
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQuinoline-triazole hybrids inhibit falcipain-2 and arrest the development of Plasmodium falciparum at the trophozoite stage†
Anju Singh‡
a,
Md Kalamuddin‡b,
Asif Mohmmedb,
Pawan Malhotra*b and
Nasimul Hoda *a
*a
aDrug Design and Synthesis Lab., Department of Chemistry, Jamia Millia Islamia, Jamia Nagar, New Delhi-110025, India. E-mail: nhoda@jmi.ac.in; Fax: +91-11-26985507; Tel: +91-9910200655
bInternational Centre for Genetic Engineering and Biotechnology (ICGEB), Aruna Asaf Ali Marg, New Delhi-110067, India. E-mail: pawanm@icgeb.res.in; Tel: +91-11-267423
First published on 29th November 2019
Abstract
Falcipain-2 (FP2) is a papain family cysteine protease and a key member of the hemoglobin degradation pathway, a process that is required at erythrocytic stages of Plasmodium falciparum to obtain amino acids. In this study, we report a set of 10 quinoline-triazole-based compounds (T1–T10) which exhibit a good binding affinity for FP2, inhibit its catalytic activity at micromolar concentrations and thereby arrest the parasite growth. Compounds T4 and T7 inhibited FP2 with IC50 values of 16.16 μM and 25.64 μM respectively. Both the compounds T4 and T7 arrested the development of P. falciparum at the trophozoite stage with an EC50 value 21.89 μM and 49.88 μM. These compounds also showed morphological and food-vacuole abnormalities like E-64, a known inhibitor of FP2. Our results thus identify the quinoline-triazole-based compounds as a probable starting point for the design of FP2 inhibitors and they should be further investigated as potential antimalarial agents.
1. Introduction
Malaria is a major life-threatening disease caused by Plasmodium parasites. According to the WHO 2018 report, Plasmodium parasites affect 216 million people each year. In 2017, 445![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 people, mostly children, died due to malaria. Malaria is caused by different species of Plasmodium protozoan, among which P. falciparum is responsible for causing the most deaths due to malaria, especially among children under the age of 5 in Africa. The malaria parasite follows a complex life cycle that includes many formative stages in the human host and mosquito vector, which contributes greatly to the challenge of its treatment.1–4
000 people, mostly children, died due to malaria. Malaria is caused by different species of Plasmodium protozoan, among which P. falciparum is responsible for causing the most deaths due to malaria, especially among children under the age of 5 in Africa. The malaria parasite follows a complex life cycle that includes many formative stages in the human host and mosquito vector, which contributes greatly to the challenge of its treatment.1–4
Artemisinin in combination with other drugs (artemisinin-combination therapy, ACT) is the first-choice treatment for malaria worldwide. However, recently resistance to most of the antimalarial drugs including ACTs is being reported in different geographical areas.5 Therefore, identification of novel multistage drug targets with new modes of action is required to develop new antimalarials against these resistant parasites.
Plasmodium proteases play crucial roles at all stages of the parasite life cycle and are thus potential antimalarial targets.6 Falcipains (FPs), majorly FP2 and FP3 are unique food vacuole Plasmodium cysteine proteases that rapidly cleave hemoglobin, an essential step in asexual blood stage parasite survival. FP2 is also involved in degradation of erythrocyte-membrane cytoskeletal proteins mainly ankyrin and band 4.1.7,8
With due course of time and advancement in this field of research, scientific community has developed diverse range of antimalarial drug molecules targeting inhibition of FP2. Gallinamide A (Fig. 1a) leads to swelling of parasite food vacuole when dosed upon cultured P. falciparum and cause parasite death.9,10 “Gallinamide A” analogues have been found to exhibit effective inhibiting ability for FP2, FP3 and for the development of P. falciparum in vitro.11 Along with this, E64 (Fig. 1b) an epoxide and an important irreversible inhibitor against general cysteine proteases and displayed a significant outcome on growth, adherence and viability of parasite trophozoites.12 A representative of benzoxaboroles, AN3661 (Fig. 1c), has been found to markedly reduce the FP2 transcripts in malarial parasites.13 A new class of peptidomimetic inhibitor of FP2 having 1,4-benzodiazepine scaffold (Fig. 1d) joint with vinyl ester, evidenced to be a highly potent and selective FP2 inhibitor.14 Derivatives of some heteroaryl nitrile with the 5-substituted-2-cyanopyrimidine (Fig. 1e) displayed a potential FP2 inhibition and therefore substantial antiparasitic effect.15 Natural product hybrids like lactone–chalcone and isatin–chalcone have also been reported to inhibit FP2 through probing the S2 active site pocket of the enzyme.16 Stilbene–chalcone hybrids have been reported to cause to cause apoptosis in malaria parasite.17 4-phenylchalcone based compounds have been observed to inhibit cathepsins B, H and L.18 In addition, coumarin-chalcone have also been reported to be potential bioactive agents.19 Although plentiful active and efficient FP2 inhibitors in in vivo models of malaria have been reported, but none has yet been progressed into human clinical trials.20–22
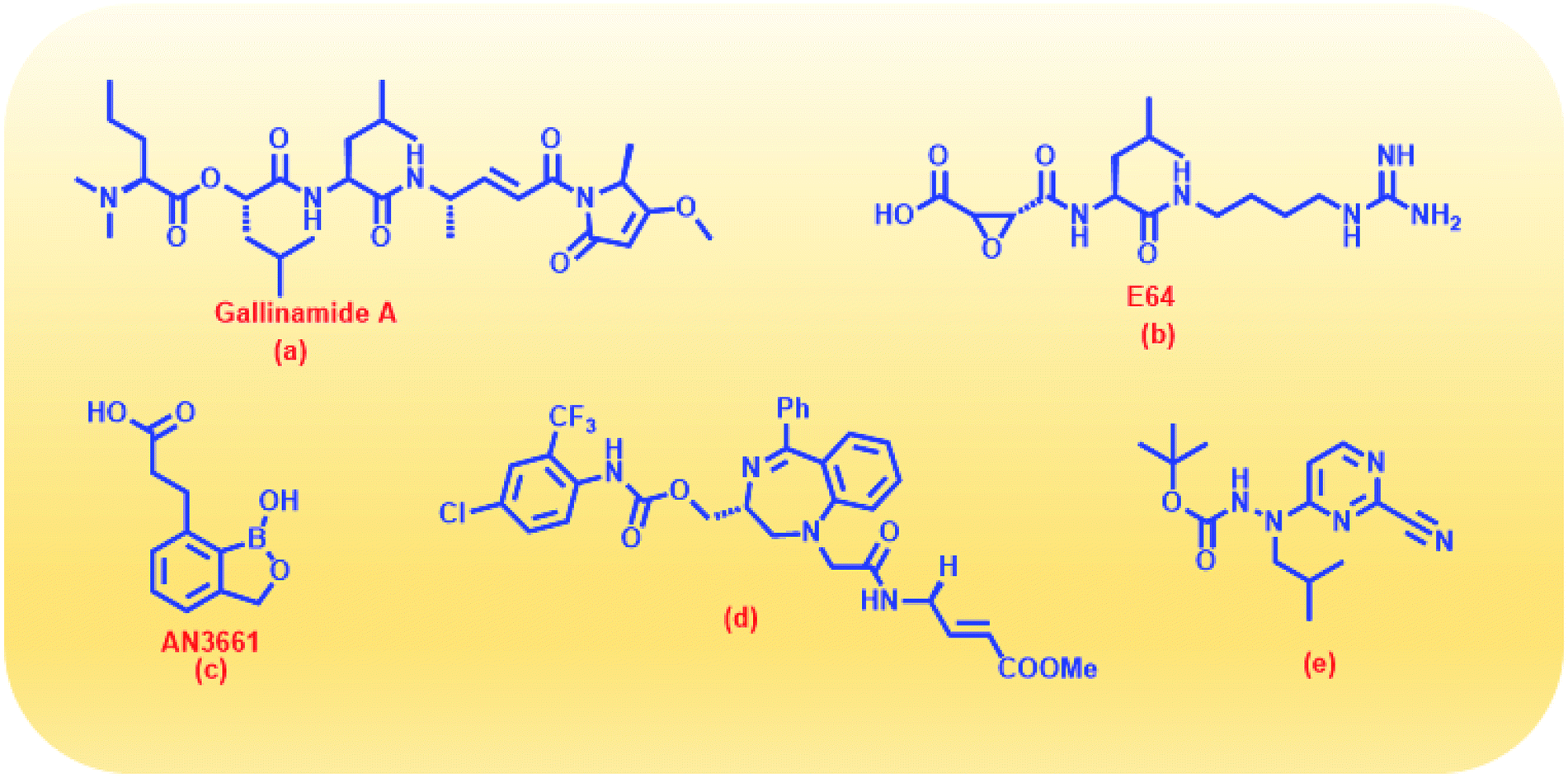 | ||
| Fig. 1 Structures of some reported FP2 inhibitors. | ||
Triazoles are 5-membered heterocyclic class of organic molecules, which exist in two forms, the 1,2,4-triazole and 1,2,5-triazole based on the position of nitrogen atoms. Even though the triazoles have a bright history in pharmacology, it gained more popularity by the copper catalyzed click chemistry. Triazole besides acting as a passive linker also participates as a dominant scaffold to enhance biological activity of drug molecules. Its acceptability as a bioactive scaffold is related to its ability to bind with target receptors by means of H-bonding, π–π stacking and dipole–dipole interactions. In addition, its ability to survive both moderate to acid–base conditions as well as its resistance to metabolic degradation make it an attractive pharmacophore for drug design.23–25 1,2,3-Triazole derivatives have been shown to have numerous medicinal values such as antihypertensive, anticancer, antitubercular, anti-inflammatory, antifungal, anticholinergic, antibacterial and antiviral properties.26–29 These compounds are also known for their anti-MRSA potency30 over and above powerful antimalarial, anticancer and antidiabetic activities.31 Fluconazole, voriconazole and itraconazole are some of the important triazole core moiety-based drugs. Recently, admirable triazole-based FP2 inhibitors have been developed32–34 among which a few are given in (Fig. 2). Such molecules have been the main source of consideration to design new potential FP2 inhibitors.
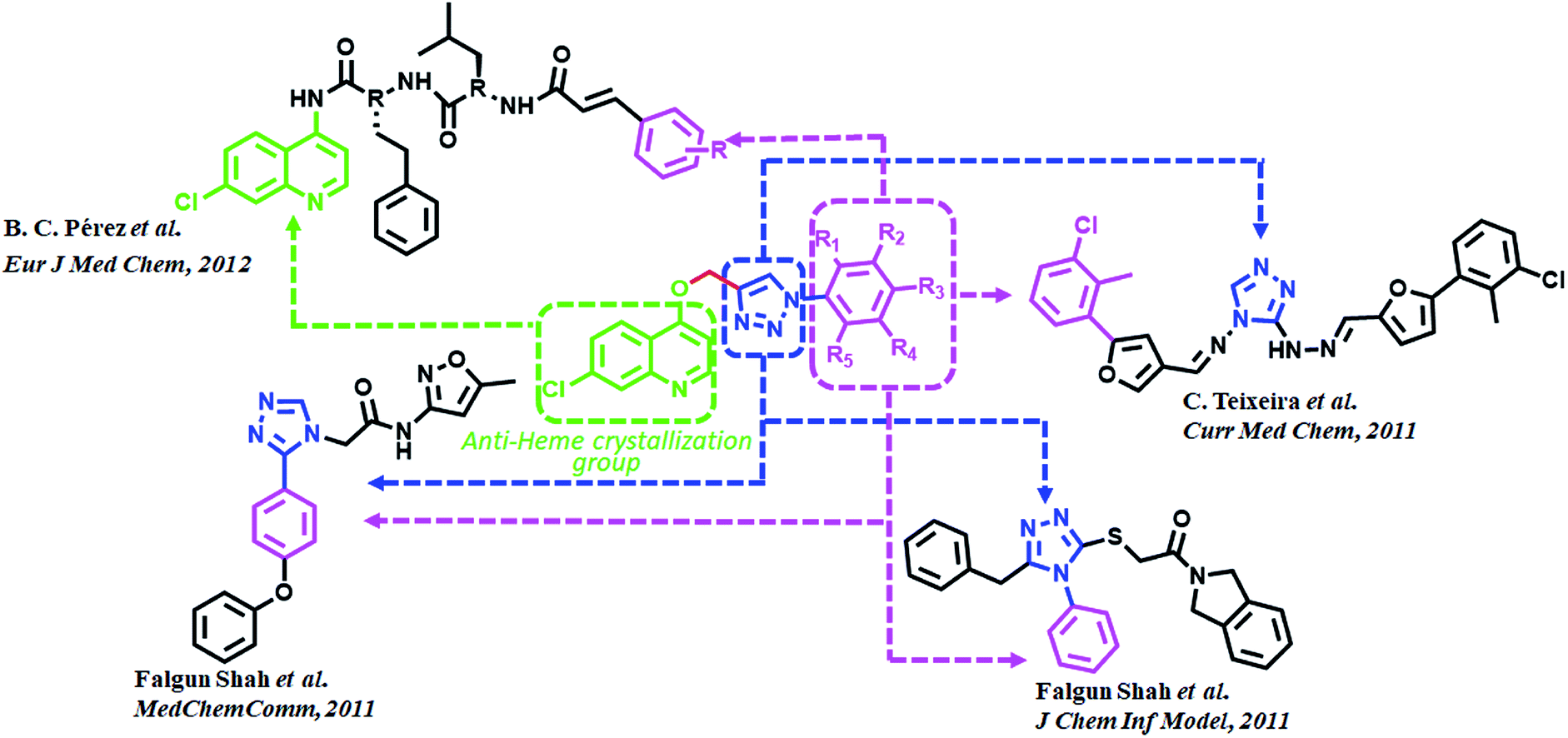 | ||
| Fig. 2 Rationale for the design and synthesis of triazole-quinoline derivatives as FP2 inhibitors. | ||
Quinoline and its derivatives, have been reported as rich bioactive heterocyclic compounds, which show antimalarial, antibiotic, anticancerous and anti-inflammatory properties.35–39 Many current antimalarials such as chloroquine, amodiaquine, mefloquine and primaquine have quinoline as core moiety. Ester containing quinoline compounds have also a bright history for antimalarial effect.40 Quinoline analogues are well known for inhibiting hemozoin formation (heme crystallization).41–46 These analogues bind with precrystalline forms of heme that can conceivably bring about such inhibition and have been characterized by various spectroscopic methods.47–49 N-(7-chloroquinolinyl-4-aminoalkyl)arylsulfonamides have been found to inhibit hemozoin formation and display promising antimalarial activity.50 Potential antiplasmodial effect has been reported from short peptide-based compounds with IC50 values ranging between 1.4–4.7 mg mL−1.51 B. C. Pérez and his co-workers reported a set of chloro quinoline containing cinnamic acids as excellent inhibitors of FP2.52 (Fig. 2).
Based on the previously reported excellent pharmacokinetic characteristics and favorable safety profile of triazole and quinoline derivatives,53–58 we synthesized a series of chloroquinoline triazole analogues as antimalarial agents. Indeed, we employed the archetypical click reaction: the Huisgen [3 + 2] cycloaddition between alkynes and azides catalyzed by copper(I). Thus, the removal of dynamic proton has helped us to come out with triazoles, which predominately exist in only one isomeric form.
2. Results and discussion
2.1. In silico design of potential FP2 inhibitors
The docking results predict the binding affinity (−ΔG in kcal mol−1) between the ligand and the active site of FP2 (PDB ID 3BPF). Noncovalent interactions like π–π interactions, allyl–π interactions and H-bond interactions were studied. It was found that the alkyl or aryl groups with varying hydrophobicity and electronic environment decided the binding affinity of the ligands to the drug target. The binding free energy (−ΔG) of the designed molecules with FP2 is enlisted in Table 1. The computational binding free energy of complexation of T7 with the target enzyme was found to be more favorable (−7.91 kcal mol−1 as per ParDOCK), indicating its better binding affinity towards the enzyme. From the docking pose of compound T7 shown in (Fig. 3a), the aromatic π electron cloud of triazole core structure was engaged with Trp43 through π–π stacking interaction. In addition, Cys39 was observed to form strong H-bond with oxygen of nitrobenzene group while as Asn81 formed a H-bond with nitrogen of triazole system. However, the standard FP2 inhibitor: E64 merely displayed two H-bonds with Asn173 and Gly83 as shown in (Fig. 3b). Since E64 lacks an aromatic system and is unable to take part in π–π interactions, may be the reason for its less binding affinity with the enzyme than that of T7. From various molecular modelling studies, we observed that our designed molecules fix well into the active site grove of FP2 as visualized in (Fig. 3c). However, the standard molecule: E64 was not observed to fix appropriately into the active site groove of the enzyme (Fig. 3d).| Compounds | Structures | Computational binding free energy (kcal mol−1) using ParDOCK | Computational binding free energy (kcal mol−1) using AutoDock |
|---|---|---|---|
| T1 |  |
−6.31 | −5.7 |
| T2 |  |
−6.21 | −6.8 |
| T3 |  |
−6.43 | −6.6 |
| T4 | 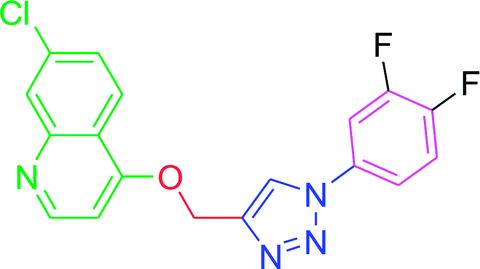 |
−6.15 | −6.9 |
| T5 | 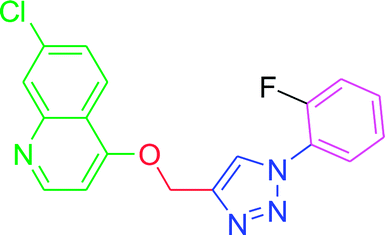 |
−6.32 | −6.4 |
| T6 | 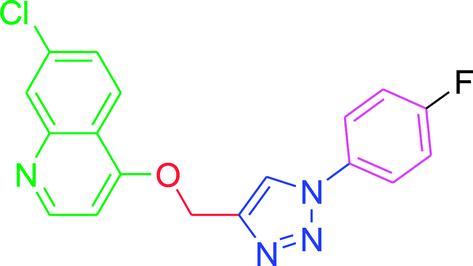 |
−6.31 | −6.7 |
| T7 | 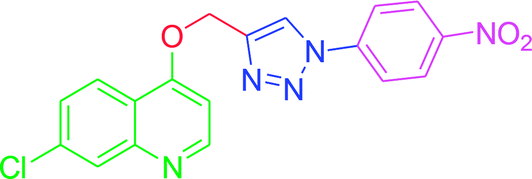 |
−7.91 | −6.7 |
| T8 | 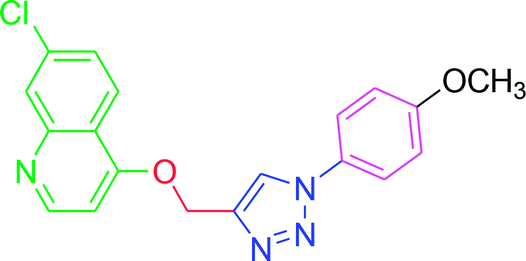 |
−7.52 | −6.5 |
| T9 | 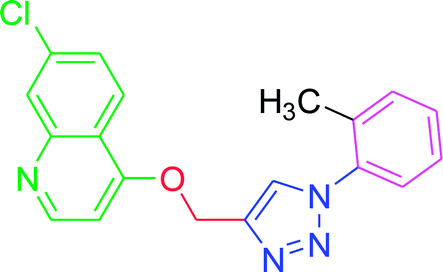 |
−6.13 | −6.1 |
| T10 | 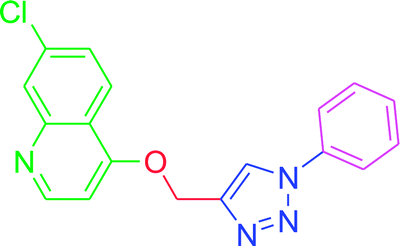 |
−5.91 | −6.5 |
| E64 | 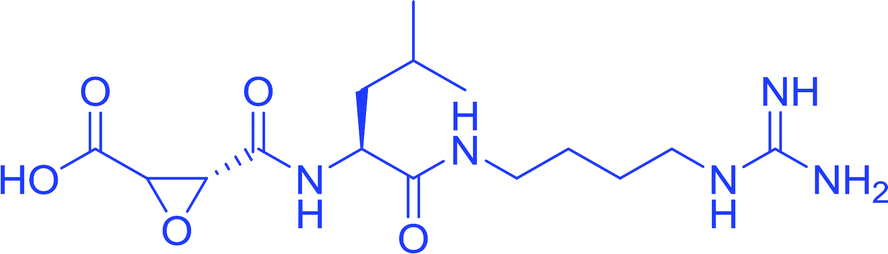 |
−6.14 | −6.9 |
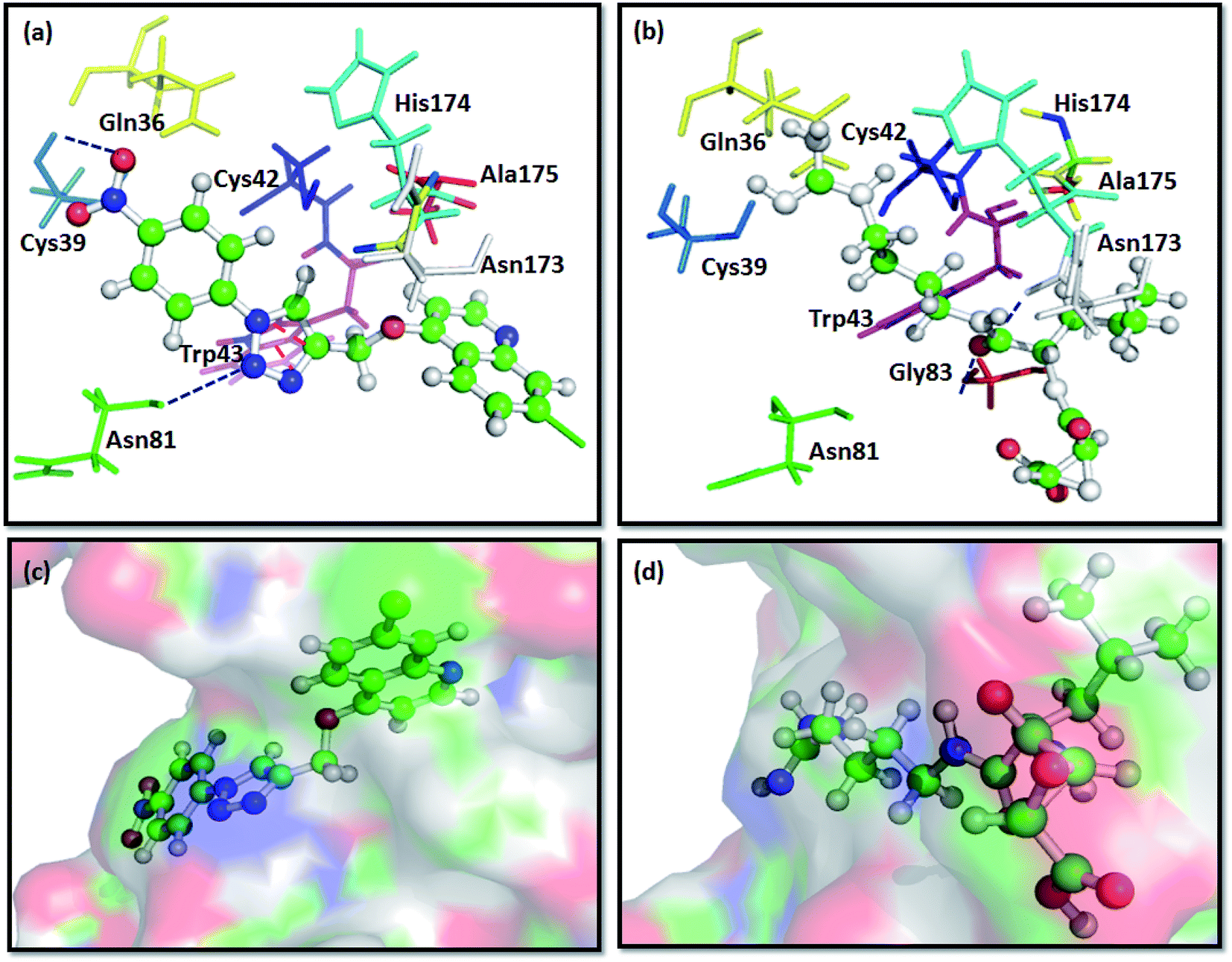 | ||
| Fig. 3 Docking interactions of T7 and E64, a known standard FP2 inhibitor with FP2:PDB ID 3BPF; (a) binding interaction of compound T7 with FP2 showing π–π stacking between Trp43 and aromatic triazole ring (represented with red coloured dashes) and two H-bonds between the ligand and Cys39 and Asn81 of FP2 (represented with dark blue coloured dashes). (b) Showing H-bond interaction of E64 with the Gly83 and Asn173 of FP2 (represented with dark blue coloured dashes). (c) Surface view of T7 into the active site groove of the enzyme: (d) Surface pose of E64 with FP2. | ||
These in silico results were also validated with in vitro FP2 inhibition and showed a notable agreement; hence such molecules could be used as lead to develop effectual FP2 inhibitors.
2.2. Chemical synthesis
The development of target molecules was accomplished via synthetic route shown in Schemes 1–3. In Scheme 1, differently substituted azides (3) were synthesized from differently substituted anilines (1). In the Scheme 2, 4,7-dichloroquinoline was made to react with propargyl alcohol in presence of sodium hydride and dry dimethylformamide at room temperature to obtain 7-chloro-4-(prop-2-ynyloxy)quinoline (6) according to the literature method. In Scheme 3, compounds 3(a-j) were coupled with 7-chloro-4-(prop-2-ynyloxy)quinoline complex (6) in presence of CuSO4·5H2O, sodium ascorbate, (THF:H2O, 1:2), under the reflux conditions 80 °C for 8 h to obtain our target compounds 7(a-j).61 The structures of these compounds were confirmed through various analytical and spectroscopic methods: (1H, 13C NMR and mass spectra).
| Compound | R1 | R2 | R3 | R4 | R5 |
|---|---|---|---|---|---|
| T1 | H | CF3 | H | H | H |
| T2 | H | Cl | F | H | H |
| T3 | F | H | F | H | H |
| T4 | H | F | F | H | H |
| T5 | F | H | H | H | H |
| T6 | H | H | F | H | H |
| T7 | H | H | NO2 | H | H |
| T8 | H | H | OCH3 | H | H |
| T9 | CH3 | H | H | H | H |
| T10 | H | H | H | H | H |
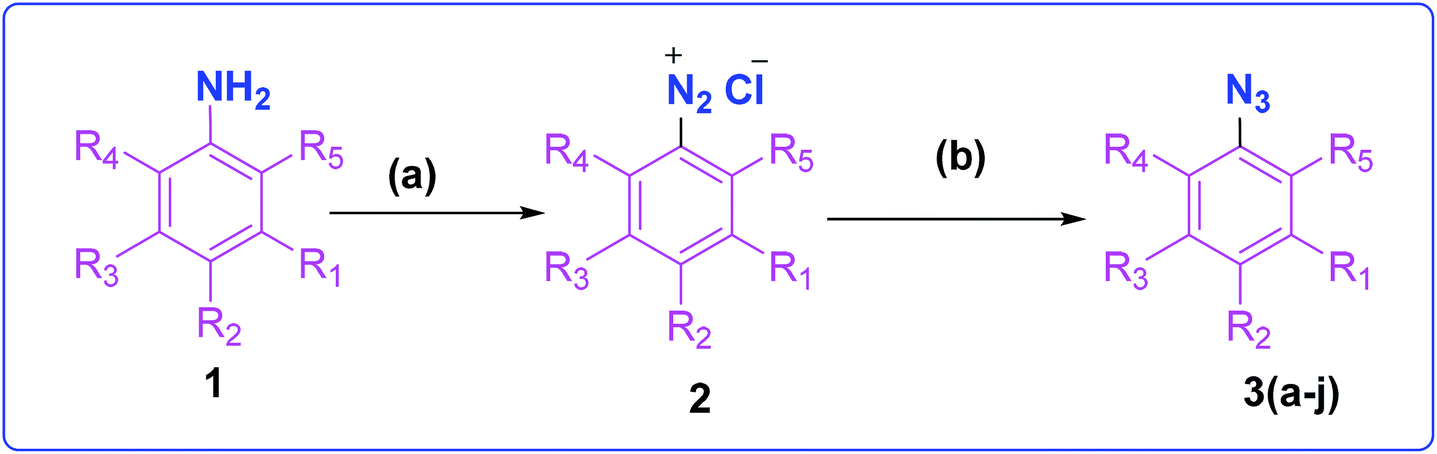 | ||
| Scheme 1 Synthesis of different aromatic azides. Reagents and conditions: (a) NaNO2, HCl (3 N), ice bath (b) NaN3, water, room temperature, 4 h, 83–90% yield. | ||
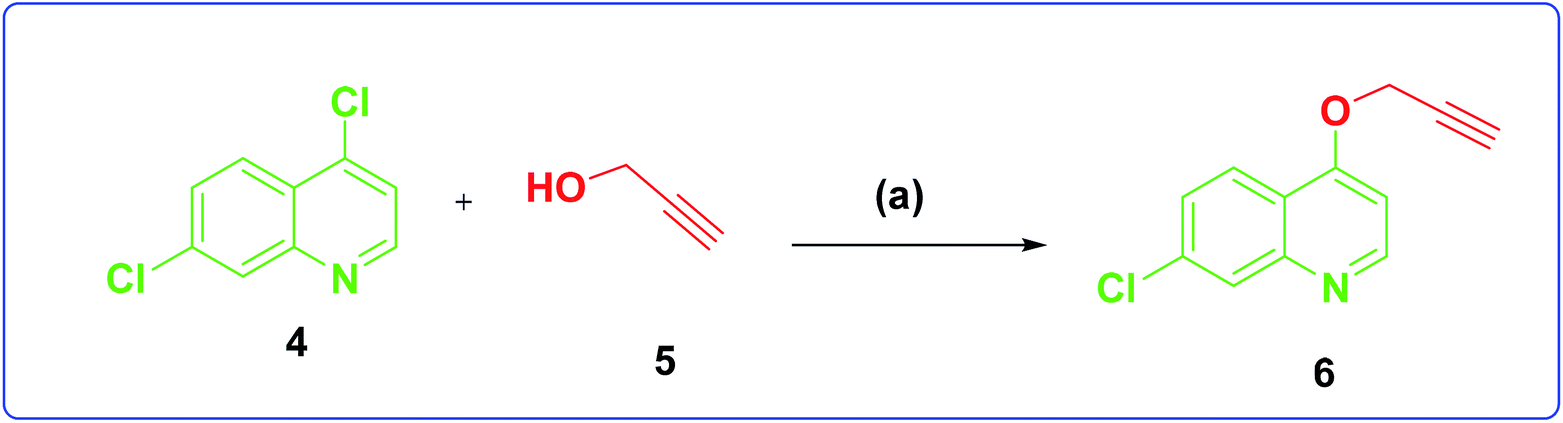 | ||
| Scheme 2 Synthesis of 7-chloro-4-(prop-2-yn-1-yloxy)quinoline. Reagents and conditions: (a) NaH, dry DMF, propargyl, alcohol room temperature, 4 h, 70% yield. | ||
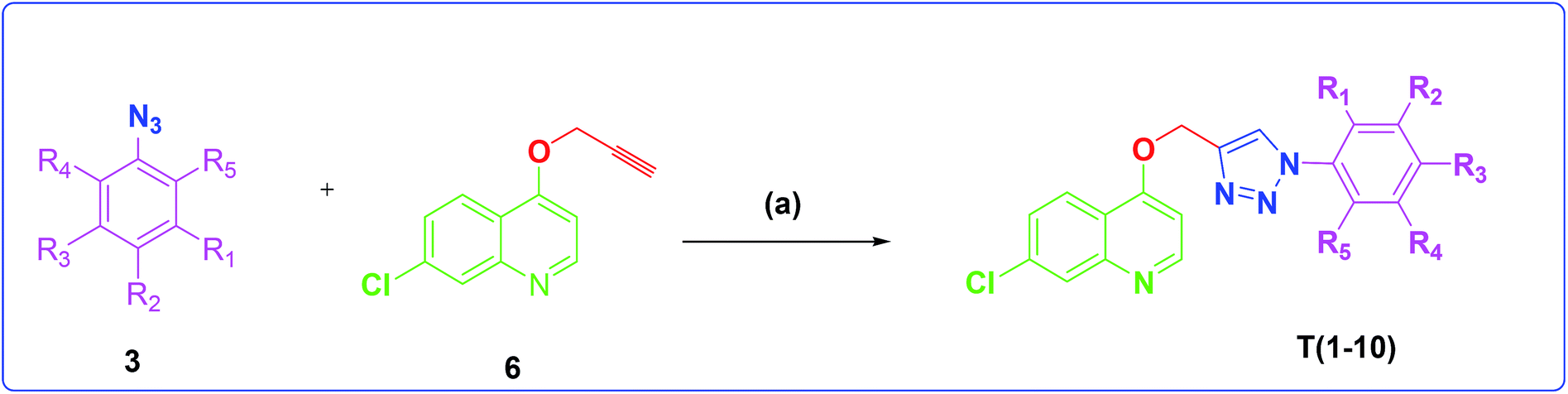 | ||
| Scheme 3 Synthesis of different quinoline-triazole-based target compounds. Reagents and conditions: (a) CuSO4·5H2O, sodium ascorbate, (THF–H2O, 1:2), 80 °C, 8 h. | ||
2.3. Anti-malarial screening of the synthesized compounds
| S. no. | Compound | Inhibition of FP2 IC50 (μM) | Std. error | Inhibition of P. falciparum EC50 (μM) | Std. error |
|---|---|---|---|---|---|
| 1 | T1 | 26.59 | 5.45 | 49.50 | 10.54 |
| 2 | T2 | 45.08 | 9.46 | 405.64 | 398.80 |
| 3 | T3 | 25.38 | 3.18 | 159.22 | 150.16 |
| 4 | T4 | 25.64 | 4.13 | 21.89 | 4.51 |
| 5 | T5 | 107.32 | 135.24 | 66.57 | 45.08 |
| 6 | T6 | 45.78 | 18.98 | 57.86 | 183.93 |
| 7 | T7 | 16.16 | 3.03 | 49.88 | 7.41 |
| 8 | T8 | 25.37 | 1.94 | 51.71 | 8.57 |
| 9 | T9 | 20.9 | 4.15 | 146.98 | 345.29 |
| 10 | T10 | 58.46 | 19.07 | 57.63 | 14.02 |
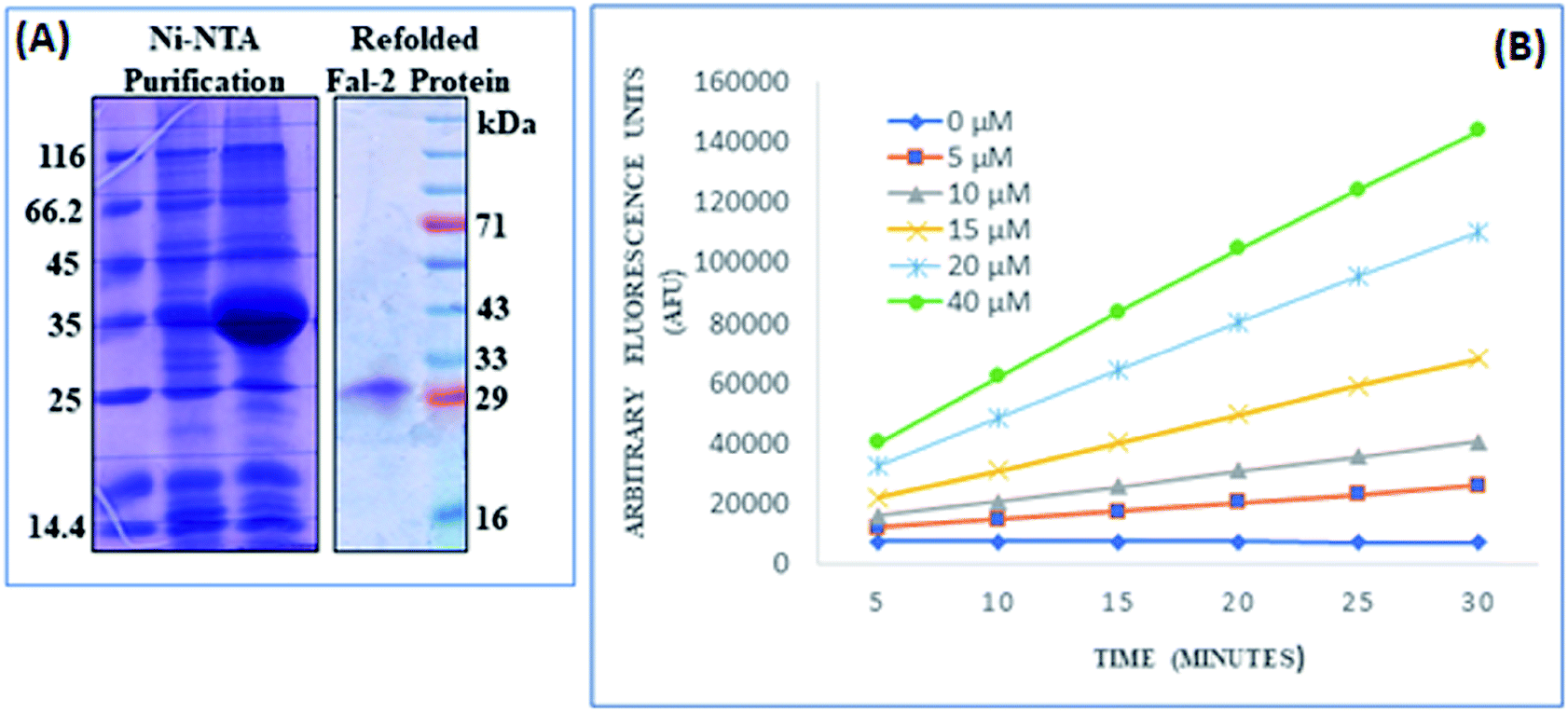 | ||
| Fig. 4 Recombinant FP2 protein expression and activity analysis: (A) FP2 protein was expressed in E. coli cells and purification was done with Ni2+-NTA column followed by refolding process. (B) Activity analysis of refolded FP2 using ZFR-AMC as substrate on dose dependent manner over a period of 30 min was measured at pH 5.5. | ||
All the synthesized compounds were subsequently analyzed for their antimalarial activities in an in vitro P. falciparum 3D7 culture. As shown in Table 2, compound T4 containing difluoro benzene ring attached to the triazole core moiety, inhibited P. falciparum with an EC50 value 21.89 μM. Furthermore, compound T1 exhibited second best inhibition of P. falciparum 3D7 culture with EC50 49.50 μM. Almost similar results EC50 49.88 μM were found for T7 containing nitro group at para position in the benzene ring. Compound T10 with unsubstituted benzene ring inhibited the parasite growth in the range of 57.63 μM. Similar EC50 value ∼57.86 were observed for T6 compound containing p-fluoro benzene attached to the main trizole moiety. T5 having o-fluoro benzene group, displayed the parasite development inhibition, EC50 49.50 μM. T9, T3 and T2 disclosed lowest ability to inhibit the parasite development with EC50 values 146.98 μM, 159.22 μM and 405.64 μM, respectively.
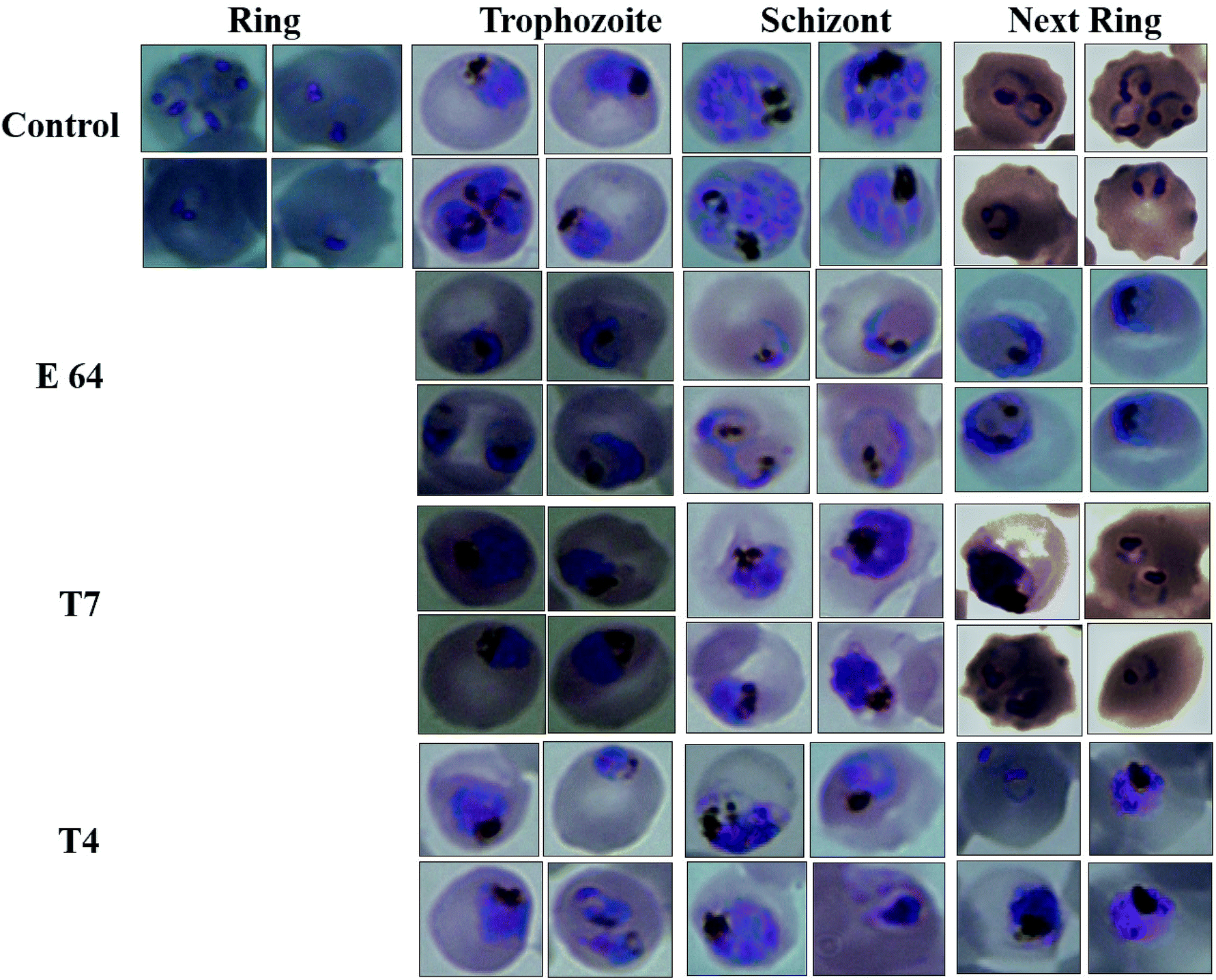 | ||
| Fig. 5 Effect of E-64, compounds T7 and T4 on the growth and development of P. falciparum. Light microscopy images of parasitized red blood cells at different time points after the treatment with E64, T7 and T4. | ||
3. Conclusion
In the present study, a set of 10 triazole-quinoline derivatives (T1–T10) were computationally designed, synthesized and evaluated for FP2 inhibition and growth and development of P. falciparum. ParDOCK was used for docking and scoring purposes. The designed compounds were synthesized in the wet lab using click chemistry approach. Since the triazole scaffold is extremely versatile and has been featured in several clinically used drugs hence it once again evidenced to be a better drug candidate in the malaria treatment. In addition, quinoline moiety also verified to be a potential anti-malarial moiety. Generally, fluoro substituted triazole-quinoline molecules exhibited better enzyme inhibition as well as parasite growth and development inhibition. Computationally, compound T7, containing nitrobenzene attached to the triazole core structure exhibited the more negative binding free energy (−7.91 kcal mol−1) with the target enzyme FP2 in the series, while as the standard inhibitor of FP2, E64 exhibited binding energy of (−6.14 kcal mol−1). The aromatic π electron cloud of triazole of T7 was observed to interact with Trp43 via π–π stacking. The nitrogen of triazole formed a H-bond with Asn81 and oxygen of nitrobenzene of T7 showed a H-bond with Cys39 with bond length of 2 Å. Compound T7 displayed best inhibition of FP2 while as, T4 containing difluoro benzene attached to the triazole ring arrested the development of P. falciparum at the trophozoite stage and showed morphological and food-vacuole abnormalities. The in silico and experimental results correlate to each other. These results make a clear clue for the usage of triazole and quinoline moieties in the development of future drugs to eradicate malaria completely.4. Experimental section
4.1. Docking protocol
Protein Data Bank (http://www.rcsb.org)64 was used as a source for obtaining the crystal structure of FP2 (PDB ID 3BPF) in pdb format which was further submitted along with the ligand molecules to ParDOCK module of Sanjeevini65,66 for molecular docking and scoring determinations. It is important to mention that ParDOCK is an inclusive drug design suite which functions on physicochemical descriptors by integrating several protocols and deals protein and the ligands at the atomic level. The ligand was produced in its input pose for the calculation. Addition of hydrogen to the ligand was accomplished using xleap of AMBER67 and geometry optimization and partial charge calculations were performed via AM1-BCC procedure. The docked structures are energy minimized using the sander module of AMBER and scored using scoring function Bappl68 which is an all atom-based scoring function consisting electrostatics, van der Waals hydrophobicity, loss of conformational entropy of protein side chains after complexation. In addition to ParDOCK, AutoDock Vina program60 (version 1.5.6) was used to validate the docking results and detailed information is provided in the ESI section.†PyMOL, a molecular visualizing software was used to visualize the molecular interactions. Weak non-covalent interactions like π–π stacking interaction and H-bonding were observed between the active site residues of FP2 and the ligand molecules lying within a range of 2 Å. In addition, the binding free energy of the molecules was calculated based on which the molecules were screened.
4.2. Chemistry
The chemicals, reagents and solvents used during the synthesis and characterization of the compounds were brought from the commercial vendors like Sigma Aldrich, Alfa-Aesar and Spectrochem Pvt. Ltd. (India). 1H NMR and 13CNMR spectra were observed on Bruker 75 and 300 MHz spectrophotometer using deuterated solvents (CDCl3, δ 7.26; DMSO-d6 δ 2.54) and multiplicities of NMR signals are designated as s (singlet), d (doublet), dd (double doublet), t (triplet), q (quartet), m (multiplet, for unresolved lines). Chemical shifts were showed in δ (ppm) and tetramethylsilane was used as the internal standard. Mass spectra were recorded on UPLC XEVO G2-XS QTOF Spectrometer (HRMS) instrument. Thin layer chromatography was performed with Merck silica gel (60–120 and F254) aluminum-coated sheets of 0.25 mm thickness. Spots on these were observed in short (254 nm) with ultraviolet light and long (365 nm) wavelengths. Elemental analyses were obtained on Elementar Vario analyser. Elemental analyses of the compounds were found to be within ±0.4% of the theoretical values. The purity of tested compounds was >95%.:1 v/v, isolated by filtration and air dried to obtain pure compounds 3a–3j.:80. The pure compound obtained was white colored powder.:2). A freshly prepared aqueous solution of sodium ascorbate (0.12 mmol, 0.2 mL) was added followed by freshly prepared aqueous solution of CuSO4·5H2O (0.04 mmol, 0.2 mL). The yellowish reaction mixture was stirred at 80 °C. After the completion of the reaction, the reaction mixture was directly dried under vacuum and the crude material was purified by column chromatography ethyl acetate–hexane = 50:50. Yield of the desired product was found to be in the range of 60 to 80%.
4.2.3.1. 7-Chloro-4-({1-[3-(trifluoro methyl)phenyl]-1H-1,2,3-triazol-4yl}methoxy) quinoline (T1). Brown solid (Rf = 0.3 in pure ethyl acetate) mp 161–163 °C, 70% yield isolated after column chromatography. 1H NMR (300 MHz, CDCl3) δ 8.79 (d, J = 5.4 Hz, 1H), 8.24 (s, 1H), 8.13 (d, J = 9.00 Hz, 1H), 8.06 (d, J = 1.9 Hz, 1H), 8.00 (d, J = 7.5 Hz, 1H), 7.72 (m, J = 2H), 7.44 (dd, J = 8.7, 2.1 Hz, 1H), 7.27 (s, 1H), 6.99 (d, J = 5.4 Hz, 1H), 5.55 (s, 2H). 13C NMR (75 MHz, CDCl3) δ 174.86, 161.24, 151.85, 148.63, 143.58, 137.04, 136.32, 133.09, 132.65, 132.30, 131.76, 130.67, 126.92, 123.61, 123.43, 121.60, 119.50, 117.49, 101.47, 62.18. HRMS m/z: calcd for C19H12ClF3N4O [M + H]+ 404.0652 found 405.0716 elemental anal.: calcd: C, 56.47; H, 2.98; Cl, 8.57; F, 14.17; N, 13.75; O, 3.86.
4.2.3.2. 7-Chloro-4-{[1-(3-chloro-4-fluorophenyl)-1H-1,2,3-triazol-4-yl]methoxy} quinoline (T2). Yellow solid (Rf = 0.3 in pure ethyl acetate) mp 185–187 °C, 60% yield obtained after column chromatography. 1H NMR (400 MHz, CDCl3) δ 8.76 (d, J = 5.6 Hz, 1H), 8.13 (s, 1H), 8.09 (d, J = 8.8 Hz, 1H), 8.03 (d, J = 2.0 Hz, 1H), 7.85 (dd, J = 9.2, 2.0 Hz, 1H), 7.64 (m, 1H), 7.41 (dd, J = 8.9, 2.0 Hz, 1H), 7.34–7.27 (m, 1H), 6.96 (d, J = 5.2 Hz, 1H), 5.50 (s, 2H). 13C NMR (101 MHz, CDCl3) δ 175.78, 161.30, 152.05, 148.86, 143.64, 136.50, 127.12, 127.08, 123.50, 123.28, 121.63, 120.55, 120.48, 119.64, 118.00, 117.77, 101.58, 62.27. HRMS m/z: calcd for C18H11Cl2FN4O [M + H]+ 388.0294 found 389.0302 elemental anal.: calcd: C, 55.37; H, 2.76; Cl, 18.13; F, 4.79; N, 14.31; O, 4.02.
4.2.3.3. 7-Chloro-4-{[1-(2,4-difluorophenyl)-1H-1,2,3-triazol-4-yl]methoxy} quinoline (T3). White chalk (Rf = 0.5 in pure ethyl acetate) mp 173–175 °C, 70% yield obtained after column chromatography. 1H NMR (400 MHz, CDCl3) δ 8.75 (d, J = 5.2 Hz, 1H), 8.19 (d, J = 2.4 Hz, 1H), 8.11 (d, J = 9.2 Hz, 1H), 8.01 (d, J = 1.6 Hz, 1H), 7.98–7.92 (m, 1H), 7.41 (dd, J = 9.2, 2.4 Hz, 1H), 7.10–7.04 (m, 2H), 6.96 (d, J = 5.2 Hz, 1H), 5.51 (s, 2H). 13C NMR (101 MHz, CDCl3) δ 160.91, 152.53, 149.78, 143.22, 136.04, 127.94, 126.85, 126.26, 124.54, 124.46, 123.50, 119.75, 113.03, 113.00, 112.81, 112.77, 101.54, 62.15 HRMS m/z:calcd C18H11ClF2N4O [M + H]+ 372.7559 found 373.0706 elemental anal.:calcd: C, 58.00; H, 2.88; Cl, 9.33; F, 10.28; N, 15.12; O, 4.38.
4.2.3.4. 7-Chloro-4-{[1-(3,4-difluorophenyl)-1H-1,2,3-triazol-4-yl]methoxy} quinoline (T4). Brown solid (Rf = 0.5 in pure ethyl acetate) mp 177–179 °C, 80% yield obtained after column chromatography. 1H NMR (300 MHz, CDCl3) δ 8.78 (d, J = 5.4 Hz, 1H), 8.15 (s, 1H), 8.11 (d, J = 5.1 Hz, 1H), 8.04 (s, 1H), 7.73–7.66 (m, 1H), 7.51 (d, J = 9.0 Hz, 1H), 7.44 (d, J = 8.7 Hz, 1H), 7.36 (d, J = 8.4 Hz, 1H), 6.97 (d, J = 5.4 Hz, 1H), 5.53 (s, 2H). 13C NMR (75 MHz, CDCl3) δ 160.70, 152.44, 149.76, 143.81, 135.81, 133.06, 127.93, 126.74, 123.32, 121.37, 119.63, 118.64, 118.40, 116.59, 110.95, 110.66, 101.45, 62.11. HRMS m/z: calcd C18H11ClF2N4O [M + H]+ 372.7559 found 373.0706. Elemental anal.: calcd: 47.11; H, 2.88; Cl, 9.42; F, 10.37; N, 15.21; O, 4.47.
4.2.3.5. 7-Chloro-4-{[1-(2-fluorophenyl)-1H-1,2,3-triazol-4-yl]methoxy} quinoline (T5). White (Rf = 0.5 in pure ethyl acetate) mp 180–182 °C, 80% yield isolated yield after flash column chromatography. 1H NMR (300 MHz, CDCl3) δ 8.80 (d, J = 5.1 Hz, 1H), 8.15 (d, J = 9.0 Hz, 1H), 8.09 (s, 1H), 8.06 (d, J = 1.8 Hz, 1H), 7.74 (dd, J = 9.0, 4.5 Hz, 2H), 7.45 (dd, J = 9.0, 1.8 Hz, 1H), 7.29–7.23 (m, 2H), 7.01 (d, J = 5.4 Hz, 1H), 5.54 (s, 2H). 13C NMR (75 MHz, CDCl3) δ 175.35, 161.23, 152.10, 149.03, 143.35, 136.34, 133.05, 127.22, 126.98, 123.43, 122.78, 122.66, 121.59, 119.66, 117.05, 116.74, 101.55, 62.30. HRMS m/z: calcd C18H12ClFN4O [M + H]+ 354.0684 found 355.0793 HRMS m/z: calcd C18H12ClFN4O [M + H]+ 354.0684 found 355.0682. Elemental anal.: calcd: C, 60.76; H, 3.23; Cl, 10.00; F, 5.25; N, 15.79; O, 4.14.
4.2.3.6. 7-Chloro-4-{[1-(4-fluorophenyl)-1H-1,2,3-triazol-4-yl]methoxy} quinoline (T6). White, (Rf = 0.4 in pure ethyl acetate) mp 155–157 °C, 66% yield obtained after column chromatography. 1H NMR (300 MHz, CDCl3) δ 8.80 (d, J = 5.1 Hz, 1H), 8.15 (d, J = 9.0 Hz, 1H), 8.09 (s, 1H), 8.06 (d, J = 1.8 Hz, 1H), 7.74 (dd, J = 9.0, 4.5 Hz, 2H), 7.45 (dd, J = 9.0, 1.8 Hz, 1H), 7.29–7.23 (m, 2H), 7.01 (d, J = 5.4 Hz, 1H), 5.54 (s, 2H). 13C NMR (75 MHz, CDCl3) δ 175.35, 161.23, 152.10, 149.03, 143.35, 136.34, 133.05, 127.22, 126.98, 123.43, 122.78, 122.66, 121.59, 119.66, 117.05, 116.74, 101.55, 62.30. HRMS m/z: calcd C18H12ClFN4O [M + H]+ 354.0684 found 355.0793 elemental anal.: calcd: C, 60.85; H, 3.32; Cl, 9.99; F, 5.63; N, 15.97; O, 4.42.
4.2.3.7. 7-Chloro-4-{[1-(4-nitrophenyl)-1H-1,2,3-triazol-4-yl]methoxy} quinoline (T7). Light yellow solid, (Rf = 0.3 in pure ethyl acetate) mp 166–168 °C, 72% yield obtained after column chromatography. 1H NMR (300 MHz, CDCl3) δ 8.79 (d, J = 5.1 Hz, 1H), 8.53 (s, 1H), 8.44 (d, J = 8.7 Hz, 1H), 8.18 (d, J = 8.7 Hz, 1H), 8.10 (d, J = 9.0 Hz, 2H), 8.02 (s, 1H), 7.46 (d, J = 9.0 Hz, 1H), 7.38 (s, 1H), 7.03 (d, J = 5.1 Hz, 1H), 5.56 (s, 2H). 13C NMR (75 MHz, CDCl3) δ 165.53, 157.39, 148.65, 145.87, 140.32, 132.44, 131.29, 130.23, 128.52, 127.44, 125.49, 106.60, 66.82, 45.41, 44.85, 44.30. HRMS m/z: calcd C18H12ClN5O3 [M + H]+ 381.0629 found 382.0733 elemental anal.: calcd: C, 57.00; H, 3.35; Cl, 9.36; N, 18.43; O, 13.01.
4.2.3.8. 7-Chloro-4-{[1-(4-methoxyphenyl)-1H-1,2,3-triazol-4-yl]methoxy} quinoline (T8). Yellow solid powder, (Rf = 0.5 in pure ethyl acetate) mp 179–181 °C, 76% yield obtained after column chromatography. 1H NMR (300 MHz, CDCl3) δ 8.80 (d, J = 5.4 Hz, 1H), 8.15 (d, J = 9.0 Hz, 1H), 8.06 (s, 2H), 7.65 (d, J = 9.0 Hz, 2H), 7.45 (d, J = 9.0 Hz, 1H), 7.27 (s, 1H), 7.04 (d, J = 8.7 Hz, 2H), 5.53 (s, 2H), 3.88 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 175.36, 161.39, 160.10, 151.97, 148.75, 142.87, 136.33, 130.13, 126.93, 123.52, 122.29, 121.68, 119.65, 114.86, 101.57, 62.37, 55.64. HRMS m/z: calcd C19H15ClN4O2 [M + H]+ 366.0884 found 367.0927 elemental anal.: calcd: C, 62.03; H, 4.02; Cl, 10.00; N, 15.55; O, 8.54.
4.2.3.9. 7-Chloro-4-{[1-(2-methylphenyl)-1H-1,2,3-triazol-4-yl]methoxy} quinoline (T9). Snow white solid, (Rf = 0.5 in pure ethyl acetate) mp 180–182 °C, 73% yield obtained after column chromatography. 1H NMR (300 MHz, CDCl3) δ 8.80 (d, J = 5.1 Hz, 1H), 8.12 (dd, J = 16.5, 9.0 Hz, 3H), 7.62 (d, J = 8.1 Hz, 2H), 7.45 (d, J = 8.4 Hz, 1H), 7.33 (d, J = 7.9 Hz, 2H), 7.01 (d, J = 4.8 Hz, 1H), 5.53 (s, 2H), 2.43 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 175.33, 161.35, 152.04, 148.90, 142.98, 139.39, 136.30, 134.47, 130.34, 127.06, 126.94, 123.50, 121.47, 120.56, 119.75, 119.67, 101.56, 62.38, 21.05. HRMS m/z: calcd C19H15ClN4O [M + H]+ 350.0934 found 351.0924 elemental anal.: calcd: C, 65.05; H, 4.30; Cl, 10.11; N, 16.00; O, 5.01.
4.2.3.10. 7-Chloro-4-[(1-phenyl-1H-1,2,3-triazol-4-yl)methoxy] quinoline (T10). White, (Rf = 0.3 in pure ethyl acetate) mp 168–170 °C, 71% yield obtained after column chromatography. 1H NMR (300 MHz, CDCl3) δ 8.77 (d, J = 5.1 Hz, 1H), 8.15 (d, J = 5.7 Hz, 1H), 8.03 (s, 1H), 7.76 (d, J = 7.8 Hz, 2H), 7.57–7.43 (m, 4H), 7.27 (s, 1H), 6.99 (d, J = 5.4 Hz, 1H), 5.53 (s, 2H). 13C NMR (75 MHz, CDCl3) δ 160.87, 152.44, 149.69, 143.31, 136.78, 135.91, 129.86, 129.15, 127.82, 126.70, 123.43, 121.44, 120.64, 119.70, 101.51, 62.27. HRMS m/z: calcd C18H13ClN4O [M + H]+ 336.0778 found 337.0790 elemental anal.: calcd: C, 64.00; H, 4.00; Cl, 10.35; N, 17.00; O, 4.90.
4.3. Procedures for biological assays
000 rpm for 45 min at 4 °C. The pellet was washed twice with native buffer. Finally, the pellet was solubilized in UB [8 M urea, 20 mM Tris, 250 mM NaCl, pH 8.0]; (5 mL g−1 of inclusion body pellet) at room temperature for 60 min with gentle stirring. The insoluble material was separated by centrifuging at 15000 rpm for 60 min at 4 °C. Added imidazole to 10 mM, Triton X-100 to 1% supernatant. In order to purify of recombinant protein, supernatant was incubated overnight at a temperature of 4 °C with a nickel-nitrilotriacetic acid (Ni-NTA) resin. The resin was loaded on the column and it was washed with 10 bed volumes each of UB. Finally, the bound protein was eluted in UB with 250 mM imidazole and measured by the bicinchoninic acid assay. For refolding, the fractions containing FP2 protein were pooled in ice-cold refolding buffer and diluted 100-fold using Tris–HCl (100 mM), 1 mM EDTA, 20% glycerol, 250 mM L-arginine, 1 mM GSH, 1 mM GSSG, pH 8.0. The mixture was incubated with moderate stirring at 4 °C for 24 h, and concentrated to 25 mL using a stirred cell with a 10 kDa cut-off membrane (Pellicon XL device, Millipore) at 4 °C. The sample was then filtered using a 0.22 mm syringe filter. The purified and concentrated protein was quantified using bicinchoninic acid assay.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. S. would like to acknowledge University Grants Commission, Government of India for RGNF Senior Research Fellowship.References
- J. N. Burrows, E. Burlot, B. Campo, S. Cherbuin, S. Jeanneret, D. Leroy, T. Spangenberg, D. Waterson, T. N. Wells and P. Willis, Parasitology, 2014, 141, 128–139 CrossRef PubMed.
- E. L. Flannery, A. K. Chatterjee and E. A. Winzeler, Nat. Rev. Microbiol., 2013, 11, 849–862 CrossRef CAS PubMed.
- A. R. Renslo, ACS Med. Chem. Lett., 2013, 4, 1126–1128 CrossRef CAS PubMed.
- M. A. Phillips, J. N. Burrows, C. Manyando, R. H. van Huijsduijnen, W. C. Van Voorhis and T. N. C. Wells, Nat. Rev. Dis. Primers, 2017, 3, 17050 CrossRef PubMed.
- D. E. Goldberg, A. F. Slater, A. Cerami and G. B. Henderson, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 2931–2935 CrossRef CAS PubMed.
- E. Deu, FEBS J., 2017, 284, 2604–2628 CrossRef CAS PubMed.
- Cysteine Proteases of Pathogenic Organisms, ed. M. W. Robinson and J. P. Dalton, Springer US, Boston, MA, 2011, vol. 712 Search PubMed.
- M. Marco and J. M. Coterón, Curr. Top. Med. Chem., 2012, 12, 408–444 CrossRef CAS PubMed.
- S. C. Stolze, E. Deu, F. Kaschani, N. Li, B. I. Florea, K. H. Richau, T. Colby, R. A. L. van der Hoorn, H. S. Overkleeft, M. Bogyo and M. Kaiser, Chem. Biol., 2012, 19, 1546–1555 CrossRef CAS PubMed.
- P. S. Sijwali and P. J. Rosenthal, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 4384–4389 CrossRef CAS PubMed.
- A. Stoye, A. Juillard, A. H. Tang, J. Legac, J. Gut, K. White, S. A. Charman, P. J. Rosenthal, G. E. R. Grau, N. H. Hunt and R. J. Payne, J. Med. Chem., 2019, 62, 5562–5578 CrossRef CAS PubMed.
- T. B. de Carvalho, T. C. G. Oliveira-Sequeira and S. Guimaraes, Rev. Inst. Med. Trop. Sao Paulo, 2014, 56, 43–47 CrossRef PubMed.
- E. Sonoiki, C. L. Ng, M. C. S. Lee, D. Guo, Y.-K. Zhang, Y. Zhou, M. R. K. Alley, V. Ahyong, L. M. Sanz, M. J. Lafuente-Monasterio, C. Dong, P. G. Schupp, J. Gut, J. Legac, R. A. Cooper, F.-J. Gamo, J. DeRisi, Y. R. Freund, D. A. Fidock and P. J. Rosenthal, Nat. Commun., 2017, 8, 14574 CrossRef PubMed.
- R. Ettari, N. Micale, T. Schirmeister, C. Gelhaus, M. Leippe, E. Nizi, M. E. Di Francesco, S. Grasso and M. Zappalà, J. Med. Chem., 2009, 52, 2157–2160 CrossRef CAS PubMed.
- J. M. Coterón, D. Catterick, J. Castro, M. J. Chaparro, B. Díaz, E. Fernández, S. Ferrer, F. J. Gamo, M. Gordo, J. Gut, L. de las Heras, J. Legac, M. Marco, J. Miguel, V. Muñoz, E. Porras, J. C. de la Rosa, J. R. Ruiz, E. Sandoval, P. Ventosa, P. J. Rosenthal and J. M. Fiandor, J. Med. Chem., 2010, 53, 6129–6152 CrossRef PubMed.
- K. N. P. G. Allangba, M. Keita, R. Kre N'Guessan, E. Megnassan, V. Frecer and S. Miertus, J. Enzyme Inhib. Med. Chem., 2019, 34, 547–561 CrossRef CAS PubMed.
- N. Sharma, D. Mohanakrishnan, A. Shard, A. Sharma, Saima, A. K. Sinha and D. Sahal, J. Med. Chem., 2012, 55, 297–311 CrossRef CAS PubMed.
- I. Ravish and N. Raghav, RSC Adv., 2015, 5, 50440–50453 RSC.
- H. Wei, J. Ruan and X. Zhang, RSC Adv., 2016, 6, 10846–10860 RSC.
- J. E. Olson, G. K. Lee, A. Semenov and P. J. Rosenthal, Bioorg. Med. Chem., 1999, 7, 633–638 CrossRef CAS PubMed.
- P. J. Rosenthal, G. K. Lee and R. E. Smith, J. Clin. Invest., 1993, 91, 1052–1056 CrossRef CAS PubMed.
- R. Ettari, F. Bova, M. Zappalà, S. Grasso and N. Micale, Med. Res. Rev., 2010, 30, 136–167 CAS.
- G. C. Tron, T. Pirali, R. A. Billington, P. L. Canonico, G. Sorba and A. A. Genazzani, Med. Res. Rev., 2008, 28, 278–308 CrossRef CAS PubMed.
- S. G. Agalave, S. R. Maujan and V. S. Pore, Chem.–Asian J., 2011, 6, 2696–2718 CrossRef CAS PubMed.
- A. D. Moorhouse, A. M. Santos, M. Gunaratnam, M. Moore, S. Neidle and J. E. Moses, J. Am. Chem. Soc., 2006, 128, 15972–15973 CrossRef CAS.
- X.-L. Wang, K. Wan and C.-H. Zhou, Eur. J. Med. Chem., 2010, 45, 4631–4639 CrossRef CAS PubMed.
- R. P. Tripathi, A. K. Yadav, A. Ajay, S. S. Bisht, V. Chaturvedi and S. K. Sinha, Eur. J. Med. Chem., 2010, 45, 142–148 CrossRef CAS PubMed.
- X. Li, Y. Lin, Q. Wang, Y. Yuan, H. Zhang and X. Qian, Eur. J. Med. Chem., 2011, 46, 1274–1279 CrossRef CAS PubMed.
- A. Montagu, V. Roy, J. Balzarini, R. Snoeck, G. Andrei and L. A. Agrofoglio, Eur. J. Med. Chem., 2011, 46, 778–786 CrossRef CAS PubMed.
- B. Fang, C.-H. Zhou and X.-C. Rao, Eur. J. Med. Chem., 2010, 45, 4388–4398 CrossRef CAS PubMed.
- N. Devender, S. Gunjan, R. Tripathi and R. P. Tripathi, Eur. J. Med. Chem., 2017, 131, 171–184 CrossRef CAS PubMed.
- F. Shah, P. Mukherjee, J. Gut, J. Legac, P. J. Rosenthal, B. L. Tekwani and M. A. Avery, J. Chem. Inf. Model., 2011, 51, 852–864 CrossRef CAS PubMed.
- C. Teixeira, J. R. B. Gomes and P. Gomes, Curr. Med. Chem., 2011, 18, 1555–1572 CrossRef CAS PubMed.
- F. Shah, Y. Wu, J. Gut, Y. Pedduri, J. Legac, P. J. Rosenthal and M. A. Avery, MedChemComm, 2011, 2, 1201 RSC.
- Y.-Q. Hu, S. Zhang, F. Zhao, C. Gao, L.-S. Feng, Z.-S. Lv, Z. Xu and X. Wu, Eur. J. Med. Chem., 2017, 133, 255–267 CrossRef CAS PubMed.
- L.-S. Feng, M.-L. Liu, S. Zhang, Y. Chai, B. Wang, Y.-B. Zhang, K. Lv, Y. Guan, H.-Y. Guo and C.-L. Xiao, Eur. J. Med. Chem., 2011, 46, 341–348 CrossRef CAS PubMed.
- S. Zhang, Z. Xu, C. Gao, Q.-C. Ren, L. Chang, Z.-S. Lv and L.-S. Feng, Eur. J. Med. Chem., 2017, 138, 501–513 CrossRef CAS PubMed.
- Z. Xu, S. Zhang, C. Gao, J. Fan, F. Zhao, Z.-S. Lv and L.-S. Feng, Chin. Chem. Lett., 2017, 28, 159–167 CrossRef CAS.
- X.-D. Jia, S. Wang, M.-H. Wang, M.-L. Liu, G.-M. Xia, X.-J. Liu, Y. Chai and H.-W. He, Chin. Chem. Lett., 2017, 28, 235–239 CrossRef CAS.
- S. K. Puri and G. P. Dutta, Trans. R. Soc. Trop. Med. Hyg., 1990, 84, 759–760 CrossRef CAS PubMed.
- A. P. Gorka, J. N. Alumasa, K. S. Sherlach, L. M. Jacobs, K. B. Nickley, J. P. Brower, A. C. de Dios and P. D. Roepe, Antimicrob. Agents Chemother., 2013, 57, 356–364 CrossRef CAS PubMed.
- D. C. Warhurst, J. C. Craig, I. S. Adagu, R. K. Guy, P. B. Madrid and Q. L. Fivelman, Biochem. Pharmacol., 2007, 73, 1910–1926 CrossRef CAS PubMed.
- A. C. Chou, R. Chevli and C. D. Fitch, Biochemistry, 1980, 19, 1543–1549 CrossRef CAS.
- A. F. G. Slater and A. Cerami, Nature, 1992, 355, 167–169 CrossRef CAS PubMed.
- T. J. Egan, D. C. Ross and P. A. Adams, FEBS Lett., 1994, 352, 54–57 CrossRef CAS PubMed.
- D. J. Sullivan, I. Y. Gluzman, D. G. Russell and D. E. Goldberg, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 11865–11870 CrossRef CAS PubMed.
- L. B. Casabianca, J. B. Kallgren, J. K. Natarajan, J. N. Alumasa, P. D. Roepe, C. Wolf and A. C. de Dios, J. Inorg. Biochem., 2009, 103, 745–748 CrossRef CAS PubMed.
- L. B. Casabianca, D. An, J. K. Natarajan, J. N. Alumasa, P. D. Roepe, C. Wolf and A. C. de Dios, Inorg. Chem., 2008, 47, 6077–6081 CrossRef CAS PubMed.
- A. Dorn, S. R. Vippagunta, H. Matile, C. Jaquet, J. L. Vennerstrom and R. G. Ridley, Biochem. Pharmacol., 1998, 55, 727–736 CrossRef CAS.
- S. Verma, S. Pandey, P. Agarwal, P. Verma, S. Deshpande, J. K. Saxena, K. Srivastava, P. M. S. Chauhan and Y. S. Prabhakar, RSC Adv., 2016, 6, 25584–25593 RSC.
- A. Mahindra, R. P. Gangwal, S. Bansal, N. E. Goldfarb, B. M. Dunn, A. T. Sangamwar and R. Jain, RSC Adv., 2015, 5, 22674–22684 RSC.
- B. C. Pérez, C. Teixeira, M. Figueiras, J. Gut, P. J. Rosenthal, J. R. B. Gomes and P. Gomes, Eur. J. Med. Chem., 2012, 54, 887–899 CrossRef PubMed.
- M. C. Lombard, D. D. N'Da, J. C. Breytenbach, N. I. Kolesnikova, C. Tran Van Ba, S. Wein, J. Norman, P. Denti, H. Vial and L. Wiesner, Eur. J. Pharm. Sci., 2012, 47, 834–841 CrossRef CAS PubMed.
- R. Kharb, P. C. Sharma and M. S. Yar, J. Enzyme Inhib. Med. Chem., 2011, 26, 1–21 CrossRef CAS PubMed.
- D. K. Yadav, R. Rai, N. Kumar, S. Singh, S. Misra, P. Sharma, P. Shaw, H. Pérez-Sánchez, R. L. Mancera, E. H. Choi, M. Kim and R. Pratap, Sci. Rep., 2016, 6, 38128 CrossRef CAS PubMed.
- D. E. V. Pires, T. L. Blundell and D. B. Ascher, J. Med. Chem., 2015, 58, 4066–4072 CrossRef CAS PubMed.
- H. Miyakoshi, S. Miyahara, T. Yokogawa, K. Endoh, T. Muto, W. Yano, T. Wakasa, H. Ueno, K. T. Chong, J. Taguchi, M. Nomura, Y. Takao, A. Fujioka, A. Hashimoto, K. Itou, K. Yamamura, S. Shuto, H. Nagasawa and M. Fukuoka, J. Med. Chem., 2012, 55, 6427–6437 CrossRef CAS.
- P. Filippakopoulos, J. Qi, S. Picaud, Y. Shen, W. B. Smith, O. Fedorov, E. M. Morse, T. Keates, T. T. Hickman, I. Felletar, M. Philpott, S. Munro, M. R. McKeown, Y. Wang, A. L. Christie, N. West, M. J. Cameron, B. Schwartz, T. D. Heightman, N. La Thangue, C. A. French, O. Wiest, A. L. Kung, S. Knapp and J. E. Bradner, Nature, 2010, 468, 1067–1073 CrossRef CAS PubMed.
- A. Gupta, A. Gandhimathi, P. Sharma and B. Jayaram, Protein Pept. Lett., 2007, 14, 632–646 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CAS.
- S. Kumar, A. Saini, J. Gut, P. J. Rosenthal, R. Raj and V. Kumar, Eur. J. Med. Chem., 2017, 138, 993–1001 CrossRef CAS.
- B. R. Shenai, P. S. Sijwali, A. Singh and P. J. Rosenthal, J. Biol. Chem., 2000, 275, 29000–29010 CrossRef CAS PubMed.
- A. Kumar, P. V. N. Dasaradhi, V. S. Chauhan and P. Malhotra, Biochem. Biophys. Res. Commun., 2004, 317, 38–45 CrossRef CAS.
- V. M. S. Gil and N. C. Oliveira, J. Chem. Educ., 1990, 67, 473 CrossRef CAS.
- B. Jayaram, T. Singh, G. Mukherjee, A. Mathur, S. Shekhar and V. Shekhar, BMC Bioinf., 2012, 13, S7 CrossRef PubMed.
- T. Singh, D. Biswas and B. Jayaram, J. Chem. Inf. Model., 2011, 51, 2515–2527 CrossRef CAS PubMed.
- D. A. Pearlman, D. A. Case, J. W. Caldwell, W. S. Ross, T. E. Cheatham, S. DeBolt, D. Ferguson, G. Seibel and P. Kollman, Comput. Phys. Commun., 1995, 91, 1–41 CrossRef CAS.
- T. Jain and B. Jayaram, FEBS Lett., 2005, 579, 6659–6666 CrossRef CAS.
- W. Trager and J. B. Jensen, Science, 1976, 193, 673–675 CrossRef CAS PubMed.
- C. Lambros and J. P. Vanderberg, J. Parasitol., 1979, 65, 418–420 CrossRef CAS PubMed.
- M. Smilkstein, N. Sriwilaijaroen, J. X. Kelly, P. Wilairat and M. Riscoe, Antimicrob. Agents Chemother., 2004, 48, 1803–1806 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra06571g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |