
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMesoporous cerium oxide for fast degradation of aryl organophosphate flame retardant triphenyl phosphate
Jakub
Ederer
 *a,
Martin
Šťastný
b,
Marek
Došek
a,
Jiří
Henych
ab and
Pavel
Janoš
a
*a,
Martin
Šťastný
b,
Marek
Došek
a,
Jiří
Henych
ab and
Pavel
Janoš
a
aFaculty of Environment, University of Jan Evangelista Purkyně, Králova Výšina 7, 400 96 Ústí nad Labem, Czech Republic. E-mail: jakub.ederer@ujep.cz; Fax: +420-475-284-158; Tel: +420-475-284-111
bInstitute of Inorganic Chemistry of the Czech Academy of Sciences, Řež, 25068, Czech Republic
First published on 8th October 2019
Abstract
Cerium oxide nanoparticles were prepared by calcination of basic cerous carbonate (as a precursor) obtained by precipitation from an aqueous solution. Prepared samples were characterized by X-ray diffraction (XRD), infrared spectroscopy (FTIR), high resolution scanning electron microscopy (HRSEM), BET (Brunauer–Emmett–Teller) surface area and porosity measurement. Prepared cerium oxide was applied as a destructive sorbent for the fast and safe degradation of organophosphorus flame retardant triphenyl phosphate (TPP). It was shown that cerium dioxide was effective in the decomposition of TPP by cleavage of the P–O–aryl bond in the flame retardant molecule. A degradation mechanism for TPP on the ceria surface was proposed. The degradation is governed by conversion of TPP via diphenyl phosphate (DPP) to the final product identified as phenol (Ph). The key parameter increasing the degradation efficiency of CeO2 is the temperature of calcination. At optimum calcination temperature (500 °C), the produced ceria retains a sufficiently high surface area and attains an optimum degree of crystallinity (related to a number of crystal defects, and thus potential reactive sites). The fast and efficient degradation of organophosphorus flame retardant TPP was observed in a polar aprotic solvent (acetonitrile) that is miscible with water.
1. Introduction
Organophosphorus compounds are commonly used as pesticides (parathion-methyl, fenchlorphos), or flame retardants (triphenyl phosphate, tris(2-chloroethyl) phosphate, tris(2-ethylhexyl) phosphate).1 The organophosphorus flame retardants (PFRs) are widely used in industrial polymeric materials such as lubricants, electrical and electronic equipment2 and hydraulic fluids.3,4 Furthermore, PFRs are also used as additive chemicals because they are not covalently bound to the polymeric materials, however they are able to migrate into the environment5 and most of the flame-retardant products eventually become waste. Municipal waste is generally disposed of by incineration or landfilling. Incineration of flame-retardant products can produce various toxic compounds, including halogenated dioxins and furans further polluting water, air and soil.6 In the environment, PFRs may have a direct adverse effect on aquatic organisms and can contaminate drinking water.4 Triphenyl phosphate (TPP) is representative organophosphorus compound widely used as a flame retardant component in various industries.2–4 TPP is also used as antioxidant and stabilizer in phenolic, PVC, polyurethane, and other resins, and is considered as a neurotoxic and a new type of allergen in polyvinylchloride gloves7 which can cause allergic contact dermatitis.8 TPP degradation products and its fate in soil were investigated by Anderson et al.3 under different conditions (aerobic and anaerobic) or by Kawagoshi et al.9 in leachate from sea based solid waste disposal site. The degradation of TPP in soil and water is relatively a slow process.3 Therefore, the demands for remediation technologies that would be able to quickly and efficiently remove surpluses of these pollutants in the environment increase rapidly nowadays.The metal oxides are often used due to their excellent catalytic properties in petroleum, environmental and chemical industries as catalysts or catalysts support. Mixed metal oxides are also extensively used as fuel cells, gas sensors, and other applications. Nanostructured metal oxides can be also applied as reactive adsorbents as we have shown elsewhere.10 Nevertheless, to the best of our knowledge the decomposition of TPP by nanocrystalline oxide was not widely investigated.
Cerium oxide (CeO2) is considered, due to extraordinary thermal and chemical stability, as the most important rare-earth oxide. One of the most important applications of cerium oxide is as a catalyst promoter, catalyst or catalysts supports.11,12 Depending on the intended use, several methods have been developed for preparing cerium oxide, such as chemical vapor deposition,13 electrochemical synthesis,14 template synthesis,15 biological synthesis,16etc. Li et al.17 compared some of the methods for the preparation of CeO2, namely the sol–gel method, precipitation method, and homogeneous hydrolysis. However, owing to the advantages of simple process, easy scale-up and low cost, the precipitation technique has attracted the most extensive attention; cerium dioxide is most often prepared by the precipitation of sparingly soluble cerous oxalates or carbonates and subsequent calcination. Cerium carbonates with variable composition and morphology may be precipitated from an aqueous solution of alkaline or ammonium carbonate/bicarbonate18,19 or by a gaseous mixture of carbon dioxide and ammonia.
Although CeO2 particles prepared by the precipitation technique have been extensively studied, most of the previous reports were focused on effects of cerium precursors, ligands and additives in the reaction media. Less information was reported on the influence of the reaction temperature. However, the temperature during calcination plays a crucial role in the surface properties (specific surface area, the presence of surface active sites), crystallinity, and other physicochemical characteristics.17,20,21
In this work, we have tested the unique properties of cerium (III, IV) dioxides for fast and safe degradation of organophosphorus flame retardants in the environment using triphenyl phosphate (TPP) as a model compound. The sorbents prepared by our developed procedure are inexpensive and efficient in degradation of organophosphorus compounds.9–11 As we have shown,10 nanocrystalline cerium oxide can be used for degradation of organophosphorus pollutants, but it can be also used in catalysis,22 fuel cells,11,23 as polishing material24,25 and in biomedical applications.26 The precipitated ceria samples were annealed at different temperatures (200; 500; 800; and 1100 °C) and characterized by scanning electron microscopy (SEM), Brunauer–Emmett–Teller (BET) surface area, X-ray diffraction (XRD) and infrared spectroscopy (FTIR). The main TPP degradation products diphenyl phosphate (DPP) and phenol (Ph) were identified and quantified using high-performance liquid chromatography with DAD detection (HPLC-DAD). The mechanism of the degradation of TPP on the ceria in a polar aprotic solvent (acetonitrile) was proposed and discussed.
2. Materials and methods
2.1 Chemicals and preparation of sorbents
All chemicals were obtained from commercial sources and were used without further purification. Triphenyl phosphate (TPP), diphenyl phosphate (DPP) and phenol (Ph) were purchased from Sigma Aldrich (Steinheim, Germany). All purchased chemicals were of analytical grade (p.a.). TPP stock solution (5 g L−1), DPP and Ph stock solutions (equimolar to TPP) were prepared in acetonitrile (ACN). HPLC-grade organic solvents and deionized water from a GORO Pharmapur system (Goro, Prague, Czech Republic) were used to prepare the solutions including mobile phases for liquid chromatography. Cerous nitrate, Ce(NO3)3·6H2O was obtained from Sigma-Aldrich (Steinheim, Germany) as reagent grade chemicals with purity 99.9%; ammonium bicarbonate, NH4HCO3, 99.5%, was obtained from the same supplier. The destructive sorbent for pesticide degradation was prepared by the procedure described elsewhere.10 Briefly, the carbonate precursor was prepared by precipitation of an aqueous solution of Ce(NO3)3 (0.2 mol L−1) with an excess of NH4HCO3 (0.5 mol L−1) under stirring. After the precipitation process, the agitation continued for one more hour and the precipitate was left in the solution until the next day. The precipitate was separated by filtration, washed with deionized water and dried overnight at 110 °C. The CeO2 samples were prepared from carbonate precursor by calcination at selected temperatures for two hours in a muffle furnace. Titanium dioxide (TiO2, Degusa), nickel oxide (NiO, nanopowder < 50 nm), zinc oxide (ZnO, nanopowder < 100 nm) and iron (II, III) oxide (Fe3O4, nanopowder < 50 nm) were purchased from Sigma Aldrich. Eqn (1)–(5) illustrated reaction during preparation of carbonate precursor and subsequent calcination.Ce(NO3)3·6H2O(s) → ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Ce(aq)3++3NO(aq)3− + 6H2O(aq), Ce(aq)3++3NO(aq)3− + 6H2O(aq), | (1) |
| NH4HCO3(s) → NH4(aq)+ + H(aq)+ + CO3(aq)−, | (2) |
| 2Ce(aq)3+ + 2CO3(aq)− + nH2O(aq) → Ce2O(CO3)2·nH2O(s)↓, | (3) |
 | (4) |
 | (5) |
2.2. Characterization methods
The diffraction pattern was collected with diffractometer Bruker D2 equipped with conventional X-ray tube (Cu Kα radiation, 30 kV, 10 mA) and the LYNXEYE 1-dimensional detector. The primary divergence slit module width 0.6 mm, Soler Module 2.5, Airscatter screen module 2 mm, Ni Kbeta-filter 0.5 mm, step 0.00607° and time per step 0.3 s were used. Qualitative analysis was performed with the DiffracPlus Eva software package (Bruker AXS, Germany) using the ICDD PDF database.The surface area of the sample was determined from nitrogen adsorption–desorption isotherm at liquid nitrogen temperature using a Coulter SA3100 instrument with 15 min outgas at 150 °C. The Brunauer–Emmett–Teller (BET) method was used for surface area calculation.
The FTIR spectra of as prepared CeO2 samples were measured by diffuse reflectance Nicolet Impact 400D spectrophotometer (Thermo Nicolet, Madison) in diffuse reflectance mode in the 4000–500 cm−1 range at 128 scans per spectrum and expressed in a transmittance mode (%T). Raw FTIR data were processed utilizing the OMNIC 7.3 software.
High-resolution scanning electron microscopy (HRSEM) analysis was conducted on an FEI Nova NanoSEM scanning electron microscope equipped with an Everhart-Thornley detector (ETD), Through Lens detector (TLD), and accelerating voltage 4–30 kV. The samples were plated on the carbon holder.
2.3. Degradation of organophosphorus flame retardants
For testing of stoichiometric degradation of TPP a new testing procedure was developed, because the degradation procedure published elsewhere10 was found to be inappropriate in our case. 800 mg of each tested sorbent was weighted into the 25 mL glass vials with magnetic stirrer, and 4 mL of acetonitrile (ACN) and 12 mL of TPP stock solution in ACN (5 g L−1) was added. In corresponding time intervals (0.5; 8; 16; 32; 64; 96 and 128 minutes) 0.5 mL aliquots were taken and added into 4 mL vials containing 0.5 mL of formic acid (1:1; v/v) and filled with mobile phase (75% methanol and 25% formic acid (1%)). Vials were centrifuged at 4000 RPM for 4 min and the supernatant was transferred into the 25 mL volumetric flask. The extraction process of degradation products of TPP from sorbents surfaces with a mobile phase was done three times. All the supernatants were combined into one volumetric flask, made up to the mark with the mobile phase, and analyzed immediately by liquid chromatography (HPLC). Preparation of standard solution of TPP, DPP, and phenol were made by the same procedure without adding the sorbent. All experiments were performed at a laboratory temperature 22 ± 1 °C.
2.4. Analysis and data evaluation
Ultra-high performance liquid chromatography (U-HPLC) Dionex UltiMate 3000 system was used to determine the TPP concentrations and for the detection of degradation products. The liquid chromatograph consisted of the high-pressure pump, the Rheodyne 7125 injection valve with 20 μL sampling loop, vacuum degasser, and the variable wavelength DAD detector operating at a wavelength of 261 nm (absorption maximum for TPP), 265 nm (absorption maximum for DPP) and 275 nm (absorption maximum for Ph). Chromatographic separations were carried out on the Phenomenex column 150 × 4.6 mm packed with pentafluorophenyl stationary phase Kinetex, PFP, 5 μm (USA) with mobile phase consisting of 75% methanol and 25% formic acid (1%) (v/v); the flow rate was 1 mL min−1. MS Excel and OriginPro 8.5 (OriginLab Corp., USA) software were used for calculations and data evaluations.3. Results and discussion
3.1 Characterization of prepared sample
Fig. 1 shows the results of powder X-ray diffraction (XRD). The XRD analysis identified a single crystalline phase in the calcination products obtained in the temperature range of 200–1100 °C. In all samples was identified cerium dioxide with its characteristic face-centered cubic fluorite-type structure with corresponding diffraction lines (111), (200), (220), (311), (222), (400), (331) and (420) located at 28.535°, 33.080°, 47.495°, 56.348°, 59.102°, 69.427°, 76.710°, 79.073° and 88.447° (ICDD PDF 34-0394). From the individual diffractograms are evident constriction and increase of intensity of diffraction lines with increasing calcination temperature, as a consequence of increasing of the crystallite size. The crystallite size was calculated from the diffraction line extension using the Scherrer eqn (6) and the data are summarized in Table 1 (second column): | (6) |
 | ||
| Fig. 1 XRD patterns of cerium carbonate (precursor) and cerium oxides annealed at 200, 500, 800, 1100 °C and cerium oxide 00-004-0593 (from top to bottom). | ||
| Sample (annealing temperature, °C) | Crystallite size (nm) | BET (m2 g−1) | Total pore volume (cm3 g−1) |
|---|---|---|---|
| CeO2_200 | 12 | 102 | 0.05 |
| CeO2_500 | 28 | 85 | 0.07 |
| CeO2_800 | 43 | 18 | 0.04 |
| CeO2_1100 | 51 | 6 | 0.05 |
Using the X-ray diffraction (XRD) analysis, a crystalline phase of the Ce2O(CO3)2·nH2O type was identified (see eqn (3)). Destructive sorbents were prepared from this carbonate precursor by calcination at pre-determined temperatures in the range of 200–1100 °C for 2 h; possible chemical reactions are described by eqn (5), which suggests that a non-stoichiometric cerium oxide (CeO2−x) may occur as a result of the changes of the surface to volume ratio.27 The increasing annealing temperature cause the increase of nanocrystallites size, and decrease in the surface-to-volume ratio of nanocrystallites. It reduces the formation energy of oxygen vacancies positioned close to a crystallite surface. Vacancy creation leads to a change in electronic density since the electrons that are left behind are led to oxygen atom occupy 4f electron energy levels of cerium ions in the vicinity of a newly created vacancy and reduce Ce4+ ions to the Ce3+ state.28,29
SEM images of the ceria powders prepared by the chemical precipitation method with various calcination temperature namely 200; 500; 800; and 1100 °C are shown in Fig. 2.
 | ||
| Fig. 2 SEM images of the prepared samples (a) carbonate precursor; (b) particle-size distribution curve with pore diameter about 3–4 nm and nitrogen adsorption–desorption pattern (inset) of sample CeO2 annealed at 500 °C; (c) cerium carbonate annealed at 200 °C; (d) cerium carbonate annealed at 500 °C; (e) cerium carbonate annealed at 800 °C; and (f) cerium carbonate annealed at 1100 °C. | ||
From the SEM images, it was found that all the nanoparticles exhibit a flake-like morphology and clusters of thin plates of irregular shapes with a characteristic diameter of several micrometers. During calcination, the plates were broken down into smaller submicron particles that remained assembled in larger clusters. The specific surface area of the as-prepared samples, calculated by the multi-point Brunauer–Emmett–Teller (BET) method ranges from 6 to 102 m2 g−1 and total pore volume was in range 0.04–0.07 cm3 g−1 (Table 1). Barrett–Joyner–Halenda pore-size distribution plot and nitrogen adsorption/desorption isotherms (inset) of the as-prepared representative sample annealed at 500 °C is shown in Fig. 2(b). According to IUPAC notation,30 microporous materials have pore diameters of less than 2 nm and macroporous materials have pore diameters of greater than 50 nm; the mesoporous category thus lies in the middle. The mean pore size is around ∼3–4 nm and the pore size distribution being relatively narrow. All samples have a type IV isotherm, which is characteristic of mesoporous materials with type H2 hysteresis, which is a characteristic of large-pore mesoporous materials and can be ascribed to capillary condensation in mesopores. The high steepness of the hysteresis indicates the high order of mesoporosity.
Fig. 3 shows the FTIR spectra of the ceria nanopowders calcinated at various temperatures. The absorption peak at around 1300–1400 cm−1 band was identified by its intense vibrations NO (1384.23 cm−1) due to the presence of unreacted nitrate.31
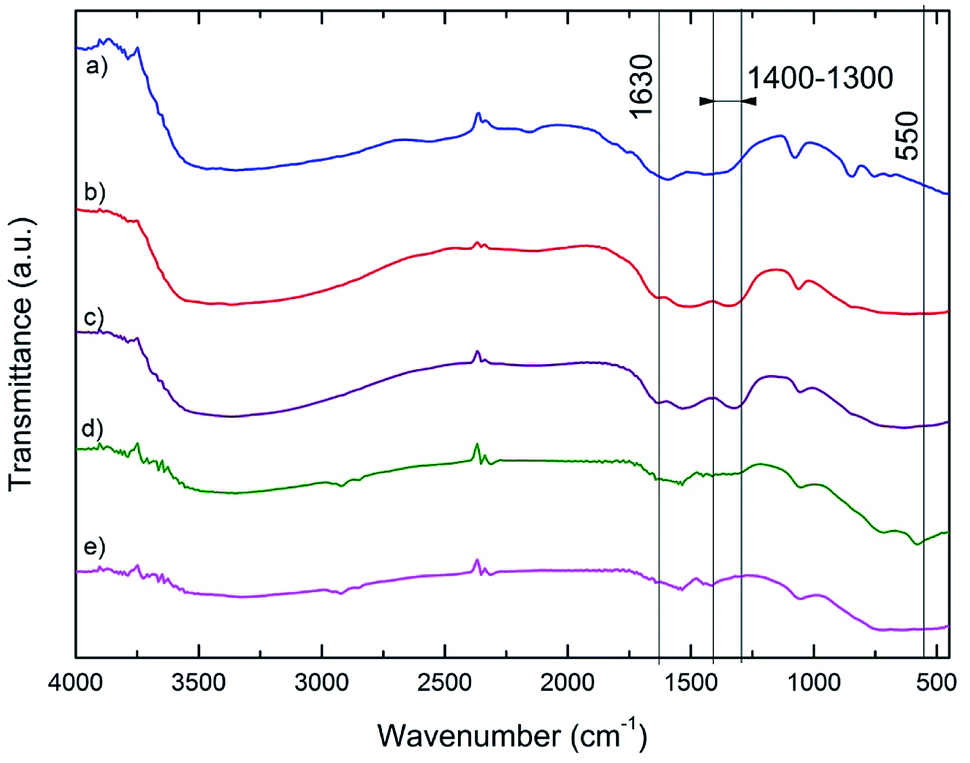 | ||
| Fig. 3 FTIR spectra of annealed CeO2 powders prepared by the chemical precipitation method: (a) carbonate precursor, (b) 200 °C, (c) 500 °C, (d) 800 °C and (e) 1100 °C. | ||
The broad absorption peak at 3000–3600 cm−1 and peak at around 1630 cm−1 correspond to hydroxyl groups υ(O–H) of water on the surface of the sorbent. In the as-prepared and annealed samples the residual water and hydroxyl groups were detected and further heat treatment is causing their elimination. FTIR spectra of cerium carbonate and annealed sample (200 °C) show small absorption due to the carbon–oxygen double bond (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) at about 2500 cm−1.32,33 It was also identified weak absorption band corresponding to the valence vibration Ce–O (550.55 cm−1).34
O) at about 2500 cm−1.32,33 It was also identified weak absorption band corresponding to the valence vibration Ce–O (550.55 cm−1).34
3.2 Degradation mechanism of TPP on CeO2 surface
Some transition metal oxides (such as MgO, CaO, Al2O3, etc.) exhibit unique ability to adsorb and decompose organophosphates from the group of organophosphorus pesticides, chemical warfare agents (CWA's) and also aryl organophosphate flame retardants.35–38The time dependence of the TPP degradation was measured and the experimental data were fitted to the Guldberg–Waage form equations for describing of consecutive reactions more than a single reaction (i.e., a reaction network):39
| qTPP = q0TPPexp(−k1τ) | (7) |
 | (8) |
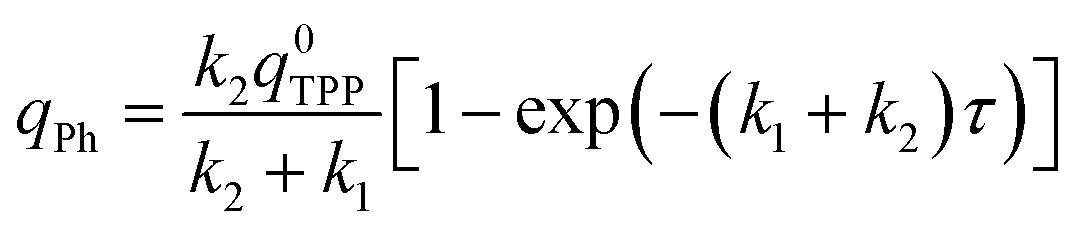 | (9) |
| Sample | Triphenyl phosphate (TPP) | Diphenyl phosphate (DPP) | Phenol (Ph) | ||||
|---|---|---|---|---|---|---|---|
| Degree of removal (%) | k 1 (min−1) | k 1 (min−1) | k 2 (min−1) | Degree of conversion (%) | k 1 (min−1) | k 2 (min−1) | |
| CeO2_200 | 51.8 | 0.119 | ND | ND | 40.9 | 0.056 | 0.102 |
| CeO2_500 | 70.2 | 0.459 | 0.036 | 0.069 | 115.3 | 0.043 | 0.082 |
| CeO2_800 | 16.7 | 0.043 | 0.018 | 0.133 | 5.95 | 1.858 | 1.978 |
| CeO2_1100 | 3.10 | 0.011 | ND | ND | ND | ND | ND |
 | ||
| Fig. 4 Degradation of TPP on cerium oxide annealed at 200 °C, 500 °C, 800 °C and 1100 °C. | ||
Organophosphates rapidly degrade in the environment by hydrolysis. The hydrolysis of organophosphate esters generally takes place either through a trigonal bipyramidal hydroxyphosphorane as intermediate (with the expansion of the coordination number of phosphorus from four to five).40 Similarly, on metal oxides surface the organophosphate species can be adsorbed and chemically decomposed that is often attributed to the proportion lot of reactive sites. Therefore, they can be effectively used as industrial adsorbents,41 catalysts,42 and several other potential decontamination applications.43 Due to their high surface area, a large number of highly reactive edges, corner defects sites, defects in the crystal lattice (Lewis acid attributed to metal cations) and reactive sites (such as surface hydroxyl groups), they may chemically degrade toxic compounds down to non-toxic or only slightly toxic organic compounds.44 Janoš and co-workers (2014) found that in nanocrystalline CeO2 are irregularities in the lattice near the surface such as Ce3+ that allows adsorption and stoichiometric degradation of toxic organophosphorus pesticides (e.g. parathion methyl, chlorpyrifos, etc.).20 As we shown in this work by XPS, annealing temperature have negligible effect on Ce3+ content, and hence Ce3+/Ce4+ ratio. The results suggested that Ce3+/Ce4+ ratio could not be one of the main parameters involved in CeO2 reactivity and irregularities, different particles morphology also participate in reactivity. Zhang et al. showed that CeO2 nanoparticles with similar morphology, nevertheless with different oxygen vacancies concentration showed different catalytic activity in photocatalytic water oxidation.45
Similarly to pesticides, other organophosphates like triphenyl phosphate (TPP) can degrade on the surface of CeO2. The reaction mechanism, shown in Fig. 6, suggest that the reactions are similar to hydrolysis reactions proceeding in the environment. The surface hydroxyls (−OH) and surface physisorbed water can act as strong nucleophiles and attack the electrophile phosphorus center.46
Using a simplified degradation test (fixed reaction time 32 min), the degradation efficiency of the cerium oxides annealed at the temperatures of 200 to 1100 °C was compared with the degradation efficiency of some other transition metal oxides. As can be seen, the other tested oxides are not suitable for decomposition of TPP or other organophosphorus compounds. The cerium oxide samples annealed at 200 and 500 °C exhibits the highest TPP activity (see Fig. 5). TiO2 has shown relatively highest activity compared to other transition metal oxides which could be probably ascribed to the slight influence of photocatalytic properties of TiO2 that could be subject for further research.
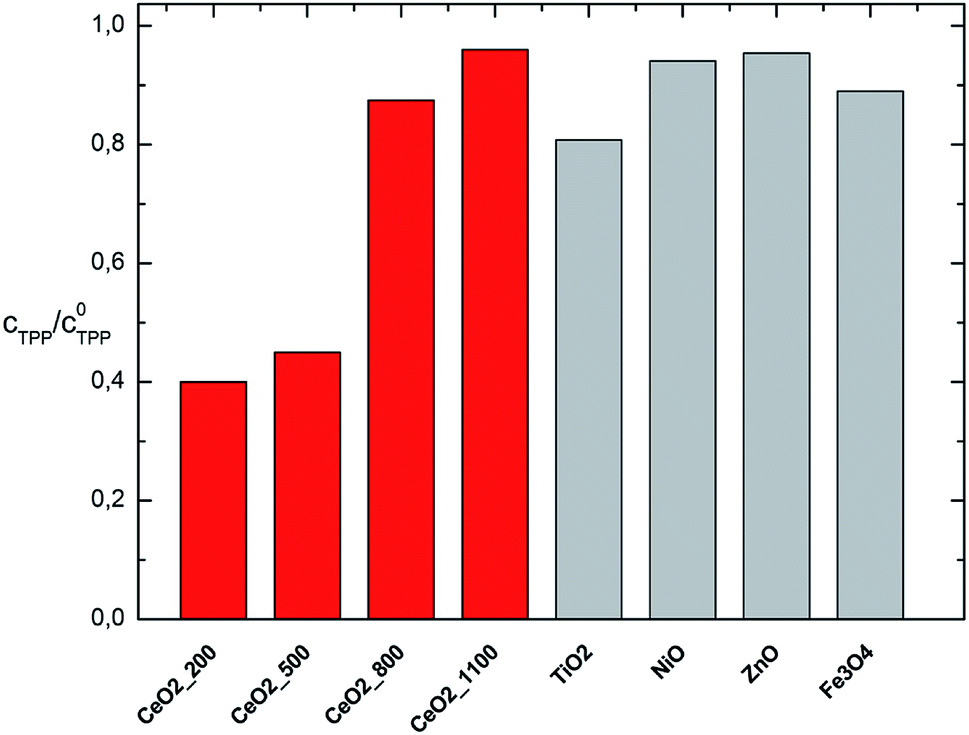 | ||
| Fig. 5 TPP decomposition activity of CeO2 annealed at 200, 500, 800 and 1100 °C compared with other metal oxides (TiO2, NiO, ZnO, and Fe3O4) after 32 min reaction. | ||
In polar media (acetonitrile), triphenyl phosphate (TPP) leads to the formation of phenol (Ph) as the main degradation product via intermediate diphenyl phosphate (DPP). It has been hypothesized that TPP rapidly adsorbs and decomposes through the phosphoryl oxygen on the surface of CeO2 at Lewis acid (metal atom) or at Brønsted acid (surface hydroxyl) sites.47,48 The mechanism of interaction (Fig. 6) on the CeO2 surface, suggested that, at first, TPP is adsorbed molecularly through the phosphoryl oxygen to the surface at an acid site (metal cation Ce4+), followed by elimination of the phenoxy group. This phenoxy group subsequently combines with surface hydrogen atoms to release phenol into the solvent as the main degradation product, which can be immediately analyzed with chromatography (e.g. HPLC-MS).
 | ||
| Fig. 6 Hydrolysis reactions of TPP on the surface of nanostructured CeO2: R1, R2, R3 show phenyl groups. | ||
The kinetics of triphenyl phosphate (TPP) hydrolysis was investigated on CeO2 powders prepared at various calcination temperatures. On the CeO2 (500 and 800 °C) surface were found hydrolysis products diphenyl-phosphate (DPP) and phenol (Ph). Further hydrolysis of diphenyl-phosphate probably proceed much slower and no other intermediate (e.g. monophenyl phosphate) was not found in these experiments.
4. Conclusion
Cerium oxide is very perspective reactive adsorbent for the rapid destruction of aryl organophosphate flame retardants, alternatively, other organophosphates compounds (OPs). CeO2 samples were synthesized by the simple chemical precipitation of cerous nitrate with an excess of ammonium bicarbonate in aqueous solution. The results showed that prepared CeO2 samples were effective in the decomposition of triphenyl phosphate (TPP) by cleavage of the P–O–aryl bond in the retardant molecule in a polar aprotic solvent (acetonitrile). These findings could lead to the future use of cerium-based reactive sorbents for practical applications in decontamination nanotechnologies such as polar “water-compatible” degradation strategy in connection with the technologies supporting bioremediation techniques for bio-removal of phenolic compounds.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Additional financial support was obtained from the Internal Student Grant Agency (Grant No. SPS 984599) of the University of Jan Evangelista Purkyně in Ústí nad Labem. The financial support of the Czech Scientific Foundation (grant no. 19-07460S) is acknowledged. Michaela Slušná from the Materials Chemistry Department, Institute of Inorganic Chemistry AS CR v.v.i., Řež, is thanked for measurements of the surface areas and porosity of samples.References
- A. Möller, R. Sturm, Z. Xie, M. Cai, J. He and R. Ebinghaus, Environ. Sci. Technol., 2012, 46, 3127–3134 CrossRef.
- C. C. Zhang and F. S. Zhang, Chem. Eng. J., 2014, 240, 10–15 CrossRef CAS.
- C. Anderson, D. Wischer, A. Schmieder and M. Spiteller, Chemosphere, 1993, 27, 869–879 CrossRef CAS.
- K. Lin, Environ. Chem. Lett., 2009, 7, 309–312 CrossRef CAS.
- S. H. Brandsma, J. De Boer, P. E. G. Leonards, W. P. Cofino and A. Covaci, TrAC, Trends Anal. Chem., 2013, 43, 217–228 CrossRef CAS.
- H. Meyer, M. Neupert, W. Pump and B. Willenberg, Kunstst. Ger. Plast., 1993, 83, 3–6 Search PubMed.
- K. Suuronen, M. Pesonen, M. L. Henriks-Eckerman and K. Aalto-Korte, Contact Dermatitis, 2013, 68, 42–49 CrossRef CAS.
- T. P. Keogh, J. Am. Dent. Assoc., 1999, 130, 474–476 CrossRef CAS.
- Y. Kawagoshi, S. Nakamura and I. Fukunaga, Chemosphere, 2002, 48, 219–225 CrossRef CAS.
- P. Janoš, P. Kuráň, M. Kormunda, V. Štengl, T. M. Grygar, M. Došek, M. Štastný, J. Ederer, V. Pilařová and L. Vrtoch, J. Rare Earths, 2014, 32, 360–370 CrossRef.
- Y. Zhou and J. a. Switzer, J. Alloys Compd., 1996, 237, 1–5 CrossRef CAS.
- A. Bumajdad, J. Eastoe and A. Mathew, Adv. Colloid Interface Sci., 2009, 147–148, 56–66 CrossRef CAS.
- K. D. Pollard, H. A. Jenkins and R. J. Puddephatt, Chem. Mater., 2000, 12, 701–710 CrossRef CAS.
- Y. Zhou, R. J. Phillips and J. A. Switzer, J. Am. Ceram. Soc., 1995, 78, 981–985 CrossRef CAS.
- K. S. Lin and S. Chowdhury, Int. J. Mol. Sci., 2010, 11, 3226–3251 CrossRef CAS.
- B. M. Gel, G. S. Priya, A. Kanneganti, K. A. Kumar, K. V. Rao and S. Bykkam, International Journal of Scientific and Research Publications, 2014, 4, 1–4 Search PubMed.
- M. Li, Z. Liu, Y. Hu, Z. Shi and H. Li, Colloids Surf., A, 2007, 301, 153–157 CrossRef CAS.
- M.-Y. Cho, K.-C. Roh, S.-M. Park, H.-J. Choi and J.-W. Lee, Mater. Lett., 2010, 64, 323–326 CrossRef CAS.
- Y. Zhai, S. Zhang and H. Pang, Mater. Lett., 2007, 61, 1863–1866 CrossRef CAS.
- P. Janoš, T. Hladík, M. Kormunda, J. Ederer and M. Šťastný, Adv. Mater. Sci. Eng., 2014, 2014, 1–12 CrossRef.
- J. Luňáček, O. Životský, P. Janoš, M. Došek, A. Chrobak, M. Maryško, J. Buršík and Y. Jirásková, J. Alloys Compd., 2018, 753, 167–175 CrossRef.
- P. Janoš, J. Ederer and M. Došek, Nova Biotechnol. Chim., 2014, 13, 148–161 Search PubMed.
- K. Eguchi, T. Setoguchi, T. Inoue and H. Arai, Solid State Ionics, 1992, 52, 165–172 CrossRef CAS.
- P. Janoš, P. Kuráň, J. Ederer, M. Šastný, L. Vrtoch, M. Pšenička, J. Henych, K. Mazanec and M. Skoumal, Adv. Mater. Sci. Eng., 2015, 2015, 1–8 CrossRef.
- P. Janoš, J. Ederer, V. Pilařová, J. Henych, J. Tolasz, D. Milde and T. Opletal, Wear, 2016, 362–363, 114–120 CrossRef.
- S. M. Hirst, A. S. Karakoti, R. D. Tyler, N. Sriranganathan, S. Seal and C. M. Reilly, Small, 2009, 5, 2848–2856 CrossRef CAS.
- S. Deshpande, S. Patil, S. V. Kuchibhatla and S. Seal, Appl. Phys. Lett., 2005, 87, 133113 CrossRef.
- L. Minervini, Solid State Ionics, 1999, 116, 339–349 CrossRef CAS.
- S. Aškrabia
![[c with combining umlaut]](https://www.rsc.org/images/entities/char_0063_0308.gif) , Z. Dohčevia-Mitrovia, A. Kremenovia, N. Lazarevia, V. Kahlenberg and Z. V. Popovia, J. Raman Spectrosc., 2012, 43, 76–81 CrossRef.
, Z. Dohčevia-Mitrovia, A. Kremenovia, N. Lazarevia, V. Kahlenberg and Z. V. Popovia, J. Raman Spectrosc., 2012, 43, 76–81 CrossRef. - F. Caruso, W. V. Gmbh and W. Isbn, Colloids and Colloid Assemblies, 2004, vol. 14 Search PubMed.
- M. Chelliah, J. B. B. Rayappan and U. M. Krishnan, J. Appl. Sci., 2012, 12, 1734–1737 CrossRef CAS.
- M. Mecozzi, E. Pietrantonio, M. Amici and G. Romanelli, Analyst, 2001, 126, 144–146 RSC.
- N. Kovac, J. Faganeli, O. Bajt, B. Orel and A. Surca Vuk, Mater. Geoenvirnment, 2005, 52, 81–85 CAS.
- A. a. Ansari, P. R. Solanki and B. D. Malhotra, J. Biotechnol., 2009, 142, 179–184 CrossRef CAS.
- V. Štengl, V. Houšková, S. Bakardjieva, N. Murafa, M. Maříková, F. Opluštil and T. Němec, Mater. Charact., 2010, 61, 1080–1088 CrossRef.
- S. Rajagopalan, O. Koper, S. Decker and K. J. Klabunde, Chem. - Eur. J., 2002, 8, 2602–2607 CrossRef CAS.
- T. H. Mahato, G. K. Prasad, B. Singh, K. Batra and K. Ganesan, Microporous Mesoporous Mater., 2010, 132, 15–21 CrossRef CAS.
- G. W. Wagner, O. B. Koper, E. Lucas, S. Decker and K. J. Klabunde, J. Phys. Chem. B, 2000, 104, 5118–5123 CrossRef CAS.
- R. W. Missen, C. A. Mims and B. A. Saville, Introduction to Chemical Reaction Engineering, Wiley, 1998 Search PubMed.
- S. Bondarenko and J. Gan, Environ. Toxicol. Chem., 2004, 23, 1809 CrossRef CAS.
- S.-Y. Lee and S.-J. Park, J. Ind. Eng. Chem., 2014, 23, 1–11 Search PubMed.
- C. L. Carnes and K. J. Klabunde, J. Mol. Catal. A: Chem., 2003, 194, 227–236 CrossRef CAS.
- T. H. Mahato, G. K. Prasad, B. Singh, J. Acharya, a. R. Srivastava and R. Vijayaraghavan, J. Hazard. Mater., 2009, 165, 928–932 CrossRef CAS.
- N. Sharma and R. Kakkar, Adv. Mater. Lett., 2013, 4, 508–521 CrossRef.
- Y.-C. Zhang, Z. Li, L. Zhang, L. Pan, X. Zhang, L. Wang, Fazal-e-Aleem and J.-J. Zou, Appl. Catal., B, 2018, 224, 101–108 CrossRef CAS.
- Y. Rao and D. M. Antonelli, J. Mater. Chem., 2009, 19, 1937 RSC.
- M. B. Mitchell, V. N. Sheinker and E. a Mintz, J. Phys. Chem. B, 1997, 101, 11192–11203 CrossRef CAS.
- E. Lucas, S. Decker, A. Khaleel, A. Seitz, S. Fultz, A. Ponce, W. Li, C. Carnes and K. J. Klabunde, Chem. - Eur. J., 2001, 7, 2505–2510 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |