
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn situ K2S activated electrospun carbon nanofibers with hierarchical meso/microporous structures for supercapacitors†
Hua Liu *a,
Weiguo Songb and
Aihua Xing*a
*a,
Weiguo Songb and
Aihua Xing*a
aNational Institute of Clean-and-Low-Carbon Energy, Beijing 102211, P. R. China. E-mail: hua.liu.w@chnenergy.com.cn; aihua.xing@chnenergy.com.cn
bBeijing National Laboratory for Molecular Sciences (BNLMS), Key Laboratory of Molecular Nanostructure and Nanotechnology, Institute of Chemistry, Chinese Academy of Sciences, Beijing, 100190, P. R. China
First published on 18th October 2019
Abstract
Porous electrospun carbon nanofibers (CNFs) can be produced by a more advantageous ‘in situ activation’ method by electrospinning polyacrylonitrile (PAN) with an activation agent. However, most in situ activated electrospinning processes yield porous CNFs with rather limited surface area and less porosity due to the inappropriately selected activation agents. Here we found K2S could perfectly meet both compatibility and reactivity requirements of PAN electrospinning to generate hierarchical meso/micropores inside electrospun CNFs. During the whole fabrication process, K2S experiences a phase evolution loop and the hierarchical pore structures are formed by the reaction between K2S oxidative derivatives and the as-formed carbon during heat treatment. The hierarchical meso/microporous CNFs not only showed a large surface area (835.0 m2 g−1) but also exhibited a high PAN carbonization yield (84.0 wt%) due to improved cyclization of PAN's nitrile group during the pre-oxidation stage. As an electrode material for supercapacitors, the corresponding electrodes have a capacitance of 210.7 F g−1 at the current density of 0.2 A g−1 with excellent cycling durability. The hierarchically porous CNFs produced via in situ activation by K2S combine the advantages of interconnected meso/micropores and are a promising candidate for electrochemical energy conversion and storage devices.
1. Introduction
Electrochemical double-layer capacitors (EDLCs) store charge and release energy based on the formation of oppositely charged layers at the interface of the electrolyte and electrodes.1–6 The reversible fast-surface-charge-storage process of ions between an electrolyte and high accessible surface area electrodes (e.g., carbon, transition metal oxides or hydroxides and conducting polymers) endows supercapacitors with high power density and long cycling lifetime that are widely employed as electric energy storage devices. Up until now, supercapacitors have still been plagued by their insufficient energy density, hindering their scope of applications, especially when a high-energy output is required.7 One of the critical issues is the implementation of ideal electrode materials, which require a high conductivity, large ion-accessible specific surface area (SSA) and appropriate pore size distribution,8–12 because the electric double-layer formation is determined by surface chemistry and apertures of pores upon carbon materials.13–16 The electrode materials need to possess both high specific surface area and appropriate pore size distribution that combines narrow micropores suitable for ion accommodation with mesopores favorable for ion transport.Electrospun PAN based carbon nanofibers can offer large surface area accessible to the electrolyte, efficient electron and ion transport, and structural stability to prevent the electrode from destruction due to swelling and shrinkage, which is very crucial to real application in supercapacitors.7 Carbon nanofibers with hierarchical meso/microporous structure are highly desirable for the improved performance of supercapacitors.17–23 While micropores provide a large accessible surface area for ion charge accommodation, mesopores could offer low-resistant channels for fast ion transportation. However, conventional electrospinning technique can only produce solid carbon nanofibers, and the resultant electrospun CNFs possess nonporous structure with very low SSA of 10–30 m2 g−1 which greatly hindered the utilization of electrospun CNFs as electrode materials in supercapacitors. Great efforts have been made to enrich the pore structure of electrospun CNFs to improve the reversible fast-surface-charge-storage process of ions between an electrolyte and high accessible surface area electrodes in EDLCs. Pore generating methods such as utilizing sacrificial agents (PMMA,24–26 PTA,27 SiO2 nanoparticles,28 etc.) and post activation by steam,29–32 KOH33,34 and CO2,35–37 had been utilized to produce porous electrospun CNFs. However, most of these methods suffered from disadvantages of complicated manufacturing process with high temperature, high cost, or environmental destructiveness, and more than half of carbon precursor mass of sacrificial polymer agents are consumed to develop porous structure during the activation process resulting in high price, significantly limiting their further application in most supercapacitors.
Porous electrospun CNFs can also be produced in a simpler manner that enables the formation of porous structure under the presence of an additional activated phase in PAN/dimethylformamide (DMF) electrospinning solution during heat treatment processes. The porous structured carbon nanofibers were formed due to in situ catalytic behaviors of the second activated phase in the carbon nanofibers, such as organic compound (Fe(acac)2,38 nano-CaCO3,39,40 etc) and inorganic compounds (ZnCl2,41 CoCl2,42 Co(NO3)2 6H2O,43 etc). Unfortunately, using the above activation agents generally yield porous carbon nanofibers with rather limited surface area and less porosity (less than 550 m2 g−1 and 0.4 cm3 g−1) and the as-prepared porous CNFs contain large amount of doped metal which have to be removed by acid rinsing. All of these unfavorable aspects could be mainly ascribed to the inappropriately selected activation agents. Hence, one has to find a more effective activation agent in the electrospinning solution system for the fabrication of porous CNFs with high SSA and porosity.
There are two key requirements for a second phase activation agent for the successful synthesis of PAN-based porous CNFs: (1) the compatibility and electrospinnability of an activation agent in PAN/DMF solution; (2) the reactivity between activation agent and the as-formed carbon nanofibers. In this study, we found for the first time that K2S could perfectly meet both requirements to generate hierarchical meso/micropores inside electrospun CNFs. 1D carbon nanofibers composed of hierarchical meso- and microporous structures were produced by electrospinning PAN and K2S followed by thermal treatment and water washing. During the electrospinning and preoxidative processes, K2S transformed into its oxidative derivatives (e.g. polysulfide, thiosulfate, pyrosulfate and sulfate state) which served as real activation species reacting with the as-formed carbon nanofibers to generate porous structure. The sample not only show a large SSA (835.0 m2 g−1), but also exhibited a high PAN carbonization yield of (84.0 wt%) which is due to improved cyclization of nitrile group during the pre-oxidation stage with the addition of K2S. The sample combines the advantages of interconnected meso- and micropores and has shown excellent performance as an electrode material for supercapacitors.
2. Experimental
2.1 Fabrication of PAN–K2S composite nanofibers
A common electrospinning method was used to fabricate the composite precursor nanofibers. In a typical procedure, calculated amount of K2S was dissolved in 10 g of DMF (Alfa) by magnetic stirring for 30 min, followed by adding 1.0 g of PAN (150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, Aldrich) into the above solution at 80 °C and stirring for 2 h to form a homogeneous suspension, all steps are operated in N2 atmosphere to prevent K2S from oxidizing to K2S2 under the air. The violet suspension was transferred into a plastic syringe equipped with a 17 gauge stainless needle which was connected to a high-voltage supply. The distance from the needle to the grounded aluminium foil was set at 15 cm and the voltage was adjusted at 25 kV with solution feeding rate of 1.5 mL h−1. The as-prepared pale yellow PAN–K2S composite nanofibers were peeled off from the aluminium foil collector and was denoted as As_PAN_x, where x% indicates the content of K2S in the electrospun PAN–K2S nanofiber sample.
000, Aldrich) into the above solution at 80 °C and stirring for 2 h to form a homogeneous suspension, all steps are operated in N2 atmosphere to prevent K2S from oxidizing to K2S2 under the air. The violet suspension was transferred into a plastic syringe equipped with a 17 gauge stainless needle which was connected to a high-voltage supply. The distance from the needle to the grounded aluminium foil was set at 15 cm and the voltage was adjusted at 25 kV with solution feeding rate of 1.5 mL h−1. The as-prepared pale yellow PAN–K2S composite nanofibers were peeled off from the aluminium foil collector and was denoted as As_PAN_x, where x% indicates the content of K2S in the electrospun PAN–K2S nanofiber sample.
2.2 Preparation of porous carbon nanofiber
The nonwoven mat was put into a horizontal tube furnace and was heated to 280 °C in air (2 °C min−1) with subsequent pre-oxidative stabilization for 2 h. For carbonization and in situ activation, the sample was heated up to 800–1000 °C (4 °C min−1) and kept for 2 h under argon gas flow (60 mL min−1). After cooling under Ar atmosphere, the porous carbon nanofibers were obtained by washing with deionized water three times followed by drying at 120 °C for 8 h. The pre-oxidative sample were denoted as Pre_PAN_x and the obtained carbon nanofibers were designated as Car[y]_CNF_x, where x% represent the weight percentage of K2S in the electrospun PAN–K2S nanofiber sample and y stands for carbonization temperature.2.3 Characterization
The electrospinning solution parameters were determined by using AR-2000 rheometer and Accumet Excel XL50 for viscosity and conductivity measurements, respectively. Surface tensions of solutions were measured using interfacial tensiometer (CSC-Denouy 70545). The structures and morphologies of the products were characterized by transmission electron microscope (TEM, JEOL-1011) and scanning electron microscope (SEM, JEOL 6701F). The nitrogen adsorption and desorption isotherms were collected on Quantachrome Autosorb AS-1 Instrument. The X-ray diffraction (XRD) patterns of samples were recorded on Bruker X-ray diffractometer using Cu Kα radiation (40 kV, 40 mA). Raman spectra were measured using LabRam HR-800 micro-Raman spectroscopy system.Thermal behavior of pre-oxidation fibers were studied using a differential scanning calorimeter (NETZSCH STA449F3) with a heating rate of 10 °C min−1 under air in the range of 30–360 °C. Thermal decomposition behavior were investigated by thermo-gravimetric and differential thermal analysis (TG/DTA 6300), which were performed between ambient and 1000 °C with heating rate of 10 °C min−1 under flowing air. The mass carbonization yield of CNFs was calculated based on corresponding TG curves according to eqn (1):
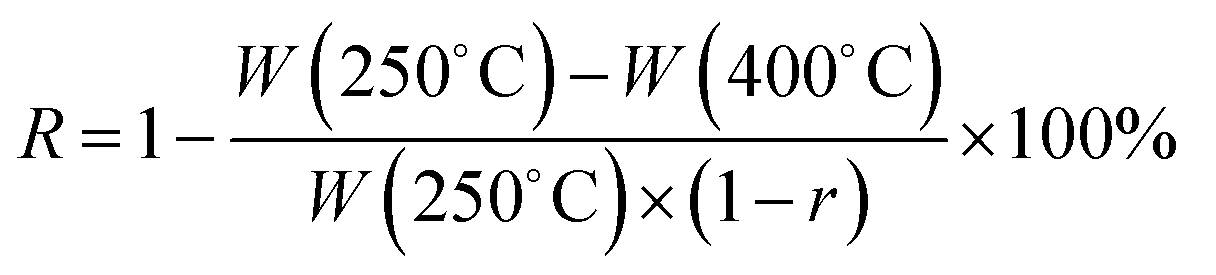 | (1) |
Infrared spectra were recorded on Bruker VERTEX in attenuated total reflectance (ATR) mode. The spectra were collected at room temperature in the scanning range of 650–4000 cm−1 (2 cm−1 resolution) using Hg–Cd–Te (MCT) detector with averaging 64 scans. The X-ray photoelectron spectroscopy (XPS, ESCALAB 250Xi) was used to evaluate the elemental compositions. The binding energy scales were calibrated in reference to the C 1s peak position (284.6 eV).
2.4 Electrochemical measurements
Two pieces of porous CNF electrodes with a separator was assembled in to a symmetrical two-electrode coin to evaluate the capacitive performance in an KOH aqueous system (electrolyte: 6 M KOH). Both the electrode materials and separator were previously immersed in 6 M KOH solution under vacuum for 24 h to achieve sufficient electrolyte saturation before assembly. Cyclic voltammogram (CV) measurement (scanning rates varying from 5 to 200 mV s−1) was conducted on an electrochemistry workstation (Princeton PARSTAT 2273). Galvanostatic charge/discharge (GCD) measurements were conducted with a charge–discharge tester (Arbin 2000) with current density varying from 0.2 to 20 A g−1. Electrochemical impedance spectroscopy (EIS) measurements were recorded on Princeton PARSTAT 2273 in the frequency region of 100 kHz to 0.1 Hz. A three-electrode system were also employed for the measurements, with a 1 cm × 1 cm Pt sheet and SCE (saturated calomel electrode) as counter and reference electrodes respectively. The working electrode was assembled by pressing the carbon nanofiber mat and nickel foam (used as the current collector) together without any additional binders or conductive additives.3. Results and discussion
K2S can be easily dissolved in DMF (Fig. S1†) and it has shown an excellent compatibility with PAN, ensuring a homogeneous dark green solution without any phase separation in DMF solution. Besides, K2S possesses high melting point (m. p. 840 °C) and high boiling point (b. p. 912 °C) and can undergo reaction with air to form various oxidative states to react with the as-formed carbon. All of the above properties of K2S allowed us to anticipate that K2S would be an interesting in situ activation agent for the fabrication of porous carbon nanofibers. Verification experiments of this assumption mainly consisted of three steps to prepare K2S activated carbon nanofibers: (1) electrospinning of K2S–PAN composite nanofibers; (2) preoxidative stabilization of K2S–PAN composite nanofibers in air at 280 °C; (3) carbonization of electrospun K2S–PAN composite nanofibers and in situ activation under high temperature.Fig. 1a shows the N2 adsorption/desorption isotherms of as-prepared carbon nanofibers with 0 wt%, 10.0 wt%, 20.0 wt%, and 30.0 wt% addition of K2S in the K2S–PAN composite nanofibers, and remarkable difference was observed among these samples. The as-prepared carbon nanofiber with the K2S addition above 20 wt% possesses massive adsorption capacity in low relative pressure (P/P0 < 0.05) and a hysteresis loop (type H4) at a relative pressure range of 0.45–0.95, indicating that the as-formed carbon nanofibers features both massive micropores and mesopores inside the carbon nanofibers. On the other hand, for carbon nanofiber prepared with the K2S addition below 10 wt%, neither adsorption in low relative pressure nor hysteresis loop at high relative pressure was observed, indicating nonporous structure inside the carbon nanofibers. This had confirmed our assumption that K2S was surely an excellent activation agent for the fabrication of porous carbon nanofiber. Table 1 further illustrates the detailed difference of pore structures among these samples. The total surface area of sample Car[900]_CNF_30 reached up to 835.0 m2 g−1 with 78.5% of microporous surface area (655.5 m2 g−1) and the micropore volume (0.387 cm3 g−1) occupied 57.1% of the total pore volume (0.678 cm3 g−1) (Table 1). Sample Car[900]_CNF_20 with less addition of K2S exhibited much less total surface area (325.6 cm3 g−1) and pore volume (0.314 cm3 g−1) than Car[900]_CNF_30. For carbon nanofiber with K2S less than 10 wt%, the total surface area of Car[900]_CNF_10 and Car[900]_CNF_0 were only 7.0 and 10.0 m2 g−1 respectively, indicating no micro/mesoporous structures were formed, which suggested that a threshold amount of K2S was required to generate meso-/microporous structure during the activation process.
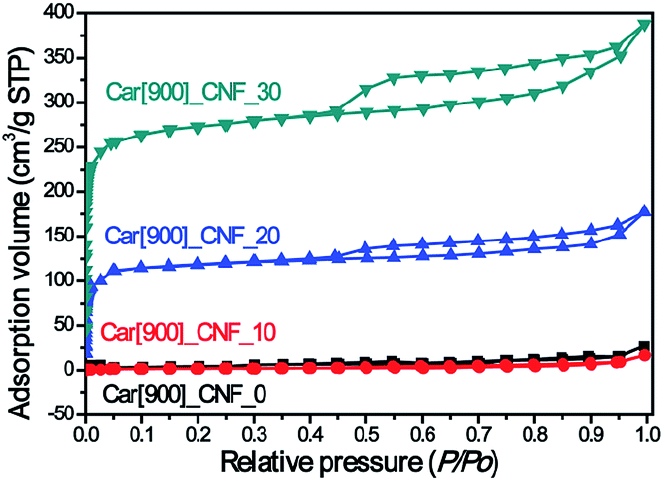 | ||
| Fig. 1 N2 adsorption/desorption isotherms of as-prepared carbon nanofibers under 900 °C. | ||
| Sample | K2S content (wt%) | SBET (m2 g−1) | Smeso (m2 g−1) | Smicro (m2 g−1) | Vtotal (cm3 g−1) | Vmeso (cm3 g−1) | Vmicro (cm3 g−1) | dHK (nm) | dBJH (nm) |
|---|---|---|---|---|---|---|---|---|---|
| Car[900]_CNF_0 | 0 | 10.0 | — | — | 0.032 | — | — | — | — |
| Car[900]_CNF_10 | 10 | 7.0 | — | — | 0.021 | — | — | — | — |
| Car[900]_CNF_20 | 20 | 325.6 | 123.2 | 201.4 | 0.314 | 0.104 | 0.210 | 0.557 | 3.90 |
| Car[900]_CNF_30 | 30 | 835.0 | 316.2 | 518.8 | 0.618 | 0.234 | 0.387 | 0.563 | 3.81 |
| Car[800]_CNF_30 | 30 | 655.2 | 320.9 | 334.3 | 0.486 | 0.238 | 0.248 | 0.541 | 3.97 |
| Car[1000]_CNF_30 | 30 | 614.8 | 282.7 | 332.1 | 0.435 | 0.200 | 0.235 | 0.601 | 4.06 |
The morphologies of electrospun PAN and K2S/PAN composite nanofibers with different K2S contents were recorded by using SEM method. As shown in Fig. 2, all the samples exhibit continuous and long 1D nanofiber interconnecting to form 3D nonwoven membrane under the same electrospun conditions. While the diameter of pure PAN nanofiber was 382.1 nm, all K2S/PAN composite nanofibers demonstrate lower fiber diameter with 214.9 nm, 200.5 nm and 250.3 nm for As_PAN_10, As_PAN_20 and As_PAN_30, respectively. This is due to that the diameter of electrospun nanofiber is governed by a complex set of parameters of electrospinning solution, including viscosity, surface tension and electrical conductivity.42 The decreased diameter of K2S/PAN composite nanofibers with the addition of K2S is mainly due to the enhanced conductivity of the electrospun solution (Table 2), which facilitate concentration of charge on the tip surface and help overcoming the surface tension under electrostatic force.44 Further increasing K2S addition did not follow this rule as we didn't observe an decreasing fiber diameter with continuing to increase K2S content, probably due to K2S interacting with PAN that cause the increased viscosity of the electrospun solution (Table 2). After carbonization, the fiber diameter of all samples further shrunk to half of its original diameter due to the removal of volatile species during thermal treatment. Instead of nonporous structure in the neat CNFs, all K2S activated CNFs exhibit porous structure insides fiber matrix (Fig. S2†).
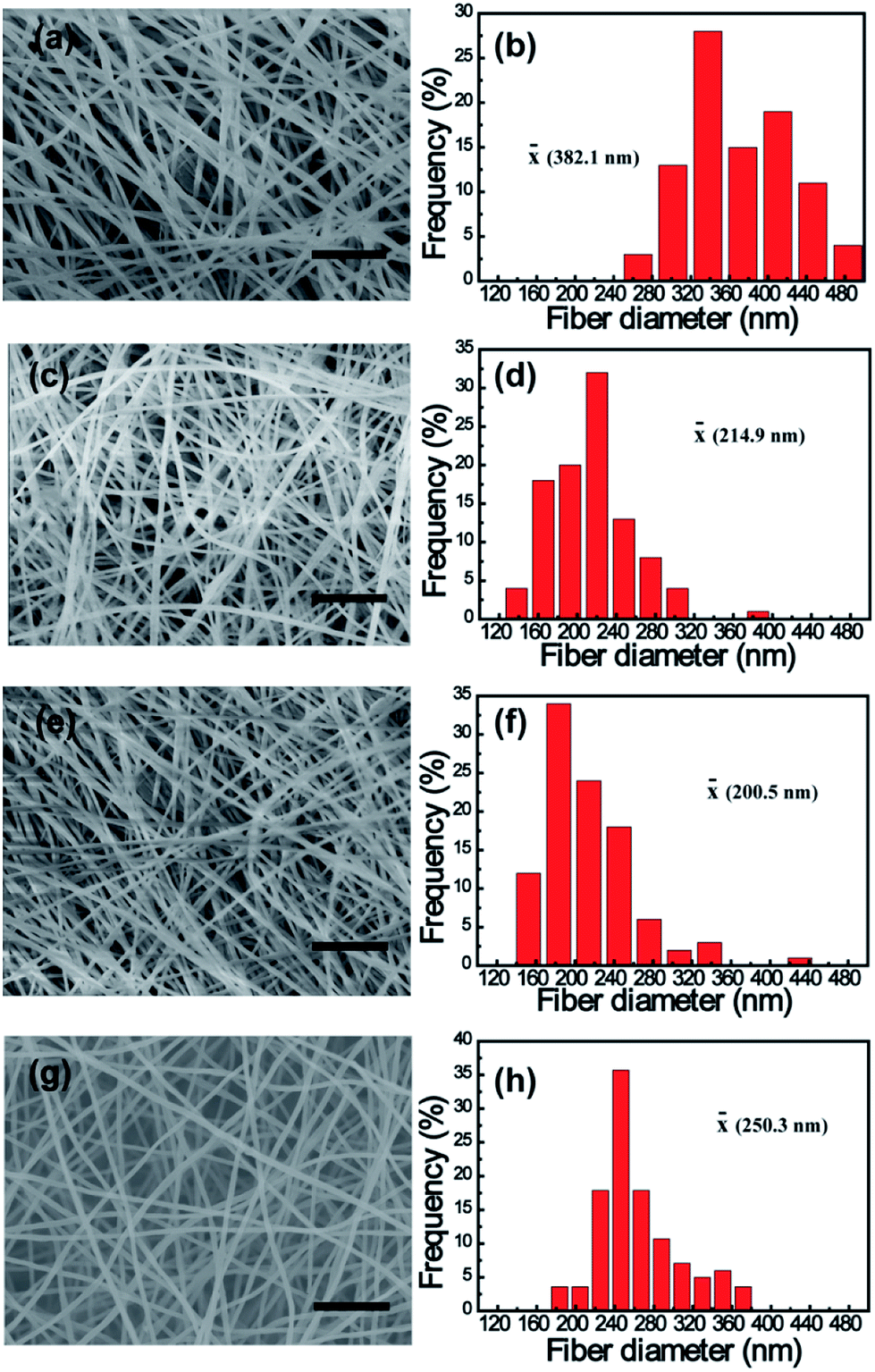 | ||
| Fig. 2 SEM images and diameter distribution of as-prepared electrospun K2S/PAN composite nanofibers with different K2S contents: (a and b) 0 wt% (pure PAN nanofibers), (c and d) 10 wt%, (e and f) 20 wt% and (g and h) 30 wt%. Scale bar denotes 5 μm. | ||
| K2S content (wt%) | Viscosity (Pa s) | Surface tension (mN m−1) | Conductivity (μs cm) | K2S/PAN diameter (nm) | CNF diameter (nm) |
|---|---|---|---|---|---|
| 0 | 0.82 | 34.7 | 53.0 | 382.1 ± 50.3 | 187.0 ± 19.1 |
| 10 | 0.88 | 35.1 | 325.1 | 214.9 ± 30.7 | 120.5 ± 20.6 |
| 20 | 0.91 | 34.4 | 846.7 | 200.5 ± 28.8 | 137.9 ± 17.3 |
| 30 | 1.05 | 35.0 | 1643.5 | 250.3 ± 25.1 | 122.2 ± 10.3 |
As PAN nanofibers converted to carbon nanofibers, oxidative stabilization is considered to be the most decisive step for developing dimensional stability during thermal treatment, since it significantly governs the desired structure with an infusible stable ladder polymer structure that do not melt at high temperature processing and avoid excessive volatilization of elemental carbonaceous material in carbonization step.45 In order to determine the influence of K2S on the oxidative stabilization process, we performed TG/DTG and DSC on electrospun K2S/PAN composite nanofibers in air. As shown in Fig. 3a, all As_PAN samples experienced two different periods of transformation: oxidative stabilization at ca. 300 °C and consumption of carbon in air at temperature above 500 °C. Pure PAN nanofibers exhibited a strong DTG peak (Fig. S3†) corresponding to complex and multiple chemical reactions (e.g., cracking, dehydration, cyclization, dehydrogenation, cross-linking).40,41,46 Two important reactions, i.e. dehydrogenation and cyclization reaction, occur during this period under the oxidative atmosphere. Since cyclization of nitrile groups to form this stable ladder polymer structure does not involve weight loss, the weight loss during oxidative process is mainly due to the C–C dissociation of PAN forming low molecular weight volatile.47 The lower bond energy for the C–C dissociation of PAN was reported at 297 kJ mol−1 in the main PAN chain, corresponding to 330 °C for 30 min half weight loss temperature.48 With increasing K2S content in the as-prepared K2S/PAN composite nanofibers, the DTG peak shifted to higher temperature and declined rapidly with a broadening peak wing (Fig. S3†). When the K2S concentration exceeded 10 wt%, the DTG peak diminished completely. Some residual remained above 800 °C in the case of the K2S/PAN nanofiber samples owing to the presence of K2S, while for pure PAN nanofiber sample, nanofibers were completely decomposed. It is noteworthy that the carbonization yield of carbon nanofiber is greatly improved with the increased K2S content determined from eqn (1), suggesting that K2S might catalyze the oxidative stabilization process, promoting the cyclization reaction and decreasing the cracking reaction. DSC curves of K2S/PAN composite nanofibers containing different K2S contents were recorded at temperatures ranging from 50 to 350 °C to confirm this presumption. As shown in Fig. 3b, all four nanofiber samples exhibit a relatively large and sharp exothermic peak centered at ca. 180 to 300 °C. Pure electrospun PAN nanofibers demonstrates a strong exothermic peak centered at ca. 294 °C, while the presence of K2S in the nanofibers significantly lowers this exothermic peak (ca. 238 °C), and further increasing K2S content (20 wt% and 30 wt%) results in continuous lower exothermic temperatures of 223 and 194 °C respectively. The DCS results indicate that K2S acted as a catalyst to promote the cyclization of nitrile group during the pre-oxidation stage, causing this lower cyclization temperature compared with that of pure PAN nanofibers. More stable ladder polymer structure are formed under the oxidative process with the increase of K2S addition, and this will significantly alleviate the C–C dissociation of PAN main chain from causing poor quality of carbon nanofibers. Therefore, the yield of carbonization rises and less weight loss occurred during the oxidative stabilization process with the increasing addition of K2S.
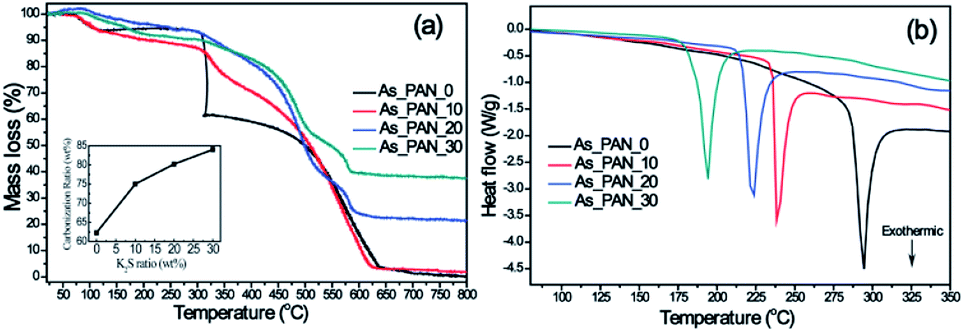 | ||
| Fig. 3 (a) TGA thermograms and the calculated carbonization ratio curve (inset), (b) DSC curves of as-prepared K2S/PAN composite nanofibers with different K2S contents. | ||
ATR-FTIR for all samples was recorded to understand the effect of K2S on the structure change of PAN during different stages (Fig. 4). For the as-prepared K2S/PAN nanofibers (Fig. 4a), the peak at 2240 cm−1 in the infrared spectra was assigned to the –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N stretching of the acrylonitrile unit in the polymer chains, and the peak at 2940 cm−1 and 2870 cm−1 correspond to the asymmetric and symmetric stretching of –CH2– groups respectively.49 1453 cm−1 can be ascribed to bending –CH2 and 1360 cm−1 correspond to CH bending and –CH2 wagging adsorption. Upon addition of K2S, a new adsorption peak at 1673 cm−1 emerged, which can be ascribed to C
N stretching of the acrylonitrile unit in the polymer chains, and the peak at 2940 cm−1 and 2870 cm−1 correspond to the asymmetric and symmetric stretching of –CH2– groups respectively.49 1453 cm−1 can be ascribed to bending –CH2 and 1360 cm−1 correspond to CH bending and –CH2 wagging adsorption. Upon addition of K2S, a new adsorption peak at 1673 cm−1 emerged, which can be ascribed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching, indicating that K2S or its oxidative compounds can react with the cyan groups of PAN to form CN group. The small stretching vibration peaks of CN at the range of 2170–1943 cm−1 corresponds to inorganic CN groups formed with addition of K2S. Upon exposure to oxidative atmosphere during the preoxidative stage (Fig. 4b), –CN stretching (2240 cm−1) of the acrylonitrile was largely preserved along with the appearance of a new conjugated –CN (2200 cm−1) group, and both of which gradually decreased with increasing addition of K2S. Another sharp vibration adsorption peak at 2050 cm−1 emerged with addition of K2S during this stage, and this peak can be ascribed to –CN stretching of –S–CN group, indicating that K2S or its derivatives reacted with cyan group of PAN to form –S–CN group. After carbonization process (Fig. 4c), all samples without water washing exhibit similar IR pattern, and all CN and –S–CN stretching vibration disappeared. Small peaks with similar intensity were still observable at 1673 cm−1 indicating that CN group were still preserved in the carbon nanofiber matrix.
N stretching, indicating that K2S or its oxidative compounds can react with the cyan groups of PAN to form CN group. The small stretching vibration peaks of CN at the range of 2170–1943 cm−1 corresponds to inorganic CN groups formed with addition of K2S. Upon exposure to oxidative atmosphere during the preoxidative stage (Fig. 4b), –CN stretching (2240 cm−1) of the acrylonitrile was largely preserved along with the appearance of a new conjugated –CN (2200 cm−1) group, and both of which gradually decreased with increasing addition of K2S. Another sharp vibration adsorption peak at 2050 cm−1 emerged with addition of K2S during this stage, and this peak can be ascribed to –CN stretching of –S–CN group, indicating that K2S or its derivatives reacted with cyan group of PAN to form –S–CN group. After carbonization process (Fig. 4c), all samples without water washing exhibit similar IR pattern, and all CN and –S–CN stretching vibration disappeared. Small peaks with similar intensity were still observable at 1673 cm−1 indicating that CN group were still preserved in the carbon nanofiber matrix.
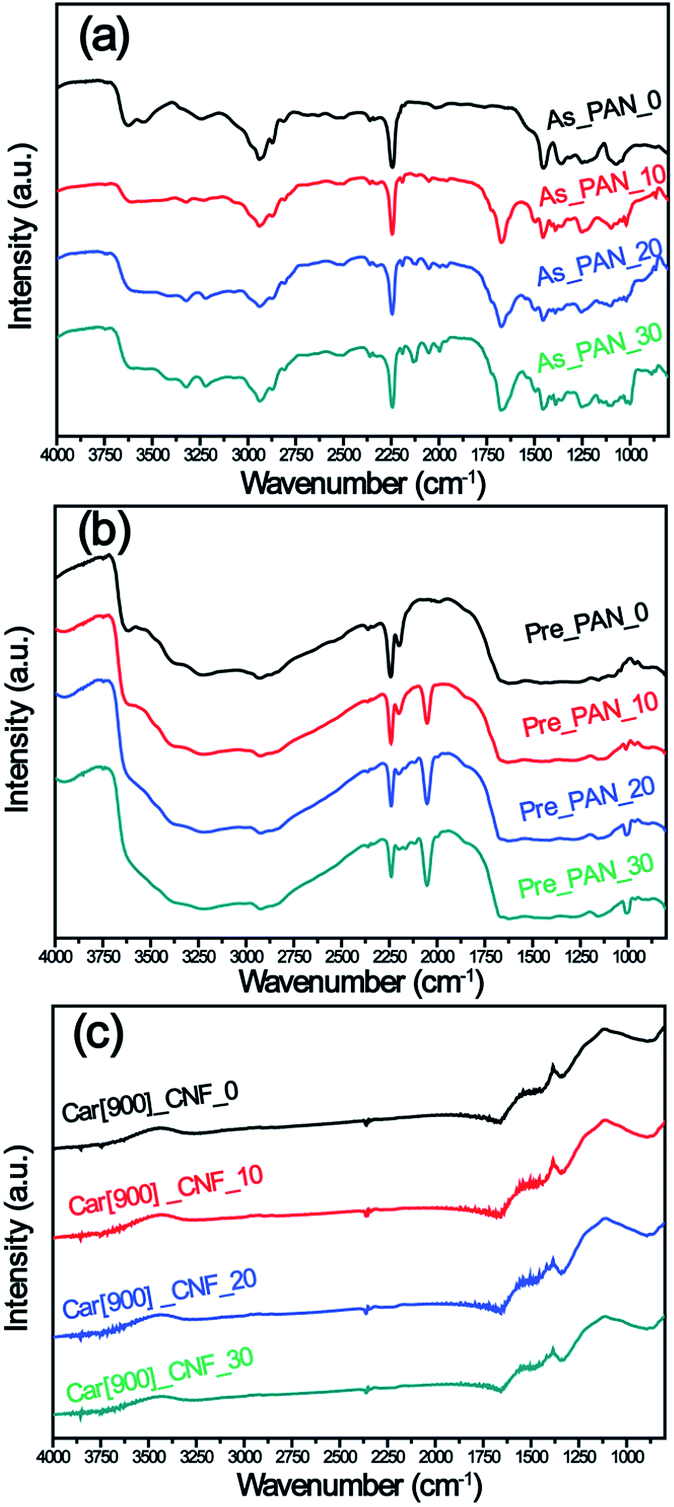 | ||
| Fig. 4 ATR-FTIR spectra of (a) K2S/PAN composite nanofibers, (b) preoxidative K2S/PAN composite nanofibers and (c) carbon nanofibers with different K2S contents. | ||
To better understand the activation mechanism of K2S during the formation of porous CNFs, XRD were recorded to detect the phase evolution of K2S and PAN throughout the whole processes. As can be seen in Fig. 5a, freshly prepared PAN nanofibers feature two typical diffraction peaks at 2θ = 16.5° and 2θ = 29.0° which correspond to the crystal plane (100) reflecting the spacing of molecular chain and (110) crystal plane of the nearly parallel molecular pieces.50 Upon addition of K2S into PAN matrix, the substantially decreased characteristic diffraction peaks of PAN suggested that the addition of K2S significantly influenced the long-range order of PAN matrix resulting in this disordered orientation and decreased crystallinity of PAN matrix. As for the phase transformation of K2S, situation became much more complex as the unstable K2S under oxidative environment transformed into its oxidative derivatives with more than one phase, which included oxidative sulfide mixtures of KS, K2S3, K2S4 and K2S5, sulfur, and sulfate mixtures of K2SO3, K2SO5, K2SO7, and K2SO4, etc. From Fig. 5a, we could identify that the oxidative extent of K2S is determined by the amount of K2S added within PAN matrix, the sulfide mixtures constitutes high content with more K2S addition in K2S/PAN composite nanofiber.
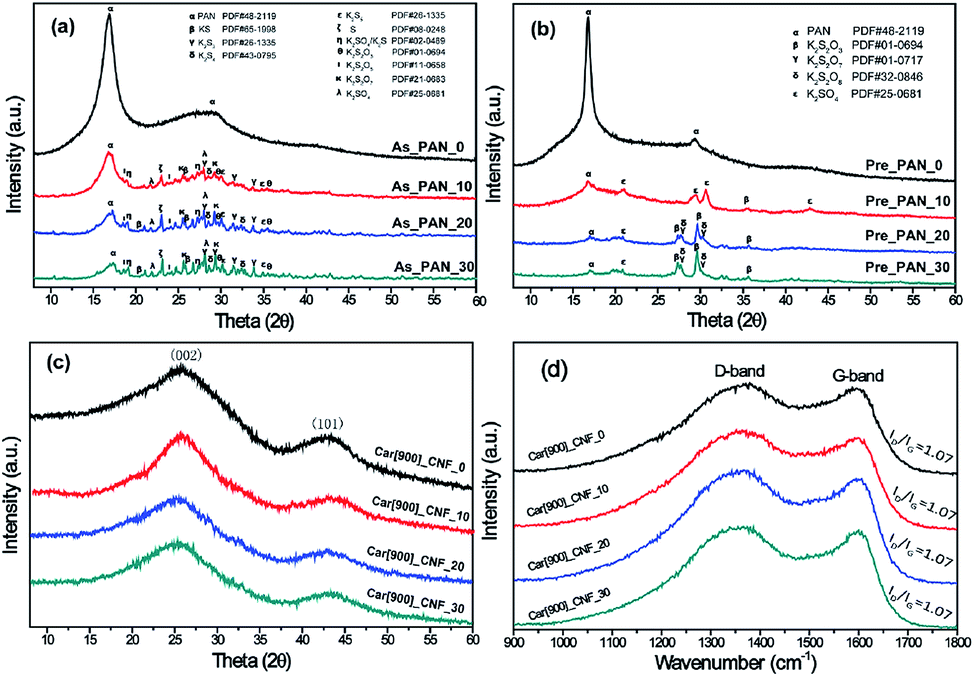 | ||
| Fig. 5 XRD patterns of (a) as prepared K2S/PAN samples, (b) pre-oxidative K2S/PAN samples, (c) as-prepared carbon nanofibers prepared at 900 °C without wash and (d) the corresponding Raman spectra. | ||
Fig. 5b depicts the XRD patterns of K2S/PAN composite nanofibers treated in air. Upon experiencing preoxidation where cyclization reaction typically occurs, all K2S/PAN composite nanofiber shows a drastic decrease of the diffraction peak at 16.9° compared to PAN nanofibers, which is due to the improved cyclization reaction with the help of K2S. Meanwhile, K2S in Pre_PAN samples transformed into its sulfate form during the oxidative stabilization process. As for sample Pre_PAN_10 with 10 wt% K2S, the diffraction peaks match well with standard diffraction peaks of K2SO4 (JCPDF 25-0681) suggesting that K2S was fully oxidized into its highest oxidation state during the pre-oxidative stage. When the K2S content was increased to 20 wt%, the diffraction peaks of the transformed K2S in sample Pre_PAN_20 could be attributed to compounds of sulfate and thiosulfate form. For the carbonization process at 900 °C, all the four CNF samples exhibited similar diffraction pattern assigned to the (002) and (101) diffraction of hexagonal carbon material (JCPDS card No. 75-1621),51 indicating the crystalline nature of carbon with very small particle size.52 It is worth noted that all K2S-related compound diffraction peaks disappeared among all the samples, this is probably because the as-formed thiosulfate and sulfate compounds reacted with carbon under the reducing atmosphere to form the terminal K2S compounds which could be easily escape from carbon nanofiber under the temperature near its boiling point (912 °C, K2S). The structural perfections of carbon nanofibers were investigate by Raman spectroscopy in Fig. 5d. All samples possess identical intensity ratios (R = ID/IG), indicating that the perfectness of structurally ordered graphite phases in the carbon nanofibers was not affected by the addition of K2S.
From the above results, we could conclude that it was the mixtures of potassium thiosulfate, potassium sulfate and potassium pyrosulfate that were reacting with the as-formed carbon nanofiber resulting in such porous structure in carbon nanofiber. The possible K2S evolution during the whole process and the activation mechanism is proposed as follows:
| K2SO4 + 4C = K2S + 4CO | (2) |
| K2SO4 + 2C = K2S + 2CO | (3) |
| K2S2O7 + 4C = SO3 + K2S + 4CO | (4) |
| K2S2O3 + 3C = S + K2S + 3CO | (5) |
During the electrospinning process, K2S within the PAN matrix transformed into as-formed polysulfide and thiosulfate state, and these newly generated mixtures were further transformed into deeper oxidation state (e.g. pyrosulfate and sulfate) during the preoxidative process. Under the carbonization process, the as-formed potassium sulfate, potassium pyrosulfate and potassium thiosulfate consumed the as-formed carbon in the CNF matrix forming porous structure according to the proposed eqn (2)–(5), and this process also accompanied with the formation of the terminal product of K2S at the end of this activation process. When the carbonization temperature rises above the boiling point of K2S, the formed K2S escapes out of the CNF matrix (Scheme 1) leaving only porous structures in the CNF matrix.
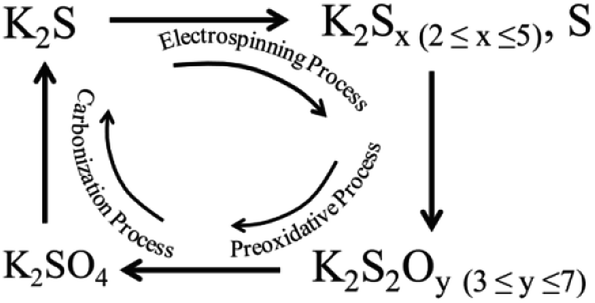 | ||
| Scheme 1 Schematic diagram of the K2S evolution during the whole processes. | ||
800 °C, 900 °C and 1000 °C were chosen to investigate the effect of carbonization temperature on the pore structure of micro/mesoporous carbon nanofibers based on the fact that the melting point (840 °C) and boiling point (912 °C) of K2S were in the range of 800–1000 °C. N2 adsorption/desorption isotherms of as-prepared porous carbon nanofibers with the 30% K2S addition were shown in Fig. 6. All three samples exhibit similar hysteresis loop (type H4) at a relative pressure range of 0.45–0.95, indicating that these three samples possesses similar porous structure with the mesopore volume being 0.238 cm3 g−1 for Car[800]_CNF_30, 0.234 cm3 g−1 for Car[900]_CNF_30 and 0.200 cm3 g−1 for Car[1000]_CNF_30, respectively (Table 1). Significant different adsorption capacities in low relative pressure (P/P0 < 0.05) were observed among these three samples. Carbonization of K2S/PAN composite nanofibers at 900 °C yielded the most abundant microporosity with 0.387 cm3 g−1 of micropore volume and 518.8 m2 g−1 of microporous surface area for Car[900]_CNF_30. Whereas for carbonization temperature at 800 °C and 1000 °C, only 0.248 cm3 g−1 and 0.235 cm3 g−1 of micropore volume were observed for Car[800]_CNF_30 and Car[1000]_CNF_30 respectively, which is much less than that of Car[900]_CNF_30 (Table 1). This is probably because K2S was at a molten state under the temperature of 900 °C and can improve the activation with the local as-formed carbon nanofiber more efficiently than that under 800 °C, thus generating more pores inside carbon nanofibers. When the carbonization temperature reached 1000 °C, which is beyond the boiling point of K2S, part of K2S will evaporate from the local carbon nanofiber and will thus inevitably lower the effectiveness of activation. Therefore, carbonization temperature of 900 °C would be optimum activation temperature for the synthesis of porous CNFs.
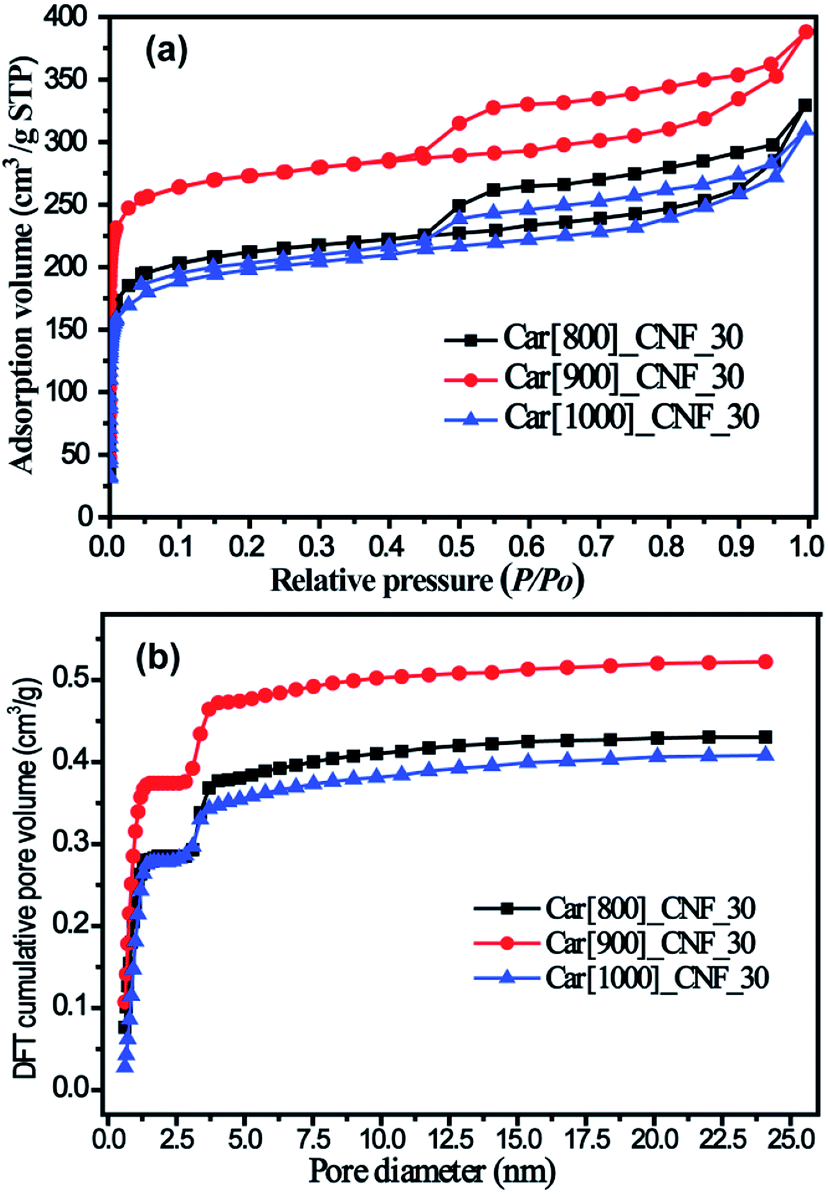 | ||
| Fig. 6 (a) N2 adsorption/desorption isotherms of as-prepared carbon nanofibers under various temperature and (b) corresponding DFT cumulative pore volume curves. | ||
Surface functionalization is important for the electrochemical performance of carbon electrode materials. Table 3 shows the composition of all as-prepared carbon nanofiber samples. The overall C and N contents of K2S activated carbon nanofibers are a little lower than that of neat CNFs (Car[900]_CNF_0) suggesting that the cyan group can be abstracted from the PAN matrix with the help of K2S, which is in accordance with FT-IR results (Fig. 4b). The overall oxygen contents are enhanced from 9.48% to 12–14 wt% for K2S activated carbon nanofibers due to oxidation by K2S oxidative derivates. Note the sample were thoroughly washed with DI water prior to XPS characterization, no sulfur and potassium signals were determined indicating all sulfur and potassium can be removed simply by water washing. N 1s spectrum of carbon nanofiber samples could be fitted into three component peaks at 396.6 eV, 397.4 eV, and 399.2 eV (Fig. 7), corresponding to three types of nitrogen functional groups, i.e. N-6 (pyridinic N), N-5 (pyrrolic N), and N-Q (quaternary N), respectively.13,53–56 For carbon nanofiber samples obtained at 900 °C, all samples exhibited three types of nitrogen with different relative amounts. Upon increasing K2S contents, the intensity of N-Q gradually decreases and its proportion is becoming smaller, while both N-5 and N-6 are increasing in terms of intensity and proportion. These results indicate that the activation agent tends to induce nitrogen atom of PAN into formation of polypyrrole (pentagonal ring, N-5) and polypyridinic (six-membered ring, N-6) structure rather than the formation of N-Q during carbonization process. It is also reported that among these three types of commonly observed N-doped atoms, N-6 and N-5 could offer more active sites to enhance the pseudocapacitance, resulting in improved the power density of supercapacitors.54,57,58 Hence, it is reasonable to believe that Car[900]_CNF_30 of high amount of N-5 and N-6 structures, combined with high surface area and porosity, possess the necessity for high capacitive performance of supercapacitors.
| Sample | C 1s (at%) | O 1s (at%) | N 1s (at%) | Nitrogen functional groups (at%) | |
|---|---|---|---|---|---|
| N-Q | N-5 + N-6 | ||||
| Car[900]_CNF_0 | 84.04 | 9.48 | 6.46 | 3.94 | 2.50 |
| Car[900]_CNF_10 | 81.46 | 13.41 | 5.13 | 3.11 | 2.02 |
| Car[900]_CNF_20 | 81.01 | 13.71 | 5.26 | 3.00 | 2.26 |
| Car[900]_CNF_30 | 81.68 | 12.59 | 5.71 | 3.22 | 2.49 |
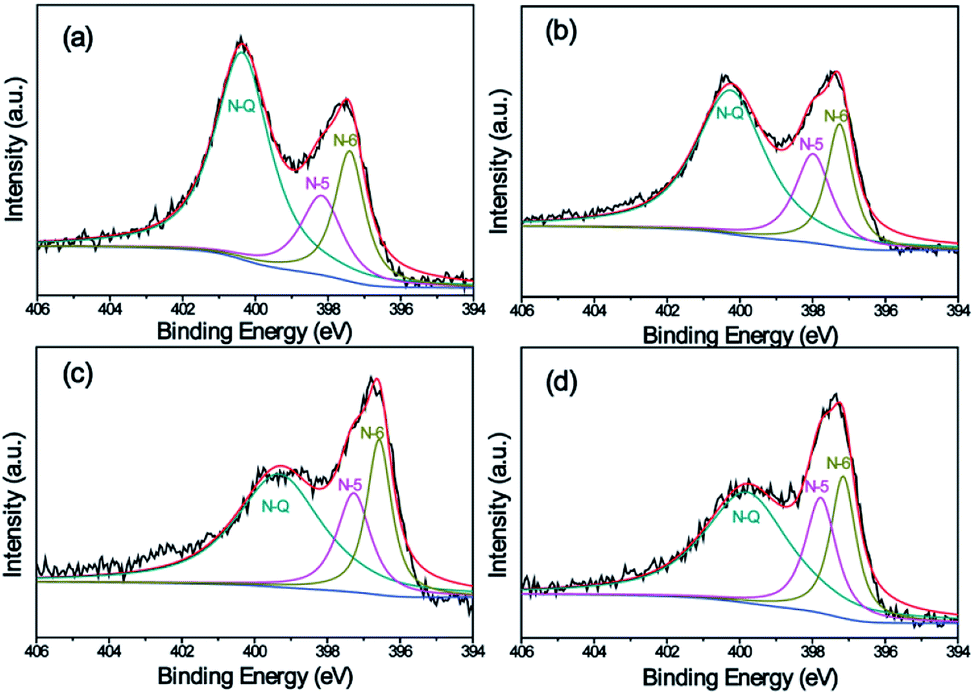 | ||
| Fig. 7 High-resolution XPS spectra of the N 1s peak of samples: (a) Car[900]_CNF_0, (b) Car[900]_CNF_10, Car[900]_CNF_20, and (d) Car[900]_CNF_30. | ||
The supercapacitive performance of the samples as electrode materials were evaluated based on a two-electrode configuration in a liquid electrolyte of 6 M KOH at room temperature. Fig. 8a shows the comparative CV plots at a scan rate of 5 mV s−1 of all samples with different K2S mass addition. The areas surrounded by the CV curves of Car[900]_CNF_30 and Car[900]_CNF_20 are much larger than that of Car[900]_CNF_10 and Car[900]_CNF_0, indicative of much higher specific capacitance for carbon nanofiber electrode prepared with K2S addition exceeding 20 wt%. Car[900]_CNF_30, which possess the highest surface area of 835.0 m2 g−1, has the highest specific capacitance (Cs) of 217.8 F g−1. Fig. 8b shows the CV measurement results of Car[900]_CNF_30 at different scanning rates from 5 to 200 mV s−1. Notwithstanding the pseudo-rectangular shape of CV curves on the two-electrode configuration (Fig. 8a and b) which is usually taken for pure capacitive behavior, the CV curve of Car[900]_CNF_30 in three-electrode system shows evidently distorted CV profile (Fig. S4a†), especially at negative potentials, indicating pseudocapacitive contribution due to the side reactions in charge and discharge process by the appreciable contents of O and N groups. The corresponding calculated specific capacitances in the three-electrode system determined from CV curves are shown in Fig. S4b,† which presents a similar situation as in the two-electrode system.
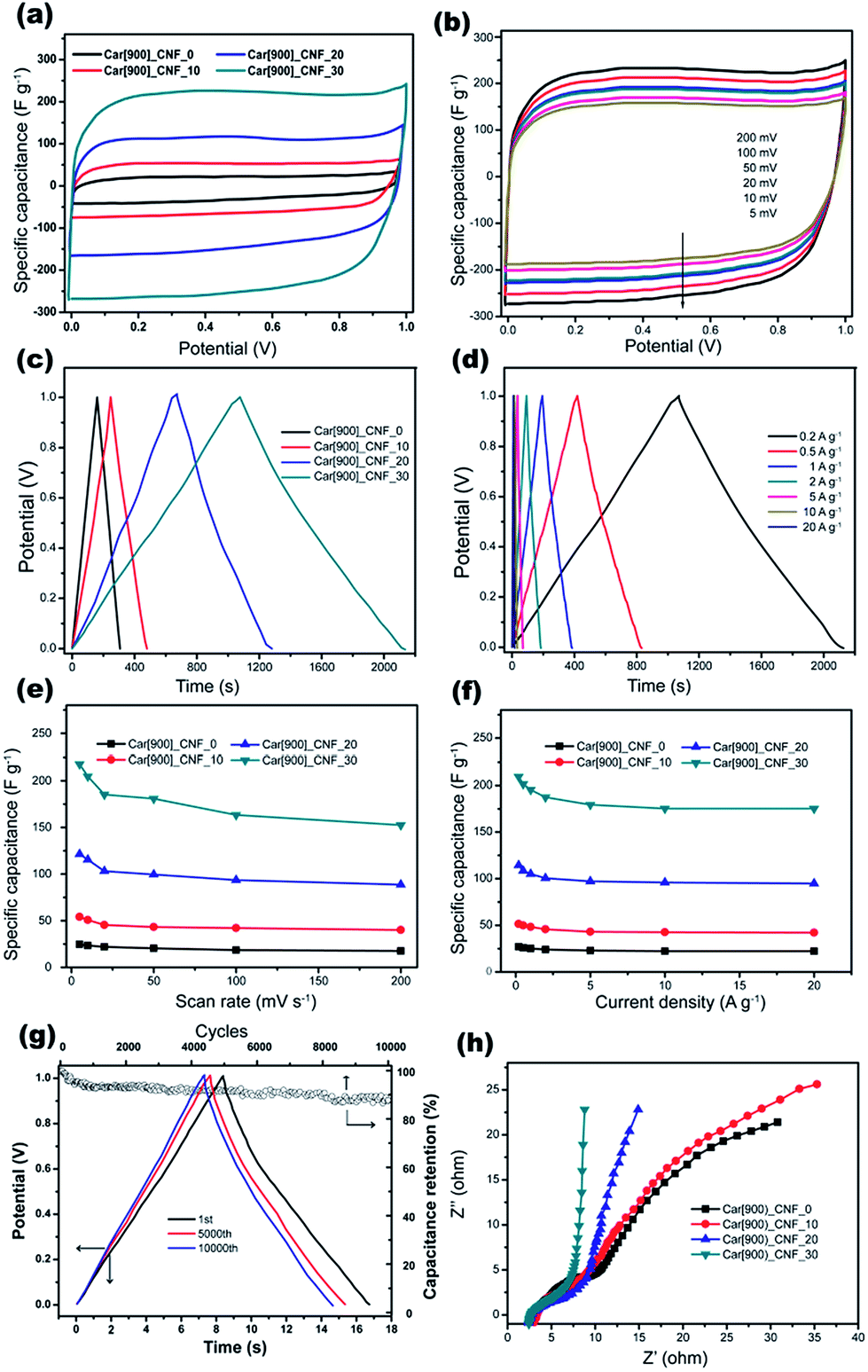 | ||
| Fig. 8 Electrochemical properties of carbon nanofiber samples in a two-electrode system of 6 M KOH solution. CV curves for (a) all samples and (b) different scanning rates with Car[900]_CNF_30, GCD curves for (c) all samples and (d) different current density with Car[900]_CNF_30, specific capacitance of all samples calculated from (e) CV curves and (f) GCD methods, (g) cycling stability of Car[900]_CNF_30 and (h) Nyquist plots of all samples. | ||
Fig. 8c shows the GCD curves of CNFs samples with an applied current density of 0.2 A g−1, the highest specific capacitance of 210.7 F g−1 was also obtained by using Car[900]_CNF_30 and the Cs sequence among all samples was in accordance with the results of the cyclic voltammetry. The GCD curves at different current densities are shown in Fig. 8d and f. The typical GCD profiles of Car[900]_CNF_30 demonstrate highly symmetry and relatively linear indicating ideal capacitive behavior. When the current density raised up to 20 A g−1, the specific capacitance stays at 83% of the highest Cs value, reflecting the high rate capability of Car[900]_CNF_30. The specific capacitances of all samples against the scanning rate calculated by CV curves are displayed in Fig. 8e. The Cs values clearly show that an increase of scanning rate from 5 to 200 mV s−1 decreases the capacitance value, which is commonly happened on carbon materials and caused by the short time available for ion diffusion and adsorption inside micropores.59 Car[900]_CNF_30 demonstrated high Cs retention capability (70% of the highest Cs) at high scanning rate of 200 mV s−1, indicating high power handling capability due to the open pore structure (inter-fiber spacing) in nanofiber mats resulting in faster electrolyte/ion diffusion. As for the electrochemical stability in charge/discharge cycles for supercapacitors, 89% of the initial capacitance is retained after 10000 cycles in the electrochemical stability test equipped with the Car[900]_CNF_30 at a current density of 20 A g−1 (Fig. 8g).
The Nyquist plots of different samples were compared in Fig. 8h. At high frequencies, a semi-circular arc is related to intrinsic resistance of active material and the contact resistance between working electrode and electrolyte.60,61 The smaller diameter of semicircle in the EIS spectrum of Car[900]_CNF_30 indicates lower resistance which is associated with better electrolyte pore accessibility.41 In the low frequency region, Car[900]_CNF_30 demonstrates the most steep line suggesting more ideal capacitive behavior. Calculated from the GCD curve at a current density of 1 A g−1 (Fig. 8d), Car[900]_CNF_30 delivered a high energy density of 27.2 W h kg−1 with a power density of 508.7 W kg−1, and the energy density is 24.3 W h kg−1 even at the high power density of 10.4 kW kg−1 indicating high energy storage capability under high current density. Compared with those published on electrospun carbon fibers and activated carbons (Table S1†), the performance of Car[900]_CNF_30 is competitive among the electrode carbons reported, and this further demonstrates that the formation of hierarchical porous structures could effectively improve the power density of the device.
As it has been proposed that energy storage of electric double layer is mainly dependent on electrode/electrolyte interface performance, it is crucial for the electrode materials to have suitable pore size, reasonable proportion of micropores, high surface area and surface functionalities which can improve the EDL and pseudocapacitance. The superior capacitive performance of Car[900]_CNF_30 can be due to the nanofibrous structure that offers excellent accessibility for electrolyte ions, and the hierarchical porosity (0.618 cm3 g−1) with rational ratio of micropores to mesopores (3:2) and high SSA (835.0 m2 g−1) induced by the activation of K2S during carbonization stage, which facilitate the accessibility of electrolyte ions to the interior surface of the porous carbon resulting in improved formation of electric double layer. Therefore, porous electrospun carbon nanofibers prepared via K2S in situ activation could be promising candidates for supercapacitors.
4. Conclusions
In summary, for the first time we found K2S is a perfect activation agent for the successful fabrication of hierarchical porous CNFs with high SSA and porosity by employing in situ activation electrospinning method. During the whole fabrication process, K2S experiences a phase evolution loop including oxidation and reduction process. The former process involves the transformation of K2S to its oxidative derivatives and the improved cyclization of PAN nitrile groups during the electrospinning and preoxidative stages, the latter includes the K2S oxidative derivatives transforming back to K2S accompanied with the activation of CNFs during carbonization stage. The as-prepared hierarchical porous CNFs not only show a large SSA (835.0 m2 g−1), but also exhibited a high PAN carbonization yield of (84.0 wt%). The hierarchical porous CNFs combine the advantages of interconnected meso/micropores and are a promising candidate for electrochemical energy conversion and storage devices.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Analysis & Characterization Center of National Institute of Clean-and-Low-Carbon Energy (NICE) for the help of sample characterization.References
- Z. Wu, L. Li, J. M. Yan and X. B. Zhang, Adv. Sci., 2017, 4, 1600382 CrossRef.
- P. Simon and Y. Gogotsi, Acc. Chem. Res., 2013, 46, 1094–1103 CrossRef CAS.
- E. Frackowiak, K. Metenier, V. Bertagna and F. Beguin, Appl. Phys. Lett., 2000, 77, 2421–2423 CrossRef CAS.
- S. Li, C. Zhao, K. W. Shu, C. Y. Wang, Z. P. Guo, G. G. Wallace and H. K. Liu, Carbon, 2014, 79, 554–562 CrossRef CAS.
- G. Lota, K. Fic and E. Frackowiak, Electrochem. Commun., 2011, 13, 38–41 CrossRef CAS.
- S. T. Senthilkumar, R. K. Selvan, Y. S. Lee and J. S. Melo, J. Mater. Chem. B, 2013, 1, 1086–1095 RSC.
- J. J. Xue, T. Wu, Y. Q. Dai and Y. N. Xia, Chem. Rev., 2019, 119, 5298–5415 CrossRef CAS.
- R. Hou, G. S. Gund, K. Qi, P. Nakhanivej, H. Liu, F. Li, B. Y. Xia and H. S. Park, Energy Storage Materials, 2019, 19, 212–241 CrossRef.
- Y. M. Tan, C. F. Xu, G. X. Chen, Z. H. Liu, M. Ma, Q. J. Xie, N. F. Zheng and S. Z. Yao, ACS Appl. Mater. Interfaces, 2013, 5, 2241–2248 CrossRef CAS PubMed.
- B. Liu, H. Shioyama, H. L. Jiang, X. B. Zhang and Q. Xu, Carbon, 2010, 48, 456–463 CrossRef CAS.
- M. Biswal, A. Banerjee, M. Deo and S. Ogale, Energy Environ. Sci., 2013, 6, 1249–1259 RSC.
- P. Hao, Z. H. Zhao, J. Tian, H. D. Li, Y. H. Sang, G. W. Yu, H. Q. Cai, H. Liu, C. P. Wong and A. Umar, Nanoscale, 2014, 6, 12120–12129 RSC.
- L. J. Hou, Z. G. Hu, X. T. Wang, L. L. Qiang, Y. Zhou, L. W. Lv and S. S. Li, J. Colloid Interface Sci., 2019, 540, 88–96 CrossRef CAS.
- M. J. Bleda-Martinez, J. A. Macia-Agullo, D. Lozano-Castello, E. Morallon, D. Cazorla-Amoros and A. Linares-Solano, Carbon, 2005, 43, 2677–2684 CrossRef CAS.
- D. Lozano-Castello, D. Cazorla-Amoros, A. Linares-Solano, S. Shiraishi, H. Kurihara and A. Oya, Carbon, 2003, 41, 1765–1775 CrossRef CAS.
- Y. T. Li, L. Liu, Y. Z. Wu, T. Wu, H. Y. Wu, Q. P. Cai, Y. T. Xu, B. R. Zeng, C. H. Yuan and L. Z. Dai, J. Mater. Chem. B, 2019, 7, 13154–13163 RSC.
- T. Kim, G. Jung, S. Yoo, K. S. Suh and R. S. Ruoff, ACS Nano, 2013, 7, 6899–6905 CrossRef CAS.
- S. Dutta, A. Bhaumik and K. C. W. Wu, Energy Environ. Sci., 2014, 7, 3574–3592 RSC.
- Y. M. He, W. J. Chen, C. T. Gao, J. Y. Zhou, X. D. Li and E. Q. Xie, Nanoscale, 2013, 5, 8799–8820 RSC.
- S. J. Song, F. W. Ma, G. Wu, D. Ma, W. D. Geng and J. F. Wan, J. Mater. Chem. B, 2015, 3, 18154–18162 RSC.
- X. B. Yan, Z. X. Tai, J. T. Chen and Q. J. Xue, Nanoscale, 2011, 3, 212–216 RSC.
- L. J. Xie, G. H. Sun, F. Y. Su, X. Q. Guo, Q. Q. Kong, X. M. Li, X. H. Huang, L. Wan, W. Song, K. X. Li, C. X. Lv and C. M. Chen, J. Mater. Chem. B, 2016, 4, 1637–1646 RSC.
- Y. L. Wen, L. P. Zhang, J. Liu, X. Wen, X. C. Chen, J. L. Ma, T. Tang and E. Mijowska, Nanotechnology, 2019, 30, 295703 CrossRef CAS.
- G. Li, T. S. Xie, S. L. Yang, J. H. Jin and J. M. Jiang, J. Phys. Chem. C, 2012, 116, 9196–9201 CrossRef CAS.
- W. H. Li, Z. Z. Yang, Y. Jiang, Z. R. Yu, L. Gu and Y. Yu, Carbon, 2014, 78, 455–462 CrossRef CAS.
- R. Zhang, L. Wang, J. Zhao and S. W. Guo, ACS Sustainable Chem. Eng., 2019, 7, 632–640 CrossRef CAS.
- H. Liu, C. Y. Cao, F. F. Wei, P. P. Huang, Y. B. Sun, L. Jiang and W. G. Song, J. Mater. Chem. B, 2014, 2, 3557–3562 RSC.
- L. W. Ji, Z. Lin, A. J. Medford and X. W. Zhang, Carbon, 2009, 47, 3346–3354 CrossRef CAS.
- Z. Y. Guo, J. T. Huang, Z. H. Xue and X. M. Wang, Chem. Eng. J., 2016, 306, 99–106 CrossRef CAS.
- S. Y. Kim and B. H. Kim, J. Power Sources, 2016, 328, 219–227 CrossRef CAS.
- D. Nan, Z. H. Huang, F. Y. Kang and W. C. Shen, New Carbon Materials, 2016, 31, 393–398 Search PubMed.
- M. X. Wang, Z. H. Huang, T. Shimohara, F. Y. Kang and K. M. Liang, Chem. Eng. J., 2011, 170, 505–511 CrossRef CAS.
- C. Ma, Y. Song, J. L. Shi, D. Q. Zhang, X. L. Zhai, M. Zhong, Q. G. Guo and L. Liu, Carbon, 2013, 51, 290–300 CrossRef CAS.
- C. Ma, R. R. Wang, Z. Y. Xie, H. X. Zhang, Z. Y. Li and J. L. Shi, J. Porous Mater., 2017, 24, 1437–1445 CrossRef CAS.
- Q. Dong, G. Wang, B. Q. Qian, C. Hu, Y. W. Wang and J. S. Qiu, Electrochim. Acta, 2014, 137, 388–394 CrossRef CAS.
- C. H. Kim, J. H. Wee, Y. A. Kim, K. S. Yang and C. M. Yang, J. Mater. Chem. B, 2016, 4, 4763–4770 RSC.
- S. Zhang, Q. Zhang, H. Y. Wang, Y. H. Ni and Z. B. Zhu, Int. J. Hydrogen Energy, 2014, 39, 17913–17920 CrossRef CAS.
- J. S. Cho, J. S. Park and Y. C. Kang, Nano Res., 2017, 10, 897–907 CrossRef CAS.
- L. J. Zhang, Y. Z. Jiang, L. W. Wang, C. Zhang and S. X. Liu, Electrochim. Acta, 2016, 196, 189–196 CrossRef CAS.
- H. Liu, C.-Y. Cao, F.-F. Wei, Y. Jiang, Y.-B. Sun, P.-P. Huang and W.-G. Song, J. Phys. Chem. C, 2013, 117, 21426–21432 CrossRef CAS.
- C. Kim, B. T. N. Ngoc, K. S. Yang, M. Kojima, Y. A. Kim, Y. J. Kim, M. Endo and S. C. Yang, Adv. Mater., 2007, 19, 2341–2346 CrossRef CAS.
- Y. Aykut, ACS Appl. Mater. Interfaces, 2012, 4, 3405–3415 CrossRef CAS.
- Y. Liu, J. Y. Zhou, L. L. Chen, P. Zhang, W. B. Fu, H. Zhao, Y. F. Ma, X. J. Pan, Z. X. Zhang, W. H. Han and E. Q. Xie, ACS Appl. Mater. Interfaces, 2015, 7, 23515–23520 CrossRef CAS PubMed.
- X. H. Qin, E. L. Yang, N. Li and S. Y. Wang, J. Appl. Polym. Sci., 2007, 103, 3865–3870 CrossRef CAS.
- M. S. A. Rahaman, A. F. Ismail and A. Mustafa, Polym. Degrad. Stab., 2007, 92, 1421–1432 CrossRef CAS.
- K. K. Karthikeyan and P. Biji, Microporous Mesoporous Mater., 2016, 224, 372–383 CrossRef CAS.
- Q. Ouyang, L. Cheng, H. Wang and K. Li, Polym. Degrad. Stab., 2008, 93, 1415–1421 CrossRef CAS.
- G. Henrici-Olivé and S. Olivé, The chemistry of carbon fiber formation from polyacrylonitrile, Berlin, Heidelberg, 1983, pp. 1–60 Search PubMed.
- E. Cipriani, M. Zanetti, P. Bracco, V. Brunella, M. P. Luda and L. Costa, Polym. Degrad. Stab., 2016, 123, 178–188 CrossRef CAS.
- M. M. Qiao, H. J. Kong, X. M. Ding, Z. F. Hu, L. W. Zhang, Y. Z. Cao and M. H. Yu, Polymers, 2019, 11, 403 CrossRef PubMed.
- F. Su, C. K. Poh, J. S. Chen, G. Xu, D. Wang, Q. Li, J. Lin and X. W. Lou, Energy Environ. Sci., 2011, 4, 717–724 RSC.
- F. Zheng, Y. Yang and Q. Chen, Nat. Commun., 2014, 5, 5261 CrossRef CAS.
- W. Lei, L. L. Han, C. J. Xuan, R. Q. Lin, H. F. Liu, H. L. L. Xin and D. L. Wang, Electrochim. Acta, 2016, 210, 130–137 CrossRef CAS.
- R. Singhal and V. Kalra, J. Mater. Chem. B, 2015, 3, 377–385 RSC.
- J. Tan, Y. L. Han, L. He, Y. X. Dong, X. Xu, D. N. Liu, H. W. Yan, Q. Yu, C. Y. Huang and L. Q. Mai, J. Mater. Chem. B, 2017, 5, 23620–23627 RSC.
- T. Le, Y. Yang, Z. H. Huang and F. Y. Kang, J. Power Sources, 2015, 278, 683–692 CrossRef CAS.
- W. R. Li, D. H. Chen, Z. Li, Y. F. Shi, Y. Wan, J. J. Huang, J. J. Yang, D. Y. Zhao and Z. Y. Jiang, Electrochem. Commun., 2007, 9, 569–573 CrossRef CAS.
- U. B. Nasini, V. G. Bairi, S. K. Ramasahayam, S. E. Bourdo, T. Viswanathan and A. U. Shaikh, J. Power Sources, 2014, 250, 257–265 CrossRef CAS.
- T. Le, Y. Yang, Z. Huang and F. Kang, J. Power Sources, 2015, 278, 683–692 CrossRef CAS.
- A. Garcia-Gomez, P. Miles, T. A. Centeno and J. M. Rojo, Electrochim. Acta, 2010, 55, 8539–8544 CrossRef CAS.
- C. Portet, P. L. Taberna, P. Simon and C. Laberty-Robert, Electrochim. Acta, 2004, 49, 905–912 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Digital photograph K2S/DMF, TEM, DTG and CV profiles. See DOI: 10.1039/c9ra06847c |
| This journal is © The Royal Society of Chemistry 2019 |