au/Pt/Cu in the nanostar is 1:3.27:2.79 (labelled as Au@Pt1.2Cu due to surface chemical formula close to Pt1.2Cu), which is remarkably higher than the molar ratio (nPt:nCu = 1:3) between Pt and Cu salt precursors fed in the synthesis (see Table S1†).
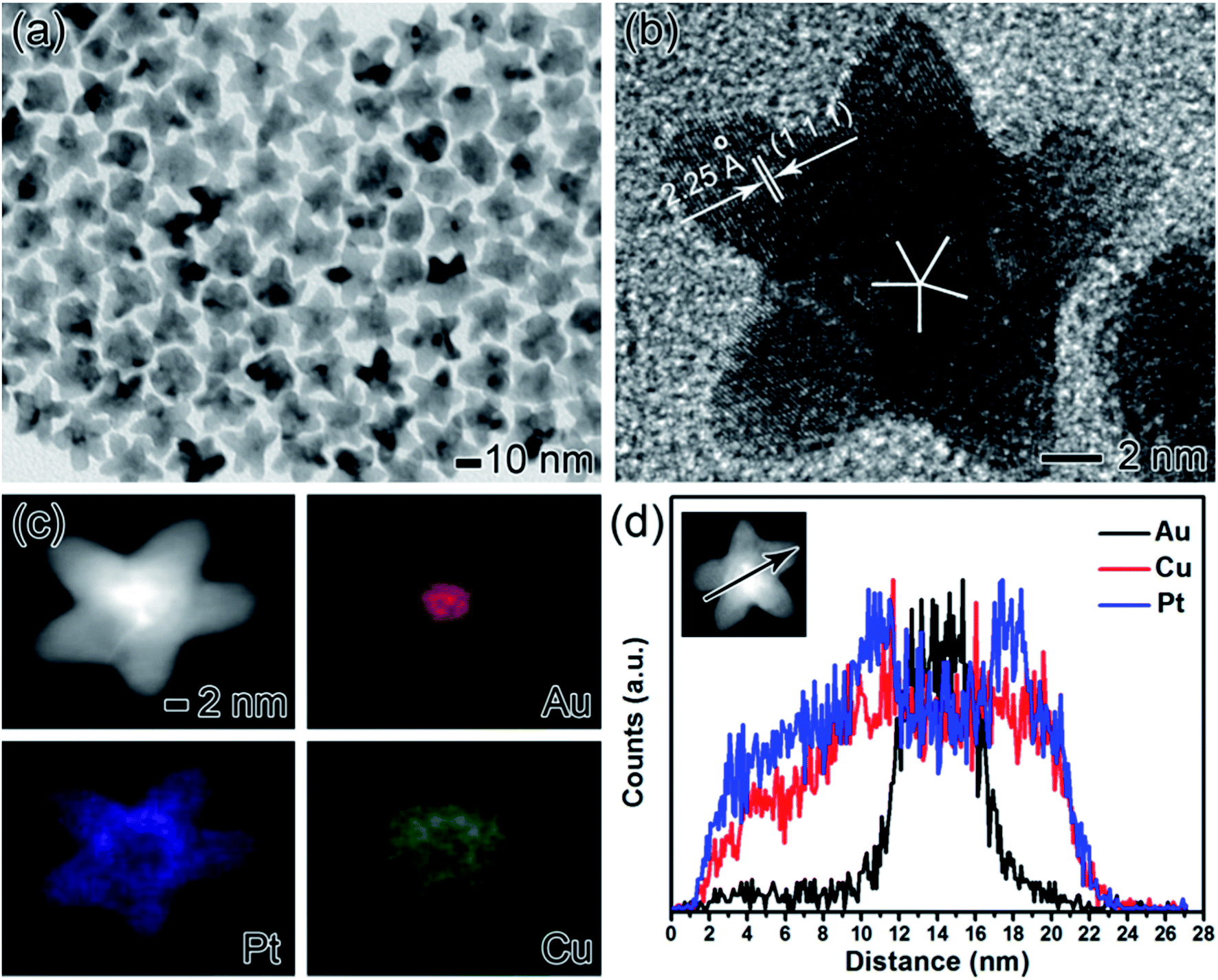 | ||
| Fig. 1 Morphological, structural, and compositional characterizations of the Au@PtCu nanostars prepared using the standard procedure. (a) TEM image, (b) HRTEM image, (c) HAADF-STEM-EDX image, and (d) EDX line-scan profile. | ||
X-ray photoelectron spectroscopy (XPS) was used to characterize the electronic state and surface chemistry of Au, Pt and Cu elements in the Au@Pt1.2Cu nanostars. In Au 4f XPS spectra for Au@Pt3 NPs, two peaks at 84.08 eV (Au 4f7/2) and 87.75 eV (Au 4f5/2) could be assigned to zerovalent state of Au0 (Fig. S3a†). Impressively, the above pair peaks in Au@Pt1.2Cu nanostars shift to 83.88 and 87.55 eV, respectively (Fig. S4†). In the Pt 4f spectrum of Au@Pt1.2Cu nanostars, the two strong peaks located at 70.8 and 74.1 eV can be assigned to the 7/2 and Pt 4f5/2 of metallic Pt0. While, the weak peaks at 71.9 and 75.23 eV are attributed to the bivalent state of Pt(II) (Fig. 2a). Similarly, in Pt 4f XPS spectra for Au@Pt3 NPs, two peaks at 71.17 and 74.5 eV could be assigned to Pt0. Besides, the minor peaks assigned to Pt2+ (emerged at 71.97 and 75.3 eV) (Fig. S3b†). The Pt 4f spectra of Au@Pt1.2Cu nanostars apparently shift to lower binding energies compared with the as-prepared Au@Pt3 NPs due to the alloying with Cu element. As shown in Fig. 2b, the Cu 2p spectrum can be split into two groups of peaks associated with metallic Cu0 (931.97 eV and 951.6 eV) and bivalent Cu(II) (935.12 eV and 955.02 eV). The satellite peak also confirms the presence of a small amount of Cu(II) in the nanostars. According to the XPS data, the shift of the binding energies provides a strong evidence of the electrons that transfer from Cu to Pt and cause a downshift of d-band center of Pt, which is beneficial for improving the MOR properties.
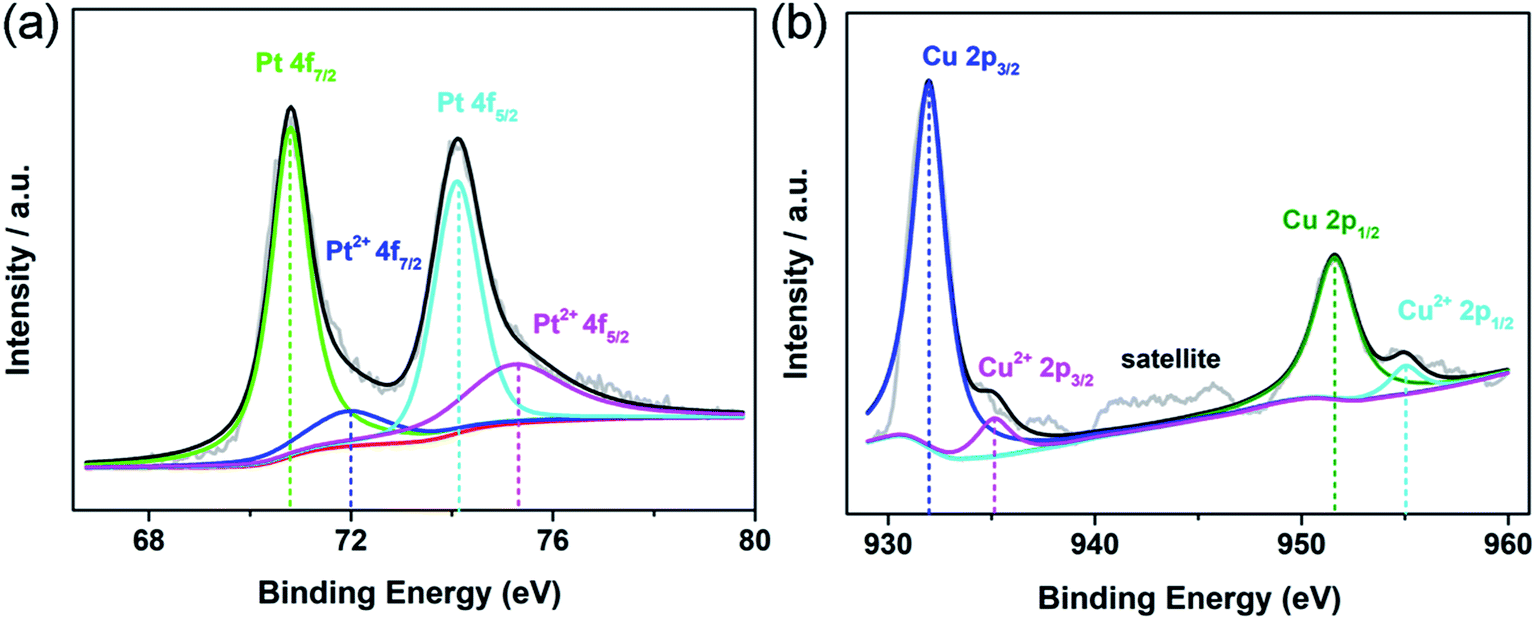 | ||
| Fig. 2 XPS spectra of the Au@Pt1.2Cu nanostars for (a) Pt 4f and (b) Cu 2p orbitals, respectively. | ||
In order to clarify the formation mechanism of the Au@PtCu nanostars, the morphological evolution of the nanocrystals at different reaction time was investigated by TEM analysis (Fig. 3). In the initial stage (Fig. 3a, t = 5 min), a large number of nearly spherical nanocrystals were obtained. When the reaction time increased to 30 min (Fig. 3b), the Au@PtCu nanocrystals presented an irregular polyhedral shape, together with few star-shaped nanostructures, implying that Pt and Cu atoms were gradually deposited on the Au core. As the reaction proceeded for 60 (Fig. 3c) and 120 min (Fig. 3d), the decahedral Au seeds were encased by PtCu shells with five tiny tips. When the reaction time increased to 180 min, the five-fold Au@PtCu core–shell nanostars with rich twin defects were formed by preferential epitaxial overgrowth of PtCu shells on decahedral Au seeds. Since Pt and Cu have a higher surface energy than Au, such overgrowth will increase the total surface energy.32 In addition, decahedron is one kind of highly strained structure, consisting of five single-crystalline domains and five twin boundaries.33 To release the extra strain energy, most of the Pt and Cu atoms tend to be deposited at five single-crystalline domains of Au decahedra with only a few atoms being added to five twin boundaries, thereby promoting the formation of star-shaped decahedra.
 | ||
| Fig. 3 TEM images of the Au@PtCu nanocrystals prepared using the standard procedure, except for different period of the reactions: (a) 5, (b) 30, (c) 60, and (d) 120 min. | ||
The composition of Pt and Cu in the Au@PtCu nanostars was tuned by simply varying the feeding ratio of Pt to Cu precursors while keeping the amount of Au seeds constant. On the basis of ICP-AES measurements (see Table S1†), Pt3.2Cu, Pt2.3Cu, Pt1.1Cu, Pt1.2Cu, Pt1.2Cu, Pt1.2Cu, and Pt1.2Cu alloyed shells were eventually obtained when the molar ratios of Pt and Cu precursors varied from 1/0.3, 1/0.6, 1/1, 1/2, 1/3, 1/4 to 1/5, respectively. Interestingly, Cu proportion in the alloyed shell cannot exceed 50%, regardless of the initial amount of Cu precursor fed in the synthesis, which is in accordance with previous result.34 The molar ratios of Pt to Cu precursors have also an influence on the morphology of the final product, as shown in Fig. S5.† When the molar ratios of Pt and Cu precursors were 1/0.3 and 1/0.6, a large number of nearly spherical particles were formed (Fig. S5a and b†). As the molar ratios of Pt and Cu precursors decreased to 1/1 and 1/2, the nanocrystals with tiny bumps were generated (Fig. S5c and d†). High-quality star-shaped decahedra were synthesized only when the molar ratio of Pt and Cu precursors reached 1/3. Further decreasing such molar ratio to 1/5 promoted the growth of branched arms (Fig. S5e†). Finally, Au@PtCu core–shell nanodendrites were fabricated when the molar ratio of Pt to Cu precursors decreased to 1/6 (Fig. S5f†). In the absence of Cu precursors or Au decahedral seeds, Au@Pt3 core–shell NPs and Pt1.2Cu nanobranches were generated, respectively (Fig. S6†). Fig. S7† compares X-ray diffraction (XRD) patterns of the Au@Pt1.2Cu nanostars with Pt1.2Cu nanobranches and Au@Pt3 NPs. All diffraction peaks of Pt1.2Cu nanobranches are located at the position similar to the standard value of PtCu (JCPDS Card No: 48-1549), indicating the formation of PtCu alloy. Compared to Au@Pt3 NPs, Au@Pt1.2Cu nanostars exhibited diffraction peaks with a slight shift to higher angles due to the alloying of Pt atoms with smaller Cu atoms. The shift of peaks confirms the lattice shrinkage, which is beneficial to the improvement of electronic structure.35
Besides composition, reaction temperature also played a crucial role in controlling the shape of Au@PtCu nanocrystals, as shown in Fig. S7.† At 160 °C, only small rounded nanoparticles less than 10 nm in size were produced (Fig. S8a†). In contrast, increasing the temperature to 180 °C led to the formation of polyhedral nanocrystals. As the temperature further increased to 200 and 220 °C (Fig. S8c and d†), star-shaped Au@PtCu decahedra were gradually formed. High-quality nanostars exposed with {111} facets were successfully generated only when the reaction temperature exceeded 240 °C. These temperature-dependent experiments further confirmed the growth of Au@PtCu nanostars under a thermodynamic control.
In general, OAm can serve as the solvent, reducing agent, and stabilizer for the synthesis of metal nanocrystals.36 For example, amine group in OAm played an important role in facilitating the epitaxial growth of Pt on Au seeds by dramatically decreasing the surface energy of Pt due to the strong adsorption in our previous report.37 Moreover, we believe that underpotential deposition (UPD) was responsible for the co-reduction of the Pt and Cu precursors in OAm. Such a Cu UPD-assisted mechanism was also observed in our previous studies for the synthesis of the PtCu concave nanocubes.38 In addition, Br− ions (derived from CTAB) could serve as a capping agent to promote the formation of Au@PtCu nanostars due to their preferential chemisorption on {111} surfaces. This result was supported by the control experiments with different amounts of CTAB being added. In the absence of CTAB, uneven polyhedra were obtained, as shown in Fig. S9a.† When the amount of CTAB increased to 100 mg (Fig. S9b†), the nanocrystals with a star-like shape began to appear. Only when the amount of CTAB exceeded 300 mg, high-quality nanostars were eventually obtained. The role of Br− ions derived from CTAB can be further confirmed by replacing it with the same amount of ammonium bromide (Fig. S10†).
3.2 Catalytic activities of Au@PtCu nanostars
The Au@PtCu nanostars with rich defects were then evaluated as electrocatalysts towards MOR in the acid media. The cyclic voltammograms (CVs) of the Au@Pt1.2Cu star-shaped and Au@Pt3.2Cu rounded decahedra were first obtained in N2-purged 0.1 M HClO4 solutions with the Au@Pt3 NPs, Pt1.2Cu nanobranches, and commercial Pt/C as the references, as shown in Fig. 4a. Their electrochemically active surface areas (ECSAs) were calculated by integrating the charge transfer during the hydrogen adsorption/desorption process, assuming a value of 210 μC cm−2 for the adsorption of a hydrogen monolayer.24 To be specific, the ECSA of commercial Pt/C was measured to be 63.1 m2 gPt−1, which is significantly higher than those of Au@Pt3.2Cu (15.3 m2 gPt−1), Au@Pt1.2Cu (16.8 m2 gPt−1), Au@Pt3 (46.5 m2 gPt−1), and Pt1.2Cu (11.7 m2 gPt−1). The lower ECSA of the Au@PtCu electrocatalysts can be attributed to their large nanoparticle size and the residual organic species (e.g., OAm and CTAB) on their surfaces. Fig. 4b and c compares the CV curves of these five electrocatalysts towards MOR that performed in N2-saturated 0.1 M HClO4 solution containing 1 M CH3OH at a scan rate of 50 mV s−1. As observed, the specific activities of such electrocatalysts followed a sequence of Au@Pt1.2Cu nanostars > Au@Pt3.2Cu decahedra > Pt1.2Cu nanobranches > Pt/C > Au@Pt3 NPs. Specifically, the Au@Pt1.2Cu nanostars achieved the highest specific activity (1.06 mA cm−2, Fig. 4d) for MOR, which were 2.0, 6.9, 4.0 and 5.9 times higher than those of the Au@Pt3.2Cu decahedra, Au@Pt3 NPs, Pt1.2Cu nanobranches and commercial Pt/C, respectively. And the mass activities (calculated in Pt) of these five catalysts were summarized in Fig. 4d and Table S2,† indicating that the Au@Pt1.2Cu nanostars are the best MOR electrocatalysts. As shown in Fig. S11,† Au@Pt1.2Cu nanobranches achieved lower specific (0.68 mA cm−2) and mass (0.11 mA mgPt−1) activities, indicating that more PtCu nanobranches on the Au core will hinder the MOR. We think the possible reason is that too much PtCu dendritic structures will weaken the influence of twins and Au core. Moreover, such a MOR activity for Au@Pt1.2Cu nanostars was superior to or comparable to those reported for Pt-based MOR electrocatalysts as summarized in Table S3.† In addition, the If/Ib ratio, in which If and Ib representing the forward and backward current density, respectively, is generally used to evaluate poisoning tolerance of the catalyst to CO.16 Compared to the commercial Pt/C (1.2), the Au@Pt1.2Cu nanostars showed higher If/Ib value (2.0), implying better CO tolerance.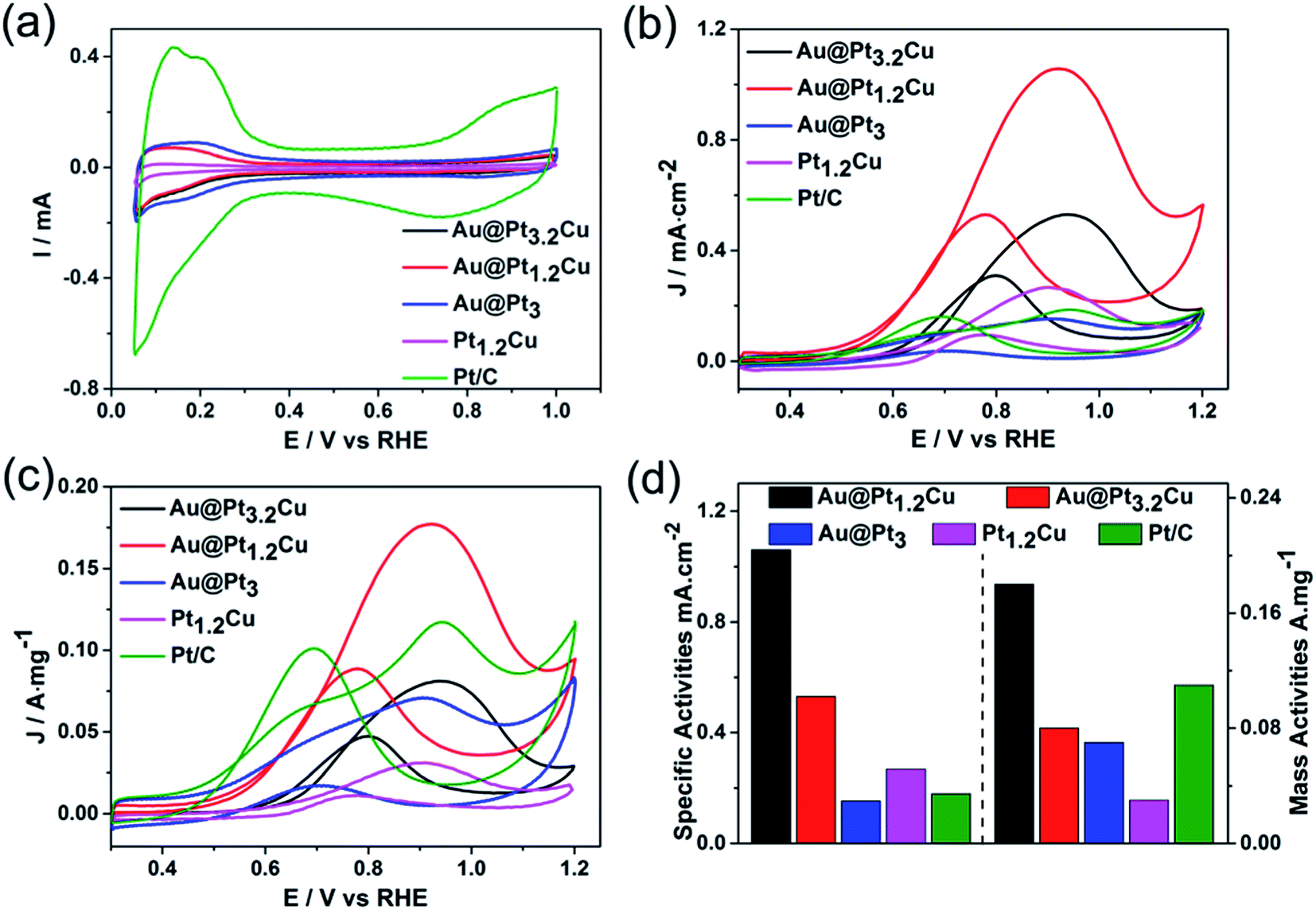 | ||
| Fig. 4 (a) Cyclic voltammograms (CVs) recorded at room temperature with a sweep rate of 50 mV s−1 in N2-saturated 0.1 M HClO4 for Au@Pt3.2Cu decahedra, Au@Pt1.2Cu nanostars, Au@Pt3 NPs, Pt1.2Cu nanobranches and commercial Pt/C catalysts. (b and c) CVs of these five catalysts in a 0.1 M HClO4 + 1 M CH3OH solution at a scan rate of 50 mV s−1 for MOR normalized by surface area (is, mA cm−2Pt) and Pt mass (im, mA μgPt−1), respectively. (d) Specific and mass activities at the peak position of forward curve. | ||
To further evaluate the stability of the Au@Pt1.2Cu electrocatalysts toward MOR, CV curves were obtained at a sweep rate of 100 mV s−1 for 1500 cycles with commercial Pt/C as a reference (Fig. 5). As observed, the peak current density of the Au@Pt1.2Cu nanostars only decreased by ∼5%, while a significant loss of ∼28% in current density was obtained for commercial Pt/C, indicating the superior durability of the Au@Pt1.2Cu nanostars. The superior catalytic stability of Au@Pt1.2Cu nanostars was also supported by chronoamperometric measurement (Fig. S12†). The current density of Au@Pt1.2Cu nanostars remained to be 26.5 mA mg−1 and the Pt/C remained 10.2 mA mg−1, which is about 3 times higher than the Pt/C. In order to better explain the role of Au in Au@PtCu nanostars, we compared the decay of current with the stars-like PtCu/XC-72 reported by Tian et al.39 According to the chronoamperometric curves, the current density of stars-like PtCu/XC-72 catalysts remained to be 21.1 mA mg−1 with a loss of ∼97% (vs. initial current) and the Au@Pt1.2Cu nanostars showed a loss of ∼82%, indicating that adding an Au core would enhance the stability. Furtherly, the If/Ib value of Au@Pt1.2Cu nanostars is also better than stars-like PtCu. From TEM images in Fig. S13a and b,† the Au@Pt1.2Cu nanostars were still well-dispersed on the carbon support before and after 1500 cycles CV tests with part of the Pt and Cu atoms being dissolved at five corners of the star-shaped decahedra, which is responsible for a slight drop in MOR properties. There are two possible reasons for improving the electrocatalytic activity of the Au@Pt1.2Cu nanostars. First, these twinned structures can offer significant benefits toward MOR because they have plenty of defects and {111} facets exposed on the surface.28 In addition, alloying Pt with Cu can cause a small compressive strain in the out layer, leading to the modified electronic structure and enhanced activity. Moreover, the Cu component with an oxophilic feature can facilitate CO oxidation by catalyzing water to generate hydroxyl species such as OHads, and thus enhance the CO tolerance.40 As such, all of these advantages are beneficial for the enhancement in catalytic activity of the Au@PtCu nanostars relative to the commercial Pt/C.
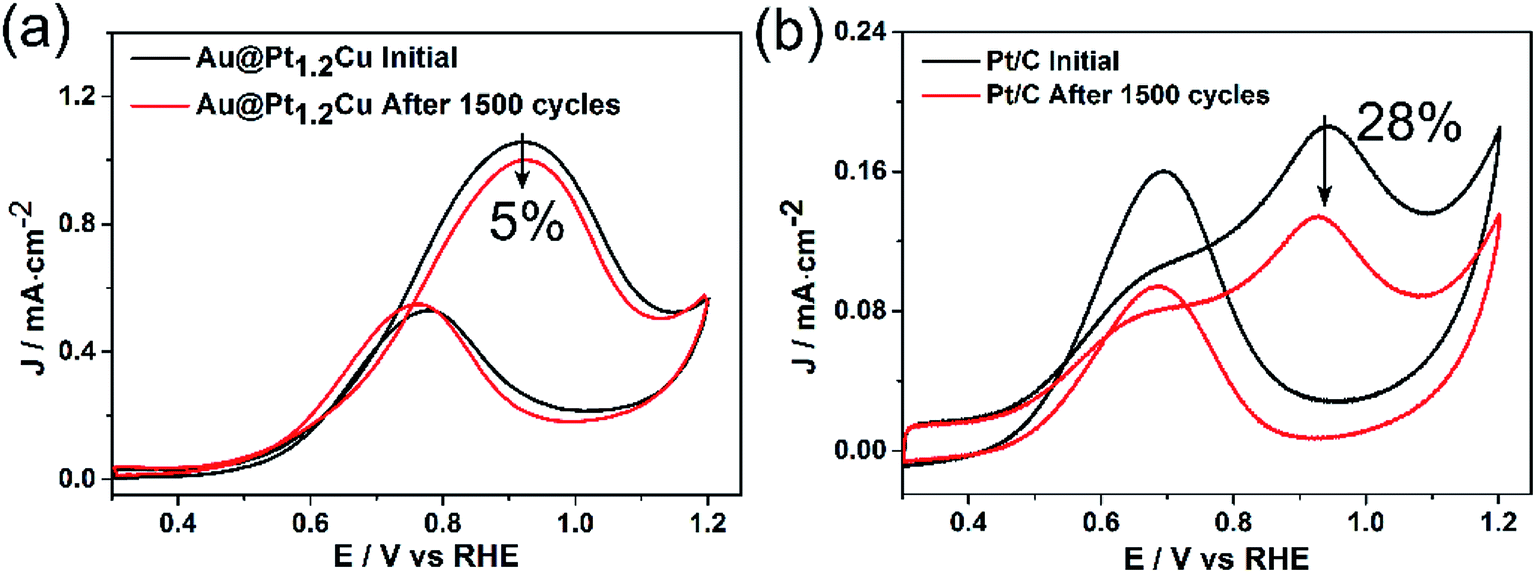 | ||
| Fig. 5 CVs of the (a) Au@Pt1.2Cu nanostars and (b) commercial Pt/C catalysts before and after 1500 potential cycles between 0.3 and 1.2 V in a 0.1 M HClO4 + 1 M CH3OH solution at a sweep rate of 100 mV s−1. | ||
4. Conclusions
In summary, high quality five-fold-twinned Au@PtCu nanostars with rich defects on the surface were successfully prepared by a seed-mediated approach. With the composition optimization, the Au@Pt1.2Cu catalysts showed the best electrocatalytic performances toward MOR in an acid electrolyte with specific and mass activities being 5.9 and 1.6 times higher than those of commercial Pt/C. In addition, the Au@Pt1.2Cu catalysts exhibited excellent durability with a 5% decay in specific activity after 1500 potential cycling, comparing to a ∼28% loss for commercial Pt/C. This enhancement can be attributed to their unique twinned structure and possible bifunctional mechanism between Pt and Cu elements. This work provides a facile and effective strategy to synthesize multi-metallic Pt-based catalysts with rich defects for various significant reactions in electrocatalysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledged financial support by the Natural Science Foundation of Jiangsu (BK20170580), National Science Foundation of China (51901088), China Postdoctoral Science Foundation (2017M611745), Jiangsu Postdoctoral Science Foundation (1701114B), Natural Science Foundation of Jiangsu (17KJB480002), Zhenjiang Key Laboratory of Marine Power Equipment Performance (SS2018006), Postgraduate Foundation (SJCX19_0615). The work on electron microscopy was carried out in the Center of Electron Microscopy and the Analysis Platform of Department of Chemistry, Zhejiang University.References
- X. M. Guo, C. Qian, R. H. Shi, W. Zhang, F. Xu, S. L. Qian, J. Zhang, H. Yang, A. H. Yuan and T. Fan, Small, 2019, 15, 1804855 CrossRef.
- Y. Li, X. L. Zhong, K. Luo and Z. P Shao, J. Mater. Chem. A, 2019, 7, 15593–15598 RSC.
- L. T. Zhang, Z. L. Cai, Z. D. Yao, L. Ji, Z. Sun, N. H. Yan, B. Zhang, B. B. Xiao, J. Du and X. Q. Zhu, J. Mater. Chem. A, 2019, 7, 5626–5634 RSC.
- Z. Lin, W. Chen, Y. Jiang, T. Bian, H. Zhang, J. Wu, Y. Wang and D. Yang, Nanoscale, 2016, 8, 12812–12818 RSC.
- J. Sheng, J. Kang, H. Ye, J. Xie, B. Zhao, X. Z. Fu, Y. Yu, R. Sun and C. P. Wong, J. Mater. Chem. A, 2018, 6, 3906–3912 RSC.
- H. H. Li, Q. Q. Fu, L. Xu, S. Y. Ma, Y. R. Zheng, X. J. Liu and S. H. Yu, Energy Environ. Sci., 2017, 10, 1751–1756 RSC.
- S. Q. Lu, H. M. Li, J. Y. Sun and Z. B. Zhuang, Nano Res., 2018, 11, 2058–2068 CrossRef CAS.
- X. Cui, P. P. Xiao, J. Wang, M. Zhou, W. L. Guo, Y. Yang, Y. He, Z. Wang, Y. Yang and Y. Zhang, Angew. Chem., Int. Ed., 2017, 56, 4488–4493 CrossRef CAS PubMed.
- S. I. Choi, S. Xie, M. Shao, J. H. Odell, N. Lu, H. C. Peng, L. Protsailo, S. Guerrero, J. Park, X. Xia, J. Wang, M. J. Kim and Y. Xia, Nano Lett., 2013, 13, 3420–3425 CrossRef CAS.
- T. Y. Kwon, M. K. Jun, H. Y. Kim, A. Oh, J. S. Park, H. S. Baik, S. H. Joo and K. Lee, Adv. Funct. Mater., 2018, 28, 1706440 CrossRef.
- K. Wang, R. R. Sriphathoorat, S. P. Luo, M. Tang, H. Y. Du and P. K. Shen, J. Mater. Chem. A, 2016, 4, 13425–13430 RSC.
- A. X. Yin, X. Q. Min, W. Zhu, W. C. Liu, Y. W. Zhang and C. H. Yan, Chem.–Eur. J., 2012, 18, 777–782 CrossRef CAS.
- P. Strasser, S. L. Koh, T. L. Anniyev, J. Greeley, K. More, C. F. Yu, Z. Liu, S. Kaya, D. Nordlund and H. Ogasawara, Nat. Chem., 2010, 2, 454–460 CrossRef CAS.
- D. Xu, S. Y. Bliznakov, Z. P. Liu, J. Y. Fang and N. L. Dimitrov, Angew. Chem., 2010, 122, 1304–1307 CrossRef.
- T. Bian, H. Liu, B. Sun, B. B. Xiao, Y. Jiang, C. Jin, A. H. Yuan, H. Zhang and D. Yang, J. Alloys Compd., 2019, 788, 1334–1340 CrossRef CAS.
- B. Y. Xia, H. B. Wu, X. Wang and X. W. Lou, J. Am. Chem. Soc., 2012, 134, 13934–13937 CrossRef CAS.
- W. Hong, J. Wang and E. K. Wang, Nano Res., 2015, 8, 2308–2316 CrossRef CAS.
- F. S. Saleem, Z. C. Zhang, B. Xu, X. B. Xu, P. L. He and X. Wang, J. Am. Chem. Soc., 2013, 135, 18304–18307 CrossRef CAS.
- Y. N. Xia, Y. J. Xiong, B. K. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2009, 48, 60–103 CrossRef CAS.
- J. B. Wu, L. Qi, H. J. You, A. Gross, J. Li and H. Yang, J. Am. Chem. Soc., 2012, 134, 11880–11883 CrossRef CAS PubMed.
- Q. F. Xu, W. L. Chen, Y. C. Yan, Z. M. Wu, Y. Jiang and J. J. Li, Sci. Bull., 2018, 63, 494–501 CrossRef CAS.
- H. Y. An, N. T. Khi, J. Yoon, H. Lee, H. Baik, J. H. Sohn and K. Lee, Nanoscale, 2015, 7, 8309–8314 RSC.
- X. H. Sun, K. Z. Jiang, N. Zhang, S. J. Guo and X. Huang, ACS Nano, 2015, 9, 7634–7640 CrossRef CAS PubMed.
- Z. C. Zhang, Z. M. Luo, B. Chen, C. Wei, L. Zhao, J. Z. Chen, X. Zhang, Z. Lai, Z. Fan, C. Tan, M. Zhao, Q. Lu, B. Li, Y. Zong, C. Yan, G. Wang, Z. J. Xu and H. Zhang, Adv. Mater., 2016, 28, 8712–8717 CrossRef CAS.
- Y. C. Yan, H. Shan, G. Li, F. Xiao, Y. Y. Jiang, Y. Y. Yan, C. Jin, H. Zhang, J. Wu and D. Yang, Nano Lett., 2016, 16, 7999–8004 CrossRef CAS.
- M. B. Gawande, A. Goswami, T. Asefa, H. Guo, A. V. Biradar, D. L. Peng, R. Zboril and R. S. Varma, Chem. Soc. Rev., 2015, 44, 7540–7590 RSC.
- Y. L. Xiong, Y. L. Ma, J. J. Li, J. B. Huang, Y. C. Yan, H. Zhang, J. Wu and D. Yang, Nanoscale, 2017, 9, 11077–11084 RSC.
- T. Bian, H. Zhang, Y. Jiang, C. Jin, J. Wu, H. Yang and D. R. Yang, Nano Lett., 2015, 15, 7808–7815 CrossRef CAS.
- C. Wang, D. Van Der Vliet, K. L. More, N. J. Zaluzec, S. Peng, S. Sun, H. Daimon, G. Wang, J. Greeley and J. Pearson, Nano Lett., 2010, 11, 919–926 CrossRef.
- J. Zhang, K. Sasaki, E. Sutter and R. Adzic, Science, 2007, 315, 220–222 CrossRef CAS.
- X. L. Sun, D. G. Li, Y. Ding, W. L. Zhu, S. J. Guo, Z. L. Wang and S. Sun, J. Am. Chem. Soc., 2014, 136, 5745–5749 CrossRef CAS.
- L. Vitos, A. V. Ruban, H. L. Skriver and J. Kollár, Surf. Sci., 1998, 411, 186–202 CrossRef CAS.
- Y. Y. Ma, J. Zeng, W. Y. Li, M. McKiernan, Z. X. Xie and Y. N. Xia, Adv. Mater., 2010, 22, 1930–1934 CrossRef CAS.
- Y. Q. Jiang, Y. Y. Jia, J. W. Zhang, L. Zhang, H. Huang, Z. Xie and L. Zheng, Chem.–Eur. J., 2013, 19, 3119–3124 CrossRef CAS.
- D. Wang, Y. C. Yu, H. L. Xin, R. Hovden, P. Ercius, J. A. Mundy, H. Chen, J. H. Richard, D. A. Muller and F. J. DiSalvo, Nano Lett., 2012, 12, 5230–5238 CrossRef CAS PubMed.
- Z. C. Xu, C. M. Shen, Y. L. Hou, H. J. Gao and S. H. Sun, Chem. Mater., 2009, 21, 1778–1780 CrossRef CAS.
- B. H. Wu, N. F Zheng and G. Fu, Chem. Commun., 2011, 47, 1039–1041 RSC.
- Y. Qi, T. Bian, S. I. Choi, Y. Y. Jiang, C. H. Jin, M. S. Fu, H. Zhang and D. Yang, Chem. Commun., 2014, 50, 560–562 RSC.
- D. H. Chen, Y. C. Zhao, X. L. Peng, X. Wang, W. J. Hu, C. Jing, S. S. Tian and J. N. Tian, Electrochim. Acta, 2015, 177, 86–92 CrossRef CAS.
- Z. L. Liu, X. Y. Ling, X. D. Su and J. Y. Lee, J. Phys. Chem. B, 2004, 108, 8234–8240 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra06893g |
| This journal is © The Royal Society of Chemistry 2019 |