
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly stereoselective gram scale synthesis of all the four diastereoisomers of Boc-protected 4-methylproline carboxylates†
Kehuan
Sun‡
ac,
Cheng
Tao‡
bcd,
Bohua
Long
 *cd,
Xiaobin
Zeng
*e,
Zhengzhi
Wu
*cd and
Ronghua
Zhang
*a
*cd,
Xiaobin
Zeng
*e,
Zhengzhi
Wu
*cd and
Ronghua
Zhang
*a
aCollege of Traditional Chinese Medicine, Jinan University, Guangzhou 510632, China
bIntegrated Chinese and Western Medicine Postdoctoral Research Station, Jinan University, Guangzhou 510632, China
cThe First Affiliated Hospital of Shenzhen University, Shenzhen 518035, China. E-mail: bhlong121@163.com
dShenzhen Institute of Geriatric Medicine, Shenzhen, 518035, China
eCentral Center Lab of Longhua Branch, Department of Infectious Disease, Shenzhen People's Hospital, 2nd Clinical Medical College of Jinan University, Shenzhen 518120, Guangdong Province, China
First published on 8th October 2019
Abstract
A general and practical synthetic process for all the four diastereoisomers of Boc-protected 4-methylproline carboxylates has been developed with essentially complete stereoselectivity on the gram scale, which represents the most diastereoselective preparation of 4-methylproline derivatives to date. This synthesis features an Evans asymmetric alkylation to elegantly establish the challenging stereochemistry of the 4-methyl group, providing valuable insights for the diastereoselective preparation of 4-substituted prolines.
4-Methylprolines (Fig. 1) are one of the most important structural motifs found in numerous natural products and biologically active compounds. For example, (2S,4S)-4-methylproline (1) is the crucial component of the dimerized pentapeptides nazumazoles A–C which have been found to exhibit potential cytotoxicity against P388 murine leukemia cells;1 the trans-isomers (2 and 4) are valuable precursors of various natural compounds, such as methylgriselimycin,2a monamycin D1,2b and cyclopeptide FR235222;3 furthermore, (2R,4R)-4-methylproline (3) has found wide application in therapeutic agents such as the N14-desacetoxytubulyisn analogue with potential applications in clinics.4 In this regard, development of a general and highly diastereoselective route to 4-methylprolines would greatly facilitate the synthesis of various relevant natural products and medicines. We wish to develop a general and practical synthetic process of all the four diastereoisomers of 4-methylprolines for our long-term efforts on the total synthesis of nazumazoles A–C and other 4-methylproline-containing bioactive compounds.
 | ||
| Fig. 1 Selective relevant natural products and bioactive compounds containing 4-methylprolines (1–4). | ||
During the last few decades, several strategies have been employed to the diastereoselective preparation of 4-methylprolines. In 1991, Beaulieu and co-workers reported the synthesis of Boc-protected 4-methyl proline carboxylates by utilizing the intramolecular radical cyclization approach.5 In 2003, Goodman and co-workers realized the diastereoselective synthesis of Boc-protected 4-methyl prolines employing a divergent asymmetric hydrogenation strategy.6 In 2010, Willis and co-workers developed a chemoenzymatic route toward the 4-methylprolines by taking advantage of two biotransformations to establish the absolute configuration.7 However, most of these methods suffer from the poor control of the stereochemistry of the 4-methyl group and often use expensive catalysts or toxic regents. Furthermore, the harsh conditions coupled with expensive reagent cost would impede the application of the current methods to multigram-scale production.
From a viewpoint of synthetic application, the Boc-protected carboxylate form of 4-methylprolines (8, 8′, ent-8 and ent-8′) were more practical and operationally simple than 4-methylprolines (1–4), because the Boc-protected 4-methylproline carboxylates can be easily purified and stored, and they also can be further elaborated to 4-methylprolines (1–4) and other valuable intermediates according to literature procedure.8
As part of our long-term efforts on the total synthesis of nazumazoles A–C and other 4-methylproline-containing bioactive compounds, we present herein a general and practical synthetic process for all the four diastereoisomers of Boc-protected 4-methylproline carboxylates with essentially complete stereoselectivity on gram scale (Scheme 1).
 | ||
| Scheme 1 Diastereoselective synthesis of the 4-methylproline derivatives. | ||
We envisaged that the main synthetic challenge of 4-methylprolines is that of exercising control of the absolute configuration at C4, and this vexing problem could be addressed by employing the asymmetric alkylation approach developed by Evans.9 With this idea in mind, we start our retrosynthetic analysis. As the retrosynthetic analysis strategy was also suit for other three diastereoisomers, we just take one isomer as an example (Scheme 2).
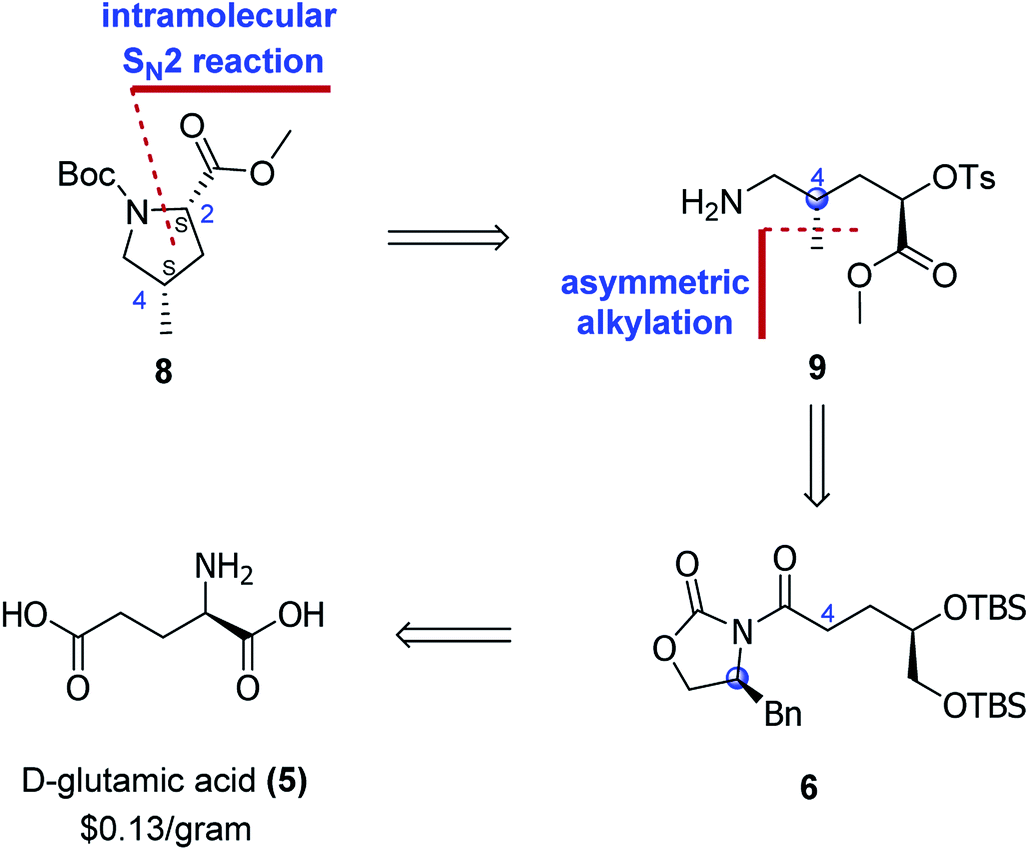 | ||
| Scheme 2 Retrosynthetic analysis. | ||
As for the Boc-protected (2S,4S)-4-methylproline carboxylate (8), the C2 stereocenter would be introduced via an intramolecular SN2 reaction of amine 9. The C4 stereocenter could be installed by utilizing the asymmetric alkylation reaction, which traced the origin of amine 9 to Evans' chiral auxiliary 6. The Evans' chiral auxiliary 6 could be easily prepared from the cheap commercial chiral glutamic acid. In such case, choosing the different chiral glutamic acid (D-glutamic acid or L-glutamic acid) and different chiral N-acyloxazolidinone auxiliarie ((S)-4-benzyl-2-oxazolidinone or (R)-4-benzyl-2-oxazolidinone) would control the configuration of the generated stereogenic center at C4. As such, this would provide a general and facile synthetic protocol for all the four diastereoisomers of 4-methylprolines.
Our strategy for rapid access to the Evans' chiral auxiliary 6 commenced with the chiral D-glutamic acid (5) (Scheme 3). Compound 10 was prepared from D-glutamic acid via a known two-step process10 in 80% yield. Then a chemoselective reduction of the ester group adjacent to the hydroxyl group was achieved with borane-dimethyl sulfide complex to afford diol 11 in 90% yield.11 Then protection of diol 11 followed by hydrolysis of the methyl ester with lithium hydroxide gave acid 12, which was directly coupled with chiral (S)-oxazolidinone auxiliary in the presence of pivaloyl chloride and triethyl amine to afford Evans' chiral auxiliary 6 (72% yield, three steps).
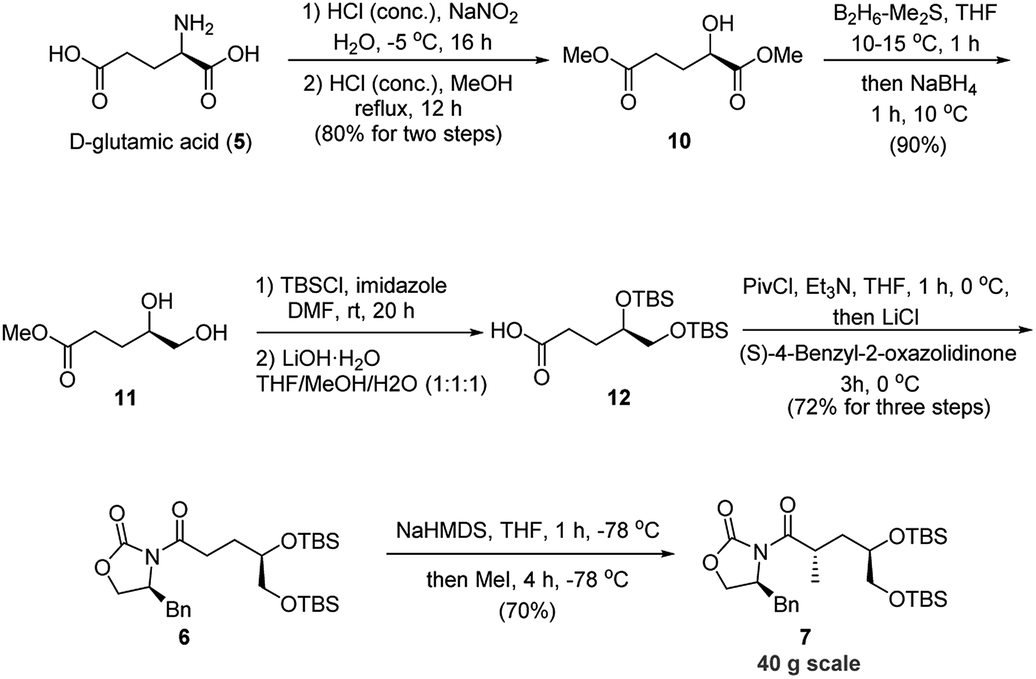 | ||
| Scheme 3 Establishment of the C4 stereochemistry. | ||
With the Evans' chiral auxiliary 6 in hand, the crucial asymmetric alkylation was then explored. The methylation proceeded successfully when using NaHMDS as the base followed by treatment with MeI to give desired product 7 in 70% yield. Gratifyingly, the desired product 7 was obtained as a single stereoisomer on a 40 g scale. As such, the C4 configuration was established with essentially complete stereoselectivity. As we haven't observed and isolated any isomers of 7 in this reaction, the 70% yield may be caused by certain unknown non-isomeric byproducts of unwanted side-reactions, which is common in the Evan's methylation reactions.9
With the access to C4 configuration realized, we next turned our attention to establishment of the requisite C2 stereocenter and the completion of the diastereoselective synthesis of Boc-protected 4-methylproline carboxylates.
As shown in Scheme 4, reductive cleavage of the Evans auxiliary by utilizing sodium borohydride generated alcohol 13 in 90% yield.12 Then the hydroxyl group in compound 13 was directly transformed into the azido group by treatment with DPPA and DBU in dry toluene under reflux condition in 85% isolated yield along with 10% recovered starting material. Much to our delight, when xylene was used as solvent, the azide 14 was forged in 92% yield.
 | ||
| Scheme 4 Synthesis of the Boc-protected (2S,4S)-4-methylproline carboxylate. | ||
With the azide 14 in hand, selective cleavage of the primary TBS-ether was then explored. A variety of traditional methods including CSA/MeOH,13 TsOH/MeOH,14 and conc. HCl/CHCl3 (ref. 15) were examined, and many of our efforts were fruitless, leading to incomplete conversion or deprotection of the two TBS-ether. In contrast, treatment of 14 with NH4F (50 eq.) in MeOH at room temperature for 6 h provided the desired primary alcohol 15 in 75% yield. Unfortunately, oxidation of the resulting hydroxyl group to carboxylic acid with NaIO4 in the presence of catalytic amounts of RuCl3 (ref. 16) was not achieved, leading to decomposition of the starting material. Therefore, the transformation was operated in a two-step process. Alcohol 15 was initially subjected to oxidation with IBX in DMSO at 30 °C, and the resulting aldehyde 16 was subsequently transformed into the corresponding methyl ester 17 by adding NIS and potassium carbonate in MeOH at room temperature17 in 90% overall yield. However, considering NIS was an expensive reagent, we were not satisfied with the result and were eager to find a more cheap procedure for the preparation of compound 17 to make it more accessible. Gratifyingly, when aldehyde 16 was further oxidized with KOH/I2/MeOH system,18 the desired methyl ester 17 was easily and economically synthesized under the mild condition (95% yield, two steps, 13 g scale).
Then TBS ether 17 was deprotected with CSA to afford the corresponding alcohol 18. Tosylation of alcohol 18 was carried out with p-toluenesulfonyl chloride in the presence of base such as pyridine, Et3N or DMAP, leading to less than 50% yield. Fortunately, the yield was significantly increased to 90% when DABCO (2.0 eq.) was used as the base.19
Finally, the two-step azide reduction/intramolecular SN2 cyclization procedure was performed as a tandem one-pot process successfully under the hydrogenation condition. Then the resulting cyclization product was directly protected as its Boc derivative by adding NaHCO3 and Boc2O to afford the desired Boc-protected (2S,4S)-4-methylproline carboxylate (8) in 90% yield. It should be noted that, the present turned out to be very practical, and the desired product (8) can be routinely prepared on a 8.5 g scale with full stereochemical control. The conversion of 8 into (2S,4S)-4-methylproline (1) has been previously reported.8
As shown in Scheme 5, we then accomplished the gram-scale synthesis of the other three diastereoisomers 8′ (15 steps, 13%), ent-8 (15 steps, 12%), ent-8′ (15 steps, 17%) by utilizing the same strategy described above. Because a large amount of 8 was needed for our current program on the total synthesis of nazumazoles A–C, 8 was prepared on a 8.5 g scale (15 steps, 17%). We thought that obtaining the other three isomers in gram scale was enough to illustrate the high synthetic practicality of the present route, so we haven't attempted bigger quantities of the other three isomers. In the key Evan's methylation reactions,9 the desired methylation products 7′, ent-7, ent-7′ were obtained in yields of 66%, 73%, 73%, respectively and without any isomeric methylation products observed, which highlights the essentially complete selectivity of our route. For experimental details, see ESI.†
 | ||
| Scheme 5 Gram scale synthesis of the other three diastereoisomers of Boc-protected 4-methylproline carboxylates. | ||
Conclusions
In conclusion, a general and practical synthetic route for all the four diastereoisomers of Boc-protected 4-methylproline carboxylates has been developed with essentially complete stereoselectivity on gram scale. To the best of our knowledge, our present route represents the most diastereoselective preparation of 4-methylproline derivatives to date. Other features of the synthesis include the accessibility of simple starting materials, cheap synthetic cost, and mild reaction conditions, all of which highlight the high synthetic practicality of the present route. This concise strategy offers a general and facile synthetic protocol for chiral 4-methylprolines and 4-substituted prolines. On the basis of the current work, the investigation of the dimerized pentapeptides nazumazoles A–C and other complex natural products containing 4-methylprolines is ongoing in the laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Health and Family Planning Commission of Shenzhen Municipality (201601053), Shenzhen Science and Technology Program (JCYJ20160428110124165, JCYJ20160428110235852, JCYJ20170413161352000), NSFC (81673837, 81574038), Guangdong Science and Technology Program (2016A020226039, 2015B020211001), and Sanming Project of Medicine in Shenzen (SZSM201612049) for financial support.Notes and references
- K. Fukuhara, K. Takada, S. Okada and S. Matsunaga, Org. Lett., 2015, 17, 2646 CrossRef CAS.
- (a) P. Lukat, Y. Katsuyama, S. wenzel, T. binz, C. König, W. Blankenfeldt, M. Brönstrup and R. Müller, Chem. Sci., 2013, 1 Search PubMed; (b) H. J. Hale, N. Jogiya and S. Manaviazar, Tetrahedron Lett., 1998, 39, 7163 CrossRef.
- W. Xie, B. Zou, D. Pei and D. Ma, Org. Lett., 2005, 7, 2775 CrossRef CAS.
- D. Toader, F. Wang, L. Gingipalli, M. Vasbinder, M. Roth, S. Mao, M. Block, J. Harper, S. Thota, M. Su, J. Ma, V. Bedian and A. Kamal, J. Med. Chem., 2016, 59, 10781 CrossRef CAS.
- F. Soucy, D. Wernic and P. Beaulieu, J. Chem. Soc., Perkin Trans. 1, 1991, 2885 RSC.
- J. R. Del Valle and M. Goodman, J. Org. Chem., 2003, 68, 3923 CrossRef CAS.
- S. R. Crosby, R. B. Sessions and C. L. Willis, Synlett, 2010, 4, 539 Search PubMed.
- (a) X. Gao, Y. Liu, S. Kwong, Z. Xu and T. Ye, Org. Lett., 2010, 12, 3018 CrossRef CAS; (b) J. Goodreid, E. Silveira dos Santos and R. Batey, Org. Lett., 2015, 17, 2182 CrossRef CAS.
- (a) D. A. Evans, M. D. Ennis and D. J. Mathre, J. Am. Chem. Soc., 1982, 104, 1737 CrossRef CAS; (b) M. Heravi, V. Zadsirjan and B. Farajpour, RSC Adv., 2016, 6, 30498 RSC; (c) Y. Lu and P. W. Schiller, Synthesis, 2001, 11, 1639 CrossRef; (d) S. Das and R. K. Goswami, J. Org. Chem., 2014, 79, 9778 CrossRef CAS; (e) S. Guchhait, S. Chatterjee, R. S. Ampapathi and R. K. Goswami, J. Org. Chem., 2017, 82, 2414 CrossRef CAS.
- Y. Geng, M. Zheng, J. Li, D. Zou, Y. Wu and Y. Wu, Tetrahedron Lett., 2017, 58, 3966 CrossRef CAS.
- M. Fox, M. Jackson, I. C. Lennon and R. McCague, J. Org. Chem., 2005, 70, 1227 CrossRef CAS.
- M. Prashad, D. Har, H. Y. Kim and O. Repic, Tetrahedron Lett., 1998, 39, 7067 CrossRef CAS.
- R. S. Ghogare, S. B. Wadavrao and A. V. Narsaiah, Helv. Chim. Acta, 2016, 99, 247 CrossRef CAS.
- P. V. Ramachandran, A. Srivastava and D. Hazra, Org. Lett., 2007, 9, 157 CrossRef CAS.
- S. Tripathy and A. Chattopadhyay, Tetrahedron: Asymmetry, 2012, 23, 1423 CrossRef CAS.
- B. Long, S. Tang, L. Chen, S. Qu, B. Chen, J. Liu, A. R. Maguire, Z. Wang, Y. Liu, H. Zhang, Z. Xu and T. Ye, Chem. Commun., 2013, 49, 2977 RSC.
- C. McDonald, H. Holcomb, K. Kennedy, E. Kirkpatrick, T. Leathers and P. Vanemon, J. Org. Chem., 1989, 54, 1213 CrossRef CAS.
- S. Yamada, D. Morizono and K. Yamamoto, Tetrahedron Lett., 1992, 33, 4329 CrossRef CAS.
- J. Hartung, S. Hünig, R. Kneuer, M. Schwarz and H. Wenner, Synthesis, 1997, 1433 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra06827a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |