
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of graphene via electrochemical exfoliation in different electrolytes for direct electrodeposition of a Cu/graphene composite coating†
Xinyu Maoa,
Liqun Zhua,
Huicong Liu a,
Haining Chena,
Pengfei Jub and
Weiping Li*a
a,
Haining Chena,
Pengfei Jub and
Weiping Li*a
aKey Laboratory of Aerospace Materials and Performance (Ministry of Education), School of Materials Science and Engineering, Beihang University, Beijing 100191, China. E-mail: liweiping@buaa.edu.cn; Fax: +86 1082317113; Tel: +86 1082317113
bShanghai Aerospace Equipment Manufacture, Shanghai 200245, China
First published on 1st November 2019
Abstract
Directly dispersing graphene into an electrolyte still remains a crucial difficulty in electrodepositing a graphene enhanced composite coating onto electrical contact materials. Herein, graphene was synthesized via electrochemical exfoliation in an N,N-dimethylformamide (DMF)/H2O solution containing (NH4)2SO4. The electrochemically exfoliated graphene nanosheets (GNs) were directly dispersed by sonication. In comparison with graphene synthesized from aqueous solution, the GNs electrochemically exfoliated in the DMF/H2O–(NH4)2SO4 solution exhibit a lower degree of oxidation. Cu/graphene composite coatings were subsequently electrodeposited onto Cu foils by adding Cu2+ into the as-fabricated graphene solution. The surface nanostructure of the Cu/graphene composite coatings was transformed from loose pine needles to a uniform and compact structure with an increase in the concentration of Cu2+, which indicated that the controllable synthesis of Cu/graphene composite coatings with different performances could be achieved in graphene dispersions after adding Cu2+. In order to synthesize graphene via electrochemical exfoliation and directly electrodeposit a Cu/graphene composite coating without adding CuSO4 or any other additive, an attempt was made to directly electrodeposit a Cu/graphene composite coating in CuSO4/DMF/H2O solution after electrochemical exfoliation.
1. Introduction
Composite coatings have played a significant role in past few decades owing to the rapid development of novel technologies, which require high-performance materials. Metal matrix composites containing an enhanced second phase exhibit desirable electrical conductivity, excellent ductility and significantly improved mechanical performance, since they combine the mechanical and physical properties of metals with those of the dispersed phase.1–3 Composite electrodeposition is a facile and effective method to co-deposit a second phase in the electrodeposited layer to improve material properties. Graphene, a two-dimensional material consisting of monolayered hexagonally arrayed sp2-bonded carbon atoms, has already attracted a lot of attention in the field of material science.4,5 Various graphene/metal (or metal oxide) composite coatings have been synthesized for electrical contact or electrode materials6–14 owing to an improvement in the electrical conductivity or specific surface area of the coating after the incorporation of graphene. Obstructions to the fabrication of metal/graphene composite coatings mainly arise from two aspects. First, it is difficult to achieve a homogeneous dispersion of graphene in a plating bath and metal matrix. Besides, the synthesis process may induce significant defects in graphene, which degrade its outstanding properties. The one-step method to synthesize graphene and its subsequent co-deposition with metal ions without further treatment is a promising and facile method for the deposition of metal/graphene composite coatings, because it could simplify the synthesis technique and be suitable for the large-scale fabrication of controllable coatings without using volatile solvents or reducing agents.15Layer-by-layer exfoliation is a facile and effective route to synthesize two-dimensional flakes from materials with a hierarchical structure. Some previous work on exfoliation has been reported: for example, the mass production of graphene-like ultrathin nanosheets by an intercalant-assisted thermal cleavage method.16–18 Electrochemical exfoliation has drawn a lot of attention as a promising approach for producing graphene on an industrial scale, which is efficient, economic and environmentally friendly.19 The electrolytes utilized in electrochemical exfoliation could be divided into two categories: aqueous solutions and ionic-liquid (IL)-based electrolytes. For electrochemical exfoliation conducted in aqueous solution, graphene is hard to obtain and disperse directly. Organic solvents with a low surface energy were usually employed to disperse the graphene exfoliated in an aqueous electrolyte. Liu et al. investigated a direct electrochemical exfoliation approach for the preparation of IL functionalized graphene using an IL/water electrolyte, and the exfoliated precipitate was dispersed in DMF.20 Although IL can promote green chemistry applications by reducing the utilization of toxic chemicals,21 it is relatively expensive when applied to the large-scale production of graphene.
So far, the electrolytes employed to electrochemically exfoliate the graphite anode have included sulfonate,22 sulfate,23 nitrate,24 phosphate,25 and carboxylate.26 Since the key reaction on the surface of the graphite anode during electrochemical exfoliation was caused by the anions of the electrolytes, the common cations of the electrolytes mentioned above were sodium ions, ammonium ions, potassium ions to avoid unnecessary reaction on the surface of the cathode. Nevertheless, a solution of copper phthalocyanine tetrasulfonic acid27 was employed to produce few-layered graphene materials. Now that Cu based composites can be used as electrolytes for electrochemical exfoliation, it is possible to explore a Cu based electrolyte to synthesize graphene via electrochemical exfoliation from a graphite anode and directly combine it with electrodeposition on the surface of the cathode to fabricate a Cu/graphene composite coating.
Herein, we report a DMF/H2O solution system to synthesize graphene via electrochemical exfoliation and direct dispersion for the subsequent electrodeposition process. Cu/graphene composite coatings were electrodeposited on Cu foils by adding different amounts of CuSO4 to the graphene dispersion after electrochemical exfoliation. Since the electrochemically exfoliated GNs could be directly dispersed in the DMF/H2O solution system, CuSO4 was employed as an electrolyte for electrochemical exfoliation of graphene and direct electrodeposition of the Cu/graphene composite coating without any additives. It is supposed that this facile, affordable, and scalable method to electrodeposit a Cu/graphene composite coating in electrochemically exfoliated graphene dispersions will have great potential in practical applications.
2. Experimental section
2.1 Materials
Graphite anodes (C content of 99.9970%) and Cu anodes were both purchased from the Guangzhou Etsing Plating Research Institute. DMF (99.5%) was purchased from Shanghai Aladdin Biochemical Technology Co. Ltd., China. (NH4)2SO4 (≥99.0%) and CuSO4 (≥99.0%) were obtained from Xilong Scientific Co., Ltd., China.2.2 Synthesis of graphene via electrochemical exfoliation
The electrochemical exfoliation was conducted in a two-electrode system. A graphite anode was employed as the anode and carbon source. The area of the surface of the graphite anode right against the cathode was 1 cm2 to be consistent with a platinum cathode. (NH4)2SO4 was separately added into deionized water and DMF/H2O solutions (volume ratios of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5, 1:3 and 1:1) to form solutions with a concentration of 0.2 mol L−1. The electrochemical exfoliation was conducted by applying a DC bias (+25 V) to the graphite anode for 90 min. After the electrochemical exfoliation, the dispersions formed in DMF/H2O solutions were ultrasonically treated (at a power of 250 W) for 30 min. All the dispersions were then filtered through a polytetrafluoroethylene (PTFE) membrane, and the residuals on the membrane were washed thoroughly with deionized water by vacuum filtration. The particles were vacuum dried overnight.
:5, 1:3 and 1:1) to form solutions with a concentration of 0.2 mol L−1. The electrochemical exfoliation was conducted by applying a DC bias (+25 V) to the graphite anode for 90 min. After the electrochemical exfoliation, the dispersions formed in DMF/H2O solutions were ultrasonically treated (at a power of 250 W) for 30 min. All the dispersions were then filtered through a polytetrafluoroethylene (PTFE) membrane, and the residuals on the membrane were washed thoroughly with deionized water by vacuum filtration. The particles were vacuum dried overnight.
As for the product exfoliated in an aqueous solution, 50 mg of dried flakes were dispersed in 100 mL of DMF by 30 min of sonication and kept static for 24 h to precipitate the un-exfoliated graphite flakes. The top supernatant, about two thirds of the whole dispersion, was collected for filtration. Graphene paper was prepared via vacuum filtration of the graphene dispersions, followed by peeling from the membrane and drying in a vacuum oven overnight.
CuSO4 was dissolved in the optimized DMF/H2O solution to form an electrolyte with a concentration of 20 g L−1. A graphite rod was employed as the anode while Cu foil was applied as the cathode. The electrochemical exfoliation was carried out under a DC voltage of ∼15 V. After ultrasonic treatment for 30 min, the graphene dispersion was ready for the subsequent electrodeposition process.
2.3 Electrodeposition of Cu/graphene composite coatings
Graphene dispersions were prepared in the optimized DMF/H2O solution via the electrochemical exfoliation method followed by sonication. The Pt cathode and graphite anode were then separately replaced with a Cu anode and a Cu foil cathode. To obtain a stable and constant supply of Cu2+ ions near the cathode, different amounts of CuSO4 were separately added into the graphene dispersions with the same volume as the concentrations of 20 g L−1, 40 g L−1 and 60 g L−1. The electrodeposition was conducted under a current density of 0.5 A dm−2 for 30 min. All the samples were thoroughly washed with deionized water and dried immediately.As for the graphene dispersions synthesized in CuSO4/DMF/H2O solution, the Cu/graphene composite coating was directly electrodeposited after replacing the cathode with new Cu foil. The electrodeposition was carried out under a voltage of ∼15 V for 30 min. The sample was thoroughly washed with deionized water and dried immediately.
2.4 Characterization
Raman spectra of the graphene flakes were acquired through a Renishaw inVia Raman spectrometer with a diode laser (532 nm). X-ray diffraction (XRD) patterns were obtained on Bruker D8 Discover diffractometer at 40 kV, 200 mA with Cu Kα (Kα = 0.154 nm) radiation. The morphologies and chemical composition of the Cu/graphene composite coatings were studied with a field emission scanning electron microscope (FESEM, Zeiss SUPRA 55) equipped with an energy-dispersive X-ray spectrometer (EDS). A transmission electron microscope (TEM, FEI Talos F200X) was used to investigate the microstructure of the composite coatings through high resolution TEM images with electron diffraction patterns. Fourier-transform infrared (FT-IR) spectra were characterized on a Nexus 670 spectrometer. X-ray photoelectron spectroscopy (XPS) measurements were conducted with an ESCALAB250Xi spectrometer with monochromated Al K radiation (1486.6 eV). Calibration of all XPS spectra was performed using the C 1s line at 284.8 eV and curve fitting and background subtraction were conducted with XPS PEAK Version 4.1 software. The contact electrical resistance of the polished Cu foil and Cu/graphene composite coatings were measured with a JK2511 DC low resistance tester.3. Results and discussion
3.1 Optimization of the electrochemical exfoliation parameters
A DMF/H2O solution system was employed to electrochemically exfoliate graphene. Owing to the migration channel for charge carriers provided by H2O, the graphene flakes could be electrochemically exfoliated from the graphite anode and the subsequent electrodeposition could be directly carried out in the complex solution. The abscised flakes were directly dispersed in the solution by sonication due to the existence of DMF. Consequently, to balance the sufficient velocity of electrochemical exfoliation and the dispersibility of the GNs, optimization of the volume ratio for DMF and H2O in solution is necessary. Electrochemical exfoliation was implemented in DMF/H2O solutions (volume ratios of 1:5, 1:3 and 1:1) with 0.2 mol L−1 (NH4)2SO4. For comparison, electrochemical exfoliation was conducted in aqueous solution under the same conditions.
Some previous studies simply considered the mass of product electrochemically exfoliated from the graphite electrode as the mass of prepared graphene.28,29 Since only a fraction of the exfoliated flakes eventually becomes graphene, it is inappropriate to deem the mass of graphene as equivalent to the mass of exfoliated powder. As to the electrochemical exfoliation method adopted in this paper, because the exfoliation and subsequent sonication were accomplished in the same solution, almost all the suspended flakes could be defined as graphene. Hence, in this study, the mass of synthesized graphene could be directly obtained by weighing the mass of residues after filtration. The mass of residues obtained from the DMF/H2O solutions (DMF/H2O = 1:5, 1:3 and 1:1) were 0.0012 g, 0.0019 g and 0.0022 g, respectively. More residues means that more GNs were dispersed in solution, indicating that the dispersibility of GNs is proportional to DMF content in DMF/H2O solution. Therefore, the GNs electrochemically exfoliated in the DMF/H2O solution (DMF/H2O = 1:1) were utilized for further investigation, and the subsequent electrodeposition processes were carried out in the graphene dispersions after the addition of CuSO4.
3.2 Characterization of electrochemically exfoliated graphene
The Raman spectra and a chart of the distribution of the intensity ratio of the D band and G band of the as-prepared GNs are illustrated in Fig. 1. The typical Raman spectrum of graphene presents a D peak at 1350 cm−1, a G peak at 1580 cm−1, and a 2D peak at 2680 cm−1. The prominent D peak originated from the breathing mode of the sp2 carbon atoms and is activated by the existence of defects and structural disorders. The G peak corresponds to the first-order scattering of the E2g mode of the sp2 carbon atoms. The shape and intensity of the 2D peak identify the number of layers in the GNs.30 The average number of layers in both GNs is 5, which is calculated from the value of I2D/IG. The intensity ratio of the D band to the G band (ID/IG) of the GNs obtained from aqueous solution was 0.98 while the ratio for GNs electrochemically exfoliated in DMF/H2O solution was 0.91. The Raman measurements were randomly conducted for each graphene sample, and a chart of the distribution of ID/IG is given in Fig. 1 to verify the repeatability of the experiments. | ||
| Fig. 1 Raman spectra and the distribution of ID/IG of GNs electrochemically exfoliated (a) in aqueous solution and (b) in DMF/H2O solution. | ||
The crystal structure of the GNs was investigated by XRD, and the results are shown in Fig. 2. Consistent with the typical XRD pattern of graphene, a broadened peak could be seen in the pattern of the GNs obtained from aqueous solution. As shown in the pattern of the GNs electrochemically exfoliated in DMF/H2O solution, the most prominent peak corresponds to the (002) plane and indicates the existence of graphite intercalated compounds (GICs). The low-angle peaks at 18.34°, 19.90° and 22.52° correspond to the (011), (111) and (200) planes, respectively, which means (NH4)2SO4 still exists after the sample has been thoroughly washed. Due to the existence of DMF, the oxidation reaction occurring on the surface of the graphite anode is limited. Some GICs are not adequately intercalated before being abscised from the anode, which leads to a failure of exfoliation into GNs. Thus, the GNs synthesized in the DMF/H2O solution are mixed with a few GICs.
 | ||
| Fig. 2 XRD patterns of GNs electrochemically exfoliated (a) in aqueous solution and (b) in DMF/H2O solution. | ||
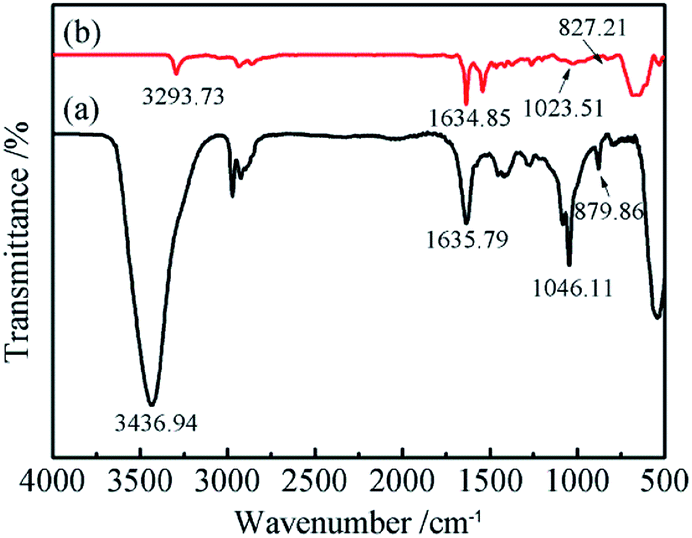
The functional groups and chemical bonding of the electrochemically exfoliated GNs were characterized via FTIR and the spectra are shown in Fig. 3. The spectrum of the GNs synthesized from aqueous solution exhibits an intensive peak at about 3437 cm−1, which corresponds to H2O molecules intercalated into the GNs. Additionally, other C–O-related functionalities, such as hydroxyl (C–OH) groups (1046 cm−1), can clearly be observed. The spectrum also shows the stretching vibration peaks corresponding to carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) groups (1636 cm−1) and epoxy (C–O–C) groups (880 cm−1), which are related to the oxidation of graphite. Compared with the GNs prepared from aqueous solution, the FTIR spectrum of the GNs electrochemically exfoliated in DMF/H2O solution shows a significantly weakened and narrowed peak at around 3293 cm−1 while the intensity of the peak at 1635 cm−1 (CO groups) becomes more intense. It can be speculated that the main type of oxygen containing functional groups are hydroxyl groups. Besides, it could be conjectured that the absence of the peak at 1046 cm−1 (C–OH) in the spectrum of GNs electrochemically exfoliated in DMF/H2O solution can be attributed to a lower degree of oxidation.
O) groups (1636 cm−1) and epoxy (C–O–C) groups (880 cm−1), which are related to the oxidation of graphite. Compared with the GNs prepared from aqueous solution, the FTIR spectrum of the GNs electrochemically exfoliated in DMF/H2O solution shows a significantly weakened and narrowed peak at around 3293 cm−1 while the intensity of the peak at 1635 cm−1 (CO groups) becomes more intense. It can be speculated that the main type of oxygen containing functional groups are hydroxyl groups. Besides, it could be conjectured that the absence of the peak at 1046 cm−1 (C–OH) in the spectrum of GNs electrochemically exfoliated in DMF/H2O solution can be attributed to a lower degree of oxidation.
 | ||
| Fig. 3 FTIR spectra of GNs electrochemically exfoliated (a) in aqueous solution and (b) in DMF/H2O solution. | ||
The atomic percentages of C and O of the GNs electrochemically exfoliated in aqueous solution and DMF/H2O solution are listed in Table 1. The XPS spectra in Fig. 4a and b show the peaks for C and O. The extent of oxidation is determined by deconvolution of the C 1s peak. The C 1s XPS spectra of both samples are further fitted by the Gaussian–Lorentzian curve-fitting method,31 as shown in Fig. 4c and d. The background spectrum is removed by using the Shirley algorithm for peak fitting. From the fitted result, the C 1s spectrum of the GNs electrochemically exfoliated in aqueous solution could be fitted into two peaks at 284.91 eV and 287.10 eV, which correspond to C1 (graphite (C–C)) and C2 (alcohol (C–OH)),32 respectively. As for the C 1s spectrum of GNs electrochemically exfoliated in DMF/H2O solution, the two fitted peaks at 284.86 eV and 286.68 eV correspond to C1 (graphite (C–C)) and C2 (alcohol (C–OH)), respectively. This indicates that the majority species of functional groups on the surface of the GNs are hydroxyl groups. Meanwhile, C/O ratios of the two samples were calculated to compare the degree of oxidation during the electrochemical exfoliation processes. The C/O ratio of the GNs electrochemically exfoliated in aqueous solution is 3.49, while the C/O ratio of the latter reaches 6.49. Since a higher value of the C/O ratio means a lower degree of oxidation, the GNs electrochemically exfoliated in DMF/H2O solution possess fewer oxygen containing functional groups, which is consistent with the FTIR spectra.
| Elements | Samples | |
|---|---|---|
| GNs electrochemically exfoliated in aqueous solution | GNs electrochemically exfoliated in DMF/H2O solution | |
| C 1s | 77.72 | 86.64 |
| O 1s | 22.28 | 13.36 |
 | ||
| Fig. 4 XPS survey spectra of GNs electrochemically exfoliated (a) in aqueous solution and (b) in DMF/H2O solution; high-resolution C 1s spectra of GNs electrochemically exfoliated (c) in aqueous solution and (d) in DMF/H2O solution. | ||
3.3 Surface morphologies of Cu/graphene composite coatings
Since the graphene was successfully synthesized and directly dispersed in DMF/H2O solution, the Cu/graphene composite coatings were separately electrodeposited onto Cu foils in graphene dispersions with various additions of CuSO4. Considering the solubility of CuSO4 in DMF/H2O solution and the investigation of Cu electrodeposition on the surface of electronic devices such as circuit boards,33 the content of added CuSO4 varied from 20 g L−1 to 60 g L−1. The evolution of the micromorphology of the Cu/graphene composite coatings with different contents of CuSO4 in the composite plating baths is shown in Fig. 5. The EDS analysis of the chemical compositions is listed in Table 2. The content of C in the Cu/graphene composite coatings increases slightly with an increase in the content of CuSO4 in the plating solution. This indicated that the greater the amount of Cu2+ added into the solution, the more GNs would be transferred to the surface of the cathode and co-deposited with Cu2+ to form a Cu/graphene composite coating. | ||
| Fig. 5 Top view SEM images of the Cu/graphene composite coatings electrodeposited in the graphene dispersions after adding different contents of CuSO4: (a) 20 g L−1; (c) 40 g L−1; (e) 60 g L−1. Cross-sectional SEM images of the Cu/graphene composite coatings electrodeposited in the graphene dispersions after adding different contents of CuSO4: (b) 20 g L−1; (d) 40 g L−1; (f) 60 g L−1. | ||
| Materials | Wt% | At% | ||
|---|---|---|---|---|
| C | Cu | C | Cu | |
| Cu/graphene composite coating (CuSO4 20 g L−1) | 5.16 | 94.84 | 22.35 | 77.65 |
| Cu/graphene composite coating (CuSO4 40 g L−1) | 6.32 | 93.68 | 26.31 | 73.69 |
| Cu/graphene composite coating (CuSO4 60 g L−1) | 6.99 | 93.01 | 28.46 | 71.54 |
The micromorphology of the surface and the cross section of Cu/graphene composite coating electrodeposited in a composite plating bath containing 20 g L−1 of CuSO4 are shown in Fig. 5a and b. The surface of the Cu/graphene composite coating exhibits pine needle hierarchical nanostructures, and the GNs are observed to be embedded into the composite coating. This indicates that the electrochemically exfoliated GNs successfully co-deposited with Cu2+ ions and formed the Cu/graphene composite coating. According to previous investigations,34,35 the formation mechanism of the pine needle nanostructure is speculated to be as follows. Partial Cu2+ ions in the plating bath coordinate with the GNs to form Cu2+–GNs complexes. During the electrodeposition process, Cu2+ ions and Cu2+–GNs complexes are transferred or driven to the region near the cathode under the electronic field force. At the initial stage of the electrodeposition, the Cu2+ ions and the Cu2+–GNs complexes are preferentially deposited onto the active sites of the cathode surface to form regular and uniformly distributed nuclei. More and more GNs or Cu2+–GNs complexes would be deposited on the formed nuclei, leading to the growth of regular spherical particles. Subsequently, spherical particles would link together to form a trunk, and new nucleation would commence on the active sites at the trunk surface, resulting in the growth of branches. Obviously, GNs play a crucial role in chelating Cu2+ ions and regulating the reaction rate of the generation of metallic Cu to promote the growth of pine needle nanostructures. In a study by Xie,34 an analogous nanostructure composite film was obtained by electrodeposition and remained stable in NaCl solution. Hence, the Cu/graphene composite coating electrodeposited in the graphene dispersions containing 20 g L−1 of CuSO4 is expected to exhibit promising corrosion resistance.
As shown in Fig. 5c and d, when the CuSO4 content of the composite plating bath increases to 40 g L−1, the micromorphology of the Cu/graphene composite coating is obviously changed. The pine needle nanostructure is replaced by spherical grains. The GNs are randomly distributed in the internal structure of the Cu/graphene composite coating. With a further increase in the CuSO4 content in the composite plating bath, the surface and cross-sectional morphology of the Cu/graphene composite coating electrodeposited in the composite plating bath containing 60 g L−1 CuSO4 is shown in Fig. 5e and f. The microstructure of the Cu/graphene composite coating becomes extremely uniform and dense. Meanwhile, the grains are obviously refined compared with previous Cu/graphene composite coatings. Although the distribution of GNs on the Cu/graphene composite coating surface is not as obvious as in the Cu/graphene composite coating prepared in the plating bath with a lower content of CuSO4, observation of the cross section proves that GNs can still be co-deposited with Cu2+ and enter into the Cu/graphene composite coating.
In order to further investigate the microstructure and composition, TEM was conducted for the Cu/graphene composite coatings electrodeposited in DMF/H2O solutions with Cu contents of 20 g L−1, 40 g L−1 and 60 g L−1. The TEM images and selected area electron diffraction (SAED) patterns are shown in Fig. 6. It can be seen that copious Cu grains are distributed on the surface of the GNs throughout Fig. 6a–c. As can be seen from the SAED patterns in Fig. 6d–f, the most prominent diffraction patterns correspond to the (002) lattice plane of the graphene mixed with a little graphite and the (111) lattice plane of Cu, respectively. This indicates that the Cu phase displays a face-centered cubic structure.
 | ||
| Fig. 6 TEM images of the Cu/graphene composite coatings electrodeposited in DMF/H2O solutions with CuSO4 contents of (a) 20 g L−1, (b) 40 g L−1 and (c) 60 g L−1; SAED patterns of the Cu/graphene composite coatings electrodeposited in DMF/H2O solution with CuSO4 contents of (d) 20 g L−1, (e) 40 g L−1 and (f) 60 g L−1. | ||
3.4 Electrical conductivity of Cu/graphene composite coatings
The measured contact electrical resistance values of polished Cu foil and Cu/graphene composite coatings are shown in Table 3. The contact electrical resistance values of all the Cu/graphene composite coatings are lower than that of polished Cu foil. For the Cu/graphene composite coatings, the contact electrical resistance values exhibit a declining trend with the content of CuSO4 in the graphene dispersions increasing from 20 g L−1 to 60 g L−1. The Cu/graphene composite coating electrodeposited in graphene dispersions containing 60 g L−1 of CuSO4 shows the lowest value of contact electrical resistance. This is attributed to the fact that the Cu/graphene composite coating prepared in the graphene dispersion with 60 g L−1 of CuSO4 possesses the most uniform and compact microstructure among the Cu/graphene composite coatings, which is consistent with the observed results of FESEM. This indicates that the GNs as a conducting network contribute to an enhanced flow of charge carriers in the Cu/graphene composite coatings.| Sample numbers | Materials | Contact electrical resistance (mΩ) |
|---|---|---|
| 1 | Cu foil | 0.28 |
| 2 | Cu/graphene composite coating (CuSO4 20 g L−1) | 0.27 |
| 3 | Cu/graphene composite coating (CuSO4 40 g L−1) | 0.25 |
| 4 | Cu/graphene composite coating (CuSO4 60 g L−1) | 0.22 |
3.5 Attempt at direct electrodeposition of Cu/graphene composite coating after electrochemical exfoliation
Since graphene has been synthesized via electrochemical exfoliation and directly dispersed in the DMF/H2O solution system, it is assumed that other sulfates could also be used as electrolytes for electrochemical exfoliation. Thus, an attempt was made to electrochemically exfoliate a graphite anode with CuSO4. In order to directly co-deposit the electrochemically exfoliated GNs with Cu2+ ions to fabricate a Cu/graphene composite coating, electrochemical exfoliation was carried out in a CuSO4/DMF/H2O solution with a DMF/H2O volume ratio of 1:1. During the electrochemical exfoliation process, the sulfate ions transferred to the surface of the anode to intercalate graphite and exfoliate the GICs into the GNs. In the meantime, the Cu2+ ions were driven to the surface of the cathode and electrodeposited onto the Cu foil. As time went on, the exfoliated GNs and the coordinated Cu2+ formed a Cu–GNs complex with a positive charge. The Cu–GN complexes were driven to the region near the cathode by the electric field force and co-deposited with Cu2+ ions to fabricate a Cu/graphene composite coating. Therefore, it is possible to simultaneously take advantage of the reactions occurring on the anode and cathode to synthesize GNs and directly electrodeposit a Cu/graphene composite coating in the same solution without further treatment. A schematic illustration of this method is shown in Scheme 1.
 | ||
| Scheme 1 Schematic illustration of the approach for direct electrodeposition of a Cu/graphene composite coating in CuSO4/DMF/H2O solution after electrochemical exfoliation. | ||
Raman, FTIR and XPS spectra were further applied to characterize the electrochemically exfoliated graphene flakes in CuSO4/DMF/H2O solution. As shown in Fig. 7a, the Raman spectrum of the exfoliated GNs exhibits the typical character of graphene. The ID/IG value of the GNs is calculated to be 0.84. This is slightly lower than for GNs electrochemically exfoliated in (NH4)2SO4/DMF/H2O solution. As for the FTIR spectrum exhibited in Fig. 7b, the main categories of the functional groups of the GNs are hydroxyl (C–OH) groups (1097.4 cm−1) and carbonyl (CO) groups. As shown in Fig. 7c, the C/O ratio is calculated to be 8.34. This indicates that the GNs electrochemically exfoliated in CuSO4/DMF/H2O solution presented a lower degree of oxidation than the GNs electrochemically exfoliated in (NH4)2SO4/DMF/H2O solution. Fig. 7d depicts the C 1s spectrum of the GNs, where the main peak at 284.8 eV corresponds to the sp2 bonded carbon, and the small deconvoluted peaks at 285.5 eV and 288.4 eV represent the C–O and CO bonds, respectively. Fig. 7e presents the C 1s spectrum of the Cu/graphene composite coating and Fig. 7f shows the Cu 2p spectrum of the Cu/graphene composite coating. The main peak in the Cu 2p spectrum at 932.7 eV (Cu 2p3/2) corresponds to the metallic Cu–Cu bond. In order to evaluate the degree of reduction of the GNs, the relative atomic concentrations of different carbon bonds were calculated from the corresponding peak areas in the C 1s spectrum, utilizing linear background subtraction. The result of the calculation shows that the atomic percentages of the sp2 bonded carbon, hydroxyl groups and carbonyl groups of the electrochemically exfoliated GNs are 68.61%, 27.25% and 4.14%. After being co-deposited with Cu and forming the Cu/graphene composite coating, the atomic percentages of the sp2 bonded carbon, hydroxyl groups and carbonyl groups of the electrodeposited GNs are 70.98%, 23.28% and 5.74%. Despite the atomic percentage of carbonyl being slightly increased after co-deposition with Cu, the sum of the atomic percentages of hydroxyl groups and carbonyl groups on the GN surface decreases after electrodeposition, which means the GNs are reduced at the cathode.
 | ||
| Fig. 7 Raman spectrum (a), FTIR spectrum (b), XPS survey spectrum (c) and high-resolution C 1s spectrum (d) of the GNs electrochemically exfoliated in CuSO4/DMF/H2O solution; high-resolution C 1s spectrum (e) and high-resolution Cu 2p spectrum (f) of the Cu/graphene composite coating electrodeposited in CuSO4/DMF/H2O. | ||
The electrical conductivity was characterized by measuring the contact electrical resistance value of the Cu/graphene composite coating. Compared with Cu/graphene composite coatings electrodeposited by adding CuSO4 in the graphene dispersions as mentioned above, a contact electrical resistance value of 0.24 mΩ indicates that the Cu/graphene composite coating electrodeposited in CuSO4/DMF/H2O solution shows better potential for application to electrical contact materials.
4. Conclusions
Graphene has been synthesized via an electrochemical exfoliation method in DMF/H2O solution containing (NH4)2SO4. Compared to GNs synthesized via electrochemical exfoliation in aqueous solution, the GNs electrochemically exfoliated in the DMF/H2O solution exhibited a lower ID/IG (0.91) and a higher C/O ratio (6.49). The majority species of functional groups on the surface of both GNs were hydroxyl groups. Cu/graphene composite coatings were subsequently fabricated by electrodeposition in graphene dispersions formed in DMF/H2O solution after the addition of CuSO4. The surface nanostructure of Cu/graphene composite coatings transformed from loose pine needles to a uniform and compact structure with an increase in CuSO4 concentration, indicating that the controllable synthesis of Cu/graphene composite coatings with different performances could be achieved by direct electrodeposition in dispersions of electrochemically exfoliated GNs. CuSO4 was utilized as an electrolyte to synthesize graphene via electrochemical exfoliation, which was directly used to electrodeposit a Cu/graphene composite coating without the further addition of CuSO4 or any other additive.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by National Key Research and Development Program (2018YFB2002000) and National Natural Science Foundation of China (Grant No. U1637204).Notes and references
- S. Suarez, N. Souza, F. Lasserre and F. Mücklich, Adv. Eng. Mater., 2016, 18, 1626–1633 CrossRef CAS.
- X. Zhang, C. S. Shi, E. Z. Liu, F. He, Li. Y. Ma, Q. Y. Li, J. J. Li, W. Bacsa, N. Q. Zhao and C. N. He, Nanoscale, 2017, 9, 11929–11938 RSC.
- Z. Y. Yang, L. D. Wang, Y. Cui, Z. D. Shi, M. Wang and W. D. Fei, Nanoscale, 2018, 10, 16990–16995 RSC.
- Y. N. Gong, Y. J. Ping, D. L. Li, C. Z. Luo, X. F. Ruan, Q. Fu and C. X. Pan, Appl. Surf. Sci., 2017, 397, 213–219 CrossRef CAS.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS.
- Y. Zhang, X. H. Xia, B. Liu, S. J. Deng, D. Xie, Q. Liu, Y. D. Wang, J. B. Wu, X. L. Wang and J. P. Tu, Adv. Energy Mater., 2019, 9, 1803342 CrossRef.
- Y. H. Pang, Y. Zhang, X. L. Sun, H. L. Ding, T. T. Ma and X. F. Shen, Talanta, 2019, 192, 387–394 CrossRef CAS.
- I. N. Kholmanov, S. H. Domingues, H. Chou, X. H. Wang, C. Tan, J. Y. Kim, H. F. Li, R. Piner, A. J. G. Zarbin and R. S. Ruoff, ACS Nano, 2013, 7, 1811–1816 CrossRef CAS.
- Q. M. Su, L. B. Yao, J. Zhang, G. H. Du and B. S. Xu, ACS Appl. Mater. Interfaces, 2015, 7, 23062–23068 CrossRef CAS.
- P. G. Perret, P. R. L. Malenfant, C. Bock and B. MacDougall, J. Electrochem. Soc., 2012, 159, A1554–A1561 CrossRef CAS.
- M. Hilder, O. Winther-Jensen, B. Winther-Jensen and D. R. MacFarlane, Phys. Chem. Chem. Phys., 2012, 14, 14034–14040 RSC.
- K. Jagannadham, J. Vac. Sci. Technol., B: Nanotechnol. Microelectron.: Mater., Process., Meas., Phenom., 2012, 30, 03D109 Search PubMed.
- K. Jagannadham, Metall. Mater. Trans. A, 2013, 44, 552–559 CrossRef CAS.
- J. H. Lee, X. L. Hu, A. A. Voevodin, A. Martini and D. Berman, Micromachines, 2018, 9, 169 CrossRef.
- M. Hilder, B. Winther-Jensen, D. Li, M. Forsyth and D. R. MacFarlane, Phys. Chem. Chem. Phys., 2011, 13, 9187–9193 RSC.
- J. M. Li, Appl. Phys. A, 2010, 99, 229–235 CrossRef CAS.
- J. M. Li and J. Fang, J. Mater. Chem. C, 2017, 5, 9545–9551 RSC.
- J. M. Li, ACS Appl. Mater. Interfaces, 2017, 9, 39626–39634 CrossRef CAS.
- A. M. Abdelkader, A. J. Cooper, R. A. W. Dryfe and I. A. Kinloch, Nanoscale, 2015, 7, 6944–6956 RSC.
- N. Liu, F. Luo, H. Wu, Y. Liu, C. Zhang and J. Chen, Adv. Funct. Mater., 2008, 18, 1518–1525 CrossRef CAS.
- X. Wang, P. F. Fulvio, G. A. Baker, G. M. Veith, R. R. Unocic, S. M. Mahurin, M. Chi and S. Dai, Chem. Commun., 2010, 46, 4487–4489 RSC.
- G. Wang, B. Wang, J. Park, Y. Wang, B. Sun and J. Yao, Carbon, 2009, 47, 3242–3246 CrossRef CAS.
- K. Parvez, Z. S. Wu, R. Li, X. Liu, R. Graf, X. Feng and K. Müllen, J. Am. Chem. Soc., 2014, 136, 6083–6091 CrossRef CAS.
- X. Lu and C. Zhao, Phys. Chem. Chem. Phys., 2013, 15, 20005–20009 RSC.
- F. Zeng, Z. Sun, X. Sang, D. Diamond, K. T. Lau, X. Liu and D. S. Su, ChemSusChem, 2011, 4, 1587–1591 CrossRef CAS.
- P. Khanra, T. Kuila, S. H. Bae, N. H. Kim and J. H. Lee, J. Mater. Chem., 2012, 22, 24403–24410 RSC.
- J. P. Mensing, T. Kerdcharoen, C. Sriprachuabwong, A. Wisitsoraat, D. Phokharatkul, T. Lomas and A. Tuantranont, J. Mater. Chem., 2012, 22, 17094–17099 RSC.
- A. J. Cooper, N. R. Wilson, I. A. Kinloch and R. A. W. Dryfe, Carbon, 2014, 66, 340–350 CrossRef CAS.
- S. T. Hossain and R. G. Wang, Electrochim. Acta, 2016, 216, 253–260 CrossRef CAS.
- A. C. Ferrari, J. C. Meyer, V. Scardaci, C. Casiraghi, M. Lazzeri, F. Mauri, S. Piscanec, D. Jiang, K. S. Novoselov, S. Roth and A. K. Geim, Phys. Rev. Lett., 2006, 97, 187401 CrossRef CAS.
- H. Ganegoda, D. S. Jensen, D. Olive, L. Cheng, C. U. Segre, M. R. Linford and J. Terry, J. Appl. Phys., 2012, 111, 053705 CrossRef.
- R. I. R. Blyth, H. Buqa, F. P. Netzer, M. G. Ramsey, J. O. Besenhard, P. Golob and M. Winter, Appl. Surf. Sci., 2000, 1, 99–106 CrossRef.
- D. H. Lee, T. H. Lim, H. N. Lim, H. J. Kim and O. J. Kwon, Int. J. Electrochem. Sci., 2018, 13, 11829–11838 CrossRef CAS.
- G. X. Xie, M. Forslund and J. S. Pan, ACS Appl. Mater. Interfaces, 2014, 6, 7444–7455 CrossRef CAS.
- C. C. Li and H. C. Zeng, J. Mater. Chem., 2010, 20, 9187–9192 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra06541e |
| This journal is © The Royal Society of Chemistry 2019 |