
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCobalt sulfides/carbon nanohybrids: a novel biocatalyst for nonenzymatic glucose biofuel cells and biosensors†
Zihan Lia,
Gangyong Lia,
Zhongdong Wub,
Shuqiang Jiao a and
Zongqian Hu*c
a and
Zongqian Hu*c
aState Key Laboratory of Advanced Metallurgy, University of Science and Technology Beijing, Beijing, 100083, P. R. China
bCollege of Materials Science and Engineering, Central South University of Forestry and Technology, Changsha, 410004, P. R. China
cBeijing Institute of Radiation Medicine, Beijing, 100850, P. R. China. E-mail: huzongqian@hotmail.com; Tel: +86-10-66932251
First published on 15th October 2019
Abstract
Exploring high-performance electrocatalysts is of great importance in developing nonenzymatic biofuel cells. Hybrid nanostructures with transition metal compounds and carbon nanomaterials exhibit excellent electrocatalytic activity and have emerged as promising low-cost alternatives for various electrochemical reactions. Herein, we report cobalt sulfide/carbon nanohybrids via a facile synthesis, which have excellent electrocatalytic activity for glucose oxidation and oxygen reduction reaction. The nonenzymatic glucose biofuel cells equipped with cobalt sulfide/carbon nanohybrids deliver a high open circuit voltage of 0.72 V with a maximum open power density of 88 μW cm−2, indicating that cobalt sulfide/carbon nanohybrids are high performance biocatalysts for bioenergy conversion.
1. Introduction
Enzymatic glucose biofuel cells are a special type of fuel cell that convert energetic biomass into electric energy through bioelectrochemical pathways using a bio-enzyme as a catalyst.1–5 During the anode reaction, glucose oxidase or glucose dehydrogenase is usually used for the oxidation of glucose,6 while laccase or bilirubin oxidase is adopted as the cathodic catalyst for the oxygen reduction reaction (ORR).4,7 In the past decade, a few glucose biofuel cells have been successfully implanted in organisms such as oranges,8 clams,9 snails10 and rats.11,12 However, the practical applications of glucose biofuel cells are limited due to the poor stability,13 rough immobilization technology14 and susceptibility to the operating environment of the enzymes.15To overcome the drawbacks of the enzyme, some metal-based nanomaterials (e.g. precious metals,13,16 metal complexes17) have been used as the catalysts to replace the bio-enzyme to fabricate nonenzymatic glucose biofuel cells (NGBCs). However, most of the NGBCs use precious metals as cathodic or anodic catalysts.13,16,18 Although the high cost and scarcity of these precious metals severely limit their applications.19,20 In this regard, developing non-precious metal anodes and cathodes in NGBCs is highly challenging but imperative for practical applications. Currently, transition metals and their composites have received wide attention, especially cobalt, due to its good biocompatibility, low cost and higher natural abundance compared with precious metals.21–23 Cobalt has been widely used as catalysts because of its loosely bound d electrons.24 While cobalt sulfides have been investigated as overall water splitting25 and carbon dioxide (CO2) reduction24 catalysts due to their excellent electrochemical activity among different transition metal chalcogenides.26–30 However, cobalt sulfides have still been seldomly reported as the dual electrode electrocatalysts of NGBCs. In addition, cobalt sulfides have low charge transport because of their poor electronic conductivity.25 Thus, integrating cobalt sulfides with conducting carbon nanomaterials is an important strategy to increase their electroactive area and enhance the electronic conductivity.31–33
In this work, we report a novel NGBC based on cobalt sulfides as dual electrode electrocatalysts. In the anode section, we designed a non-precious metal anode catalyst (Co3S4 and graphene hybrid, Co3S4–G) via a simple controlled method. The Co3S4–G sample showed an excellent glucose oxidation activity, high sensitivity (687.13 μA cm−2 mM−1), low detection limit (0.4 μM), fast response and good selectivity toward the detection of glucose. Alternatively, we used cobalt sulfide and a carbon nanotube hybrid (Co1−xS–CNT) as the cathode catalyst. The Co1−xS–CNT performed at an onset potential of 0.01 V (vs. Hg/HgO), which was close to that of commercial Pt/C (0.025 V) and a positive shift (20 mV) in the half wave potentials compared to commercial Pt/C. As a result, an enhanced NGBC was achieved on the basis of high glucose oxidation and oxygen reduction performance.
2. Experimental
2.1. Material synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3, then heated in a furnace filled with argon at 800 °C for 2 h (heating rate 5 °C min−1) to obtain the Co1−xS/Co9S8 and graphene hybrid (denoted as Co1−xS/Co9S8–G). Finally, the Co1−xS/Co9S8–G powder was annealed in a muffle furnace at 300 °C for 3 h (heating rate 2 °C min−1) to obtain Co3S4–G.:3, and heated in a furnace filled with argon at 800 °C for 2 h (heating rate 5 °C min−1) to obtain Co1−xS–CNT.
:3, then heated in a furnace filled with argon at 800 °C for 2 h (heating rate 5 °C min−1) to obtain the Co1−xS/Co9S8 and graphene hybrid (denoted as Co1−xS/Co9S8–G). Finally, the Co1−xS/Co9S8–G powder was annealed in a muffle furnace at 300 °C for 3 h (heating rate 2 °C min−1) to obtain Co3S4–G.:3, and heated in a furnace filled with argon at 800 °C for 2 h (heating rate 5 °C min−1) to obtain Co1−xS–CNT.2.2. Characterization
X-ray diffraction (XRD) measurements were performed by a PANalytical X'pert Pro X-ray diffractometer using Cu Kα radiation (λ = 1.5418 Å). Raman spectra were obtained using a WITecCRM200 instrument with a 532 nm laser. X-ray photoelectron spectroscopy (XPS) analysis was recorded on a Thermo Scientific Escalab 250 spectrometer by using an Al Kα X-ray source. The morphologies were characterized by a field emission scanning electron microscopy (FESEM, JSM-6701) and transmission electron microscopy (TEM, JEOL JEM-2100).2.3. Electrochemical tests
:1) was cast on the surface of the pretreated glass carbon electrode (GCE, 3 mm in diameter) and dried in air (denoted as Co3S4–G/GCE). All the electrochemical measurements were carried out on a CHI 660E electrochemical analyzer (Shanghai, China) using a three-electrode system configuration with a 0.1 M KOH solution as the electrolyte, in which the above mentioned modified glass carbon was used as the working electrode, Pt foil (surface area: 1 cm2) as the counter electrode and a Hg/HgO electrode as the reference electrode. Meanwhile, the solutions were deoxygenated with highly pure argon (99.99%) for at least 15 min before tests.:1 v/v water/isopropanol mixed solution and then ultrasonicated for an hour to get a homogeneous suspension. 4 μL of this suspension (0.1 mg cm−2) were dropped on a pre-treated glassy carbon rotating disk electrode (5 mm in diameter) and dried in air for use as the working electrode. The ORR activity was detected in O2-saturated 0.1 M KOH solution by using a rotating disk electrode (Pine Instruments). The linear sweep voltammograms (LSV) were recorded under a rotating speed of 1600 rpm at a scan rate of 5 mV s−1.For the rotating ring-disk electrode (RRDE) measurements, a rotating ring disk electrode with a glassy carbon disk and a Pt ring (Pine Instruments) was used as the working electrode. The disk electrode was scanned cathodically at a rate of 5 mV s−1 and the ring electrode's potential was constant at 1.5 V vs. RHE. The electron transfer number and % HO2− were determined by the equations:
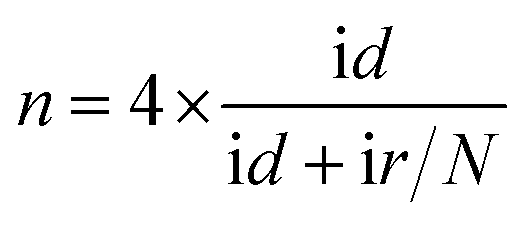 | (1) |
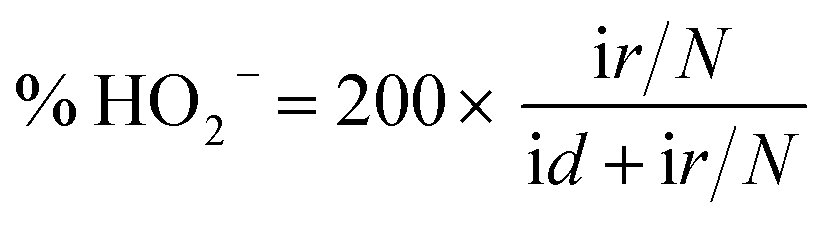 | (2) |
3. Results and discussion
The X-ray diffraction (XRD) patterns of the Co1−xS/Co9S8–G and Co3S4–G hybrid are shown in Fig. 1a. For the Co1−xS/Co9S8–G hybrid, different diffraction peaks at 2θ = 30.5°, 35.1°, 46.7°, 54.2° corresponding to the (100), (101), (102), (110) plane reflections of Co1−xS (PDF#42-0826) and different diffraction peaks at 2θ = 29.9°, 47.7°, 52.2°, corresponding to the (311), (511), (440) plane reflection of Co9S8 (PDF#75-2023) were present. The Co1−xS/Co9S8–G, Co3S4–G hybrid showed different diffraction peaks at 2θ = 26.8°, 31.5°, 38.2°, 50.4°, 55.2°, corresponding to the (220), (311), (400), (511), (440) plane reflections of Co3S4 (PDF#74-0138), respectively. However, from the XRD patterns, it was observed that some impurity exists, indicating that there is possibly a small quantity of Co3O4 doped in the Co3S4–G hybrid.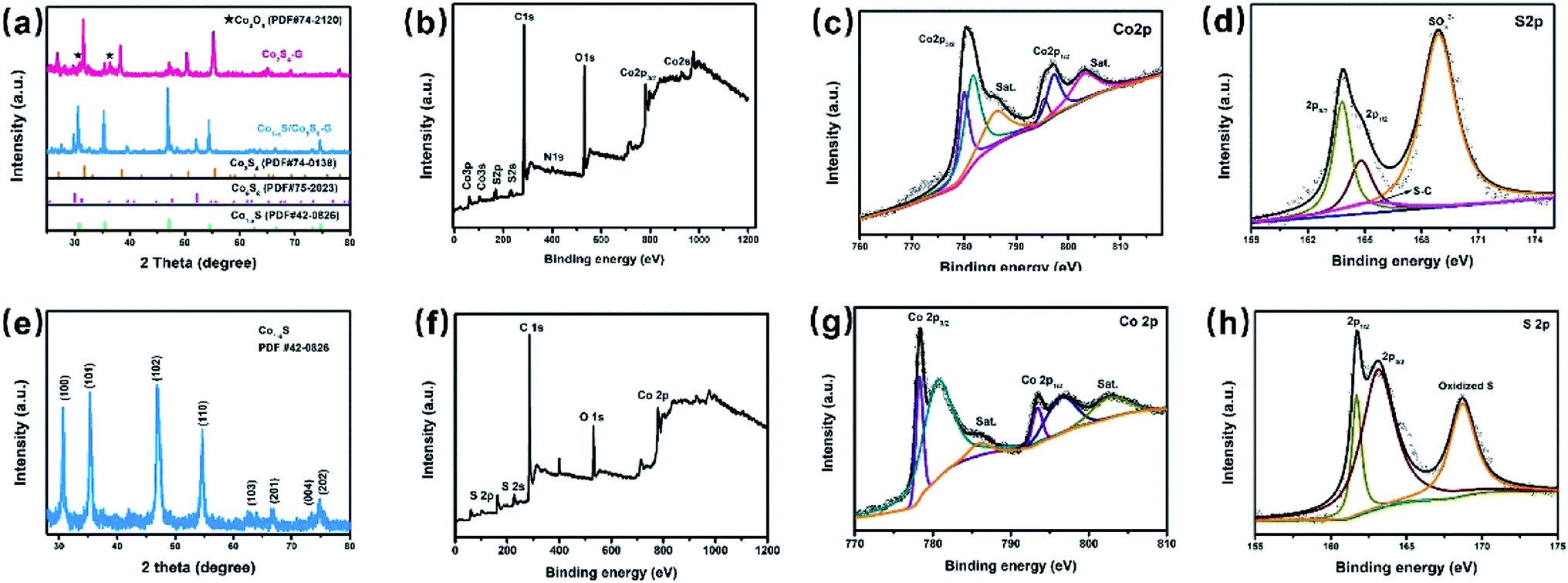 | ||
| Fig. 1 (a) XRD patterns of Co1−xS/Co9S8–G and Co3S4–G; (b) XPS spectrum of Co3S4–G; (c) Co 2p, (d) S 2p, XPS spectra of Co3S4–G; (e) XRD patterns of Co1−xS–CNT; (f) XPS spectrum of Co1−xS–CNT; (g) Co 2p (h) S 2p, XPS spectra of Co1−xS–CNT. | ||
In order to confirm the chemical composition and valence of Co3S4–G, the electronic states of the Co and S phases were identified by X-ray photoelectron spectroscopy (XPS) measurements. Fig. 1b shows the XPS spectrum of Co3S4–G. The Co 2p core-level spectra showed two predominant peaks (Co 2p3/2 and Co 2p1/2) with an energy separation of about 15.4 eV, as shown in Fig. 1c, indicating the presence of Co3S4.34 The high resolution of the S 2p spectrum (Fig. 1d) displayed peaks located at 163.7, 164.8 and 165.4 eV, which correspond to the S 2p3/2, S 2p1/2 and S–C respectively. The peak at 168.8 eV identifies the presence of S–O binding, suggesting a partial oxidation of the Co3S4 shell on the surface.35 Fig. S1a† shows that the O 1s spectrum was comprised of three components: oxygen atoms in the hydroxyl group, absorbed water, and O–Co bonds.36 The N 1s and C 1s spectrum of Co3S4–G are displayed in Fig. S1.†
Morphological and structural characterizations of the Co3S4–G hybrid and its precursors were investigated by SEM and TEM. The SEM (Fig. 2a and b) and TEM (Fig. 2c) images clearly show the formation of Co3S4 particles with an average size of ∼40 nm on graphene sheets. The graphene sheets can not only induce higher porosity to facilitate ion diffusion but also inhibit the aggregation of the composites. As shown in Fig. 2d, the ordered lattice interplanar spacings were around 0.176 nm from a high-resolution TEM image of the (311) plane of Co3S4.
 | ||
| Fig. 2 (a) (b) Different resolution SEM images of Co3S4–G; (c) TEM image of Co3S4–G; (d) HRTEM image of Co3S4–G; (e) (f) different resolution SEM images of Co1−xS–CNT; (g) TEM image of Co1−xS–CNT; (h) HRTEM image of Co1−xS–CNT. | ||
The phase composition and morphology of the Co1−xS–CNT hybrid were characterized by the same method. XRD analyses are shown in Fig. 1e, and the sharp diffraction peaks from hexagonal Co1−xS (PDF#42-0826) appear obviously, with different peaks at 2θ = 30.5°, 35.2°, 46.7°, 54.2° corresponding to the (100), (101), (102), (110) plane reflections of Co1−xS, respectively. XPS was used to further analyze the chemical composition and valence of the Co1−xS–CNT hybrid. As shown in Fig. 1g, the Co 2p1/2 and Co 2p3/2 spin-orbit coupling accompanied with weak satellite peaks can be deconvoluted from the Co 2p spectrum. The peaks located at 778.3 eV and 793.4 eV, 780.6 eV and 796.4 eV indicate the presence of Co3+ and Co2+, respectively.37 The S 2p spectrum was deconvoluted into S 2p1/2 and S 2p3/2 of S2− in Co1−xS at 161.7 eV and 163 eV as shown in Fig. 1h, and another peak at 168.6 eV corresponded to the oxidized S, possibly caused by oxidation in the air.38 The C 1s spectrum is shown in Fig. S2.†
Morphological and structural characterizations of the Co1−xS–CNT hybrid was investigated by SEM and TEM. As shown in Fig. 2e and f, irregular polygon Co1−xS particles were anchored on the surface of CNTs. The CNTs provide highly conductive pathways for electron transfer between the electrode material and the current collector to improve the overall electrical conductivity.39 The TEM images shown in Fig. 2g confirm the close relationship between the Co1−xS particles and CNTs, which is consistent with SEM characterization. The high-resolution TEM image is shown in Fig. 2h, where the crystal lattice fringe with an inter-planar distance of 0.196 nm matches well with the dominant XRD peak (101) of Co1−xS. All the characterizations above indicate that the Co3S4–G and Co1−xS–CNT hybrids were successfully synthesized.
To investigate the electrochemical catalytic activity of these cobalt sulfides based materials, the GOR and ORR performances were studied by a series of tests (described in the Experimental section). Fig. 3a shows the CVs of Co3S4–G modified GCE (Co3S4–G/GCE) and bare GCE in 0.1 M KOH in the presence of 5 mM glucose at a scan rate of 50 mV s−1. The Co1−xS–G displays two pairs of redox peaks, anodic peaks at around 0.62 V and 0.28 V, and cathodic peaks at around 0.55 V and 0.24 V. The pair of redox peaks I/II correspond to the reversible transition between Co3+ and Co4+, while peaks III/IV can be assigned to the conversion between Co2+ and Co3+.40 Notably, an obvious increase of the anodic peak current was observed after the addition of glucose, especially at peak I. The electrooxidation process was mainly carried out by Co3+ and Co4+. Co2+ was initially oxidized to Co3+ and further to Co4+. Then, the electrooxidation of glucose occured at the cost of Co4+ consumption, leading to an enhanced anodic current at peak I and a decrease in the cathodic peak current. The oxidation mechanism of Co3S4 as an active substance can be shown by the following reactions:41
| Co3S4 + OH− ↔ Co3S4OH + e− | (3) |
| Co3S4OH + OH− ↔ Co3S4O + H2O + e− | (4) |
| Co4+ + glucose → Co3+ + gluconolactone | (5) |
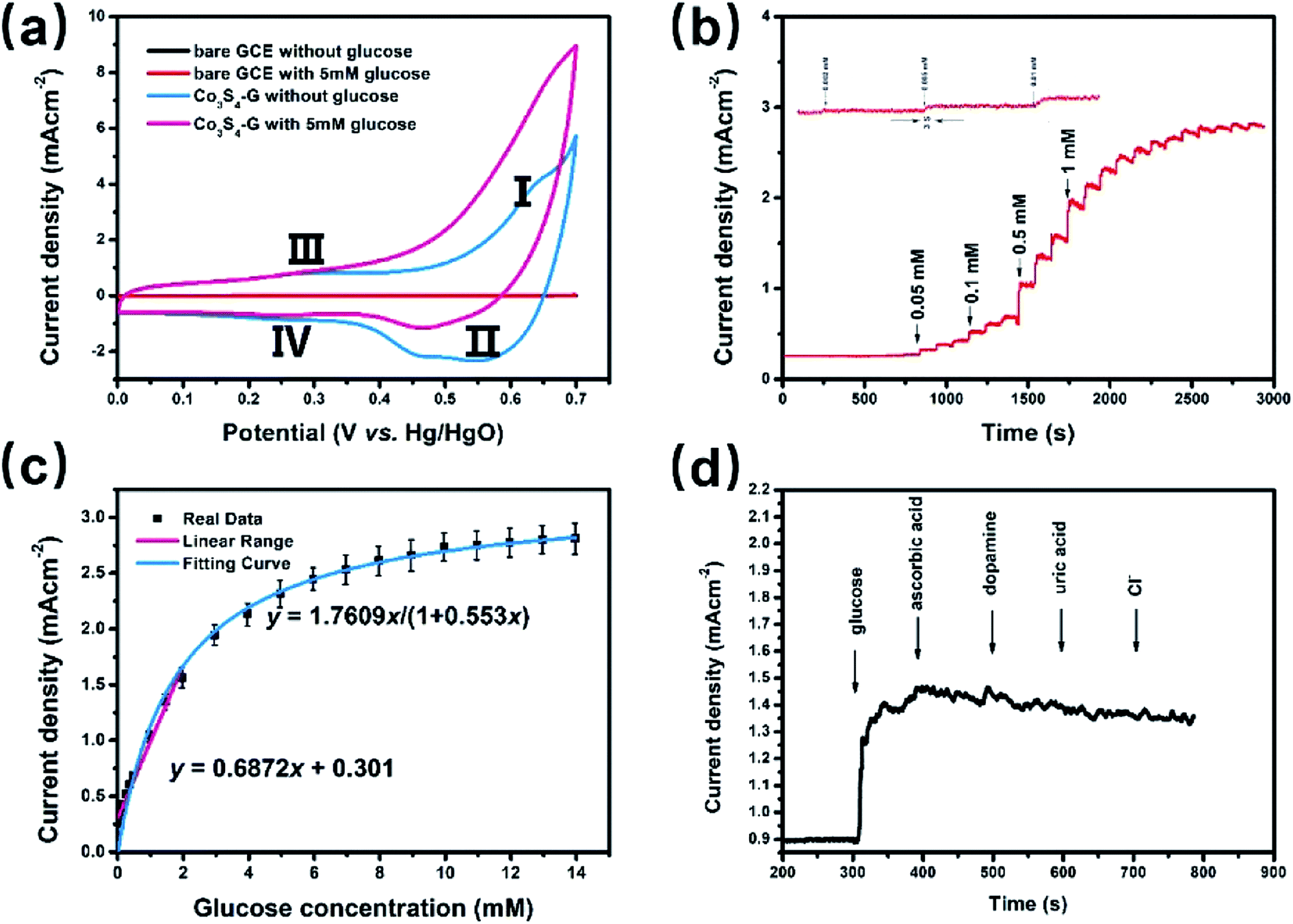 | ||
| Fig. 3 Co3S4–G hybrid as glucose oxidation catalysts. (a) CV curves of bare GCE and Co3S4–G in 0.1 M KOH with 5 mM glucose or without glucose; (b) amperometric current response of a Co3S4–G electrode in 0.1 M KOH with the continuous addition of glucose at 0.62 V; (c) calibration curve versus glucose concentration; (d) amperometric current response of a Co3S4–G electrode in 0.1 M KOH with successive addition of glucose, ascorbic acid, dopamine, uric acid and Cl−. | ||
The Co1−xS–G performance can clearly seen by the a evident catalytic current peak about 4.12 mA cm−2 in an intensity at 0.62 V (vs. Hg/HgO) in the presence of glucose, compared with that ub the absence of glucose. In contrast, bare GCE exhibits a fairly weak response toward the oxidation of glucose. As shown in Fig. 3a, the current increase with the addition of glucose at peak I (Co3+ → Co4+) was much stronger than that at peak III (Co2+ → Co3+), which may suggest that the electrooxidation of glucose is mainly mediated by Co3+/Co4+, rather than Co2+/Co3+ in an alkaline solution. Therefore, the peak I potential (+0.62 V vs. Hg/HgO) was applied for the following amperometric detection. Fig. 3b shows the amperometric current response of Co3S4–G in 0.1 M KOH with the continuous addition of glucose at 0.62 V (vs. Hg/HgO). It had a fast amperometric response toward glucose and achieved steady state current density within 3s. However, the baseline exhibited slight drift after multiple additions of glucose, which can be attribute to faster consumption of glucose than the diffusion or the adsorption of intermediates on the active sites.42 The fitting curve of this glucose sensor is shown in Fig. 3c. Langmuir isothermal theory was used to fit the curve, because the electrochemical oxidation of glucose on Co3S4–G is a surface catalytic reaction.40 From the Langmuir isothermal theory, the concentration of glucose adsorbed on the catalyst surface (Cglucose S) can be expressed as:
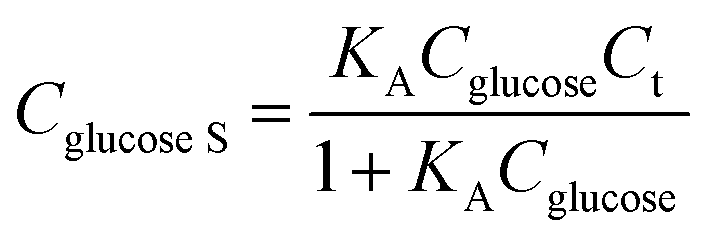 | (6) |
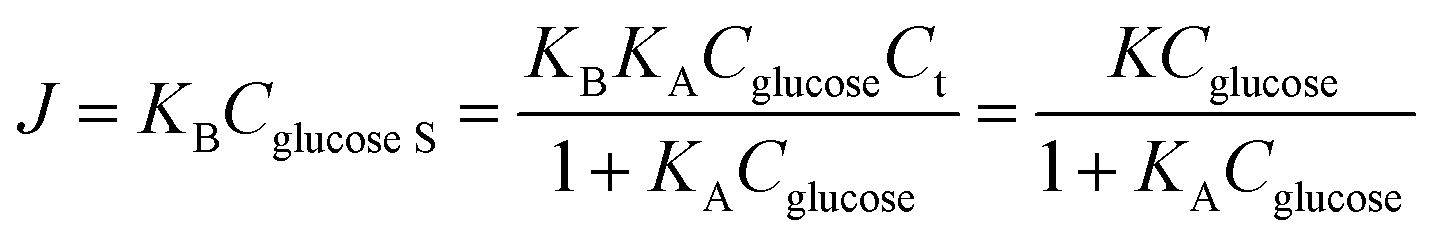 | (7) |
As shown in Fig. 3c, the K = 1.7609 and KA = 0.553 of this equation can be calculated with a sufficient fitness (R = 0.985). Thus J can be expressed as follows:
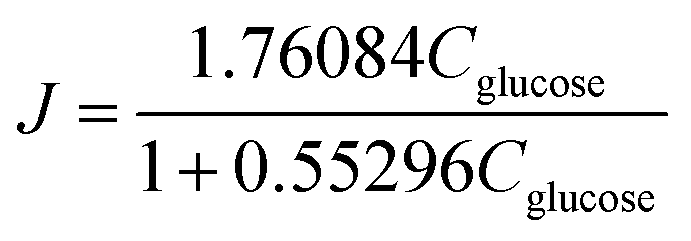 | (8) |
At lower glucose concentrations, it exhibited a linear range from 0 to 2 mM, a sensitivity of 687.2 μA cm−2 mM−1 and a detection limit of 0.4 μM (S/N = 3) was achieved. These glucose oxidation results are superior to most of the reported electrochemical glucose sensors. The stability of Co3S4–G electrode was investigated by measuring its amperometric response for a long operational period. Fig. S5a† displays the amperometric response to 0.1 mM glucose within a 7 day period. After 7 days, the final amperometric response was approximately 97% of its original counterpart. As shown in Fig. S5b,† there is nearly no loss in the current signal over a period of 3500 s for 0.1 mM glucose in 0.1 M KOH at +0.62 V, suggesting excellent stability of the Co3S4–G electrode. Selectivity performance of Co3S4–G is shown in Fig. 3d. With the successive of addition of glucose and other interferences in 0.1 M KOH, it can be seen that Co3S4–G has a remarkable response for glucose, superior to that of the interfering species. All the above results show that Co3S4–G is an excellent glucose electrochemical catalyst. According to the test above, the Co3S4–G hybrid can be considered as a high performance glucose catalyst.
The ORR activities of Co1−xS–CNT were evaluated by rotating disk electrode (RDE) measurements in 0.1 M KOH solution. Fig. 4a shows the linear scan sweep voltammetry (LSV) curves of Co1−xS–CNT, commercial Pt/C, Co1−xS and CNTs at 1600 rpm, Co1−xS–CNT exhibited an onset potential of 0.01 V (vs. Hg/HgO), which was close to that of the commercial Pt/C (0.025 V). Co1−xS–CNT showed a large positive shift in the half wave potentials of Co1−xS, CNTs and commercial Pt/C attribute to the synergistic effect of Co1−xS and CNT. The Co1−xS–CNT had a limiting current density of 5.15 mA cm−2 at −0.8 V. However, this activity is still lower than that of Pt/C (5.7 mA cm−2). To quantitatively measure the ORR catalytic pathway of the Co1−xS–CNT hybrid, the RRDE test was used to monitor the formation of peroxide species (HO2−) during the ORR process (Fig. 4b). Fig. 4c shows the measured HO2− yield was below 9% for Co1−xS–CNT and the corresponding electron transfer number (n) was above 3.85 over the potential range of −0.8 V to −0.2 V, suggesting that the Co1−xS–CNT catalyst exhibits a dominant four-electron oxygen reduction process. Furthermore, after 30000 s of long-term measurements of continuous ORR at −0.5 V in an O2-saturated 0.1 M KOH at 900 rpm, the current density remained at about 85% of the Co1−xS–CNT hybrid, whereas the attenuation for Pt/C was up to 31%. Considering the above analysis of the ORR activity test results, the Co1−xS–CNT hybrid had a high catalytic ORR activity and stability catalyst.
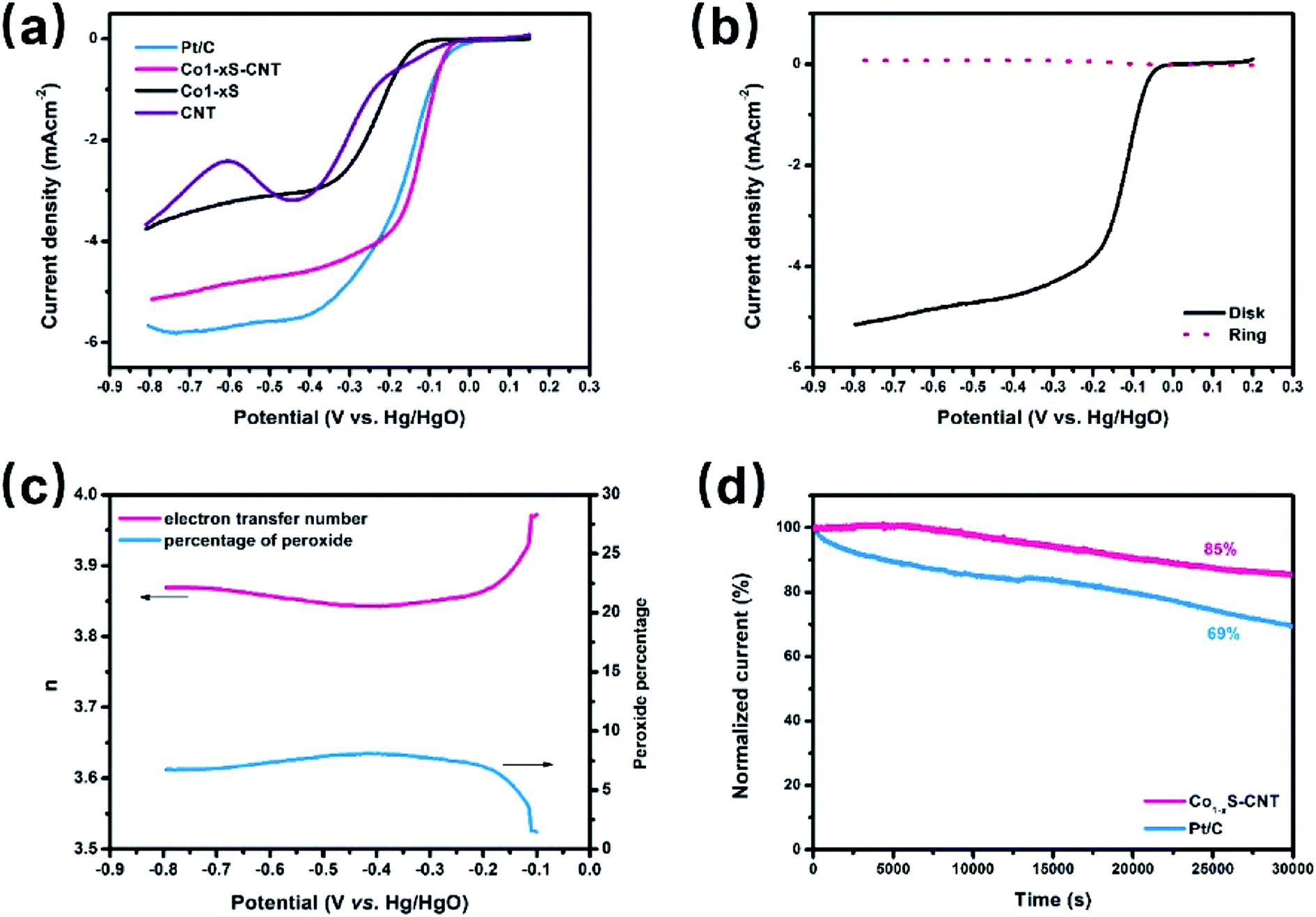 | ||
| Fig. 4 Co1−xS–CNT hybrid as oxygen reduction catalysts. (a) LSV curves of commercial Pt/C, Co1−xS–CNT, Co1−xS and CNTs in O2-saturated 0.1 M KOH at a sweep rate of 5 mV s−1; (b) RRDE experiment results of Co1−xS–CNT electrode for ORR in 0.1 M KOH at a scan rate of 5 mV s−1; (c) corresponding percentage of peroxide in the total oxygen reduction products and the electron transfer number (n) calculated by the RRDE result; (d) chronoamperometric stability tests of the commercial Pt/C and Co1−xS–CNT for 30000 s at −0.5 V (vs. Hg/HgO) in O2-saturated 0.1 M KOH at 900 rpm. | ||
The H-type glucose fuel cell was fabricated using the Co3S4–G hybrid as the anode and the Co1−xS–CNT as the cathode catalyst. Fig. 5 shows the schematic of the H-type glucose fuel cell. Glucose was oxidized by Co3S4–G at the anode, and the electrons and protons generated by the glucose oxidation reaction were transferred from the anode to the cathode through an external circuit and internal solution, respectively. At the cathode, O2 was reducted to H2O by the action of Co1−xS–CNT. As shown in Fig. 6a, the open circuit voltage (OCV) was 0.72 V, the maximum current density was ∼1700 μA cm−2, and the maximum power density was 88 μW cm−2. It had the same maximum power density, and a higher maximum current density (∼650 μA cm−2), compared with Pt/C as the cathode catalyst (Fig. S6a†). A bare electrode fabricated glucose fuel cell is shown in Fig. S6b,† which exhibited a poor maximum current density (∼4.2 μA cm−2) and maximum power density (0.5 μW cm−2), proving that the excellent performance of the battery can be attributed to our catalyst (Co3S4–G and Co1−xS–CNT). Meanwhile, the stability was a major factor to assess the performance of the fuel cell. We used electrodes that were placed for a long time after modification (in the air for one week) to fabricate the glucose fuel cell. As shown in Fig. 6b, it exhibited the same power density output (88 μW cm−2), proving that it was a stable catalyst. And long time discharge test was measured by land (CT2001A). As shown in Fig. 6c, the potential had a loss of 7% within the initial 36 h. Then loading an Rex of 100k Ω, the voltage droped to 0.5 V and then stabilized at ∼0.2 V for 24 h. Subsequently, the battery stood overnight at room temperature, and its voltage recovered to ∼0.7 V without external resistance. After 20 h, it held a voltage of ∼0.66 V. The generated voltage gradually decreased, which can be attributed to the glucose oxidizing to gluconolactone.15,43
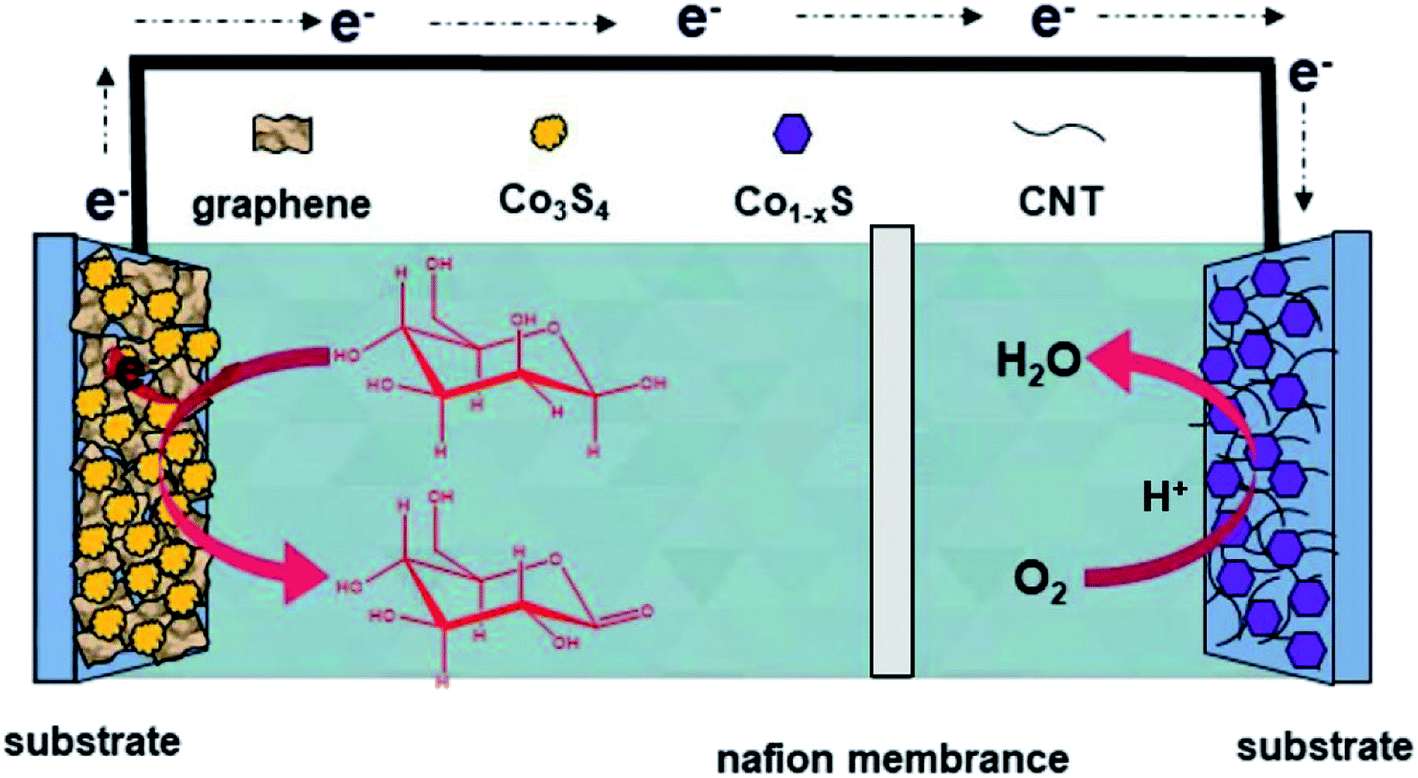 | ||
| Fig. 5 Schematic and working mechanism of the designed glucose fuel cell. | ||
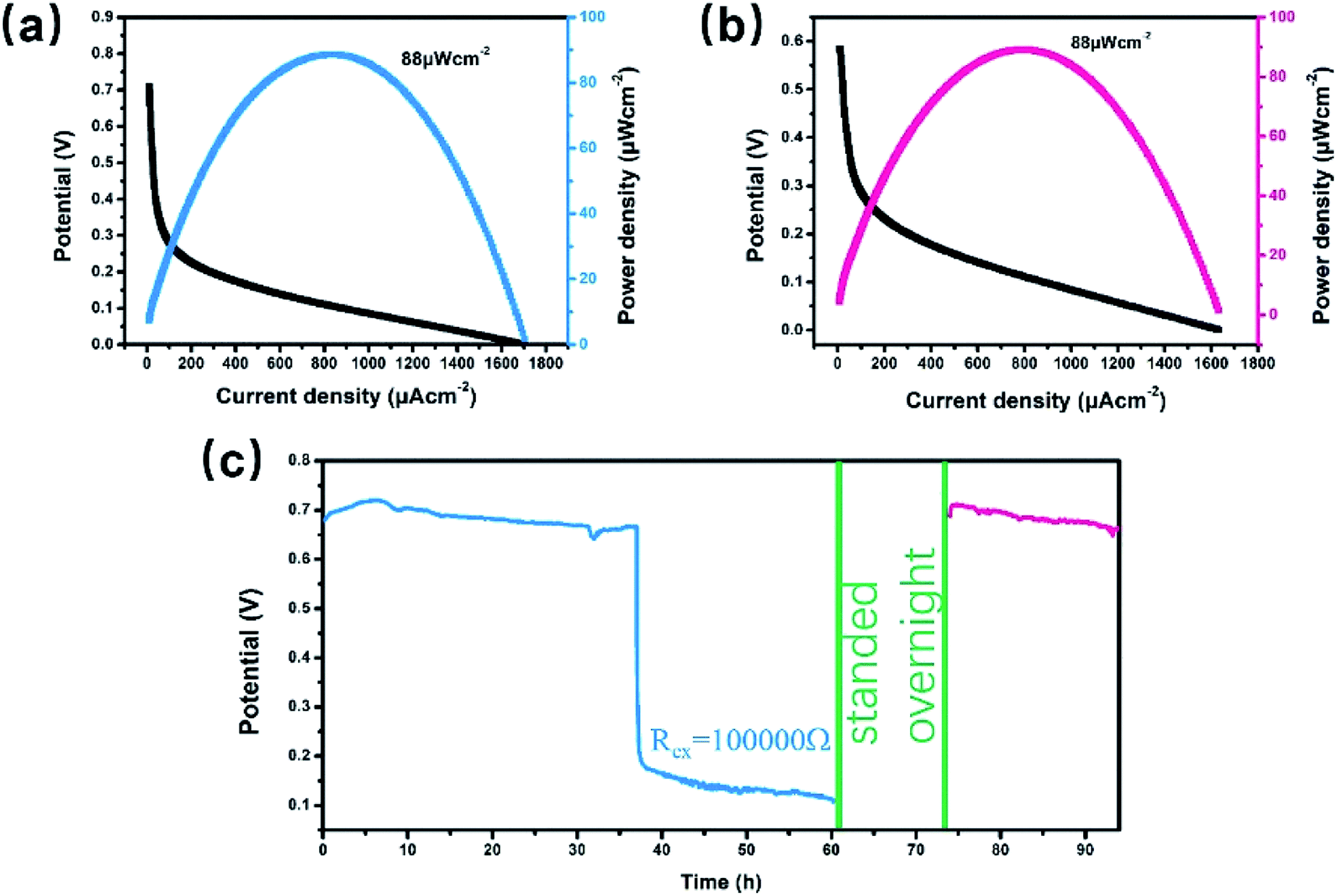 | ||
| Fig. 6 H-type glucose fuel cell tests. (a) Polarization and power density curves of the Co3S4–G/Co1−xS–CNT(a‖c)-GFC; (b) power density and current density measured with long-time placed Co3S4–G as the anode and long-time placed Co1−xS–CNT as the cathode; (c) output potential recorded for the Co3S4–G/Co1−xS–CNT(a‖c)-GFC. | ||
4. Conclusions
We successfully synthesized cobalt sulfides/carbon nanohybrids and used them in NGBCs. The Co3S4–G hybrid showed an excellent glucose electrooxidation activity, high sensitivity (687.2 μA cm−2 mM−1), low detection limit (0.4 μM), fast response, and good selectivity toward the detection of glucose, which can be used as an anodic catalyst for glucose biofuel cells. Meanwhile, the Co1−xS–CNT hybrid exhibited surprisingly high ORR activities, comparable to commercial Pt/C catalysts, and exceeded the stability of Pt/C. Therefore, Co1−xS–CNT can be used as a cathode catalyst for glucose biofuel cells. The as-designed glucose biofuel cell with Co3S4–G as the anode catalyst and Co1−xS–CNT as the cathode catalyst had advantages of being low cost, was free of enzymes, had a high open circuit voltage of 0.72 V, a maximum open power density of 88 μW cm−2, a maximum current density was ∼1700 μA cm−2 and long-term stability. The voltage even stabilized at ∼0.2 V for 24 h with loading an Rex of 100k Ω, and its voltage can recover to ∼0.7 V without external resistance. Our findings provide a new direction for the rational design or modification of dual electrode catalysts of NBGCs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work is supported by the National Natural Science Foundation of China (81301345, 81871506).References
- K. L. Jiao, Y. Jiang, Z. P. Kang, R. Y. Peng, S. Q. Jiao and Z. Q. Hu, R. Soc. Open Sci., 2017, 4, 10 CrossRef.
- Z. P. Kang, K. L. Jiao, J. Cheng, R. Y. Peng, S. Q. Jiao and Z. Q. Hu, Biosens. Bioelectron., 2018, 101, 60–65 CrossRef CAS.
- G. Y. Li, Z. H. Li, X. Xiao, Y. L. An, W. Wang and Z. Q. Hu, J. Mater. Chem. A, 2019, 7, 11077–11085 RSC.
- I. Willner, Y. M. Yan, B. Willner and R. Tel-Vered, Fuel Cells, 2009, 9, 7–24 CrossRef CAS.
- Y. X. Chen, W. H. Ji, K. Yan, J. Gao and J. D. Zhang, Nano Energy, 2019, 61, 173–193 CrossRef CAS.
- Z. P. Kang, Y. Zhang and Z. G. Zhu, Biosens. Bioelectron., 2019, 132, 76–83 CrossRef CAS PubMed.
- L. Y. Fu, J. J. Liu, Z. Q. Hu and M. Zhou, Electroanalysis, 2018, 30, 2535–2550 CrossRef CAS.
- K. MacVittie, T. Conlon and E. Katz, Bioelectrochemistry, 2015, 106, 28–33 CrossRef CAS.
- A. Szczupak, J. Halamek, L. Halamkova, V. Bocharova, L. Alfonta and E. Katz, Energy Environ. Sci., 2012, 5, 8891–8895 RSC.
- L. Halamkova, J. Halamek, V. Bocharova, A. Szczupak, L. Alfonta and E. Katz, J. Am. Chem. Soc., 2012, 134, 5040–5043 CrossRef CAS.
- J. A. Castorena-Gonzalez, C. Foote, K. MacVittie, J. Halamek, L. Halamkova, L. A. Martinez-Lemus and E. Katz, Electroanalysis, 2013, 25, 1579–1584 CrossRef CAS.
- A. Zebda, S. Cosnier, J. P. Alcaraz, M. Holzinger, A. Le Goff, C. Gondran, F. Boucher, F. Giroud, K. Gorgy, H. Lamraoui and P. Cinquin, Sci. Rep., 2013, 3, 5 Search PubMed.
- M. Chu, Y. J. Zhang, L. Yang, Y. M. Tan, W. F. Deng, M. Ma, X. L. Su, Q. J. Xie and S. Z. Yao, Energy Environ. Sci., 2013, 6, 3600–3604 RSC.
- Y. Zhao, L. Z. Fan, D. M. Gao, J. L. Ren and B. Hong, Electrochim. Acta, 2014, 145, 159–169 CrossRef CAS.
- S. Ci, Z. Wen, S. Mao, Y. Hou, S. Cui, Z. He and J. Chen, Chem. Commun., 2015, 51, 9354–9357 RSC.
- G. Slaughter and T. Kulkarni, Microelectron. Eng., 2016, 149, 92–96 CrossRef CAS.
- K. L. Jiao, Z. P. Kang, B. Wang, S. Q. Jiao, Y. Jiang and Z. Q. Hu, Electroanalysis, 2018, 30, 525–532 CrossRef CAS.
- S. Kerzenmacher, U. Kraling, T. Metz, R. Zengerle and F. von Stetten, J. Power Sources, 2011, 196, 1264–1272 CrossRef CAS.
- Y. Hou, Z. H. Wen, S. M. Cui, S. Q. Ci, S. Mao and J. H. Chen, Adv. Funct. Mater., 2015, 25, 872–882 CrossRef CAS.
- P. Y. Ren, S. Q. Ci, Y. C. Ding and Z. H. Wen, Appl. Surf. Sci., 2019, 481, 1206–1212 CrossRef CAS.
- D. H. Ge, H. B. Geng, J. Q. Wang, J. W. Zheng, Y. Pan, X. Q. Cao and H. W. Gu, Nanoscale, 2014, 6, 9689–9694 RSC.
- M. Li, C. Han, Y. F. Zhang, X. J. Bo and L. P. Guo, Anal. Chim. Acta, 2015, 861, 25–35 CrossRef CAS.
- H. M. Xu, S. Q. Ci, Y. C. Ding, G. X. Wang and Z. H. Wen, J. Mater. Chem. A, 2019, 7, 8006–8029 RSC.
- S. Gao, Y. Lin, X. C. Jiao, Y. F. Sun, Q. Q. Luo, W. H. Zhang, D. Q. Li, J. L. Yang and Y. Xie, Nature, 2016, 529, 68 CrossRef CAS.
- C. D. Bai, S. S. Wei, D. R. Deng, X. D. Lin, M. S. Zheng and Q. F. Dong, J. Mater. Chem. A, 2017, 5, 9533–9536 RSC.
- Y. H. Wang, B. H. Wu, X. Y. He, Y. Y. Zhang, H. Li, Y. Y. Peng, J. Wang and J. B. Zhao, Electrochim. Acta, 2017, 230, 299–307 CrossRef CAS.
- X. Qiao, J. Jin, H. Fan, Y. Li and S. Liao, J. Mater. Chem. A, 2017, 5, 12354–12360 RSC.
- H. L. Wang, Y. Y. Liang, Y. G. Li and H. J. Dai, Angew. Chem., Int. Ed., 2011, 50, 10969–10972 CrossRef CAS PubMed.
- X. R. Li, J. L. Wei, Q. Li, S. S. Zheng, Y. X. Xu, P. Du, C. Y. Chen, J. Y. Zhao, H. G. Xue, Q. Xu and H. Pang, Adv. Funct. Mater., 2018, 28, 7 Search PubMed.
- B. Li, Y. X. Shi, K. S. Huang, M. M. Zhao, J. Q. Qiu, H. G. Xue and H. Pang, Small, 2018, 14, 7 Search PubMed.
- J. L. Guan, Z. P. Zhang, J. Ji, M. L. Dou and F. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 30662–30669 CrossRef CAS.
- L. Tan, S. J. Li, X. T. Wu, N. Li and Z. Q. Liu, Int. J. Hydrogen Energy, 2018, 43, 10311–10321 CrossRef CAS.
- X. R. Li, S. Y. Ding, X. Xiao, J. Y. Shao, J. L. Wei, H. Pang and Y. Yu, J. Mater. Chem. A, 2017, 5, 12774–12781 RSC.
- F. Luo, D. T. Ma, Y. L. Li, H. W. Mi, P. X. Zhang and S. Luo, Electrochim. Acta, 2019, 299, 173–181 CrossRef CAS.
- X. Wang, C. Liu, Q. Li, H. Li, J. Xu, X. Chu, L. Zhang, G. Zhao, H. Li, P. Guo, S. Li and X. S. Zhao, ChemElectroChem, 2018, 5, 309–315 CrossRef CAS.
- B. Liu, D. Kong, J. Zhang, Y. Wang, T. Chen, C. Cheng and H. Y. Yang, J. Mater. Chem. A, 2016, 4, 3287–3296 RSC.
- D. He, X. Wu, W. Liu, C. Lei, C. Yu, G. Zheng, J. Pan, L. Lei and X. Zhang, Chin. Chem. Lett., 2019, 30, 229–233 CrossRef CAS.
- M. Chauhan, K. P. Reddy, C. S. Gopinath and S. Deka, ACS Catal., 2017, 7, 5871–5879 CrossRef CAS.
- X. Li, C. Hao, B. Tang, Y. Wang, M. Liu, Y. Wang, Y. Zhu, C. Lu and Z. Tang, Nanoscale, 2017, 9, 2178–2187 RSC.
- Y. Ding, Y. Wang, L. A. Su, M. Bellagamba, H. Zhang and Y. Lei, Biosens. Bioelectron., 2010, 26, 542–548 CrossRef CAS.
- A. L. Meng, L. Y. Sheng, K. Zhao and Z. J. Li, J. Mater. Chem. B, 2017, 5, 8934–8943 RSC.
- Y. W. Liu, X. Q. Cao, R. M. Kong, G. Du, A. M. Asiri, Q. Lu and X. P. Sun, J. Mater. Chem. B, 2017, 5, 1901–1904 RSC.
- E. H. Zhang, Y. Xie, S. Q. Ci, J. C. Jia and Z. H. Wen, Biosens. Bioelectron., 2016, 81, 46–53 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra06766c |
| This journal is © The Royal Society of Chemistry 2019 |