
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIr(III)-catalyzed thioether directed arene C–H alkenylation†
Chen
Li
a,
Xian
Chen‡
a,
Rui-Peng
Bao‡
b,
Dong-Li
Li
*a,
Kun
Zhang
a and
Dong-Hui
Wang
 *b
*b
aSchool of Biotechnology & Health Sciences, Wuyi University, 22 Dongcheng Vill., Jiangmen, Guangdong 529020, China. E-mail: wyuchemldl@126.com
bCAS Key Laboratory of Synthetic and Self-Assembly Chemistry for Organic Functional Molecules, Center for Excellence in Molecular Synthesis, University of Chinese Academy of Sciences, Shanghai Institute of Organic Chemistry, CAS, 345 Lingling Rd., Shanghai 200032, . E-mail: Chinadhwang@sioc.ac.cn
First published on 24th September 2019
Abstract
In this study, we demonstrate an Ir(III)-catalyzed thioether directed alkenylation of arene C–H bonds under mild reaction conditions. The selectivity for mono- or di-alkenylation is controlled by the concentration of alkene and oxidant loading. Various functional groups are tolerated, and moderate to good yields of alkenylated products are achieved.
Transition-metal catalyzed C–H functionalizations of arene compounds have attracted tremendous attention in the development of new synthetic methods, due to their innate step- and atom-economy.1 In the past decade, the directing-group strategy has witnessed great success;2 among the achievements, the majority of directing groups use N- or O-atom based directing groups, such as pyridines, oxazolines, amides, imines, carboxylic acids and their derivatives.3 The sulfur-atom is widely present in medicines and agrochemicals4 and the direct functionalization of S-containing compounds will significantly benefit the fast diversification of bioactive molecules. Herein we report an Ir(III)-catalyzed thioether directed alkenylation of arene C–H bonds under mild reaction conditions.
Though sulfur atom is strong coordinative to transition metals, and poisonous of catalysts occur in some catalytic processes, recently breakthrough have revealed that thioethers are efficient directing groups for C–H olefinations. In 2012, Zhang reported a Pd-catalyzed C–H alkenylation of thioethers (Fig. 1a). The intermediate in this reaction was isolated, which identified that sulfur was the directing-atom for the first time.5 In 2013, Shi reported Rh-catalyzed C–H alkenylation of benzylthioether (Fig. 1b).6 More recently, Miura and co-workers reported Rh-catalyzed selective C4 C–H alkenylation of indoles using 3-thioethers as directing groups (Fig. 1c).7 Other sulfur-containing functional groups, such as thiocarbonyls8 and sulfoxides,9 have also been reported in directing C–H arylations, alkenylations, alkylations, and cyclizations with Pd-, Rh-, and Ru-catalysts.10 Despite these successes, the demand is still high for S-atom directed C–H functionalization protocol that proceeds with mild reaction conditions, controllable C–H selectivity, diverse functional group tolerance, and generally applicable catalytic system. Herein, we report an Ir(III)-catalyzed thioether directed mono-alkenylation and di-alkenylation of arene C–H bonds that proceeds under mild and controllable conditions (Fig. 1d).
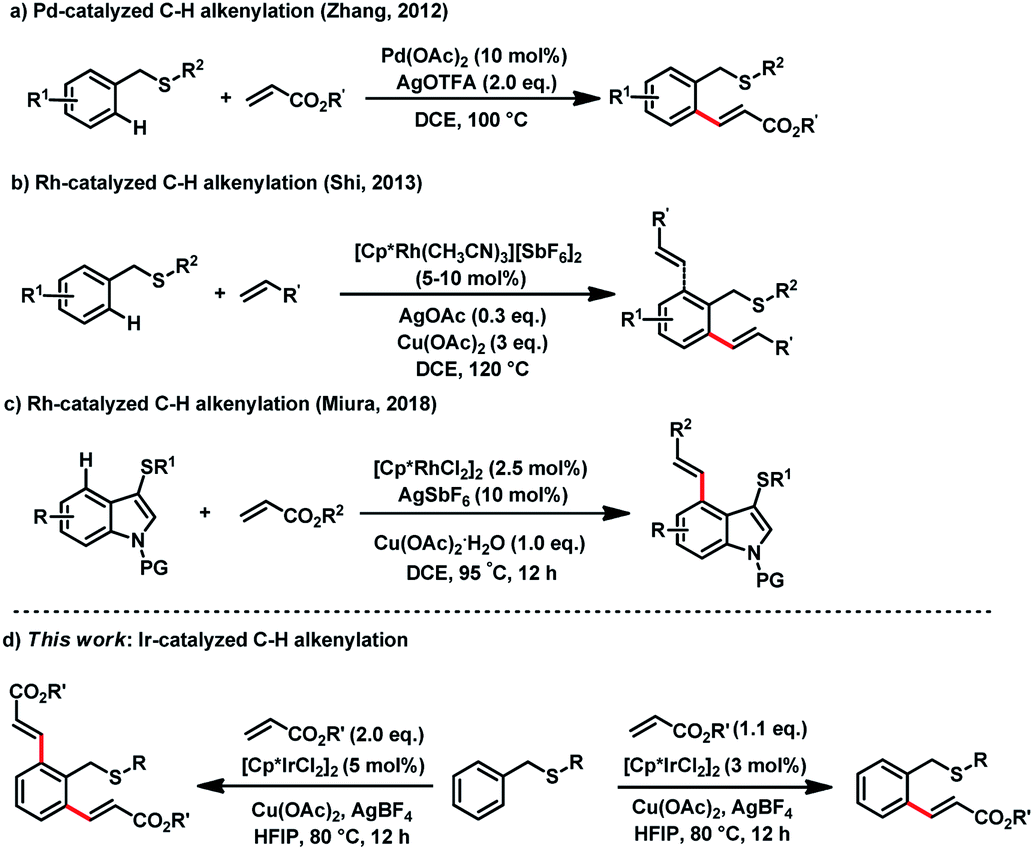 | ||
| Fig. 1 Thioethers directed ortho C–H alkenylation. | ||
We started to investigate this transformation by examining the reactivity between benzyl p-tolyl sulfide (1a) and ethyl acrylate (2a). When using 1.5 mol% of [Cp*IrCl2]2 as the catalyst, 6 mol% of AgBF4 as the activator for the Ir-catalyst, and 1.2 equiv. of Cu(OAc)2 as the oxidant in DCE at 80 °C for 12 h, the desired C–H alkenylation product 3a was obtained in 38% yield (Table 1, entry 1). After optimizing solvents, HFIP was found a superior solvent over DCE, acetone, diethyl ether, or trifluoromethylbenzene, by giving 80% of isolated yield (entries 2–5). Next, oxidants were screened. AgOAc and AgTFA provided the desired product in 72% and 70% yields, respectively (entries 6–7). While Cu(TFA)2-hydrate provided only 12% yield of 3a (entry 8). Benzoquinone (BQ) and O2 were found inefficient for the reaction, and no desired product was found from these reaction mixtures (entries 9–10). When reducing Cu(OAc)2 loading amount to 20 mol%, the yield of the desired product decreased to 40% (entry 11), which indicated that Cu2+ was critical oxidant for this catalytic reaction. Various silver salts, which worked as the activator to promote the Ir-catalyst's efficiency, was also examined, AgSbF4, AgPF6, and AgNTf2 gave the desired product in good yields (entries 12–14), but AgBF4 was superior over the others.
|
|
||||
|---|---|---|---|---|
| Entry | [Ag] | Oxidant | Solvent | Yieldb (%) |
| a Reaction conditions: 1a (0.4 mmol), 2a (2.0 equiv.), [Cp*IrCl2]2 (1.5 mol%), [Ag] (6.0 mol%), oxidant (1.2 equiv.), 0.2 M, air, 80 °C, 12 h. b Yield was determined by 1H NMR analysis of the crude reaction mixture using CH2Br2 as an internal standard. c Isolated yield. | ||||
| 1 | AgBF4 | Cu(OAc)2 | DCE | 38c |
| 2 | AgBF4 | Cu(OAc)2 | Acetone | 34 |
| 3 | AgBF4 | Cu(OAc)2 | Et20 | N.D. |
| 4 | AgBF4 | Cu(OAc)2 | PhCF3 | 38 |
| 5 | AgBF4 | Cu(OAc)2 | HFIP | 87 (80)c |
| 6 | AgBF4 | AgOAc (2.2 equiv.) | HFIP | 72 |
| 7 | AgBF4 | AgTFA (2.2 equiv.) | HAP | 70 |
| 8 | AgBF4 | Cu(TFA)2·XH2O | HFIP | 12 |
| 9 | AgBF4 | BQ | HFIP | N.D. |
| 10 | AgBF4 | O2 | HAP | N.D. |
| 11 | AgBF4 | Cu(OAc)2 (20 mol%) | HFIP | 40 |
| 12 | AgSbF6 | Cu(OAc)2 | HFIP | 62 |
| 13 | AgPF6 | Cu(OAc)2 | HFIP | 64 |
| 14 | AgNTf2 | Cu(OAc)2 | HAP | 62 |

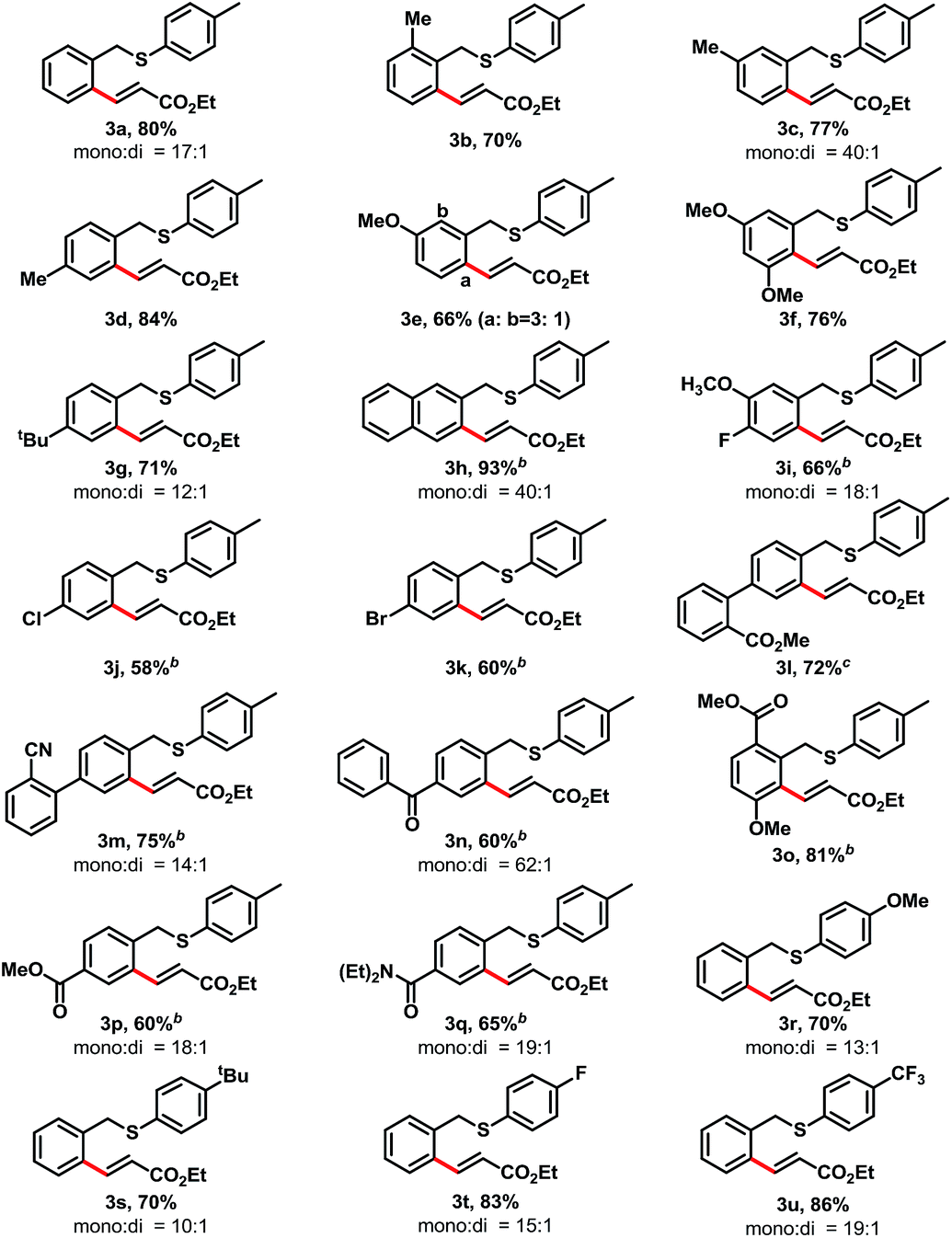
With the optimized conditions in hand, we next examined the substrate scope of thioethers for the mono-alkenylation (Table 2). Various electron-rich substrates provided the desired products in good yields when use 3 mol% of [Cp*IrCl2]2 as the catalyst. Sterically bulky substituents on the benzyl ring were well tolerated. For example, benzyl rings containing methyl groups at the ortho-, meta-, and para-positions readily provided the desired products in 70–84% yields (3b–3d). 3-Methoxybenzyl p-tolyl sulfane provided 66% yield of ortho-olefinated products at the 2-, and 6-positions with the ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3, and the less hindered 6-position was favorable (3e). Other electron-donating substituents, such as substrates containing dimethoxy and tert-butyl groups offered good yield of the desired products, 76% and 71% yields were achieved, respectively (3f–3g). The reaction of 2-naphthylmethyl p-tolyl sulfane took place selectively at the less hindered β-position and provided 3h in 93% yield. For electron-deficient substrates, the transformation showed sluggish reactivity than those electron-rich substrates. When increasing the [Cp*IrCl2]2 catalyst loading to 5 mol%, good yields were achieved. For example, halide substituents, such as fluoro-, chloro- and bromo- were compatible with the reaction conditions, and provided the desired alkenylation product in 66%, 58%, and 60% yield, respectively (3i–3k). The biaryl substrates that contain electron withdrawing groups, such as ester and cyano substituents, readily afforded the desired mono-olefinated product in 72% and 75% yield (3l, and 3m). Other carbonyl derivatives offered the desired products in good yields, too, which include, a benzoyl (3n, 60% yield), esters (3o, and 3p, 81%, and 60% yield, respectively), and an amide (3q, 65% yield). The substituents of p-tolyl sulfide were also examined. In this event, we found that the electron-deficient substituents provided higher yields of the desired products than those electron-rich ones. For example, when replacing the para-methyl group to the methoxy (–OMe, 3r) or tert-butyl (–tBu, 3s), the reaction provided moderate yields (70% for each). While replacement of the para-methyl group with fluoro (–F, 3t) or trifluoromethyl (–CF3, 3u) groups, good yields achieved (83% and 86%, respectively). Notably, in all of these samples, only benzyl C–H bonds are active for the olefination, and the arylthiol C–H bonds are inactive.
:3, and the less hindered 6-position was favorable (3e). Other electron-donating substituents, such as substrates containing dimethoxy and tert-butyl groups offered good yield of the desired products, 76% and 71% yields were achieved, respectively (3f–3g). The reaction of 2-naphthylmethyl p-tolyl sulfane took place selectively at the less hindered β-position and provided 3h in 93% yield. For electron-deficient substrates, the transformation showed sluggish reactivity than those electron-rich substrates. When increasing the [Cp*IrCl2]2 catalyst loading to 5 mol%, good yields were achieved. For example, halide substituents, such as fluoro-, chloro- and bromo- were compatible with the reaction conditions, and provided the desired alkenylation product in 66%, 58%, and 60% yield, respectively (3i–3k). The biaryl substrates that contain electron withdrawing groups, such as ester and cyano substituents, readily afforded the desired mono-olefinated product in 72% and 75% yield (3l, and 3m). Other carbonyl derivatives offered the desired products in good yields, too, which include, a benzoyl (3n, 60% yield), esters (3o, and 3p, 81%, and 60% yield, respectively), and an amide (3q, 65% yield). The substituents of p-tolyl sulfide were also examined. In this event, we found that the electron-deficient substituents provided higher yields of the desired products than those electron-rich ones. For example, when replacing the para-methyl group to the methoxy (–OMe, 3r) or tert-butyl (–tBu, 3s), the reaction provided moderate yields (70% for each). While replacement of the para-methyl group with fluoro (–F, 3t) or trifluoromethyl (–CF3, 3u) groups, good yields achieved (83% and 86%, respectively). Notably, in all of these samples, only benzyl C–H bonds are active for the olefination, and the arylthiol C–H bonds are inactive.
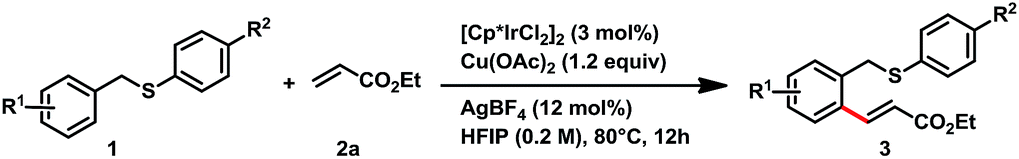
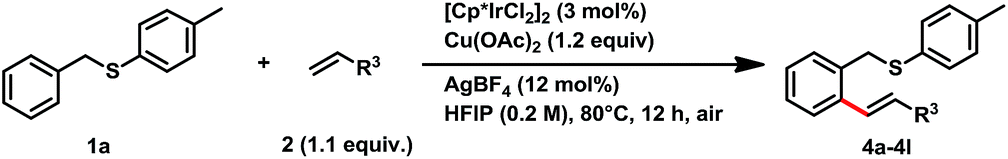
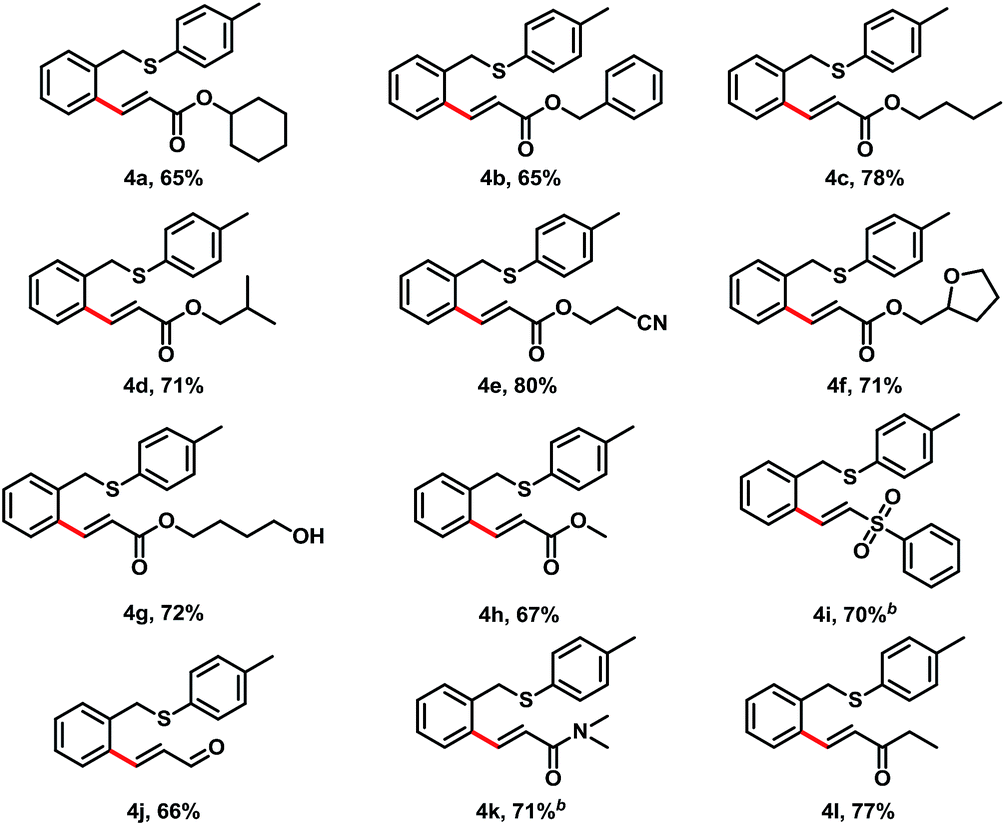
Next, we investigated the alkene scope for this transformation with benzyl p-tolyl sulfane (1a) (Table 3). Acrylates were good coupling partners for this reaction, various of alkoxyl groups on acrylate provided C–H alkenylation products in good yields, which included cyclohexyl (4a, 65% yield), benzyl (4b, 65% yield), n-butyl (4c, 78% yield), iso-butyl (4d, 71% yield), 2-cyanoethyl (4e, 80% yield), 2-furanylmethyl (4f, 71% yield), and 4-hydroxylbutyl (4g, 72% yield). Other electron-deficient alkenes also afforded the desired products in moderate to good yields, such as phenyl vinyl sulfone (4i, 70% yield), acrolein (4j, 66% yield), N,N-dimethylacrylamide (4k, 71% yield), and ethyl vinyl ketone (4l, 77% yield).
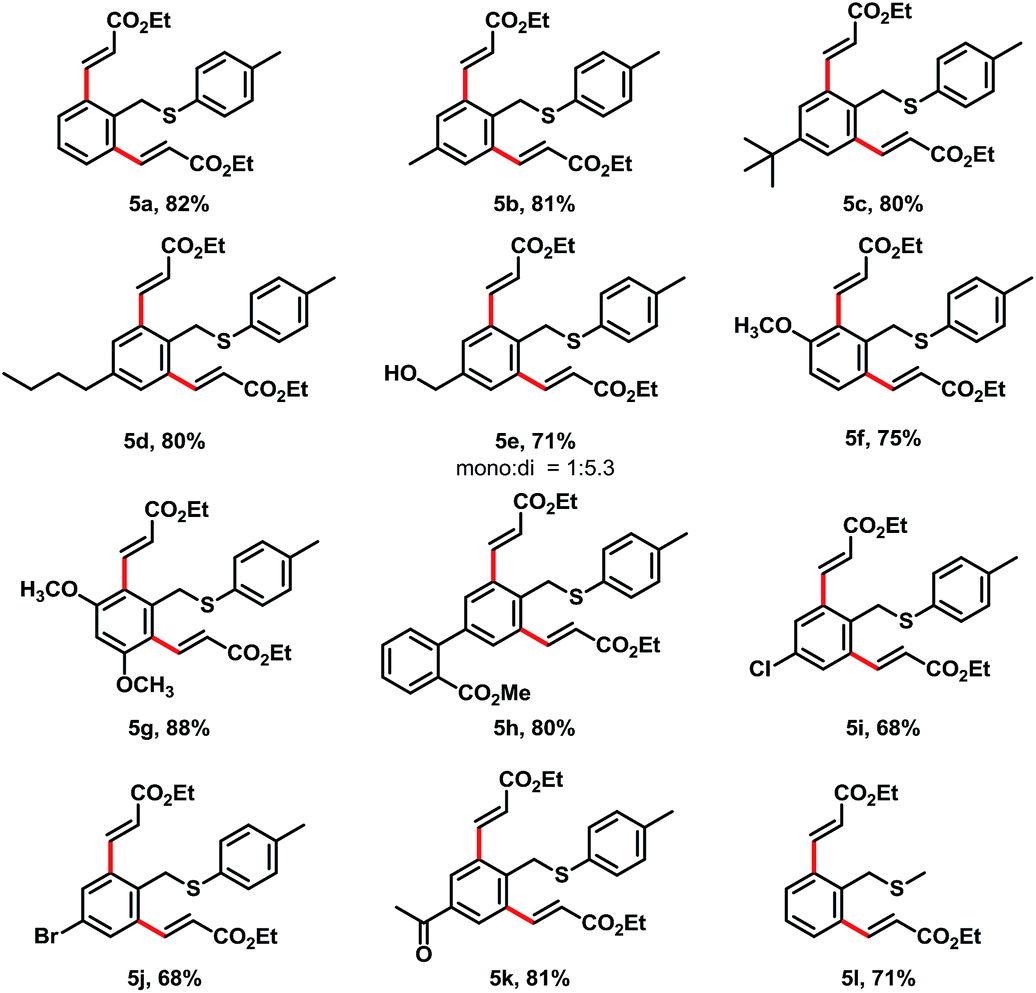
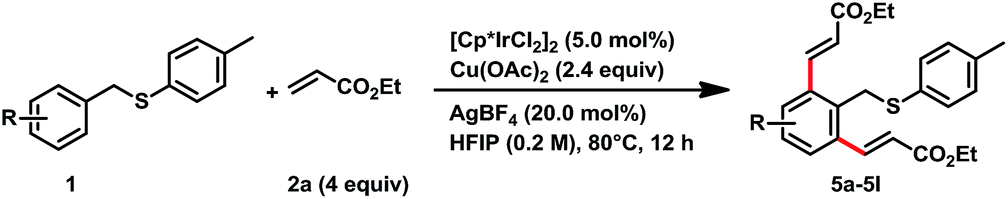
Though a high degree of mono-alkenylation products achieved from these substrates, we occasionally observed some dialkenylation products from reaction mixtures. For example, we have isolated 5a from the reaction of 1a and 2a in 12% yield (entry 6, Table 1). In order to get a high yield of dialkenylation product 5a, we optimized the reaction conditions. To our delight, when increasing Cu(OAc)2 to 2.4 equiv. and acrylate to 4 equiv., the dialkenylated product 5a was afforded in 82% yield as a single product. With these conditions, we examined the substrate scope for the dialkenylations (Table 4). Various electron-rich arenes preceded dialkenylation smoothly and provided the desired products in good to high yields. para-Methyl, tert-butyl, and n-butyl substituent arenes delivered the desired products in 81%, 80%, and 80% yields (5b–5d), respectively. para-Benzyl alcohol was well tolerated and provided the desired product 5e in 71% yield. meta-Methoxy and dimethoxy substituents were good for the transformation and provided the diolefinated products in 75% and 88% yields (5f and 5g). The electron-deficient substrates also provided the dialkenylation products in good yields. For example, ester substituents on the diphenyl substrate offered the desired product 5h in 80% yield. Chloro-, bromo- and acetal substituents at the para-position of arenes delivered the desired products in 68–81% yields (5i–5k). It was interesting that the methyl benzyl sulfane provided the desired dialkenylated product 5l in 71% yield under these conditions.
|
|
|---|
| a Reaction conditions: 1a (0.4 mmol), 2a (4.0 equiv.), [Cp*IrCl2]2 (5 mol%), Cu(OAc)2 (2.4 equiv.), AgBF4 (20 mol%), HFIP (2 mL), 80 °C, 12 h. |
|
A plausible mechanism was proposed for this Ir-catalyzed ortho-alkenylation (Fig. 2):11 [Cp*IrCl2]2 reacted with AgBF4 to provide the catalytic active Ir(III) species A. The coordination of thioether 1 with A and followed C–H activation provided iridacycle B. The coordination of olefin 2 with iridacycle B and followed insertion into Ir–C bond provided the seven-membered iridacycle C. Subsequent β-H elimination delivered the desired product. The regeneration of Ir(III) catalyst with the Cu(OAc)2 finished the catalytic cycle.
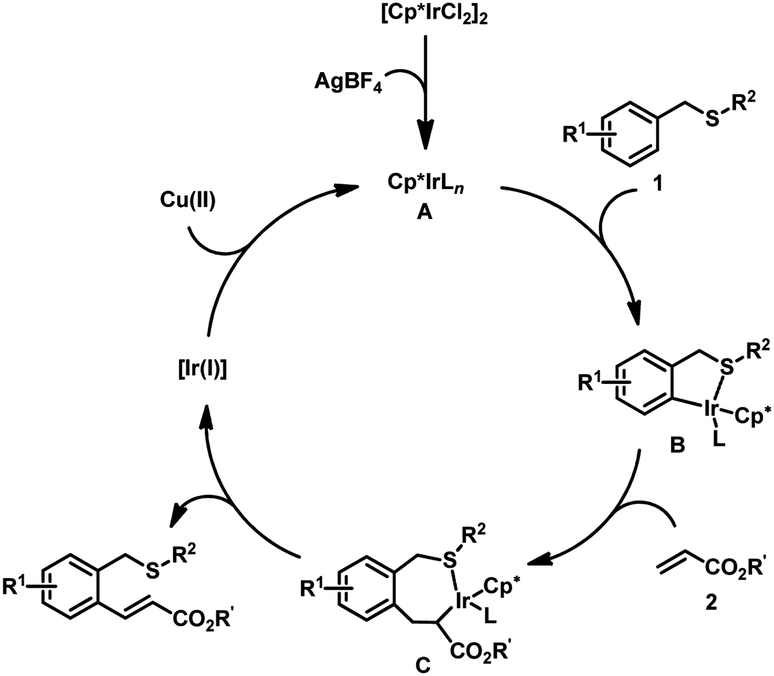 | ||
| Fig. 2 Plausible mechanism. | ||
In conclusion, we have developed an Ir-catalyzed ortho-alkenylation of arene C–H bonds using thioether as the directing group. This protocol undergo under mild and operationally simple conditions. The reaction exhibits broad functional group compatibility, and provides mono- or di-alkenylation products in good to high yields.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge support from the National Key R&D Program of China (2016YFA0202900), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB20000000), and the CNSF 21871286. X. C. thanks the Foundation from Guangdong Education Department (No. 2016KCXTD005, and 2017KSYS010).Notes and references
- For reviews, see: (a) C. Jia, T. Kitamura and Y. Fujiwara, Acc. Chem. Res., 2001, 34, 633 CrossRef CAS; (b) I. V. Seregin and V. Gevorgyan, Chem. Soc. Rev., 2007, 36, 1173 RSC; (c) J. Wencel-Delord, T. Droge, F. Liu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740 RSC; (d) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788 CrossRef CAS.
- (a) L. N. Lewis and J. F. Smith, J. Am. Chem. Soc., 1986, 108, 2728 CrossRef CAS; (b) S. Murai, F. Kakiuchi, S. Sekine, Y. Tanaka, A. Kamatani, M. Sonoda and N. Chatani, Nature, 1993, 366, 529 CrossRef CAS.
- For books, see: (a) J.-Q. Yu, L. Ackermann and Z. Shi, C–H Activation, Springer, Heidelberg, New York, 2010 CrossRef; (b) M. H. Emmert and C. J. Legacy, Arene Chemistry: Reaction Mechanisms and Methods for Aromatic Compounds, ed. J. Mortier, John Wiley & Sons, Inc, Hoboken, NJ, 2016 Search PubMed For reviews, see: (c) S. R. Neufeldt and M. S. Sanford, Acc. Chem. Res., 2012, 45, 936 CrossRef CAS; (d) G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726 CrossRef CAS; (e) R.-Y. Zhu, M. E. Farmer, Y.-Q. Chen and J.-Q. Yu, Angew. Chem., Int. Ed., 2016, 55, 10578 CrossRef CAS; (f) L. Ackermann, Acc. Chem. Res., 2014, 47, 281 CrossRef CAS; (g) O. Daugulis, J. Roane and L. D. Tran, Acc. Chem. Res., 2015, 48, 1053 CrossRef CAS PubMed; (h) C. He, W. G. Whitehurst and M. J. Gaunt, Chem, 2019, 5, 1031 CrossRef CAS.
- (a) N. Naowarojna, R. Cheng, L. Chen, M. Quill, M. Xu, C. Zhao and P. Liu, Biochemistry, 2018, 57, 3309 CrossRef CAS; (b) J. Zhao and X. Jiang, Chin. Chem. Lett., 2018, 29, 1079 CrossRef CAS; (c) B. Ashwood, M. Pollum and C. E. Crespo-Hernández, Photochem. Photobiol., 2019, 95, 33 CrossRef CAS PubMed; (d) S. Fujii, T. Sawa, H. Motohashi and T. Akaike, Br. J. Pharmacol., 2019, 176, 607 CrossRef CAS; (e) P. Putnik, D. Gabrić, S. Roohinejad, F. J. Barba, D. Granato, K. Mallikarjunan, J. M. Lorenzo and D. Bursać Kovačević, Food Chem., 2019, 276, 680 CrossRef CAS; (f) C. Zhao, K. P. Rakesh, L. Ravidar, W.-Y. Fang and H.-L. Qin, Eur. J. Med. Chem., 2019, 162, 679 CrossRef CAS.
- (a) J. Yao, M. Yu and Y. Zhang, Adv. Synth. Catal., 2012, 354, 3205 CrossRef CAS; (b) M. Yu, Y. Xie, C. Xie and Y. Zhang, Org. Lett., 2012, 14, 2164 CrossRef CAS; (c) B. Wang, Y. Liu, C. Lin, Y. Xu, Z. Liu and Y. Zhang, Org. Lett., 2014, 16, 4574 CrossRef CAS; (d) B. Wang, C. Lin, Y. Liu, Z. Fan, Z. Liu and Y. Zhang, Org. Chem. Front., 2015, 2, 973 RSC.
- (a) X.-S. Zhang, Q.-L. Zhu, Y.-F. Zhang, Y.-B. Li and Z.-J. Shi, Chem.–Eur. J., 2013, 19, 11898 CrossRef CAS; (b) X.-S. Zhang, Y.-F. Zhang, Z.-W. Li, F.-X. Luo and Z.-J. Shi, Angew. Chem., Int. Ed., 2015, 54, 5478 CrossRef CAS; (c) X.-S. Zhang, Y.-F. Zhang, K. Chen and Z.-J. Shi, Org. Chem. Front., 2014, 1, 1096 RSC.
- (a) Y. Unoh, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2015, 17, 704 CrossRef CAS; (b) Y. Unoh, Y. Yokoyama, T. Satoh, K. Hirano and M. Miura, Org. Lett., 2016, 18, 5436 CrossRef CAS; (c) C. N. Kona, Y. Nishii and M. Miura, Org. Lett., 2018, 20, 4898 CrossRef CAS; (d) M. Shigeno, Y. Nishii, T. Satoh and M. Miura, Asian J. Org. Chem., 2018, 7, 1334 CrossRef CAS; (e) S. Moon, Y. Nishii and M. Miura, Org. Lett., 2019, 21, 233 CrossRef CAS PubMed; (f) C. N. Kona, Y. Nishii and M. Miura, Angew. Chem., Int. Ed., 2019, 58, 9856 CrossRef CAS; (g) Y. Yokoyama, Y. Unoh, R. A. Bohmann, T. Satoh, K. Hirano, C. Bolm and M. Miura, Chem. Lett., 2015, 44, 1104 CrossRef CAS.
- (a) J. E. Spangler, Y. Kobayashi, P. Verma, D.-H. Wang and J.-Q. Yu, J. Am. Chem. Soc., 2015, 137, 11876 CrossRef CAS PubMed; (b) P. Jain, P. Verma, G. Xia and J.-Q. Yu, Nat. Chem., 2017, 9, 140 CrossRef CAS; (c) A. T. Tran and J.-Q. Yu, Angew. Chem., Int. Ed., 2017, 56, 10530 CrossRef CAS PubMed; (d) Z.-J. Cai, C.-X. Liu, Q. Gu and S.-L. You, Angew. Chem., Int. Ed., 2018, 57, 1296 CrossRef CAS.
- (a) R. Samanta and A. P. Antonchick, Angew. Chem., Int. Ed., 2011, 50, 5217 CrossRef CAS; (b) T. Wesch, F. R. Leroux and F. Colobert, Adv. Synth. Catal., 2013, 355, 2139 CrossRef CAS; (c) C. K. Hazra, Q. Dherbassy, J. Wencel-Delord and F. Colobert, Angew. Chem., Int. Ed., 2014, 53, 13871 CrossRef CAS PubMed; (d) B. Wang, C. Shen, J. Yao, H. Yin and Y. Zhang, Org. Lett., 2014, 16, 46 CrossRef CAS PubMed; (e) Q. Dherbassy, J.-P. Djukic, J. Wencel-Delord and F. Colobert, Angew. Chem., Int. Ed., 2018, 57, 4668 CrossRef CAS PubMed.
- (a) B. Xu, W. Liu and C. Kuang, Eur. J. Org. Chem., 2014, 2576 CrossRef CAS; (b) P. Villuendas and E. P. Urriolabeitia, Org. Lett., 2015, 17, 3178 CrossRef CAS PubMed; (c) M. Tobisu, Y. Masuya, K. Baba and N. Chatani, Chem. Sci., 2016, 7, 2587 RSC; (d) C. W. Cavanagh, M. H. Aukland, Q. Laurent, A. Hennessy and D. J. Procter, Org. Biomol. Chem., 2016, 14, 5286 RSC; (e) Y.-C. Zhu, Y. Li, B.-C. Zhang, F.-X. Zhang, Y.-N. Yang and X.-S. Wang, Angew. Chem., Int. Ed., 2018, 57, 5129 CrossRef CAS; (f) L. Jin, J. Wang and G. Dong, Angew. Chem., Int. Ed., 2018, 57, 12352 CrossRef CAS; (g) K.-X. Tang, C.-M. Wang, T.-H. Gao, L. Chen, L. Fan and L.-P. Sun, Adv. Synth. Catal., 2019, 361, 26 CrossRef CAS; (h) H. Shi and D. J. Dixon, Chem. Sci., 2019, 10, 3733 RSC; (i) Y.-H. Liu, P.-X. Li, Q.-J. Yao, Z.-Z. Zhang, D.-Y. Huang, M. D. Le, H. Song, L. Liu and B.-F. Shi, Org. Lett., 2019, 21, 1895 CrossRef CAS; (j) A. M. Romine, K. S. Yang, M. K. Karunananda, J. S. Chen and K. M. Engle, ACS Catal., 2019, 9 DOI:10.1021/acscatal.9b01471.
- J. Kim and S. Chang, Angew. Chem., Int. Ed., 2014, 53, 2203 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and compound characterization data. See DOI: 10.1039/c9ra06811b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |