
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnraveling mechanisms of the uncoordinated nucleophiles: theoretical elucidations of the cleavage of bis(p-nitrophenyl) phosphate mediated by zinc-complexes with apical nucleophiles†
Xiaoyu Zhou‡
 a,
Xue-Peng Zhang‡b,
Weikang Lia,
Jingxing Jiangc,
Huiying Xua,
Zhuofeng Kec,
David Lee Phillipsd and
Cunyuan Zhao*a
a,
Xue-Peng Zhang‡b,
Weikang Lia,
Jingxing Jiangc,
Huiying Xua,
Zhuofeng Kec,
David Lee Phillipsd and
Cunyuan Zhao*a
aMOE Key Laboratory of Bioinorganic and Synthetic Chemistry, School of Chemistry, Sun Yat-Sen University, Guangzhou 510275, P. R. China. E-mail: ceszhcy@mail.sysu.edu.cn
bSchool of Chemisty and Chemical Engineering, Shaanxi Normal University, Xi'an 710119, P. R. China
cMOE Key Laboratory of Bioinorganic and Synthetic Chemistry, School of Chemistry, School of Materials Science and Engineering, Sun Yat-sen University, Guangzhou 510275, P. R. China
dDepartment of Chemistry, University of Hong Kong, Pokfulam Road, Hong Kong, P. R. China
First published on 19th November 2019
Abstract
A theoretical approach was used to investigate the hydrolytic cleavage mechanisms of the bis(p-nitrophenyl) phosphate (BNPP−) catalyzed by Zn(II)-complexes featuring uncoordinated nucleophiles. Ligand-based and alternative solvent-based nucleophilic attack reaction models are proposed. The pKa values of the Zn(II)-bound water molecules or ligands in the [Zn(LnH)(η-H2O)(H2O)]2+ (n = 1, 2 and 3) complexes, as well as the dimerization tendency of the mononuclear Zn(II)-complexes, were found to significantly influence the reaction mechanisms. The Zn(II)-L3 complexes were found to be more favorable for the hydrolytic cleavage of the BNPP− via a ligand-based nucleophilic attack pathway. This was due to the lower pKa value for the deprotonation of the oxime ligand, the hard dimerization of the mononuclear Zn(II)-L3 species, and the presence of an uncoordinated nucleophile. The origins of the uncoordinated reactions were systematically elucidated. The theoretical results reported here are in good agreement with experimental observations and more importantly, help to elucidate the factors that influence intermolecular nucleophilic attack reactions with coordinated/uncoordinated nucleophiles.
1. Introduction
Metal-containing complexes with an ability to catalyze the cleavage of phosphate esters as artificial mimic enzymes have gained much attention over the past decade.1–8 The backbone of DNA and RNA are phosphodiesters, which are widely found in biological cells and play well-known roles in genetic information storage and metabolism processes.9 The half-life of the fission of the P–O bonds under physiological conditions is estimated to be around 30 million years at pH 7 and 25 °C.10 In contrast to this, phosphatases as well as their metal-complex based artificial mimics produce significant rate enhancements for the P–O bond cleavage.11–16 Many natural phosphatases take advantage of the metal centers, such as Ca2+, Mg2+, Fe3+, Fe2+ and Zn2+.2,17–20 Compared to other metal-complexes, the bivalent Zn(II)-containing complexes have attracted more interest due to their abundance, nontoxicity, good Lewis acidity and rapid ligand exchange.16,21–23A number of metal-complexes featuring intermolecular nucleophilic attack ability were reported during the past several decades. In mono- and dinuclear metals complexes system, following mechanisms have been proposed for metal-catalyzed hydrolysis reaction: (1) the hydroxide activation,14,23–28 in which hydroxide and alkoxy generated from external solvent molecular (water or alcohol) acts as a nucleophile; (2) the substrate activation,29–32 in which substrate itself acts as a nucleophile to attack phosphorous; especially, the ligand cooperative activation (3) is designed through the ligand structure containing the nucleophile.6,12,33–39 It should be noted that hydrolysis mechanisms (1) and (2) are reported by most of previous experimental and theoretical studies which particularly reported by Brown and coworkers,23,29 Morrow and Richard,30,31 and Gao.40 Interestingly, the hydrolysis efficiency of ligand cooperative activation is found to be higher than that of hydroxide activation in Chin and co-workers' work.39
The rapid hydrolysis cleavage of RNA was reported to involve the 2′-hydroxy group via an intramolecular nucleophilic attack reaction, but it is hard for hydrolysis DNA, and the ligand cooperative intermolecular mechanism is regarded as the more effective strategy to enhance the attack at phosphorus.41 Chin and co-workers reported that the metal alkoxide of a copper complex could efficiently cleave the phosphodiester core effectively compared to their metal-hydroxide counterparts.39 Later, a series of selected Zn(II) complexes with metal-bound alkoxide were synthesized and found to efficiently promote BNPP− (BNPP− = bis(p-nitrophenyl) phosphate) cleavage and it was suggested that this occurs via a ligand cooperative nucleophilic attack mechanism.12,34 Tonellato and co-workers reported some metal-complexes with α-nucleophiles that could promote the cleavage of a carboxylic acid ester and it was suggested their efficient catalytic activity could be attributed to the presence of the uncoordinated nucleophiles from the oxime-ligand.35 Kumar and co-workers extended the substrates to phosphate esters in cationic micellar media and an efficient hydrolytic cleavage of BNPP− was obtained from the oximate complexes when compared with hydroxamate ions.36 Later, Namrata and co-workers developed a series of similar oxime-functionalized surfactants and their kinetic studies demonstrated that the oxime moieties were the super-nucleophiles to induce the cleavage in the phosphate esters.42 Yatsimirsky et al. also reported the efficient cleavage of 4-nitrophenyl acetate mediated by either zinc(II) or cadmium(II) complexes containing a tridentate oximate ligand.43
Recently, Williams and co-workers investigated the hydrolysis of BNPP−, as a convenient model for DNA cleavage, catalyzed by a series of pyridyl Zn(II) complexes.37 They found that methylation on the pyridyl ring could substantially increase the hydrolytic activity of the Zn(II) complexes and they suggested that the reactivity of the zinc complex containing a hydrated aldehyde ligand was comparable with that of the alkoxide complexes in which the uncoordinated moiety was presumed to maintain its nucleophilicity. Later, Williams and co-workers developed new pyridyl Zn(II) complexes containing oxime that could induce the cleavage reactions under mild conditions and their experimental results suggested that the mononuclear Zn(II) complexes using the oxime as a nucleophile demonstrated the highest phosphodiester hydrolysis ability reported to date.38 They presumed that the high catalytic reactivity of the Zn(II)–oxime complexes can be ascribed to the lower pKa value for the deprotonation of the oxime. In addition, dimerization was presumed in the Zn(II)–oxime complex while this was not found in the Zn(II)–alcohol complexes. However, the influences of the pKa value of the metal-bound oxime as well as the dimerization of the mononuclear Zn(II)-complexes on the cleavage reactions of interest are not clear. Moreover, the catalytic differences between the Zn(II)–alcohol and the Zn(II)–oxime complexes are also obscure.12 Therefore, a theoretical approach was utilized here to systematically investigate the hydrolysis mechanisms of phosphodiester BNPP− mediated by three different mononuclear Zn(II)-complexes developed by Williams and co-workers37,38 (see Scheme 1). More importantly, our theoretical approach was focused on the following questions: (1) what are the pKa values of the metal-bound nucleophiles and their influence on the reactions of interest; (2) what is the dimerization tendency of the mononuclear Zn(II)–alcohol and Zn(II)–oxime complexes and their effects on the reactions of interest; (3) what are the reactive catalyst forms; (4) and last but most importantly, what are the catalytic reactivity differences between the coordinated and the uncoordinated nucleophiles, and the subsequent nucleophilic attack differences between the ligand-based and solvent-based reactions. The investigation of the overall influencing factors and alternative reaction pathways are expected to be helpful in the design of future catalysts and their industrial applications.
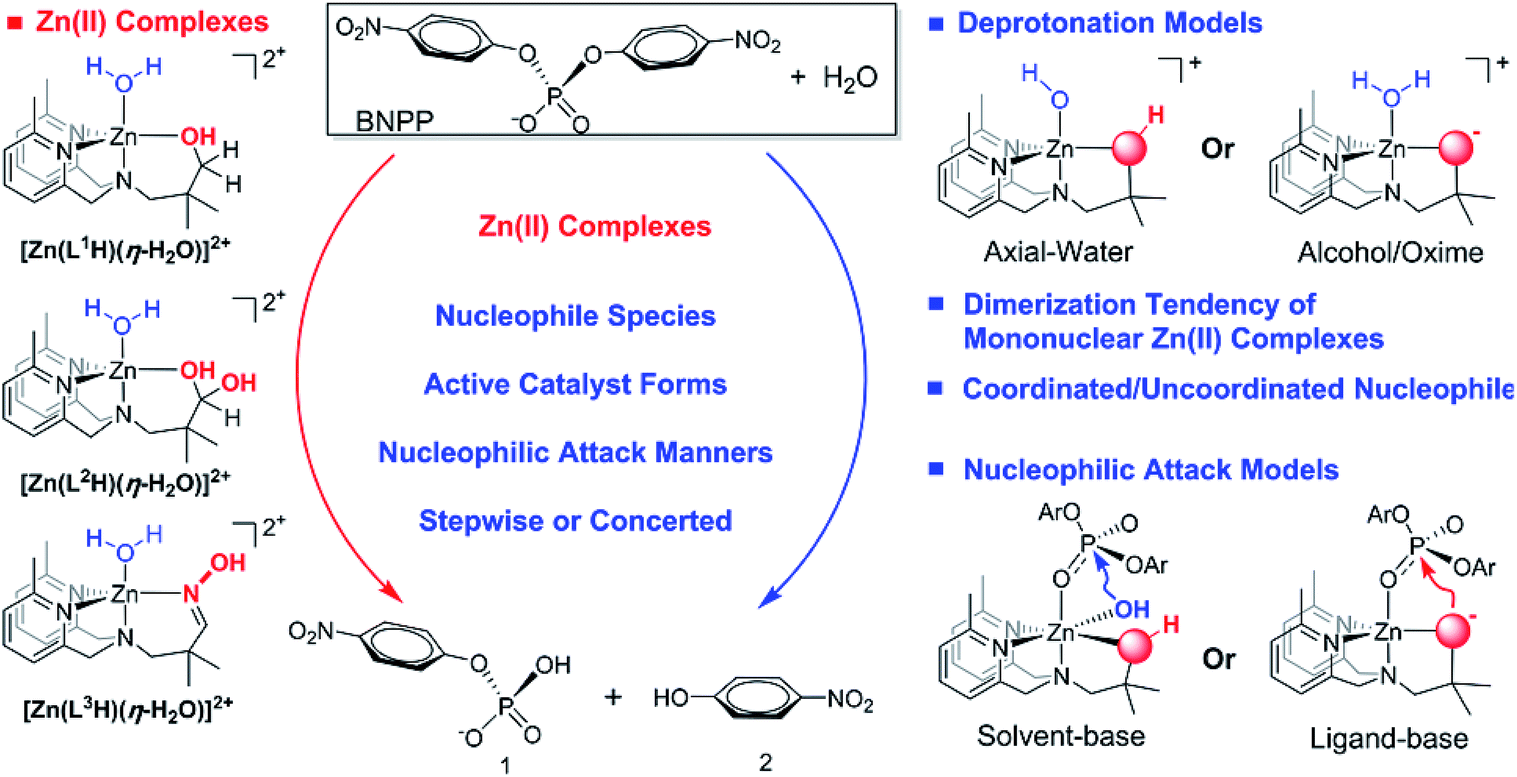 | ||
| Scheme 1 The hydrolysis mechanisms of RNA dinucleotide analogue BNPP− mediated by three different mononuclear Zn(II) complexes. | ||
2. Computational details
The theoretical calculations were performed employing the Gaussian 09D.01. program44 with the density functional theory (DFT) methodology. As demonstrated in previous reported works,19,23,32,45,46 the popular and economic hybrid B3LYP47,48 functional could rationalize the mechanisms of the phosphate transesterification processes reasonably well. The combination of the hybrid B3LYP functional and the basis sets I (denoted as BSI, 6-31G* for C and H atoms, 6-31+G* for O, N and P atoms, and Stuttgart/Dresden (SDD)49 with ECPs49,50 for Zn atoms) was utilized in the geometry optimizations, the harmonic frequency calculations as well as for the intrinsic reaction coordinate (IRC)51 calculations. The vibrational frequency calculations were conducted to firstly distinguish the transition state structures from the local minima, and secondly to obtain the thermodynamic corrections. The IRC calculations were utilized to reconfirm the transition states connecting the reaction pathway species. Furthermore, the single point energies for pKa and dimerization calculation in the solution phase were refined at the level of the B3LYP functional and the basis sets II (denoted as BSII, SDD with ECPs for the Zn atoms while 6-311++G (d, p) for the others). The larger basis sets III (6-311++G (2df, 2p) for the non-metal atoms and SDD with ECPs for the Zn atoms) with B3LYP functional was used to calculate single-point energies of optimized geometries in intermolecular nucleophilic reactions. Solvation effects were estimated by using the polarized continuum model (PCM)52,53 with the SMD54 approach. In addition, the D3 version of the Grimme's empirical dispersion correction was also considered in the solvation single point energy calculations. The natural population analysis (NPA)55 atomic partial charge was utilized to rationalize the nature of the reaction mechanisms. The 3D diagrams of optimized structures were generated using CYLView.56 The analysis of Fukui function was employing Multiwfn 3.6.573. Results and discussion
The factors influencing the pKa value and the dimerization tendency of the mononuclear Zn(II)-complexes were investigated first to determine the nucleophile species as well as the active catalyst forms. Subsequently, the Zn(II)-complexes mediated hydrolysis reaction mechanisms were investigated to rationalize the significant differences between the ligand-based and the solvent-based nucleophilic attack reactions.3.1 pKa value and the active species
The vacant coordination site of the Zn(II) center in each [Zn(LnH)]2+ (n = 1, 2 and 3) complex can be occupied by a water molecule in order to form the stable trigonal bipyramid coordination configuration of the Zn(II) center in each [Zn(LnH)(η-H2O)]2+. One extra explicit water molecule was also introduced in order to demonstrate the significant hydrogen bonds between the Zn(II)-complex and the water solvent molecules. Thereby, the [Zn(LnH)(η-H2O)(H2O)]2+ (n = 1, 2 and 3) structures were considered as starting materials. The deprotonation of either the LnH ligand or the axial Zn(II)-coordinated water molecule can take place (see in Scheme 2) and their pKa values were evaluated from the thermodynamic Born–Haber cycle of the deprotonation process58,59 (see in Scheme 3). As shown in Table 1, the pKa values of the deprotonation of the axially-coordinated water molecule to form [Zn(LnH)(η-OH)(H2O)]+ (n = 1, 2 and 3) were calculated to be 12.6, 10.3 and 11.4, respectively. Meanwhile, the pKa values to form the ligand-deprotonated [Zn(Ln)(η-H2O)(H2O)]+ (n = 1, 2 and 3) complexes were evaluated to be 14.5, 10.0 and 8.8, respectively. In addition, the relative free energy of the ligand-deprotonated [Zn(L1)(η-H2O)(H2O)]+ complex was computed to be 2.6 kcal mol−1 higher than that of the [Zn(L1H)(η-OH)(H2O)]+, while the ligand-deprotonated complex [Zn(L2)(η-H2O)(H2O)]+ or [Zn(L3)(η-H2O)(H2O)]+ was 0.4 kcal mol−1 or 3.6 kcal mol−1 lower than the axially-coordinated-water-deprotonated counterpart, respectively.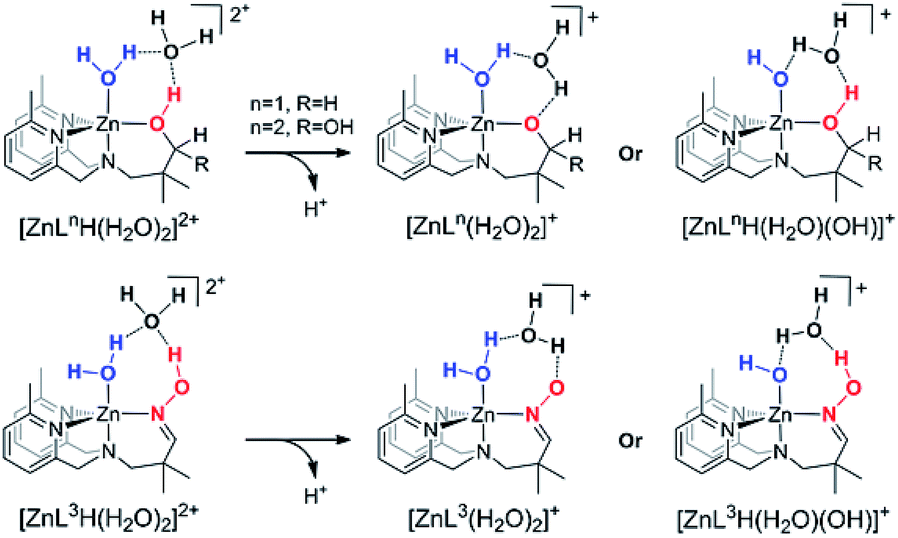 | ||
| Scheme 2 The structural variation during deprotonation processes. | ||
 | ||
| Scheme 3 The Born–Haber cycle of the deprotonation processes. | ||
| Structures | [ZnII(L1H)(H2O)2]+ | [ZnII(L2H)(H2O)2]+ | [ZnII(L3H)(H2O)2]+ |
|---|---|---|---|
| [ZnIILn(H2O)2]+ | 14.5 (0.0) | 10.0 (0.0) | 8.8 (0.0) |
| [ZnIILnH(H2O)(OH)]+ | 12.6 (−2.6) | 10.3 (0.4) | 11.4 (3.6) |
As mentioned above, for the Zn(II)–alcohol complexes, the pKa values as well as the relative free energies of the water-deprotonated [Zn(LnH)(η-OH)(H2O)]+ complexes (n = 1 and 2) and the ligand-deprotonated [Zn(Ln)(η-H2O)(H2O)]+ complexes (n = 1 and 2) were close in values, which indicated that both of the deprotonated species for the Zn(II)–alcohol complexes were active catalyst forms and either the axial Zn(II)–OH or apical Zn(II)–alkoxide would be the nucleophile. For the Zn(II)–oxime complexes, the distinctly lower pKa values as well as the relative lower energy of the ligand-deprotonated [Zn(L3)(η-H2O)(H2O)]+ complex indicated that the ligand-deprotonated Zn(II)–oxime complex was the exclusive active catalyst and the Zn(II)–oximate was the potential nucleophile. Addition, TPSS and PBE density functionals were used to calculate pKa of all the [Zn(LnH)(η-H2O)(H2O)]2+ (n = 1, 2 and 3) structures, and the results lead to similar conclusion (seen ESI Table S1†). Moreover, the Fukui function and the dual descriptor (Δf) were used to predict the ability of nucleophilic attack. The more negative value of Δf occur at oxygen atom of [Zn(Ln)(η-H2O)]+ complexes, suggesting [Zn(Ln)(η-H2O)]+ complexes are more favorable for reacting with P atom of BNPP by nucleophilic attack than that of [Zn(LnH)(η-OH)]+ complexes (exhibited in ESI Table S2†).
3.2 Dimerization tendency
As discussed above, the mononuclear complexes [Zn(L1H)(η-OH)(H2O)]+ or [Zn(L1)(η-H2O)(H2O)]+, [Zn(L2H)(η-OH)(H2O)]+ or [Zn(L2)(η-H2O)(H2O)]+, [Zn(L3)(η-H2O)(H2O)]+ were estimated to be the active catalyst forms. Possible dimerization of these active species might take place, which subsequently influences the concentration of the active species and furthermore alter their catalytic performance. Thereby, the dimerization tendency of the mononuclear active species was investigated (see Scheme 3). As shown in Scheme 4a, the dimerization between the water-deprotonated [Zn(LnH)(η-OH)(H2O)]+ (n = 1 or 2) and the ligand-deprotonated complex [Zn(Ln)(η-H2O)(H2O)]+ (n = 1 or 2) might take place to form a binuclear [Zn2(Ln)(LnH) (μ-OH)]2+ complex (n = 1 or 2). A μ-hydroxyl bridge was formed in the [Zn2(Ln)(LnH) (μ-OH)]2+ complex, and the alkoxide was coordinated with the two respective Zn(II) centers. Alternatively, as mentioned in the pKa preceding discussion, the higher pKa values were obtained for the Zn(II)–alcohol complexes, which indicated that the spontaneous deprotonation of either an axially-coordinated water molecule or a coordinated alcohol would not completely take place in aqueous solution. Thereby, dimerization between axial-water-deprotonated Zn(II)–alcohol complexes and their respective [Zn(LnH)(η-H2O)(H2O)]2+ (n = 1 and 2) starting materials were also considered and mono μ-hydroxyl bridged binuclear complexes [Zn2(LnH)2(μ-OH)]3+ (n = 1 and 2) may be formed (see Scheme 4b). Similarly, dimerization between the ligand-deprotonated [Zn(Ln)(η-OH)(H2O)]2+ (n = 1 or 2) complexes and starting materials were estimated to take place and binuclear complexes [Zn2(LnH)(Ln)]3+ (n = 1 and 2) may also be obtained (see Scheme 4c). In addition, the axially-coordinated hydroxyl can link two mononuclear Zn(II)-LnH (n = 1 or 2) complexes to form a double μ-hydroxyl bridged binuclear complex [Zn2(LnH)2(μ-OH)2]2+ (n = 1 or 2) (see Scheme 3d). Lastly but more importantly, the axially-coordinated water molecule in the ligand-deprotonated [Zn(Ln)(η-H2O)(H2O)]+ (n = 1, 2 or 3) complex can depart and the alkoxide/oximate moiety can then coordinate with the two separate Zn(II) centers to form a binuclear complex [Zn2(Ln)2]2+ (n = 1, 2 or 3) (see Scheme 4e). | ||
| Scheme 4 Possible dimerization processes for the mononuclear Zn(II)-Ln complexes of interest (the extra explicit water molecule was emitted for clarity; the red spheres represent coordinated alkoxide or oxime). | ||
The formation free energies of the binuclear complexes of interest are provided in Table 2. As shown in Table 2, the only negative formation energy (ΔGf) was observed in the dimerization process of the [Zn2(L2H)2(μ-OH)]3+ complex (−3.4 kcal mol−1) while the other positive ΔGf values indicated it is difficult to dimerization of their respective mononuclear active catalyst forms. To conclude, the dimerization tendency investigations showed that there is a weak dimerization tendency for the Zn(II)-L1 and Zn(II)-L3 mononuclear active species, while there is a slight dimerization tendency for the Zn(II)-L2 mononuclear complexes which were calculated to estimate a decrease in the concentration of the Zn(II)-L2 active species.
| Structures | [Zn2(LnH)2(μ-OH)2]2+ | [Zn2(Ln)(LnH) (μ-OH)]2+ | [Zn2(LnH)2(μ-OH)]3+ | [Zn2(Ln)(LnH)]3+ | [Zn2(Ln)2]2+ |
|---|---|---|---|---|---|
| ΔGf (L1) | 8.6 | 5.9 | 0.2 | 12.5 | 0.7 |
| ΔGf (L2) | 16.7 | 8.8 | −3.4 | 3.1 | 6.4 |
| ΔGf (L3) | — | — | — | — | 3.8 |
3.3 Catalyst–substrate bonding modes and reaction pathways
As discussed above, either the ligand-deprotonated or the axial Zn(II)–H2O deprotonated active species were obtained, in which the Zn(II) center adopts a distorted triangular bipyramid coordination sphere. The Zn(II) centers in the active species would further bind with the BNPP substrate to proceed with the hydrolysis reactions (see Scheme 5). For the Zn(II)–H2O deprotonated [Zn(LnH)(η-OH)]+ (n = 1, 2 and 3) complexes, a BNPP− substrate will bind with the Zn(II) center via a Zn⋯O(P) coordination bond and the Zn(II) center adopts an octahedron geometry. The equatorial-situated hydroxyl will then nucleophilically attack the mono-coordinated BNPP− substrate to initiate a solvent-based intermolecular hydrolysis reaction. For the ligand-deprotonated [Zn(Ln)(η-H2O)]+ (n = 1, 2 and 3) complexes, an easier ligand exchange between the axial Zn(II)-bound water molecule and a BNPP substrate will take place and the triangular bipyramid coordination sphere of Zn(II) centers remains. The apical alkoxide/oximate moiety will subsequently attack the phosphorus center of the BNPP−, which initiates a ligand-based nucleophilic attack reaction. | ||
| Scheme 5 Shown are the catalyst–substrate binding modes and the nucleophilic attack processes (the red spheres represent coordinated alkoxide or oxime). | ||
3.4 Mechanisms for the cleavage of BNPP− promoted by Zn(II) complexes
The mechanisms for the above mentioned intermolecular ligand-based and solvent-based nucleophilic attack reactions are detailed in this section. The interconversion between the active species [Zn(LnH)(η-OH)]+ and [Zn(Ln)(η-H2O)]+ (n = 1, 2 and 3) are also investigated. The free energy profiles of the proposed reaction mechanisms are provided in Fig. 1 and 2. | ||
| Fig. 1 Depicted are the free energy profiles of the proposed interconversion and ligand-based intermolecular reaction pathways by the three different mononuclear Zn(II) complexes. | ||
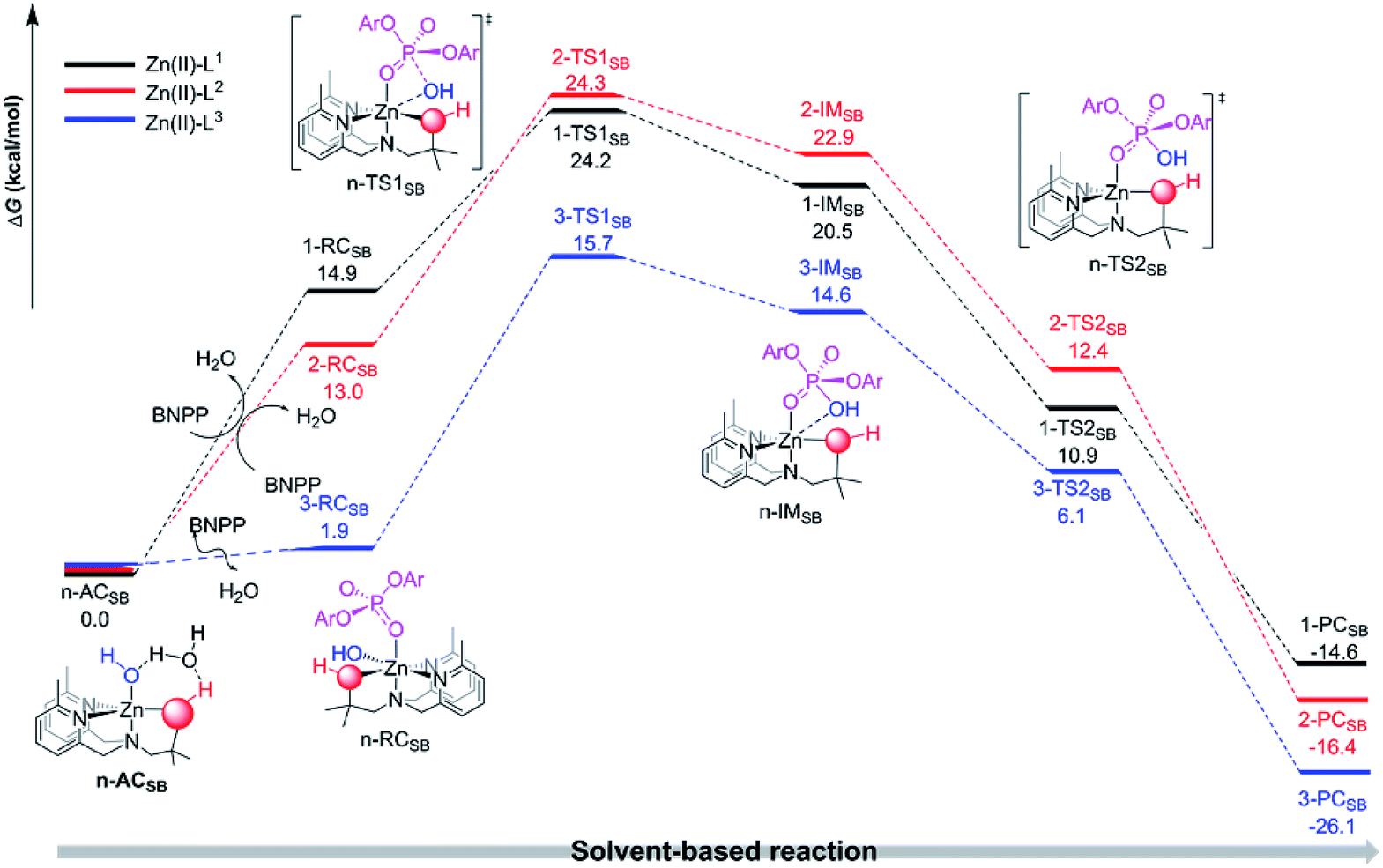 | ||
| Fig. 2 Depicted are the free energy profiles of the proposed solvent-based intermolecular reaction pathways for the hydrolysis of BNPP catalyzed by the three different mononuclear Zn(II) complexes. | ||
An extra explicit water molecule was added into the active catalyst structures in order to rationalize the roles of the solvent participation in the proton relay transfer in the interconversion between the two respective active catalyst forms in each Zn(II)-Ln entry. As shown in Fig. 1, the relative free energy barriers of the proton relay transition states n-Pre (n = 1, 2 or 3) were small which indicates there will be an easier transformation between the active catalyst of the solvent-based complexes [Zn(LnH)(η-OH)(H2O)]+ (denoted as n-ACSB) and the ligand-based complexes [Zn(Ln)(η-H2O)(H2O)]+ (denoted as n-ACLB) (n = 1, 2 or 3). The lower transformation barriers as well as the small energy gaps between the n-ACSB and n-ACLB (n = 1, 2 or 3) are in agreement with the above pKa results. For example, the distinct stability of 3-ACLB reconfirmed the exclusive ligand-deprotonated mononuclear active catalyst form.
The n-ACSB or n-ACLB can bind with a BNPP− substrate to initiate an intermolecular hydrolytic cleavage (see Fig. 1 and 2). For the solvent-based nucleophilic attack pathway (see Fig. 2), the equatorial-situated hydroxyl will nucleophilically attack the adjacent phosphorus center of the BNPP− to form the transition state n-TS1SB (n = 1, 2 and 3). The harmonic vibration of the unique imaginary frequency in each n-TS1SB (n = 1, 2 or 3) demonstrates the P⋯O(Nu) (Nu represents the nucleophile) bond forming process. The Zn⋯O(Nu) linkage in the n-TS1SB (n = 1, 2 or 3) structure retained a distorted octahedron coordination sphere for the Zn(II) center. Subsequently, the further formation of the P–O(Nu) bond was observed in the intermediate n-IMSB (n = 1, 2 and 3) and the [PO5] moiety adopted a distorted triangular bipyramid geometry with an elongated P–O(Nu) and P–O(LG) (LG represents the p-nitrophenyl leaving group) polar bonds. A notable fission of the P–O(LG) bond was found in each transition state n-TS2SB (n = 1, 2 or 3) and the leaving group finally dissociated into the solution via an exothermal reaction. As shown in Fig. 1, the relative free energy of the n-TS2SB is lower than that of the n-IMSB (n = 1, 2 or 3), which indicates that the intermolecular nucleophilic attack reaction pathway proposed here occurs via an enforced concerted mechanism.60 The distinct stability of the n-TS2SB can be ascribed to the good leaving group nature of the p-nitrophenyl moiety. To conclude, the proposed intermolecular pathway is an enforced concerted SN2-type reaction mechanism and the nucleophilic attack step is the rate-determining step.
On the other side, the ligand-based intermolecular reaction pathways were initiated via nucleophilic attacks by the Zn(II)-coordinated alkoxide/oximate moiety in n-RCLB (n = 1, 2 and 3) (see Fig. 1). Both the P–O bond formation to the nucleophile and the P–O bond fission to the leaving group were observed in the transition states n-TSLB (n = 1, 2 and 3). Finally, the transient product n-PCLB is obtained with the dissociation of the p-nitrophenyl leaving group into the solution via an exothermic process. To conclude, a concerted SN2-type ligand-based intermolecular pathway is proposed in each Zn(II)-Ln entry.
As depicted in Fig. 1, the relative free energy barriers for the ligand-based nucleophilic attack reactions (10.6, 9.8 and 4.7 kcal mol−1, respectively) for the Zn(II)-Ln entries are obviously lower than the rate-determining step barriers of their solvent-based counterparts (24.2, 24.3 and 15.7 kcal mol−1, respectively). These calculated results demonstrate that the ligand-based pathway for the nucleophilic attack is more favorable. The results is probably due to the five-coordination of ligand-based complexes is better than six-coordination of solvent-based complexes. More specifically, the Zn(II)-bound alkoxide/oximate can act as a nucleophile to directly attack the phosphodiester substrate rather than the axially-coordinated hydroxyl moiety as the nucleophile. The proposed ligand-based mechanism is consistent with the experimental results that determined that alkoxide/oximate phosphate products are obtained.37,38 In addition, the decreased energy barrier from Zn(II)-L1 to Zn(II)-L3 is in good agreement with the experimental results that found the Zn(II)–oxime complexes were better BNPP hydrolysis catalysts when compared to the Zn(II)–alcohol complexes.37,38 It is noted that, as discussed above, there is a slight dimerization tendency for the Zn(II)-L2 mononuclear complexes as estimated by the computational results here, which might hinder the catalytic processes mediated by the Zn(II)-L2 complexes. Thereby, the combination of the dimerization tendency as well as the narrow ligand-based barrier gap (0.8 kcal mol−1) between the Zn(II)-L1 and Zn(II)-L2 entries can give rise to different experimental results depending on the solution environment. As described in the Williams and coworkers experiments, the activity of the Zn(II)-L1 system is much greater than that of Zn(II)-L2 in anhydrous methanol, but in water the activity of Zn(II)-L1 is a little lower than that of Zn(II)-L2.37 The origins of the catalytic superiorities of the Zn(II)-oxime complexes are further investigated below.
3.5 Origins of the better activity for the uncoordinated ligands
It is interesting to compare how the M–L bond MLC the distinct catalytic performance for the Zn(II)–oxime complexes was estimated to be ascribed to the uncoordinated nucleophilic oxygen of the oximate. Further investigations about the behavior of the coordinated or the uncoordinated nucleophiles and their corresponding nucleophilic attack abilities are provided in this section. The L2 ligand has two hydroxyl groups and it is estimated that the coordinated/uncoordinated alkoxide/hydroxyl are able to act as nucleophiles to induce the cleavage of the BNPP− substrate. Four different nucleophilic attack modes are proposed for the Zn(II)-L2 catalyzed reactions (see Scheme 6). The Coord mode was discussed in the above mentioned ligand-based pathway, in which the coordinated alkoxide acts as a nucleophile and both the P–O(Nu) bond forming as well as the Zn–O(Nu) bond cleavage processes were observed in the 2-TSLB. Alternatively, the uncoordinated hydroxyl can nucleophilically attack the BNPP− substrate in the Uncoord1 mode. Interestingly, the P–OH(Nu) bond formation process was observed in the concerted transition state Uncoord1-TS accompanied with an energy-consuming C–OH(Nu) bond cleavage process. In the Uncoord2 mode, the nucleophile was the uncoordinated oxygen terminal of the alkoxide and both the P–O(Nu) bond formation as well as the Zn–OH(Nu) bonding cleavage processes are observed in the transition state Uncoord2-TS. All of the Coord, Uncoord1 and Uncoord2 modes are mono-deprotonation situations, the di-deprotonation case is further considered for Uncoord 3. However, the pKa value for the second ligand deprotonation of the [Zn(L2)(η-H2O)(H2O)]+ complex is calculated to be 27.3, which indicates that the second deprotonation of the L2 ligand is infeasible in neutral or weakly alkaline aqueous solution. Therefore, the Uncoord 3 mode can be considered to be not feasible. In addition, the free energy of either the Uncoord1-TS or Uncoord2-TS is found to be 21.9 kcal mol−1 or 21.0 kcal mol−1 higher than that of the 2-TSLB, which confirms that the nucleophile is the coordinated alkoxide in the Zn(II)-L2 entry. The distinct high energies for the Uncoord1-TS and Uncoord2-TS are estimated to be ascribed to the synchronous C–OH(Nu) or Zn–OH(Nu) bond cleavage processes in the nucleophilic attack steps. On the contrary, the synchronous Zn–O(Nu) bond fission and the P–O(Nu) bond formation processes in the 2-TSLB are less energy-consuming. To further understand the coordinated mechanism of Zn(II)-L2 complexes, the distortion/interaction analysis of the TSs is performed. The weaker P–O(Nu) interaction for Uncoord-TSs (1.995 Å (Uncoord1-TS), 2.026 Å (Uncoord2-TS) vs. 1.780 Å (2-TSLB)) leads the unfavored interaction energies (−12.8 and −8.5 kcal mol−1, respectively). Moreover, the length of C–O2(H) bond increase from 1.456 Å in 2-RCLB to 2.012 Å in Uncoord1-TS and 1.511 Å in Uncoord2-TS, which contributes to higher catalyst deformation energies (23.2 and 16.0 kcal mol−1, respectively). Considering the distortion/interaction energy, 2-TSLB is favor for the ligand based reaction of Zn(II)-L2 complexes. | ||
| Scheme 6 Four different nucleophilic attack modes are proposed for the Zn(II)-L2 catalyzed reactions. Key intermolecular nucleophilic attack transition states in both Zn(II)-L2 entries are also provided. Energies are in kcal mol−1. | ||
Furthermore, as depicted in Fig. 1, the energy barrier for the ligand-based intermolecular nucleophilic attack mediated by the Zn(II)–oxime complex is the lowest. In 3-RCLB, the nucleophile is the Zn(II)-coordinated oximate. The NPA atomic partial charge of the N or O in the oximate moiety are −0.257 and −0.557, respectively. The electron-sufficient oxygen terminal of the oximate gives rise to a better nucleophile. More importantly, the NPA charges on the alkoxide O terminals in the 1-RCLB and 2-RCLB are −1.007 and −0.995, respectively. The lower electron density on the N site of the oximate in 3-RCLB indicates there is a weaker electrostatic attraction between the Zn(II) center and the oximate nucleophile. Thus, as demonstrated in Fig. 1 and 2, the Zn–N(Nu) bond fission process in 3-TSLB is believed to be less energy-consuming when compared to the Zn–O(Nu) bond cleavage processes in both 1-TSLB and 2-TSLB. To conclude, the distinct catalytic performance of the Zn(II)–oxime complexes are ascribed to the uncoordinated oxygen terminal of the oximate nucleophile and more importantly, the weak electrostatic attraction between the Zn(II) center and the oximate nucleophile (more NPA charge distribution details are provided in the ESI Fig. S1†).
4. Conclusions
The hydrolysis mechanisms of the phosphodiester BNPP− mediated by Zn(II)-Ln complexes were investigated in this work using a theoretical approach.In order to investigate the nucleophile and the active species models, two deprotonation models were studied. One model involves a deprotonation in axial-water to form axial Zn(II)–OH complexes, and the other model involves a deprotonation in an alcohol/oxime ligand to form apical Zn(II)–alkoxide or Zn(II)–oximate complexes. The axial Zn(II)–OH and apical Zn(II)–alkoxide in the Zn(II)–alcohol complexes are preferred to be the nucleophile, due to their similar pKa values and the relative free energies found for the different deprotonation models. The Zn(II)–oximate appears to be the exclusive nucleophile in the Zn(II)–oxime complexes due to its lower pKa value and its lower relative free energy.
With regards to the reaction pathway, a ligand-based intermolecular and an alternative intermolecular nucleophilic attack processes have been studied. The computational results demonstrated that the ligand-based intermolecular reactions initiated by the Zn(II)–alkoxide/oximate nucleophiles (ΔG‡ = 9.2, 12.8 and 9.1 kcal mol−1) were more favorable than the solvent-based intermolecular reactions (ΔG‡ = 24.2, 24.3 and 15,7 kcal mol−1), which is agreement with experimental results. Specifically, our results here also demonstrate the significant role of an uncoordinated nucleophile. The Zn(II)-L3 complexes were found to be more favored than Zn(II)–alkoxide complexes for the hydrolytic cleavage of BNPP−. This was due to the lower pKa value for the deprotonation of the oxime ligand, the weak dimerization tendency of the mononuclear Zn(II)-L3 species, as well as the important ligand-based intermolecular nucleophilic attack conducted by the oxygen terminal of the oximate nucleophile. Moreover, the origins of the uncoordinated reactions were systematically elucidated. The metal-coordinated 2-TSLB (ΔG‡ = 12.8 kcal mol−1) is preferred than metal-uncoordinated Uncoord1-TS (ΔG‡ = 33.7 kcal mol−1) and Uncoord2-TS (ΔG‡ = 34.6 kcal mol−1) because of the lower deformation energies of substitution and weak interaction energies between substitution and catalyst.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the National Natural Science Foundation of China (Grant No. 21573292, 21773312 and 21603279). X. Z. is thankful for China Postdoctoral Science Foundation (Grant No. 2017M620496) for financial support. The research is also partially supported by the high performance grid computing platform of Sun Yat-Sen University, the Guangdong Province Key Laboratory of Computational Science, the Guangdong Province Computational Science Innovative Research Team and the Fundamental Research Funds for the Central Universities.Notes and references
- H. Lönnberg, Org. Biomol. Chem., 2011, 9, 1687–1703 RSC.
- F. Mancin and P. Tecilla, New J. Chem., 2007, 31, 800–817 RSC.
- R. S. Brown, Z.-L. Lu, C. T. Liu, W. Y. Tsang, D. R. Edwards and A. A. Neverov, J. Phys. Org. Chem., 2010, 23, 1–15 CAS.
- C. Liu, M. Wang, T. Zhang and H. Sun, Coord. Chem. Rev., 2004, 248, 147–168 CrossRef CAS.
- J. R. Morrow, Comments Inorg. Chem., 2008, 29, 169–188 CrossRef CAS.
- F. Mancin, P. Scrimin and P. Tecilla, Chem. Commun., 2012, 48, 5545–5559 RSC.
- L. R. Gahan, S. J. Smith, A. Neves and G. Schenk, Eur. J. Inorg. Chem., 2009, 2009, 2745–2758 CrossRef.
- C. Liu and L. Wang, Dalton Trans., 2009, 227–239 RSC.
- W. W. Cleland and A. C. Hengge, Chem. Rev., 2006, 106, 3252–3278 CrossRef CAS PubMed.
- C. Lad, N. H. Williams and R. Wolfenden, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 5607 CrossRef CAS PubMed.
- R. Krämer, Coord. Chem. Rev., 1999, 182, 243–261 CrossRef.
- M. Livieri, F. Mancin, G. Saielli, J. Chin and U. Tonellato, Chem.–Eur. J., 2007, 13, 2246–2256 CrossRef CAS PubMed.
- N. Singh, Y. Karpichev, A. K. Tiwari, K. Kuca and K. K. Ghosh, J. Mol. Liq., 2015, 208, 237–252 CrossRef CAS.
- S. S. Massoud, R. S. Perkins, F. R. Louka, W. Xu, A. Le Roux, Q. Dutercq, R. C. Fischer, F. A. Mautner, M. Handa, Y. Hiraoka, G. L. Kreft, T. Bortolotto and H. Terenzi, Dalton Trans., 2014, 43, 10086–10103 RSC.
- L. J. Daumann, G. Schenk, D. L. Ollis and L. R. Gahan, Dalton Trans., 2014, 43, 910–928 RSC.
- S. Enthaler, ACS Catal., 2013, 3, 150–158 CrossRef CAS.
- J. Weston, Chem. Rev., 2005, 105, 2151–2174 CrossRef CAS PubMed.
- T. Niittymäki and H. Lönnberg, Org. Biomol. Chem., 2006, 4, 15–25 RSC.
- D. Roston and Q. Cui, J. Am. Chem. Soc., 2016, 138, 11946–11957 CrossRef CAS PubMed.
- M. F. Mohamed, I. Sánchez-Lombardo, A. A. Neverov and R. S. Brown, Org. Biomol. Chem., 2012, 10, 631–639 RSC.
- X.-F. Wu, Chem.–Asian J., 2012, 7, 2502–2509 CrossRef CAS PubMed.
- X.-F. Wu and H. Neumann, Adv. Synth. Catal., 2012, 354, 3141–3160 CrossRef CAS.
- C. I. Maxwell, N. J. Mosey and R. S. Brown, J. Am. Chem. Soc., 2013, 135, 17209–17222 CrossRef CAS PubMed.
- A. Neves, M. Lanznaster, A. J. Bortoluzzi, R. A. Peralta, A. Casellato, E. E. Castellano, P. Herrald, M. J. Riley and G. Schenk, J. Am. Chem. Soc., 2007, 129, 7486–7487 CrossRef CAS PubMed.
- A. A. Neverov, C. T. Liu, S. E. Bunn, D. Edwards, C. J. White, S. A. Melnychuk and R. S. Brown, J. Am. Chem. Soc., 2008, 130, 6639–6649 CrossRef CAS PubMed.
- T. Humphry, M. Forconi, N. H. Williams and A. C. Hengge, J. Am. Chem. Soc., 2002, 124, 14860–14861 CrossRef CAS PubMed.
- X. Zhang, H. Gao, H. Xu, J. Xu, H. Chao and C. Zhao, J. Mol. Catal. A: Chem., 2013, 368–369, 53–60 CAS.
- T. Koike and E. J. Kimura, J. Am. Chem. Soc., 1991, 113, 8935–8941 CrossRef CAS.
- S. E. Bunn, C. T. Liu, Z.-L. Lu, A. A. Neverov and R. S. Brown, J. Am. Chem. Soc., 2007, 129, 16238–16248 CrossRef CAS PubMed.
- J. R. Morrow, T. L. Amyes and J. P. Richard, Acc. Chem. Res., 2008, 41, 539–548 CrossRef CAS.
- O. Iranzo, A. Y. Kovalevsky, J. R. Morrow and J. P. Richard, J. Am. Chem. Soc., 2003, 125, 1988–1993 CrossRef CAS PubMed.
- H. Gao, Z. Ke, N. J. DeYonker, J. Wang, H. Xu, Z. W. Mao, D. L. Phillips and C. Zhao, J. Am. Chem. Soc., 2011, 133, 2904–2915 CrossRef CAS PubMed.
- X.-M. Chen, S. M. Aubin, Y.-L. Wu, Y.-S. Yang, T. C. Mak and D. N. Hendrickson, J. Am. Chem. Soc., 1995, 117, 9600–9601 CrossRef CAS.
- M. Livieri, F. Mancin, U. Tonellato and J. J. C. C. Chin, Chem. Commun., 2004, 2862–2863 RSC.
- F. Mancin, P. Tecilla and U. Tonellato, Langmuir, 2000, 16, 227–233 CrossRef CAS.
- B. Kumar, M. L. Satnami, K. K. Ghosh and K. Kuca, J. Phys. Org. Chem., 2012, 25, 864–871 CrossRef CAS.
- E. Y. Tirel, Z. Bellamy, H. Adams, V. Lebrun, F. Duarte and N. H. Williams, Angew. Chem., Int. Ed., 2014, 53, 8246–8250 CrossRef CAS PubMed.
- E. Y. Tirel and N. H. Williams, Chem.–Eur. J., 2015, 21, 7053–7056 CrossRef CAS PubMed.
- M. J. Young, D. Wahnon, R. C. Hynes and J. Chin, J. Am. Chem. Soc., 1995, 117, 9441–9447 CrossRef CAS.
- Y. Fan and Y. Q. Gao, J. Am. Chem. Soc., 2007, 129(4), 905–913 CrossRef CAS PubMed.
- M. Oivanen, S. Kuusela and H. Lönnberg, Chem. Rev., 1998, 98, 961–990 CrossRef CAS PubMed.
- N. Singh, Y. Karpichev, B. Gupta, M. L. Satnami, J. Marek, K. Kuca and K. K. Ghosh, J. Phys. Chem. B, 2013, 117, 3806–3817 CrossRef CAS PubMed.
- P. Gómez-Tagle, J. C. Lugo-González and A. K. Yatsimirsky, Chem. Commun., 2013, 49, 7717–7719 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, M. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian,Inc., Wallingford CT, 2009 Search PubMed.
- X. Zhang, X. Liu, D. L. Phillips and C. Zhao, ACS Catal., 2016, 6, 248–257 CrossRef CAS.
- B. Das, H. Daver, A. Singh, R. Singh, M. Haukka, S. Demeshko, F. Meyer, G. Lisensky, M. Jarenmark, F. Himo and E. Nordlander, Eur. J. Inorg. Chem., 2014, 2014, 2204–2212 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- M. Dolg, U. Wedig, H. Stoll and H. Preuss, J. Chem. Phys., 1987, 86, 866–872 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS.
- K. Fukui, Acc. Chem. Res., 1981, 14, 363–368 CrossRef CAS.
- M. Cossi, G. Scalmani, N. Rega and V. Barone, J. Chem. Phys., 2002, 117, 43–54 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- A. E. Reed, L. A. Curtiss and F. J. C. R. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- C. Y. C. Legault, 1.0b, Universitede Sherbrooke: Québeć C. Montreal, 2009, http://www.cylview.org.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- M. D. Liptak, K. C. Gross, P. G. Seybold, S. Feldgus and G. C. Shields, J. Am. Chem. Soc., 2002, 124, 6421–6427 CrossRef CAS PubMed.
- M. D. Liptak and G. C. Shields, J. Am. Chem. Soc., 2001, 123, 7314–7319 CrossRef CAS PubMed.
- M. Nic, J. Jirat and B. Kosata, IUPAC Compendium of Chemical Terminology the“Gold Book”, 2nd edn, 2014, p. 510, http://goldbook.iupac.org Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Cartesian coordinates of all structures. See DOI: 10.1039/c9ra06737j |
| ‡ X. Zhou and X. Zhang contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |