
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemodivergent synthesis of N-(pyridin-2-yl)amides and 3-bromoimidazo[1,2-a]pyridines from α-bromoketones and 2-aminopyridines†
Yanpeng Liuab,
Lixue Lua,
Haipin Zhouc,
Feijie Xua,
Cong Ma d,
Zhangjian Huanga,
Jinyi Xu*a and
Shengtao Xu*a
d,
Zhangjian Huanga,
Jinyi Xu*a and
Shengtao Xu*a
aState Key Laboratory of Natural Medicines and Department of Medicinal Chemistry, China Pharmaceutical University, 24 Tong Jia Xiang, Nanjing 210009, China. E-mail: jinyixu@china.com
bDepartment of Chemistry, City University of Hong Kong, 83 Tat Chee Avenue, Hong Kong SAR, P. R. China
cCollege of Materials & Chemical Engineering, Chuzhou University, Chuzhou 239000, China
dState Key Laboratory of Chemical Biology and Drug Discovery, Department of Applied Biology and Chemical Technology, The Hong Kong Polytechnic University, Kowloon, Hong Kong SAR, P. R. China
First published on 28th October 2019
Abstract
N-(Pyridin-2-yl)amides and 3-bromoimidazo[1,2-a]pyridines were synthesized respectively from α-bromoketones and 2-aminopyridine under different reaction conditions. N-(Pyridin-2-yl)amides were formed in toluene via C–C bond cleavage promoted by I2 and TBHP and the reaction conditions were mild and metal-free. Whereas 3-bromoimidazopyridines were obtained in ethyl acetate via one-pot tandem cyclization/bromination when only TBHP was added, the cyclization to form imidazopyridines was promoted by the further bromination, no base was needed, and the versatile 3-bromoimidazopyridines could be further transferred to other skeletons.
Introduction
Aminopyridines serve as pharmacophores for many molecules with significant biological and therapeutic value.1 Particularly, N-(pyridin-2-yl)amides2 and imidazo[1,2-a]pyridines3 have received great attention in recent years due to their varied medicinal applications. Some examples of pharmaceutical molecules containing N-(pyridin-2-yl)amides and imidazo[1,2-a]pyridines are shown in Fig. 1. Owing to the attractive biological properties of N-(pyridin-2-yl)amides and imidazo[1,2-a]pyridines, developing convenient synthetic methods, especially by which we can synthesize these two kinds of structures respectively from the same starting materials, is very meaningful.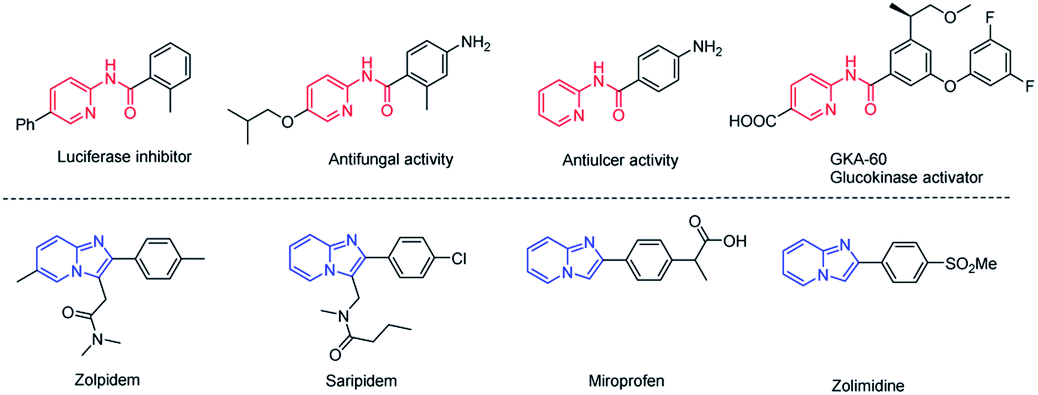 | ||
| Fig. 1 Some pharmaceutical molecules based on pyridyl-amide and imidazopyridine. | ||
Amides are ubiquitous in organic compounds, polymers, natural products, and pharmaceuticals.4 Synthesis of amides is one of the most executed reactions in organic chemistry.5 Pre-activated acyl halides or anhydrides are used as starting materials in traditional methods for constructing amide bond. Recently, many alternative substrates, such as aldehydes, alcohols, azides, aldoximes, nitriles and halides compounds had been employed for amide synthesis.6 C–C bond cleavage is another developing approach to synthesize amides, but the excellent stability of C–C bond makes this subject develop slowly, exploring new methods for constructing amides directly by C–C bond cleavage is attractive. N-(Pyridin-2-yl)amide was previously synthesized via a Cu-catalyzed dehydrogenative reaction between aldehyde and aminopyridine in the presence of I2 (Scheme 1a).7 Adimurthy's group synthesized N-(pyridin-2-yl)amides via oxidative amidation of methylarenes using TBHP in decane.8 Recently, two elegant works on Cu-catalyzed aerobic oxidative C–C bond cleavage for the preparation of N-(pyridin-2-yl)amide had been developed. Hwang et al. reported a visible-light-promoted aerobic oxidative C–N coupling between 2-aminopyridine and terminal alkynes through C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C triple bond cleavage (Scheme 1b).9 Kaliappan disclosed a biomimetic oxidation of methyl ketones for preparation of N-heterocyclic amides (Scheme 1c).10 However, these methodologies suffer from several drawbacks, such as harsh reaction conditions, long reaction time and the use of transition metal complexes. Recently, we reported a metal-free method for synthesizing N-(pyridine-2-yl)amides from ketones via oxidative cleavage of C–C bond in water.11 Herein, we will continue to report the selective synthesis of N-(pyridine-2-yl)amides from α-bromoketones and 2-aminopyridine via controlling reaction conditions.
C triple bond cleavage (Scheme 1b).9 Kaliappan disclosed a biomimetic oxidation of methyl ketones for preparation of N-heterocyclic amides (Scheme 1c).10 However, these methodologies suffer from several drawbacks, such as harsh reaction conditions, long reaction time and the use of transition metal complexes. Recently, we reported a metal-free method for synthesizing N-(pyridine-2-yl)amides from ketones via oxidative cleavage of C–C bond in water.11 Herein, we will continue to report the selective synthesis of N-(pyridine-2-yl)amides from α-bromoketones and 2-aminopyridine via controlling reaction conditions.
 | ||
| Scheme 1 Recent methods for the construction of pyridyl-amides and halosubstituted imidazopyridines. | ||
Intensive researches have been conducted to develop new and efficient strategies for constructing imidazo[1,2-a]pyridines.12 Halo-substituted imidazo[1,2-a]pyridines are important building blocks and versatile synthons for preparing more complex imidazopyridines.13 Classically, halo-substituted imidazo[1,2-a]pyri-dines are synthesized by the halogenation of imidazo[1,2-a]pyridines, such as Adimurthy's report.14 for the synthesis of the intermediate imidazo[1,2-a]pyridines, base is generally needed.15 While methods for direct synthesis of halo-substituted imidazo[1,2-a]pyridines in one-pot reaction without base were rarely reported. Jiang and co-workers reported a copper-catalyzed method for the synthesis of 2-halo-substituted imidazopyridines from haloalkynes and aminopyridines (Scheme 1d).16 3-Iodoimidazopyridines were synthesized by Zhao and co-workers in the presence of copper supported on manganese oxide-based octahedral molecular sieves OMS-2 (CuOx/OMS-2) via tandem cyclization/iodination (Scheme 1e).17 Yan and Huang's group reported copper bromide-mediated aerobic oxidative synthesis of 3-bromoimidazo[1,2-a]pyridines with pyridines and enamides (Scheme 1f).18 Besides, metal-free tandem chlorocyclization of 2-aminopyridines with carboxylic acids to synthesize chloro-substituted imidazopyridines was also described by Zeng's group.19 However, these transformations over-relied on metal catalysts or stoichiometric excess of halogen sources. From the viewpoint of green chemistry, developing an atom-economical and metal-free approach to construct halo-substituted imidazo[1,2-a]pyridine is highly desirable.
Given the importance of N-(pyridin-2-yl)amides and imidazo[1,2-a]pyridines, chemodivergent synthesis of these two skeletons from same starting materials is very appealing. Herein, we developed a method to synthesize N-(pyridin-2-yl)amides and imidazo[1,2-a]pyridines respectively from α-bromoketones and 2-aminopyridine by controlling reaction conditions (Scheme 1).
Results and discussion
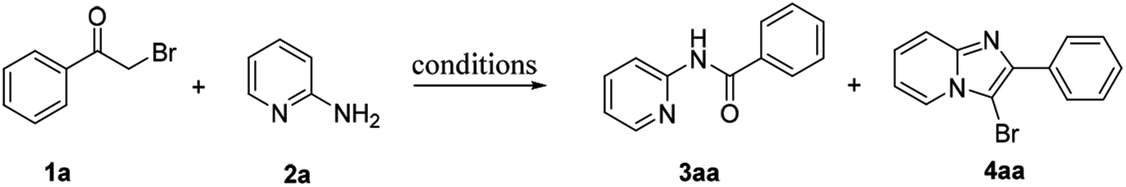
Traditionally, C–C bonds cleavage has been accomplished in the presence of transition-metal catalyst.20 Initially, various metal catalysts were screened for the preparation of amide 3aa from α-bromoacetophenone 1a and 2-aminopyridine 2a using 70% aqueous tert-butyl hydroperoxide (TBHP) as oxidant in toluene, but the yield of 3aa was not pleasurable and the highest yield was only 17% in the presence of CuI (Table 1, entry 1). Surprisingly, after extensive screening, the desired amide 3aa was obtained solely with yields of 83% and 79% respectively when I2 or KI were used as the catalyst (Table 1, entries 4, 5). This transformation was sensitive to the types of the solvents and oxidants (Table 1, entries 6–8). Finally, the optimized reaction conditions for the synthesis of amides were obtained as follows: TBHP (4 equivalents) and iodine (20 mol%) in toluene at 100 °C for 2 hours (protocol A, Table 1, entry 9). Surprisingly, the 3-bromoimidazopyridine 4aa was isolated with good yield when just using TBHP as oxidant in toluene (Table 1, entry 12). To further improve the yield of 4aa, several solvents were screened, and the results showed that ethyl acetate was the most effective solvent for the preparation of 4aa (Table 1, entry 14). Other oxidants such as H2O2 and oxygen (balloon) were also evaluated, however, the results were not pleasurable (entries 15–16). After extensive screening, 4aa was successfully obtained in yield of 93% using 2 equivalents of TBHP as oxidant in ethyl acetate at 90 °C for 3 hours (protocol B, Table 1, entry 17).
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Solvent | Oxidantb (4 eq.) | Catalyst (20 mol%) | Temp (°C) | t (h) | Yieldc (%) | |
| 3aa | 4aa | ||||||
| a Reaction conditions: 1a (0.3 mmol), 2a (0.45 mmol), oxidant (1.2 mmol), catalyst (0.06 mmol), solvent (2 mL).b 70% TBHP in water and 30% H2O2 in water were used.c Isolated yields. | |||||||
| 1 | Toluene | TBHP | CuI | 120 | 36 | 17 | 11 |
| 2 | Toluene | TBHP | CuBr | 120 | 36 | 3 | 73 |
| 3 | Toluene | TBHP | ZnBr2 | 120 | 36 | — | 5% |
| 4 | Toluene | TBHP | I2 | 120 | 2 | 83 | Trace |
| 5 | Toluene | TBHP | KI | 120 | 2 | 79 | Trace |
| 6 | DMF | TBHP | I2 | 120 | 2 | Trace | 41 |
| 7 | H2O | TBHP | I2 | 100 | 12 | 49 | 3 |
| 8 | Toluene | H2O2 | I2 | 120 | 2 | — | 28 |
| 9 | Toluene | TBHP | I2 | 100 | 2 | 84 | Trace |
| 10 | Toluene | — | — | 120 | 2 | — | — |
| 11 | Toluene | — | I2 | 120 | 2 | — | — |
| 12 | Toluene | TBHP | — | 120 | 3 | 2 | 83 |
| 13 | H2O | TBHP | — | 100 | 8 | 11 | — |
| 14 | Ethyl acetate | TBHP | — | 80 | 3 | Trace | 83 |
| 15 | Ethyl acetate | H2O2 | — | 80 | 5 | — | 36 |
| 16 | Ethyl acetate | O2 (ballon) | — | 80 | 3 | — | — |
| 17 | Ethyl acetate | TBHP (2 eq.) | — | 90 | 3 | Trace | 93 |
With the optimized reaction conditions in hand, first, the scope of the oxidative amidation approach was examined with a range of α-bromoketones and aminopyridines. As shown in Scheme 2, both the electron-rich (–Me, –OMe) and electron-withdrawing (–CF3) groups on α-haloketones were well tolerated and produced the corresponding amides in good yields. Halo-substituted α-bromoketones can also afford desired amides in moderate to good yields (3ca, 3da, 3ka, 3la). The steric factor has a slight effect on this transformation, as the meta-substituted α-bromoketones gave their corresponding products in moderate yields (3ga, 3ha). Besides, heterocycle- and alkyl-substituted α-haloketones were also tested and gave corresponding amides in 70–87% yields (3ma, 3na). Subsequently, the substrate scope of various 2-aminopyridines was investigated and the results showed that most of the substituted 2-aminopyridines could be transferred to their corresponding products in moderate to good yields.
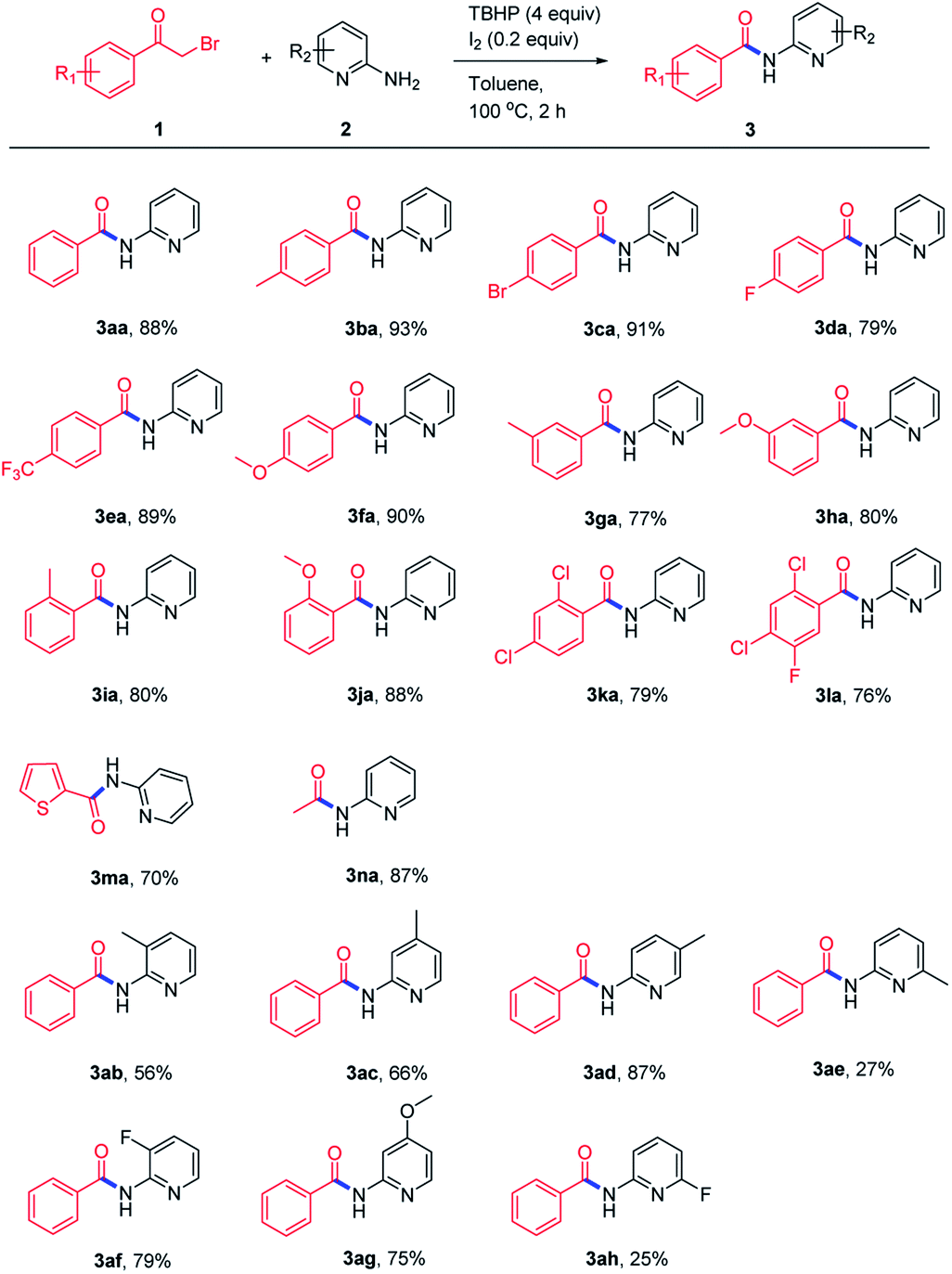 | ||
| Scheme 2 Constructing amides 3 directly from α-haloketones 1 and 2-aminopyridines 2. aReaction conditions: 1 (0.3 mmol), 2 (0.45 mmol), TBHP (1.2 mmol), I2 (0.06 mmol), toluene (2 mL), 100 °C, 2 h. | ||
Then, we evaluated various α-bromoketones 1 to examine the generality and limitations of the reaction for the synthesis of 3-bromoimidazopyridines, and the results was summarized in Scheme 3. Substrates with electron-withdrawing groups (–CF3), electron-donating groups (–Me, –OMe) or halogen groups (–F, –Cl, –Br) on the para position of aromatic ring could be transferred to the corresponding products in good to excellent yields (4ba–4ga). Furthermore, substituents at the meta or ortho positions could also provide the corresponding products with good yields (4ha–4ma). Disubstituted or trisubstituted α-bromoacetophenones were all tolerated well (4ha, 4oa). The product 4pa bearing heteroaryl moiety was also obtained in 97% yield. Some alkyl-substituted α-bromoketones, like bromoacetone and ethyl bromopyruvate, could afford the desired products in good (4qa) or moderate (4ra) yield respectively. Next, we investigated the suitability of 2-aminopyridines and their heteroaromatic analogues. The results showed that 2-aminopyridines with substituents at the C-3, C-4, C-5 position reacted smoothly to give their products in moderate yields (4ab–4ad, 4af, 4ag). This reaction is highly sensitive to the presence of a substituent at the C-6 position of pyridine ring attributed to steric hindrance (4ae).21 Besides, other ortho-amino-nitrogen-containing heteroaromatic derivatives were also investigated, and the results demonstrated that isoquinolin-1-amine was tolerated in this cascade transformation and could be transferred to products with good to excellent yields, while pyrimidin-2-amine (4ai) and 2-aminothiazole (4aj) were not suit well for this reaction.
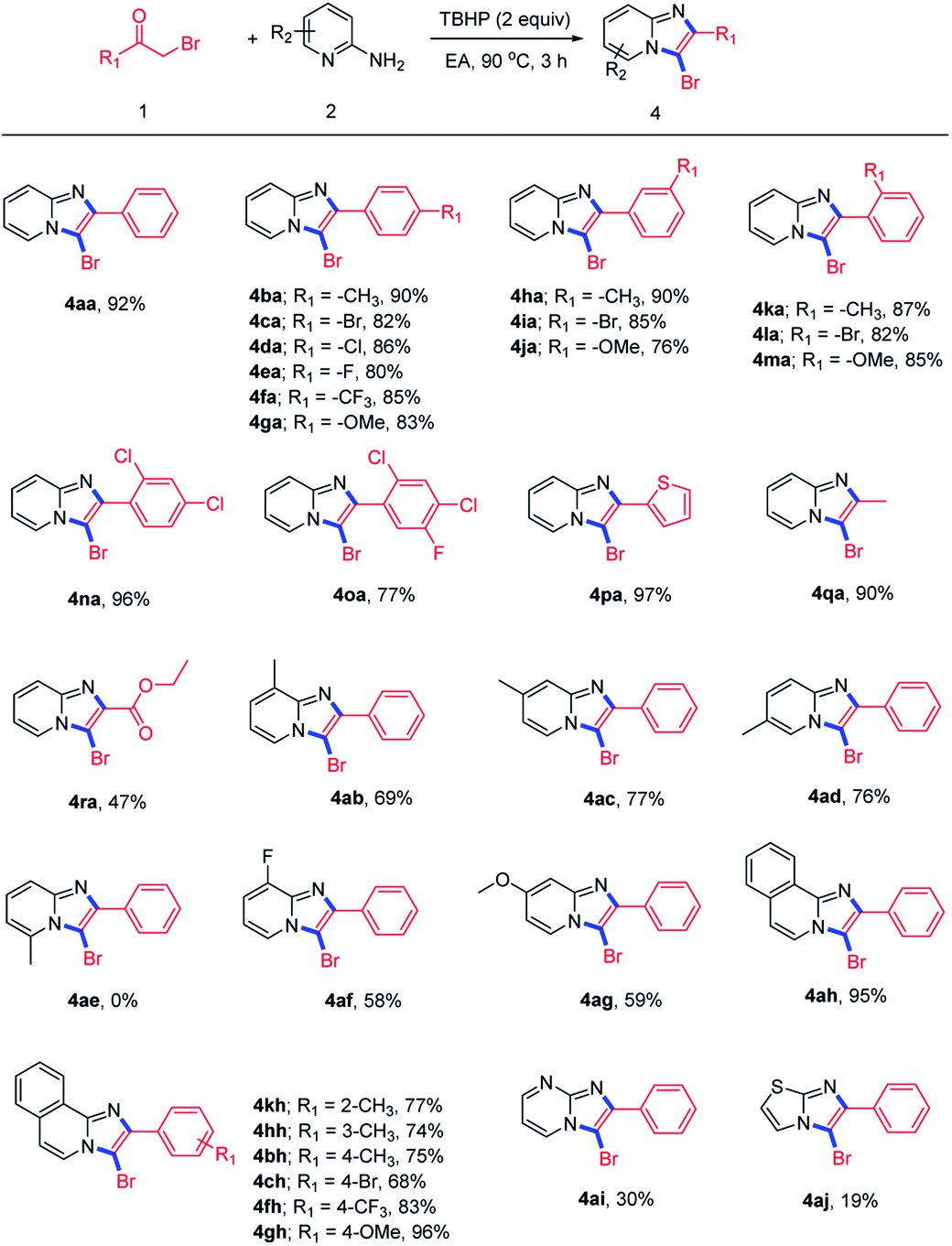 | ||
| Scheme 3 Constructing 3-bromoimidazo[1,2-a]pyridines directly from α-haloketones 1 and 2-aminopyridines 2. aReaction conditions: 1 (0.3 mmol), 2 (0.45 mmol), TBHP (0.6 mmol), EA (2 mL), 90 °C, 3 h. | ||
3-Iodoimidazo[1,2-a]pyridine could also be prepared using this methology in moderate yield (Scheme 4a). Moreover, the synthetic utility of 3-bromoimidazo[1,2-a]pyridines were explored, C3-arylation and alkynylation reactions processed smoothly through a classical Pd-catalyzed cross-coupling strategy (Schemes 4b and c). Overall, this simple, metal-free protocol could be extended as an efficient and practical method to construct more complex C-3-substituted imidazopyridines.
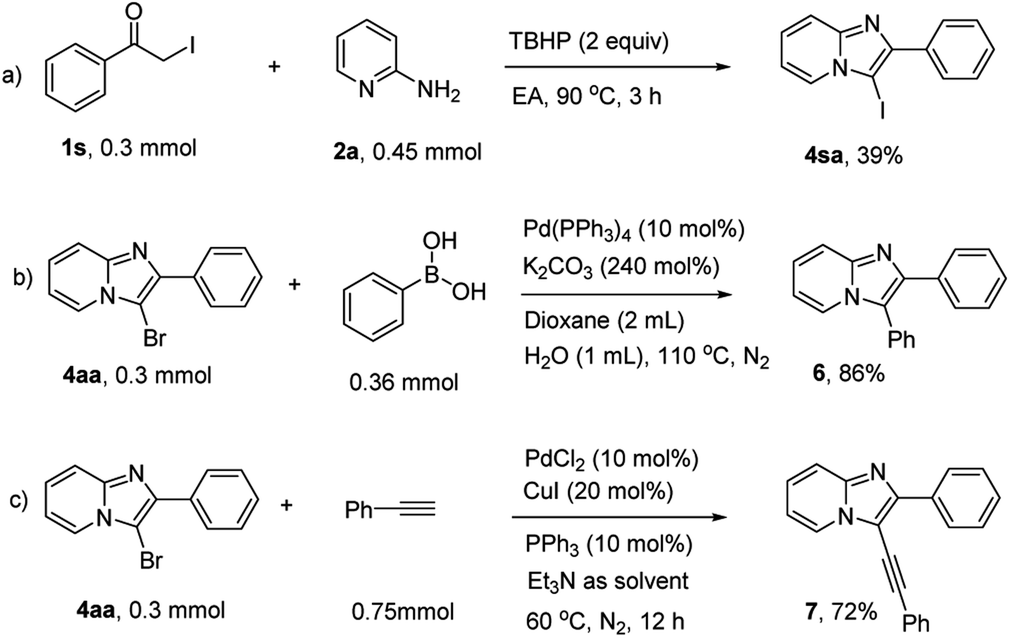 | ||
| Scheme 4 Investigation of the versatility of this strategy. | ||
To understand the mechanism of this tandem reaction, control experiments were carried out (Scheme 5). 5aa was isolated in both reactions (Scheme 5, 1). In protocol A, the reaction was suppressed remarkably when 2 equivalents of TEMPO, a radical trapping reagent, was added into the reaction system, indicating that the reaction might proceed via a free radical process (Scheme 5, 1). We speculated that 5aa was the key intermediate for these two reactions. Thus, some control experiments based on 5aa were conducted. 4aa was obtained in 42% yield when 5aa was subjected to HBr (1 equivalent) and TBHP (2 equivalents) (Scheme 5, 4) and 5aa reacted with Br2 could also form 4aa in 88% yield (Scheme 5, 5), these results showed that the bromine was formed during the synthesis of 3-bromoimidazo[1,2-a]pyridines. Furthermore, when 1-cyclohexen-1-ylbenzene was added to consume the in situ generated bromine, no desired 3-bromo-2-phenylimidazo[1,2-a]pyridine was observed (Scheme 5, 6). On the other hand, the amide product 3aa could be obtained from 5aa in I2/TBHP system (Scheme 5, 2) or only TBHP for longer time (Scheme 5, 3). Besides, no amide product (3aa) was obtained from 3-bromo-imidazopyridine (4aa) or 3-iodoimidazopyridine (4sa), which illustrated that 4aa could not be further transferred to 3aa. These results collectively indicated that there was a fierce competition between amidation and bromination, the iodine played an extremely important role in this C–C bond cleavage reaction.
 | ||
| Scheme 5 Control experiments. | ||
Based on the above results and previous knowledge, we proposed a plausible mechanism for this chemodivergent reaction.22 As shown in Scheme 6, the reaction proceeded via the initial displacement of the halogen atom by the pyridine ring nitrogen to afford pyridinium salt (8), which subjected a facile closure and provided the shared intermediate 5aa. In protocol A, t-BuO˙, which was generated in I2/TBHP system,23 attacked the double bond of 5aa to afford 9, 9 subsequently transferred to 11. Then, oxygen radical 12 was formed under t-BuO˙, 12 further transformed to 13 by C–C bond cleavage.10,20a,22e,24 Next, a tert-butyl formate was released from 13 leading to the formation of amide 14,25 which proceed an isomerization to afford the desired amide 3aa. In protocol B, the released hydrogen bromide was converted into bromine by oxidation of TBHP. The bromination of 5aa yielded the target molecule 4aa and the released hydrogen bromide could be oxidized to bromine and used for bromination again.
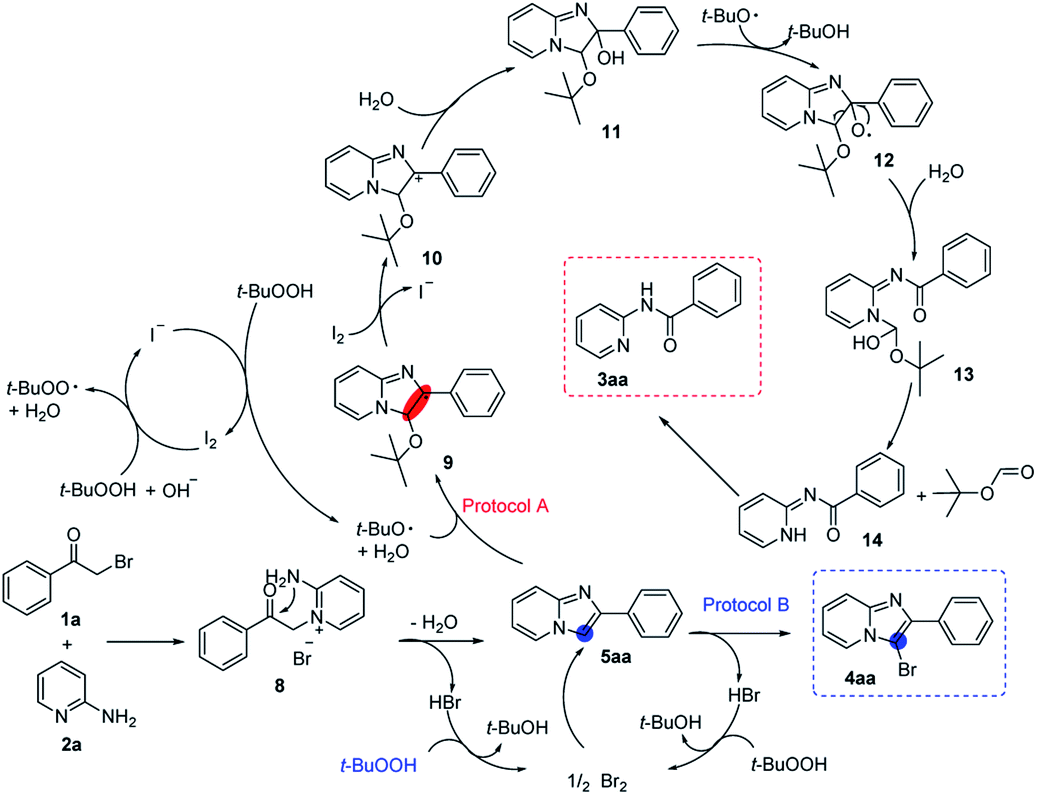 | ||
| Scheme 6 Proposed reaction mechanism. | ||
Conclusions
In conclusion, we have developed a divergent synthesis of pyridyl-amides and imidazopyridines from simple α-bromoketones and 2-aminopyridine. Various N-(pyridin-2-yl)amides or 3-bromo-imidazopyridines were obtained in moderate to good yields with high selectivity by tuning the catalyst. The one-pot tandem cyclization/bromination of α-bromoketones with 2-aminopyridine in the presence of TBHP provided versatile 3-bromo-imidazopyridines with excellent functional group tolerance and scalability. The α-haloketone acted as both the substrate and the bromo source, and the avoidance of transition-metal, fewer synthetic steps and high atom-economy make this protocol attractive. Whereas, the addition of I2 promote the oxidative C–C bond cleavage amidation afforded N-(pyridin-2-yl)amides in a metal-free condition. Less activity demonstrated by other metal catalysts suggested that I2 played a vital role in this reaction. C–C bond cleavage amidation is still a difficult and significant subject especially in modification and semisynthesis of natural medicine, which can simplify the synthetic procedure and make the semisynthesis more available. Iodine as a catalyst in C–C bond cleavage amidation is rarely reported. We believe our work will be useful for completing the C–C bond cleavage amidation field.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (No. 81673306, 81703348, 81874289), “Double First-Class” University project (No. CPU2018GY04, CPU2018GY35), China Pharmaceutical University, and the Early Career Scheme of the Research Grants Council of Hong Kong (No. 25100017).Notes and references
- (a) C. Annoni, T. Endoh, D. Hnedzko, E. Rozners and N. Sugimoto, Chem. Commun., 2016, 52, 7935 RSC; (b) T. L. Harris, C. J. Wenthur, A. Diego-Taboada, G. Mackenzie, T. S. Corbitt and K. D. Janda, Chem. Commun., 2016, 52, 4187 RSC; (c) P. Sakthivel, A. Ilangovan and M. P. Kaushik, Eur. J. Med. Chem., 2016, 122, 302 CrossRef CAS; (d) H. Y. Wang, Y. Qin, H. Li, L. J. Roman, P. Martásek, T. L. Poulos and R. B. Silverman, J. Med. Chem., 2016, 59, 4913 CrossRef CAS; (e) E. R. Baral, K. Sharma, M. S. Akhtar and Y. R. Lee, Org. Biomol. Chem., 2016, 14, 10285 RSC.
- (a) L. Ferrins, M. Gazdik, R. Rahmani, S. Varghese, M. L. Sykes, A. J. Jones, V. M. Avery, K. L. White, E. Ryan, S. A. Charman, M. Kaiser, C. A. S. Bergstrom and J. B. Baell, J. Med. Chem., 2014, 57, 6393 CrossRef CAS PubMed; (b) L. H. Heitman, J. P. D. van Veldhoven, A. M. Zweemer, K. Ye, J. Brussee and A. P. IJzerman, J. Med. Chem., 2008, 51, 4724 CrossRef CAS; (c) S. S. Kulkarni, B. Nightingale, C. M. Dersch, R. B. Rothmanb and A. H. Newman, Bioorg. Med. Chem. Lett., 2006, 16, 3371 CrossRef CAS; (d) K. Xu, Z. Y. Xiao, Y. B. Tang, L. Huang, C. H. Chen, E. Ohkoshi and K. H. Lee, Bioorg. Med. Chem. Lett., 2012, 22, 2772 CrossRef CAS.
- (a) G. Richa, L. Vijay and P. Kamaldeep, Curr. Top. Med. Chem., 2016, 16, 3590 CrossRef; (b) T. L. Kishbaugh, Curr. Top. Med. Chem., 2016, 16, 3274 CrossRef CAS; (c) C. Ravi and S. Adimurthy, Chem. Rec., 2017, 17, 1 CrossRef; (d) J. B. Xi, Y. F. Fang, B. Frett, M. L. Zhu, T. Zhu, Y. N. Kong, F. J. Guan, Y. Zhao, X. W. Zhang, H. Y. Li, M. L. Ma and W. H. Hu, Eur. J. Med. Chem., 2017, 126, 1083 CrossRef CAS; (e) J. Dam, Z. Ismail, T. Kurebwa, N. Gangat, L. Harmse, H. M. Marques, A. Lemmerer, M. L. Bode and C. B. de Koning, Eur. J. Med. Chem., 2017, 126, 353 CrossRef CAS.
- (a) R. Takise, K. Muto and J. Yamaguchi, Chem. Soc. Rev., 2017, 46, 5864 RSC; (b) Y. K. Zhang, J. J. Plattner, E. E. Easom, R. T. Jacobs, D. Guo, Y. R. Freund, P. Berry, V. Ciaravino, J. C. L. Erve, P. J. Rosenthal, B. Campo, F. J. Gamo, L. M. Sanz and J. Cao, J. Med. Chem., 2017, 60, 5889 CrossRef CAS; (c) B. M. Trost, J. J. Cregg and N. Quach, J. Am. Chem. Soc., 2017, 139, 5133 CrossRef CAS.
- (a) V. R. Pattabiraman and J. W. Bode, Nature, 2011, 480, 471 CrossRef CAS PubMed; (b) P. K. Mykhailiuk, V. Kubyshkin, T. Bach and N. Dudisa, J. Org. Chem., 2017, 82, 8831 CrossRef CAS; (c) S. Fuse, Y. Mifune, H. Nakamura and H. Tanaka, Nat. Commun., 2016, 7, 13491 CrossRef CAS; (d) H. M. Burke, L. McSweeney and E. M. Scanlan, Nat. Commun., 2017, 8, 15655 CrossRef.
- (a) R. M. de Figueiredo, J. S. Suppo and J. M. Campagne, Chem. Rev., 2016, 116, 12029 CrossRef CAS; (b) H. Lundberg, F. Tinnis, N. Selander and H. Adolfsson, Chem. Soc. Rev., 2014, 43, 2714 RSC; (c) A. Ojeda-Porras and D. Gamba-Sánchez, J. Org. Chem., 2016, 81, 11548 CrossRef CAS; (d) G. N. Papadopoulos and C. G. Kokotos, J. Org. Chem., 2016, 81, 7023 CrossRef CAS; (e) T. B. Halima, J. K. Vandavasi, M. Shkoor and S. G. Newman, ACS Catal., 2017, 7, 2176 CrossRef; (f) R. M. de Figueirede, J. S. Suppo, C. Midrier and J. M. Campagne, Adv. Synth. Catal., 2017, 359, 1963 CrossRef; (g) C. W. Cheung, M. L. Ploeger and X. Hu, Nat. Commun., 2017, 8, 14878 CrossRef.
- O. P. S. Patel, D. Anand, R. K. Maurya and P. O. Yadav, Green Chem., 2015, 17, 3728 RSC.
- V. Pappula, C. Ravi, S. Samanta and S. Adimurthy, ChemistrySelect, 2017, 2, 5887 CrossRef CAS.
- A. Ragupathi, A. Sagadevan, C. C. Lin, J. R. Hwu and K. C. Hwang, Chem. Commun., 2016, 52, 11756 RSC.
- P. Subramanian, S. Indu and K. P. Kaliappan, Org. Lett., 2014, 16, 6212 CrossRef CAS.
- Y. P. Liu, H. H. Sun, Z. J. Huang, C. Ma, A. J. Lin, H. Q. Yao, J. Y. Xu and S. T. Xu, J. Org. Chem., 2018, 83, 14307 CrossRef CAS PubMed.
- (a) Q. D. Wen, P. Lu and Y. G. Wang, Chem. Commun., 2015, 51, 15378 RSC; (b) H. Y. Zhan, L. M. Zhao, J. Q. Liao, N. Y. Li, Q. L. Chen, S. X. Qiu and H. Cao, Adv. Synth. Catal., 2015, 357, 46 CrossRef CAS; (c) D. C. Mohan, R. R. Donthiri, S. N. Rao and S. Adimurthy, Adv. Synth. Catal., 2013, 355, 2217 CrossRef; (d) A. K. Bagdi, M. Rahman, S. Santra, A. Majee and A. Hajra, Adv. Synth. Catal., 2013, 355, 1741 CrossRef CAS; (e) Z. J. Cai, S. Y. Wang and S. J. Ji, Adv. Synth. Catal., 2013, 355, 2686 CrossRef CAS; (f) C. He, J. Hao, H. Xu, Y. P. Mo, H. Y. Liu, J. J. Han and A. W. Lei, Chem. Commun., 2012, 48, 11073 RSC.
- (a) S. Kumar, N. Sharma, I. K. Maurya, A. K. K. Bhasin, N. Wangoo, P. Brandão, V. Félix, K. K. Bhasin and R. K. Sharma, Eur. J. Med. Chem., 2016, 123, 916 CrossRef CAS PubMed; (b) D. Dheer, K. R. Reddy, S. K. Rath, P. L. Sangwan, P. Das and R. Shankar, RSC Adv., 2016, 6, 38033 RSC; (c) F. Lovering, C. Aevazelis, J. Chang, C. Dehnhardt, L. Fitz, S. Han, K. Janz, J. Lee, N. Kaila, J. McDonald, W. Moore, A. Moretto, N. Papaioannou, D. Richard, M. S. Ryan, Z. K. Wan and A. Thorarense, ChemMedChem, 2016, 11, 217 CrossRef CAS.
- R. Semwal, C. Ravi, R. Kumar, R. Meena and S. Adimurthy, J. Org. Chem., 2019, 84, 792 CrossRef CAS.
- (a) H. M. Su, L. Y. Wang, H. H. Rao and H. Xu, Org. Lett., 2017, 19, 2226 CrossRef CAS PubMed; (b) H. Kim, M. Byeon, E. Jeong, Y. Baek, S. J. Jeong, K. Um, S. H. Han, G. U. Han, G. H. Ko, C. Maeng, J. Y. Son, D. Kim, S. H. Kim, K. Lee and P. H. Lee, Adv. Synth. Catal., 2019, 361, 2094 CrossRef CAS.
- Y. Gao, M. Z. Yin, W. Q. Wu, H. W. Huang and H. F. Jiang, Adv. Synth. Catal., 2013, 355, 2263 CrossRef CAS.
- X. Meng, C. Y. Yu, G. X. Chen and P. Q. Zhao, Catal. Sci. Technol., 2015, 5, 372 RSC.
- X. Q. Zhou, H. Yan, C. W. Ma, Y. Q. He, Y. M. Li, J. H. Cao, R. L. Yan and G. S. Huang, J. Org. Chem., 2016, 81, 25 CrossRef CAS.
- X. S. Xiao, Y. Xie, S. Y. Bai, Y. F. Deng, H. F. Jiang and W. Zeng, Org. Lett., 2015, 17, 3998 CrossRef CAS.
- (a) X. Q. Huang, X. Y. Li, M. C. Zou, S. Song, C. H. Tang, Y. Z. Yuan and N. Jiao, J. Am. Chem. Soc., 2014, 136, 14858 CrossRef CAS; (b) D. W. Flaherty, D. D. Hibbitts and E. Iglesia, J. Am. Chem. Soc., 2014, 136, 9664 CrossRef CAS.
- (a) M. Matsumura, Y. Sakata, A. Iwase, M. Kawahata, Y. Kitamura, Y. Murata, N. Kakusawa, K. Yamaguchi and S. Yasuike, Tetrahedron Lett., 2016, 57, 5484 CrossRef CAS; (b) Z. Fei, Y. P. Zhu, M. C. Liu, F. C. Jian and A. X. Wu, Tetrahedron Lett., 2013, 54, 1222 CrossRef CAS.
- (a) A. J. Elliott, H. Guzik and J. R. Soler, J. Heterocycl. Chem., 1982, 19, 1437 CrossRef CAS; (b) M. Jafarzadeh, E. Soleimani, H. Sepahvand and R. Adnan, RSC Adv., 2015, 5, 42744 RSC; (c) L. J. Ma, X. P. Wang, W. Yu and B. Han, Chem. Commun., 2011, 47, 11333 RSC; (d) C. D. Huo, J. Tang, H. S. Xie, Y. J. Wang and J. Dong, Org. Lett., 2016, 18, 1016 CrossRef CAS; (e) K. Yan, D. S. Yang, W. Wei, G. Q. Li, M. Y. Sun, Q. Y. Zhang, L. J. Tian and H. Wang, RSC Adv., 2015, 5, 100102 RSC; (f) W. K. Luo, X. Shi, W. Zhou and L. Yang, Org. Lett., 2016, 18, 2036 CrossRef CAS; (g) Z. J. Liu, J. Zhang, S. L. Chen, E. Shi, Y. Xu and X. B. Wan, Angew. Chem., Int. Ed. Engl., 2012, 51, 323 Search PubMed.
- G. Schmitz, Phys. Chem. Chem. Phys., 2001, 3, 4741 RSC.
- C. H. Tang and N. Jiao, Angew. Chem., Int. Ed., 2014, 53, 6528 CrossRef CAS.
- (a) F. Effenberger, M. Keill and u. E. Bessey, Chem. Ber., 1980, 113, 2110 CrossRef CAS; (b) M. Suchy, A. A. H. Elmehriki and R. H. E. Hudson, Org. Lett., 2011, 13, 3952 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra06724h |
| This journal is © The Royal Society of Chemistry 2019 |