
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSeven membered chelate Pt(II) complexes with 2,3-di(2-pyridyl)quinoxaline ligands: studies of substitution kinetics by sulfur donor nucleophiles, interactions with CT-DNA, BSA and in vitro cytotoxicity activities†
Rajesh
Bellam
 *a,
Deogratius
Jaganyi
*bc,
Allen
Mambanda
a,
Ross
Robinson
a and
Manickam Dakshinamoorthi
BalaKumaran
d
*a,
Deogratius
Jaganyi
*bc,
Allen
Mambanda
a,
Ross
Robinson
a and
Manickam Dakshinamoorthi
BalaKumaran
d
aSchool of Chemistry and Physics, University of KwaZulu-Natal, Private Bag X01, Scottsville, Pietermaritzburg 3209, South Africa. E-mail: rajeshchowdarybellam@gmail.com
bSchool of Science, College of Science and Technology, University of Rwanda, P.O. Box 4285, Kigali, Rwanda. E-mail: deojaganyi@gmail.com
cDepartment of Chemistry, Durban University of Technology, P.O. Box 1334, Durban 4000, South Africa
dDepartment of Biotechnology, Dwaraka Doss Goverdhan Doss Vaishnav College, Arumbakkam, Chennai-600106, Tamil Nadu, India
First published on 7th October 2019
Abstract
Dichloro platinum(II) complexes coordinated with 2,3-di(2-pyridyl)quinoxaline ligands which form seven-membered chelates namely, bpqPtCl2, dmbpqPtCl2 and bbqPtCl2 (where bpq, dmbpq and bbq are 2,3-di(2-pyridyl)quinoxaline, 6,7-dimethyl-2,3-di(2-pyridyl)quinoxaline and 2,3-bis(2′pyriyl)benzo[g]quinoxaline, respectively) were synthesized, characterised and their respective hydrated product complexes namely, bpqPt(OH2)22+, dmbpqPt(OH2)22+ and bbqPt(OH2)22+ were prepared by chloride metathesis. The substitution kinetics of the aquated cations by thiourea nucleophiles indicated that the two aqua ligands are substituted simultaneously according to the rate law: kobs = k2[Nu]. This is followed by a forced dechelation of the ligands from the Pt (II) to form Pt(Nu)42+ species. The dechelation step is considerably slow to be monitored reliably. The rate of substitution is marginally enhanced by introducing two methyl groups and by extending the π-conjugation on the bpq core ligand. The reactivity order increased as bpqPt(OH2)22+ < dmbpqPt(OH2)22+ < bbqPt(OH2)22+. Reactivity trends were well supported by theoretical computed DFT electronic descriptors. The interactions of the Pt(II) complexes with CT-DNA and BSA were also examined spectroscopically in tris buffers at pH 7.2. Spectroscopic and viscosity measurements suggested strong associative interactions between the Pt(II) complexes and CT-DNA, most likely through groove binding. In silico theoretical binding studies showed energetically stable poses through associative non-covalent interactions. In vitro MTT cytotoxicity IC50 values of the Pt(II) complexes on human liver carcinoma cells (HepG2) cancer cell lines revealed bbqPtCl2 as the least active. The fluorescence staining assays revealed the morphological changes suggested early apoptotic induction as well as non-specific necrosis.
1 Introduction
The discovery that cis-platin and its analogs were anti-cancer active propelled chemotherapy to the fore frontiers in the treatment of commonly occurring cancers. It is now known that upon activation by hydrolysis, cisplatin preferentially reacts with the N7 atoms of neighboring guanine bases to form 1,2-intrastrand DNA-Pt(ammine) adducts. These adducts impair the helical stability of DNA duplet. The cellular response is either to repair these adducts or to activate suicidal killing of the rapidly growing cancer cells. In spite of the therapeutic activity, these drugs have significant side effects.However, the surging interest to design new Pt(II) complexes which have better therapeutic indices than cis-platin but with reduced toxicity remains. Since, the anti-cancer mechanism of metal drugs involves covalent binding through a substitution step as well as non-covalent interactions (electrostatic association, base pair interaction, groove binding), the design of new metal-based drugs should be such that the ligands around the metal centre endow good kinetic and thermodynamic properties as well as structural aspects that favour associative non-covalent interactions. One approach in the design of these tailored anti-cancer drugs is to maintain the cis geometry of cis platin and explore further ways in which their coordination structures can bring about the increased activity so as to ultimately use small doses of the drug and thus minimise side effects. For example, the ammines of cis-platin could be replaced by planar N^N bidentate ligands which may form five, six or seven-membered chelates at the Pt metal and these confer a two-pronged mechanism of antitumor activity by new Pt(II) complexes. Thus, the structure of the ligands plays a crucial role in influencing the pharmacological properties of the complexes.
The reactivity and biological properties of complexes coordinated with N^N bidentate ligands that form mono metallic and bi metallic five-membered chelate rings with the Pt(II) ion have been reported in the literature.1,2 However, there are few reports on six-membered chelated Pt(II) complexes3 while reports for Pt(II) complexes coordinated with bidentate ligands which form seven-membered chelate rings are non-existent. Our initial goal to replace the ammines of cis-platin with planar ligands which implore a similar regime of a labializing effect, prompted us to synthesize homologs of N^N bidentate ligands which could potentially bind Pt, forming seven-membered chelates. Thus, the paper reports on Pt(II) complexes coordinated with 2,3-di(2-pyridyl)quinoxaline bidentate ligands. Due to their versatile coordination the non leaving ligands, they form unusual N^N-seven membered chelate with Pt(II) metal ion. The structures of the investigated seven-member N^N-chelated Pt(II) complexes are shown in Scheme 1. We studied the reactivity of the Pt(II) complexes in aqueous media using S-donor nucleophiles to gain insight on the potential fate of the complexes in a biological system where they can get deactivated through substitution reactions. Another important aspect of the reactivity data of Pt(II) complexes with sulphur nucleophiles is stemmed on the fact that Pt-thiolate and thioether species that forms from the reactions of the complexes with biological nucleophiles can act as temporal drug reservoirs which subsequently get substituted by N7 atoms of the purine bases of DNA.4 The later products are thermodynamically more stable. The thiourea nucleophiles which were used are strong σ-donor and π-acceptors with high aqueous solubility and bind strongly to metal ions. Thus, they are considered excellent model nucleophiles for thiolates and thioethers, which are ubiquitously present in cytosolic fluids.5 To probe on the interactions (covalent/non-covalent) of these complexes with DNA and shuttle proteins, we titrated the complexes with CT-DNA, EtBr + CT-DNA complex and BSA spectrophotometrically. To complement this data, theoretical molecular docking of complexes on B-DNA duplex was performed to ascertain the molecular mechanism of interaction of the complexes towards DNA. Furthermore, in vitro anticancer activity data of the Pt(II) complexes assessed by the MTT assay and data on nucleus morphological changes evaluated by acridine orange/ethidium bromide (AO/EB) and diamidino-2-phenylindole (DAPI) fluorescence staining assay are reported. The study provides fundamental data for understanding toxicity and development of resistance of metallodrugs in biological systems and hence is of special importance to both the pharmaceutical and biomedical research.
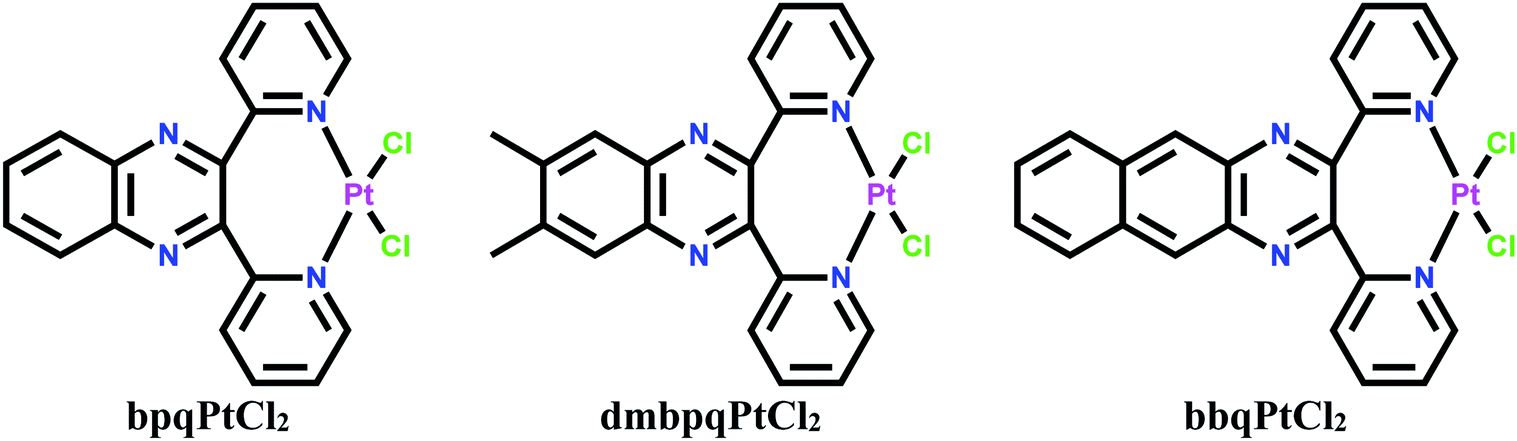 | ||
| Scheme 1 Structural formulae of the dichloro Pt(II) complexes. | ||
2 Results and discussion
2.1 pKa determination
The pKa values of diaqua P(II) complexes were determined spectrophotometrically. Owing to the poor solubility of neutral dichloro Pt(II) complexes, they were aquated through AgCl metathesis. Typical UV-Vis absorption changes in the titration of bpqPt(OH2)22+ with NaOH within pH range of 1–10 are shown in Fig. 1, (also see ESI Fig. S1a and b† for spectral changes for titrations of dmbpqPt(OH2)22+ and bbqPt(OH2)22+ with NaOH, respectively). Tight isosbestic points (four for all complexes) point to the formation of only the intermediate Pt(bpq/dmbpq/bbq)(OH)(OH2) and the final product Pt(bpq/dmbpq/bbq)(OH)2 from the diaqua complex.6 This signifies that no polymeric species were formed during the pH titration of the diaqua complexes. The pKa value of each aqua complex was determined by plotting absorbance against pH at specific wavelengths and fitting the data to standard double Boltzmann equations using the OriginPro 9.1® software.7 Inset of Fig. 1 shows absorbance changes with pH at 279 nm for the titration of bpqPt(OH2)22+. The first and second inflection points on the figure give the values of pKa1 and pKa2, respectively. The pKa values of the aquated complexes are summarised in Table 1 and the proposed deprotonation equilibria are represented in Scheme 2.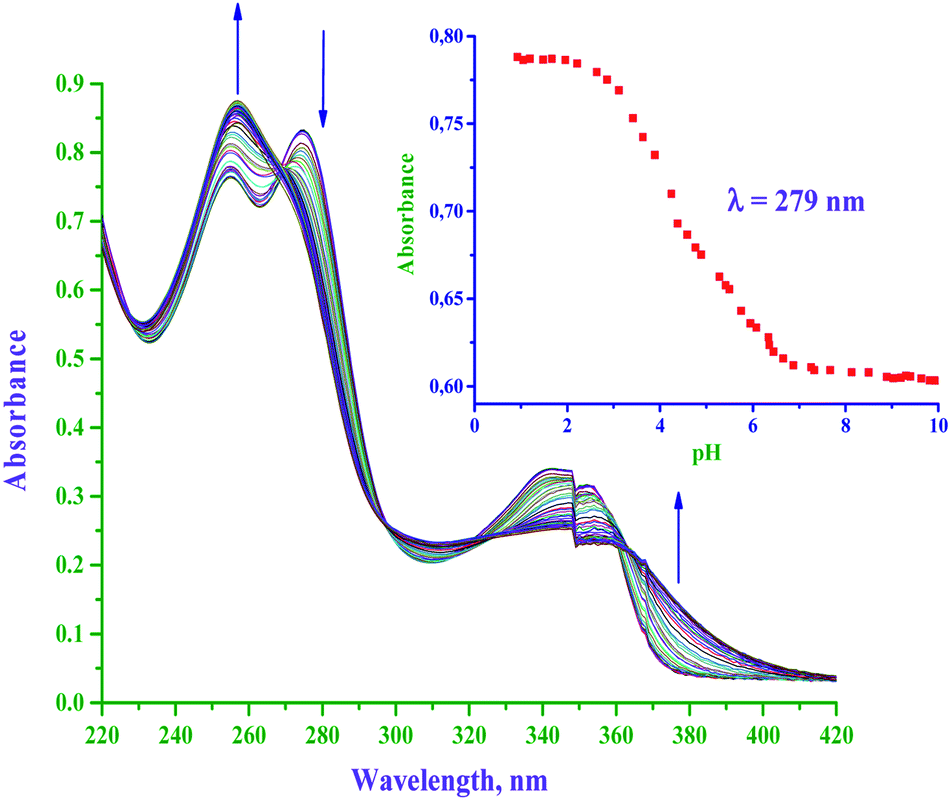 | ||
| Fig. 1 UV-Vis spectral changes of bpqPt(OH2)22+ (50 μM) with in the pH range of 1–11; arrow indicates the change in the absorbance on the addition of NaOH. Inset: plot of absorbance versus pH at the specified wavelength (279 nm); l = 0.1 M (HClO4) and T = 25 °C. | ||
| Complex | pKa | |
|---|---|---|
| pKa1 | pKa2 | |
| bpqPt(OH2)22+ | 3.91 ± 0.07 | 5.93 ± 0.12 |
| dmbpqPt(OH2)22+ | 3.62 ± 0.05 | 5.62 ± 0.10 |
| bbqPt(OH2)22+ | 3.22 ± 0.03 | 4.96 ± 0.09 |
 | ||
| Scheme 2 Proposed reaction scheme for the stepwise deprotonation and substitution of aqua molecules from diaqua Pt(II) complexes by NaOH and S-donor nucleophiles, respectively. | ||
The pKa values of the aquated complexes depend on the electrophilicity of the metal centre as determined by the type/nature of the spectator ligand on the complex.8 The acidity of the complexes depends on the extent of electron withdrawing ability from the Pt–OH2 bond. This also depends on the electronic properties of the spectator ligands. Strong π-acceptor ligands stabilise the metal centre by π-back bonding, leading to lower pKa values.9 From the trend in the pKa values (Table 1) and the structures of the complexes (Scheme 1), the addition of two methyl groups or a conjugating phenyl group on the reference ligand, 2,3-di(2-pyridyl)quinoxaline causes a decrease in the pKa values i.e., the complex dmbpqPt(OH2)22+ and bbqPt(OH2)22+ have lower pKa(1/2) than the complex bpqPt(OH2)22+ and the decreased order is bpqPt(OH2)22+ > dmbpqPt(OH2)22+ > bbqPt(OH2)22+. This is in line with a decrease in electrophilicity on the Pt(II) metal centre (see ESI Table S5†). This decrease in electron density around the Pt(II) metal centre lowered the intrinsic basicity of the complex and led to the aqua ligands becoming less basic and hence the smaller pKa values for dmbpqPt(OH2)22+ and bbqPt(OH2)22+ complexes.10 The deprotonation of the second aqua ligands and hence the pKa2 occurs at higher pH than the first deprotonation. It is well known that relatively highly charged complexes are more electrophilic/acidic than less charged complexes, and due to this, the second protonation step occurs at a higher pH because the Pt(bpq/dmbpq/bbq)(OH)2(OH)+ has lower charge.11
2.2 Species distribution of the aquated Pt(II) complexes
The pKa data was used for predicting the species distribution of the aquated species of the complexes the Specfit Global Analysis software was used to generate the distribution curves of 50 μM solution of bpqPt(OH2)22+, bpqmePt(OH2)22+ and bbqPt(OH2)22+, respectively. The distribution of the different species consecutively generated in solution as a function of pH are given in Fig. 2a–c. At pH 2.0, about 92.8, 97.4 and 98.6% of diaqua species exist in solution while about 7.2, 2.6 and 1.4% in the aqua-hydroxo species for bpqPt(OH2)22+, bpqmePt(OH2)22+ and bbqPt(OH2)22+, respectively. At physiological conditions (pH 7.2), the complexes exist predominantly as their dihydroxo and their relative amounts are 99.8, 98.4 and 88.8% while the dihydroxo species decreases to 0.2, 1.6 and 11.2% of the auqa-hydroxo species for bpqPt(OH2)22+, bpqmePt(OH2)22+ and bbqPt(OH2)22+, respectively. Thus, at pH 2.0, at which the substitution kinetics of the complexes were performed, the complexes exist mainly in their diaqua form. | ||
| Fig. 2 Distribution of the various species present in the solution for 50 μM bpqPt(OH2)22+ (a), dmbpqPt(OH2)22+ (b) and bbqPt(OH2)22+ (c) as a function of pH. | ||
2.3 Absorption spectra of chloro and aqua Pt(II) complexes
The UV-Vis absorption spectra of both the dichloro and diaqua Pt(II) complexes were also recorded and are shown in Fig. 3a and b, respectively and the data is tabulated in ESI Table S1.† The data shows the complexes dmbpqPtCl2 and bbqPtCl2 are red shifted in λmax by 3 and 29 nm in the UV and 18 and 32 nm in the visible, respectively when compared to those of bpqPtCl2. Similarly, the complexes dmbpqPt(OH2)22+ and bbqPt(OH2)22+ red shifted in λmax by 4 and 25 nm in UV and 20 and 30 nm in the visible, respectively when referenced to those of bpqPt(OH2)22+. Thus, there is electronic charge distribution from the Pt(II) 5dπ orbitals to the delocalized π* orbital located on the ligand (MLCT) and is more prominent in dmbpqPtCl2/(OH2)2 and bbqPtCl2/(OH2)2 than the reference complex, bbqPtCl2/(OH2)2.12 | ||
| Fig. 3 UV-Vis absorption spectra of chloro (a) and aqua (b) Pt(II) complexes: [bpq/mebpq/bbqPtCl2] = 8 μM and [bpq/dmbpq/bbqPt(OH2)22+] = 50 μM. | ||
2.4 The rate of substitution from the aquated Pt(II) complexes
The rate of substitution of aqua ligands from the diaqua Pt(II) complexes by S-donor nucleophiles (thiourea, TU, 1,3-dimethyl-2-thiourea, DMTU and 1,1,3,3-tetra methyl-2-thiourea, TMTU) was followed by using a conventional UV-Vis spectrophotometer, measuring the changes in absorbance at a suitable wavelength as a function of [Nu] and time. Fig. 4 shows typical spectral changes due to the reaction of the complex bpqPt(OH2)22+ with TU. The inset shows a typical kinetic trace of absorbance versus time at λ = 292 nm and T = 35 °C which was fitted to a non-linear double-exponential function. Chosen wavelengths for kinetic traces to get kobs values for the reactions of bpq/dmbpq/bbqPt(OH2)22+ complexes with S-donor nucleophiles given in ESI Table S2.†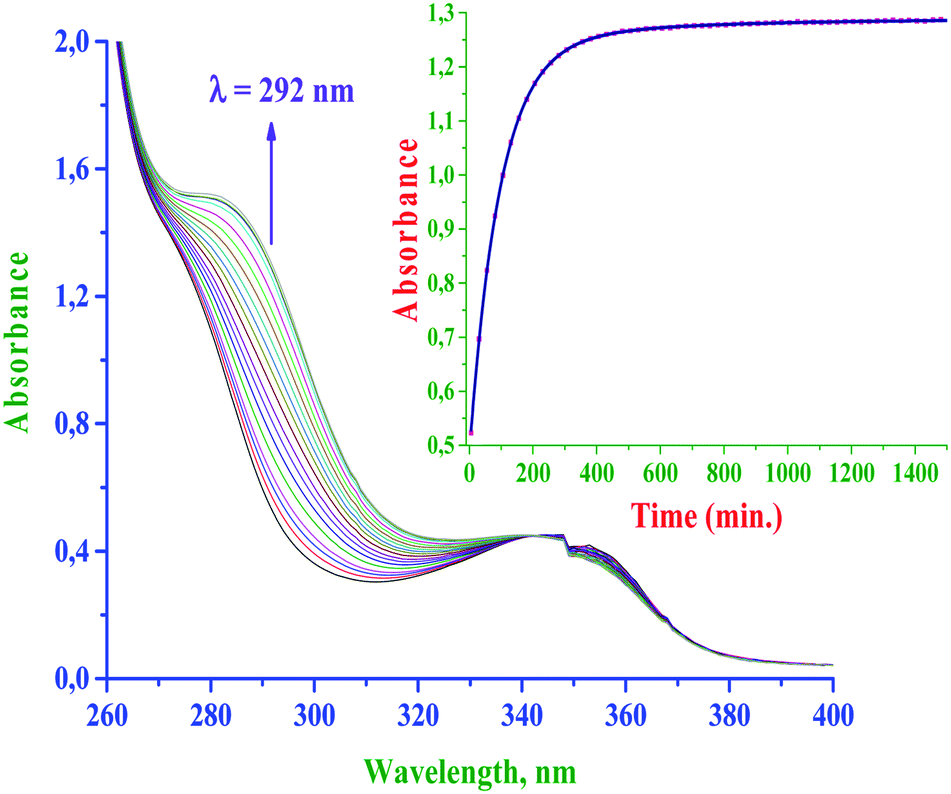 | ||
| Fig. 4 UV-Vis spectral changes for the reaction between bpqPt(OH2)22+ (50 μM) and TU (40-fold excess); inset is a typical kinetic trace of absorbance versus time at λ = 292 nm, pH = 2.0, I = 0.1 M (HClO4) and T = 35 °C. | ||
The pseudo-first-order rate constants, kobs were obtained by fitting the kinetic trace of absorbance growth to a non-linear double-exponential function using OriginPro 9.1® graphical analysis software.7 The kinetic data indicate that the substitution occurred in two consecutive steps for all reactions. The first substitution step corresponds to the simultaneous substitution of two aqua coligands by incoming Nu which occurs at the same rate. The second step is due to the dechelation of the spectator ligand from the Pt(II) metal centre. The dechelation of the ligands is confirmed by monitoring the kinetics of the reaction of one of the complexes bpqPtCl2 with excess TU using 195Pt NMR, and details are given vide infra in the product analysis section (see Fig. 5). However, the second step i.e., dechelation step is too slow to be followed accurately and hence the results were omitted for further discussion. Thus, the substitution pathway is given in Scheme 2.
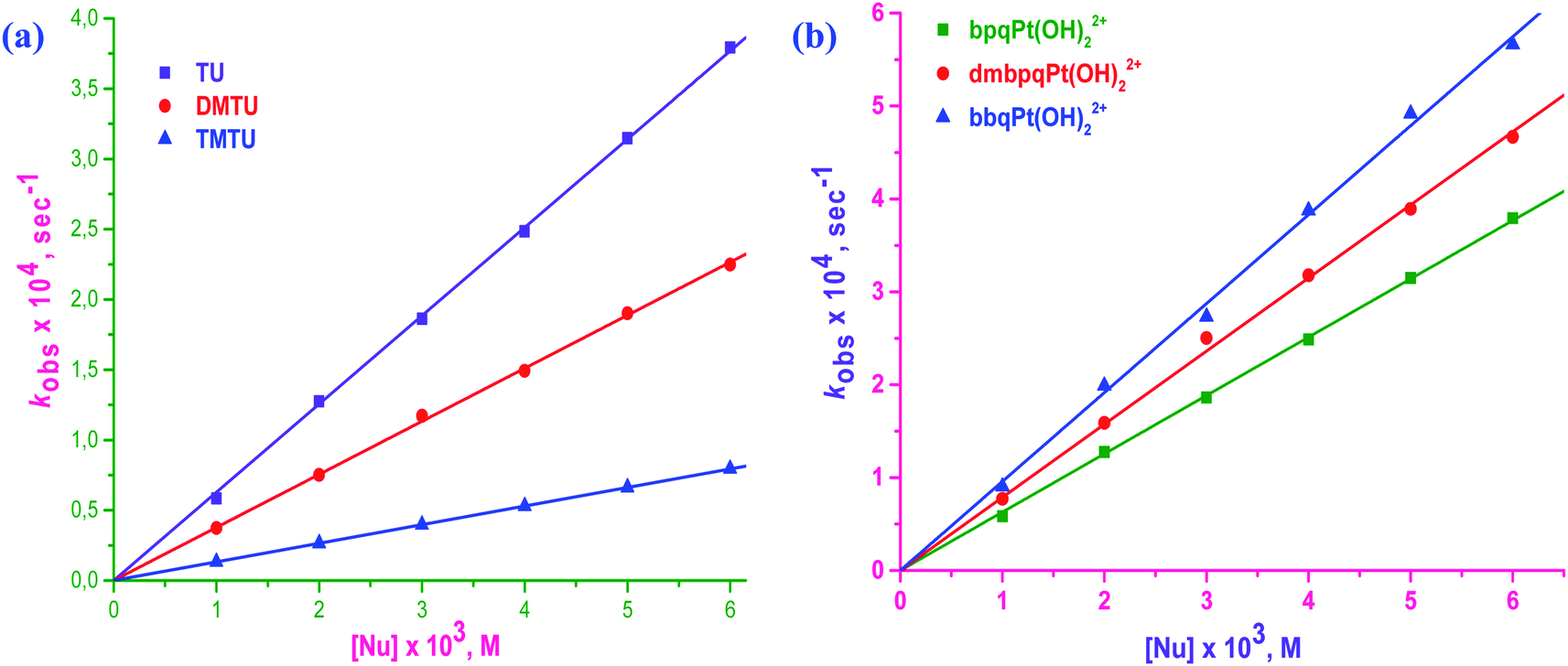 | ||
| Fig. 5 Plots of kobsversus [Nu] for the reaction of bpqPt(OH2)22+ with three S-donor nucleophiles (a), and the linear plots of kobsversus [TU] for the three Pt(II) complexes (b): [bpqPt(OH2)22+] = 50 μM, pH = 2.0, T = 35 °C and I = 0.1 M (NaClO4). | ||
Straight line plots passing through the origin were obtained when kobs values were plotted against [Nu] which gives k2 from the slopes of the plots. Linear plots for the reaction between bpqPt(OH2)22+ and Nu is given in Fig. 5a whereas the plots for the other two complexes, bpqPt(OH2)22+ and bpqPt(OH2)22+ are given in ESI Fig. S2a and b,† respectively. Plots of kobsversus [Nu] as shown in Fig. 5b shows relative reactivity of the three complexes, their reactions with TU and also see ESI Fig. S3a and b† for other reactions with DMTU and TMTU, respectively. The second order rate constants, k2 values for the simultaneous substitution of the aqua ligands by Nu at different temperatures are obtained from the slope of kobsversus [Nu] plots (measured at 25, 35, 45 and 55 °C) according to the rate law: kobs = k2[Nu]. Rate data at 35 °C is given in Table 2 and the data at other temperatures are given in ESI Table S3.†
| Complex | Nu | k 2 × 102/M−1 s−1 | ΔH#/kJ mol−1 | −ΔS#/J mol−1 K−1 | ΔG#35°C/kJ mol−1 |
|---|---|---|---|---|---|

|
TU | 6.25 ± 0.05 | 26 ± 1 | 184 ± 5 | 83 ± 2 |
| DMTU | 3.79 ± 0.03 | 34 ± 2 | 162 ± 3 | 84 ± 1 | |
| TMTU | 1.32 ± 0.02 | 41 ± 5 | 149 ± 2 | 86 ± 4 | |

|
TU | 7.95 ± 0.07 | 23 ± 1 | 192 ± 7 | 82 ± 2 |
| DMTU | 4.57 ± 0.05 | 30 ± 1 | 173 ± 4 | 84 ± 1 | |
| TMTU | 1.70 ± 0.04 | 38 ± 2 | 156 ± 2 | 86 ± 3 | |

|
TU | 9.44 ± 0.1 | 27 ± 2 | 176 ± 6 | 82 ± 2 |
| DMTU | 5.07 ± 0.08 | 32 ± 3 | 166 ± 4 | 83 ± 3 | |
| TMTU | 2.38 ± 0.05 | 43 ± 5 | 138 ± 2 | 85 ± 2 |
From the second-order rate data (k2 values) presented in Table 2, the reactivity of the complexes increases marginally by factors ranging between 1.2 to 1.5 times that of bpqPt(OH2)22+ and the order is bpqPt(OH2)22+ < dmbpqPt(OH2)22+ < bbqPt(OH2)22+. This indicates that the rate is enhanced by the introduction of the methyl groups as well as by extending π-conjugation on the reference 2,3-di(2-pyridyl)quinoxaline ligand. The addition of electron donating groups on a chelated aromatic spectator ligand usually increases electron density into the ligand which is then forwarded to the metal centre by inductive effects. As a consequence, the metal becomes less electrophilic, leading to a dampened rate of substitution with incoming nucleophiles due to decreased π-back bonding. In contrast, the rate of substitution from dmbpqPt(OH)22+ was higher than from bpqPt(OH2)22+. This is probably due to an increased ground state labialising effect through a trans σ-inductive effect. However, this influence on the leaving group is marginal, thus the enhanced rate is also marginal due to the non-planar conformation of ligands of the Pt(II) complexes. The trans influence controls the rate because of the unusual seven membered coordination chelate which assumes a puckered conformation at the Pt(II) centre. In summary, a combination of the relative strength of trans σ-effect and the structural modifications of the conjugated groups at the back of seven-membered chelate at the Pt(II) centre causes a marginal difference in the rate of substitution of the aqua ligands. Noteworth, UV-Visible spectra of dmbpqPt(OH2)22+ and bbqPt(OH2)22+ shows red shifted λmax values when compared to bpqPt(OH2)22+, suggesting the existence of more pronounced MLCT transitions from their Pt(II) 5d π-orbitals to the delocalised π*-orbitals of ligands than the reference complex, bpqPt(OH2)22+.12 This causes a significant decrease in their ΔE(π*LUMO–Pt(5dπ)–HOMO) gap compared to bpqPt(OH2)22+ (see ESI Table S5†).13 Thus, π-back bonding plays a role, be it minor on the rate of substitution from the two complexes especially in bbqPt(OH2)22+. However, the trans σ-effect appear to be a more dominant factor controlling the substitution rate.
It can also be seen from the data presented in Table 2, the rate of reaction was also enhanced by extending π-conjugation on 2,3-di(2-pyridyl)quinoxaline ligand. i.e., the bbqPt(OH2)22+ complex shows higher rates than bpqPt(OH2)22+. This is probably due to a slight enhancement of π-back bonding of electron density from the platinum 5d-orbitals into the extended π*-anti molecular orbitals of the ligand. In bbqPt(OH2)22+, the fused phenyl ring delocalised the charge away from the seven-membered puckered chelate ring making the coordinated ligand more aromatic due to a more positive charge (on Pt) in the chelate. This seven-membered platinum cycloheptanyl cation (tropylium cation) is known to be aromatic while cycloheptane or its anion is not. This improves the extent of π-back bonding relative to the other two complexes. As a result, bbqPt(OH2)22+ is less electrophilic (evidenced by an increase in positive NBO charge on Pt(II) centre) and its HOMO–LUMO energy gap is narrower (see ESI Table S5†). This makes it more reactive than bpqPt(OH2)22+. Hence, it is concluded that the rate enhancement in bbqPt(OH2)22+ is due to π-back bonding ability of the bbq ligand upon coordination to the Pt centre. This is also observed from the location of the MLCT band of bbqPt(OH2)22+ relative to that of reference complex bpqPt(OH2)22+, see Fig. 2. This band is shifted from shorter to longer wavelengths as the π-surface of the chelating ligand is increasing causing its faster reactivity discussed above.
The data in Table 2 further shows that the substitution of aqua ligands by S-donor thiourea nucleophile is depended on their bulkiness i.e., the rate of substitution decreases with increasing bulk of the incoming nucleophile.14 TMTU is the bulkiest nucleophile and offered the most steric hindrance towards nucleophilic attack of the complexes since it destabilises the bipyramidal transition state most. As a result, it substituted the aqua ligand at a significantly lower rate. The reactivity order of the nucleophiles decreased as TU > DMTU > TMTU.
2.5 Temperature effect and iso-kinetic relationship
The reactions were studied at different temperatures ranging from 25 to 55 °C with 10 °C intervals to understand the effect of temperature on the reaction rate. The temperature dependence of k2 values was used to compute activation parameters (ΔH#, ΔS# and ΔG#35°C) using the Eyring equation. Eyring plots are given in ESI Fig. S4 and S5† and the activation parameter values are presented in Table 2. The low and positive ΔH# values and large and negative ΔS# values in Table 2 support the associative substitution mechanism at the square-planar Pt(II) complexes for all reactions. The comparable magnitudes of ΔG#35°C values for the reactions of the three different Pt(II) complexes with S-donor nucleophiles, suggest that all these reactions essentially follow the same mechanism which is associative.15 The relatively smaller enthalpy of activation (ΔH#) values suggests that it is energetically favorable to form a bond in a bimolecular transition state.16 It is also well-known that relatively large negative entropy of activation (ΔS#) values signify the bimolecular nature of a transition state. Bond formation results in the formation of highly ordered and more compact transition state relative to reactants. The data show an increasing trend in ΔH# values while −ΔS# values decrease with the bulkiness of the incoming nucleophiles reacting with the complex in good correlation to a decreasing reactivity trend of the S-donor nucleophiles which supports an associative mechanism to the substitution process. Straight line plots of enthalpy of activation, ΔH#versus entropy of activation, ΔS# for all reactions showed the existence of a linear iso-kinetic free energy relationship, LFER between the activation variables of the reactions of the complexes and all nucleophiles (refer ESI Fig. S6†). The slopes and intercepts of the plot give the iso-kinetic temperatures and Gibbs free energies, ΔG# for all the substitution processes. The iso-kinetic temperature, a theoretical temperature at which all the reactions for the Pt(II) complexes and S-donor nucleophiles would proceed at equal rates of substitution was determined to be at 394 K, while the Gibbs free energy was 98 kJ mol−1. The LFER further signifies that the substitution process of all the three Pt(II) complexes occurred via the same associative mechanism.172.6 Effect of chelate size on the rate of substitution from Pt(N^N-bidentate)(OH2)2 complexes
The size of the N^N chelates made by a bidentate ligand at the metal centre can play a vital role in their reactivity. On comparing the reactivity of five,8,18 six19 and seven-membered (current study) chelates of Pt(II) diaqua complexes of the type Pt(N^N)(OH2)22+ (where N^N = aromatic bidentate), the five membered complexes are much more reactive than the six and seven-membered Pt(II) complexes. This is due to the aromaticity as well as the more acute bite angle subtended at the metal centre. Aromaticity of the chelate and the entire ligand ensure efficient π-back donating of electron density into the ligand π* MOs, provided the coordinated ligand lie in the coordination plane. Despite the free quinoxaline ligands (used in this study) being aromatic and planer, they become markedly strained as evidenced from the formed seven-membered puckered chelate ring (see crystal structure in Fig. 6vide infra). The two pyridyl rings and the rest of the ligand lie in different planes to that of PtN2(OH2)2 atoms (refer Fig. 6). This curtails efficient π-back bonding from metal centred orbitals to the strained aromatic back moieties of the ligand. If ever π-back bonding occurs, it does through the two coordinated pyridyl rings and it is not expected to be extensively delocalised into the entire ligands. However, increase π-conjugation at the back of the chelate as occurring in bpqPt(OH2)22+, turns the seven membered chelate into a tropylium cation which cause enhanced electron density withdrawal from Pt centre by π-back bonding (vide supra). Thus, compared to five-membered chelates, the seven-membered chelated Pt(II) complexes react much slowly as observed in the experimental data. While the seven-membered chelates are puckered, this allows the ligands to coordinate in a perfect square planar (bite angles near 90°, whereas five-membered rings would subtend a much smaller bite angle at the metal centre). | ||
| Fig. 6 Time-dependent 195Pt-NMR chemical shifts of bpqPtCl2 in DMSO medium with excess TU; reaction was monitored before (a), at 48 h (b) and after completion (c). | ||
Noteworthy from the literature data are the Pt(II) complexes containing N^N^N tridentate ligands which are more reactive than their corresponding Pt(II) complexes with N^N bidentate ligands.18 This is due to the more acute bite angles of the tridentate ligand and more aromaticity of the chelates that are formed at the Pt atom. This enhances the π-back donating of electron density. As a consequence, increased electrophilicity on the metal leads to faster reactivity than for complexes with N^N bidentate ligands. Pt(II) complexes with five-membered N^N^N tridentate ligand chelates react much faster than six-membered N^N^N tridentate ligand chelates.10,18,20 This can also be explained by the argument already presented.
2.7 Product analysis
The reaction of one of the complexes (bpqPtCl2) was monitored by 195Pt NMR spectroscopy in DMSO-d6. Owing to the insolubility of this dichloro Pt(II) complexes in most organic solvents, DMSO was used as a medium to run NMR kinetics (This reaction was taken into consideration as a representative reaction for product/intermediates analysis for all studied Pt(II) complexes). No chemical shift (around −2340 ppm)21 due to DMSO substituted derivative Pt(N^N) (DMSO)2 was observed. This is also supported by the single crystal X-ray diffraction structure of dmbpqPtCl2 in which DMSO co-crystallised out as a solvent molecule, given that the crystals were formed from an aged solution of a dichloro Pt(II) complex in DMSO (see Fig. 7 for molecular structure crystal of dmbpqPtCl2 with DMSO as solvent molecule). The changes in the 195Pt-NMR over time were recorded in DMSO-d6 and the array is presented in Fig. 6. | ||
| Fig. 7 Molecular structure (thermal ellipsoids at the 50%) of the dmbpqPtCl2. DMSO complex. Selected bond lengths (Å), angles(°) and torsions angles(°): Pt–Cl(1), 2.3086(6); Pt–Cl(2), 2.2985(6); Pt–N(1), 2.0328(17); Pt–N(4), 2.0254(18); Cl(1)–Pt–Cl(2), 91.23(5); Cl(1)–Pt–N(1), 89.10(5); Cl(2)–Pt–N(4), 88.64(7); N(1)–Pt–N(4), 176.37(5); N(1)–Pt–Cl(2), 178.33(5); N(4)–Pt–Cl(1), 91.23(5); N(1)–C(7)–C(8)-C(17), 51.7(3); N(4)–C(18)–C(17)–C(8), −60.1(3). The reported bond lengths are those of the major conformer. | ||
Before adding excess of TU to bpqPtCl2, the 195Pt-NMR spectrum showed Pt chemical shift at −2148 ppm, signifying the formation of Pt(N^N)Cl2 coordination species (see Fig. 6a). After addition of 10 mole equivalents of TU, a spectrum that was recorded after 48 h showed complete disappearance of the reactant peak, while a new peak at −3400 ppm is noticed (Fig. 6b). This was due to Pt(N^N)S2 spices known to occur within the range −3150 to −3550 ppm.22 Although this chemical shift is very close to that of typical PtCl2S2 species at −3440 ppm22 or Pt(dmso)2Cl2, we rule out these as possible intermediates because the N^N bidentate ligand forms quite inert bonds with metal centre due to its chelate effect. As a result we postulate that the substitution of the chlorides occur first rather than the de-coordination of the ligand. The dmso-d6 molecules (used as solvent) do not replace the N^N bidentate ligand nor the chloride from the Pt(II) centre which is evidenced by the crystal structure (vide infra). Thus, the peak around −3440 ppm cannot be due to the Pt(dmso)2Cl2/PtS2Cl2 species either. On leaving this reaction for two more days, the peak due to the intermediate also vanished while a new peak at −3980 ppm also appeared (Fig. 6c). A 195Pt(II) NMR signal in the range: −3800 to −4000 ppm is typical of a PtS4 species.23 The strong nucleophilicity of TU forced the ligand to de-chelate from the metal centre to gives a Pt(TU)42+ and a free 2,3-di(2-pyridyl)quinoxaline ligand (bpq) as a final product. This mechanism applies also to the other complexes as proposed in Scheme 2.
2.8 Crystal structure of dmbpqPtCl2
Yellow coloured block shaped crystal of dmbpqPtCl2 suitable for X-ray diffraction analyses were obtained by recrystallization of the complex from its hot DMSO solution. It crystallized out as a dmbpqPtCl2·DMSO solvent complex. None of the two labile chloride ligands were replaced by the DMSO (Fig. 7). An ORTEP view (50% probability) of the crystal structure and packing diagram are depicted in ESI Fig. S7 and S8,† respectively. Details of the data collection and refinement are given in ESI Table S4.† The Pt atom is bounded to pyridyl N-atoms in a bidentate fashion forming an unusual seven coordinate chelate. The two chlorides lie in the same plane of Pt and two pyridyl N atoms, forming a nearly perfect square planar geometry around the Pt atom. However, the pyridyl rings and the quinoxaline bridging group are out of the coordination plane. More so, it is the quinoxaline which lies approximately perpendicular to the PtN2Cl2 square planar plane and subtends a dihedral angle of about 86°. In fact, the pyridine groups had to twist out of the quinoxaline plane (dihedral angle = 85.94°) to form the seven-membered chelate with Pt. The 3D structure and hence flow of electron density from the ligand towards the metal centre resembles a butterfly, with the twisted pyridyl rings being its flight wings. The terminal methyl groups are also in a plane with the quinoxaline. The molecules of the dmbpqPtCl2 complex are disposed in a head to head to allow the parallel overlap of the quinoxaline aromatic rings while the platinum metals are facing oppositely to each other (see ESI Fig. S8†).2.9 Computational analysis
Computational calculations were performed at the DFT level to get optimised structural data of the investigated Pt(II) complexes. The geometry-optimized structures and Frontier orbital energy maps for the HOMO and LUMO are presented in ESI Fig. S9,† while the DFT calculated geometrical data are summarized in ESI Table S5.† The DFT-calculated Frontier-orbital diagrams (ESI Fig. S9†) show that most of the HOMO electron density is from the 5d-orbitals of the Pt and partly from π-orbitals of the quinoxaline moiety of the ligand molecule. However, for bbqPt(OH)22+, the HOMO electron density is distributed only on the quinoxaline. The LUMOs for all complexes are distributed mainly on the quinoxaline moiety and partially on the π*-orbitals of the entire ligand. From the Frontier orbitals distribution, it can be viewed that the transfer of electron density from HOMO (5d metal orbitals) to LUMO (ligand) can occur in these complexes. Furthermore, the UV-visible absorption spectra of both dichloro and diaqua Pt(II) complexes alludes to the existence of this MLCT which is more pronounced in the more π-conjugated complex, bbqPt(OH2)22+.24 The red shifting of the λmax (both UV and near visible regions) by the addition of methyl groups on the 2,3-di(2-pyridyl)quinoxaline of dmbpqPt(OH2)22+ as well as by extending the π conjugation in bbqPt(OH2)22+ is decreased in ΔEπ*LUMO–πHOMO compared to that of bpqPt(OH2)22+. A narrowed energy gap between πHOMO and π*LUMO makes it easier to transfer the change from metal centred 5dπ orbitals of the metal to the π* orbitals of the ligand.13,25 As a consequence of this, higher reactivity is observed for dmbpqPt(OH)22+ and bbqPt(OH)22+ than bpqPt(OH)22+.The DFT computed electrophilicity index, ω measures the degree of electron deficiency in the entire complex, the higher the value the faster the rate of substitution by the nucleophiles. The trend in ω values in ESI Table S5† supports the reactivity trend of the complexes. The trend in the dipole moment, D or polarity of the complex also correlates with the reactivity.26 The higher the polarity of the complex the faster is its substitution reactions. The complex bbqPt(OH2)22+ has the least ionisation potential and chemical hardness values while its electron affinity, chemical softness, electrophilicity index and dipole moment values are the highest had the highest reactivity. Hence, all the trends in the electronic descriptors strongly supports the observed order of reactivity trend and increases in the order: bpqPt(OH2)22+ < dmbpqPt(OH2)22+ < bbqPt(OH2)22+.
2.10 CT-DNA interactions
The interactions between metal complexes and duplex CT-DNA were monitored by following the changes in the absorbance upon addition of CT-DNA to a fixed concentration of the Pt(II) complex. The spectral titration curves showed a common hypochromic shift with an increase in CT-DNA concentration. A typical graph is given in Fig. 8 for the spectral changes due to the bpqPtCl2–CT-DNA interactions and also see ESI Fig. S10a and b† for spectral changes of other complexes. The magnitude of the shift depends on the strength of the interactions between them as measured by the intrinsic binding constant, Kb. The Kb values were calculated from the ratio of the slopes to intercepts of the Wolfe–Shimer plots and are presented in Table 3. The observed spectral changes (hypochromic shift) and the magnitude of calculated binding constants (104 M−1) clearly indicate that the Pt(II) complexes bind on to CT-DNA strongly. They do so most likely by groove binding as previously reported for similar transition metal complexes.30
 | ||
| Fig. 8 Absorption spectra of bpqPtCl2 (8 μM) in Tris–HCl/50 mM buffer at pH 7.2 upon addition of CT-DNA (0–40 μM). The arrow shows the change in absorbance upon increasing the CT-DNA concentration. Inset: plot of [CT-DNA] versus [DNA]/(εa − εf). | ||
| Complex | UV titration | Fluorescence EtBr exchange titration | ||
|---|---|---|---|---|
| K b × 10−4, M−1 | K sv × 10−3, M−1 | K app × 10−5, M−1 | k q × 10−13, M−1 s−1 | |
| bpqPtCl2 | 1.13 ± 0.04 | 2.43 ± 0.02 | 4.69 ± 0.05 | 1.06 ± 0.10 |
| dmbpqPtCl2 | 3.03 ± 0.08 | 3.26 ± 0.05 | 5.14 ± 0.07 | 1.47 ± 0.16 |
| bbqPtCl2 | 5.77 ± 0.09 | 5.49 ± 0.07 | 8.64 ± 0.10 | 2.52 ± 0.22 |
 | ||
| Fig. 9 Fluorescence emission spectra of EtBr bounded to CT-DNA in the presence of bpqPtCl2: [EtBr] = 20 μM, [CTDNA] = 20 μM and [bpqPtCl2] = 0–300 μM. The arrow shows the intensity changes upon increasing the bpqPtCl2 complex concentration. Inset: Stern–Volmer plot of Io/I versus [Q]. | ||
 | ||
| Fig. 10 Effect of increasing amounts of bpq/dmbpq/bbqPtCl2 complex (1.0–6.0 mM) and EtBr on the relative viscosities of CT-DNA in 5.0 mM Tris–HCl buffer (pH 7.2). | ||
2.11 BSA interactions
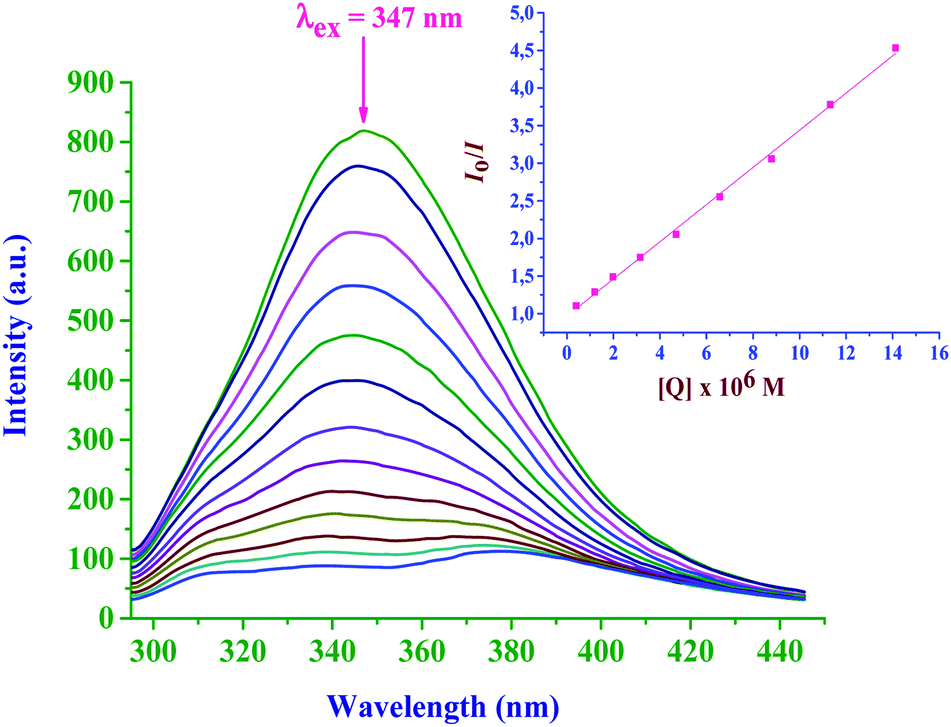 | ||
| Fig. 11 Fluorescence emission spectra of BSA in the absence and presence of bpqPtCl2: [BSA] = 1.2 μM and [bpqPtCl2] = 0–20 μM. The arrow shows the intensity changes upon increasing the bpqPtCl2 complex concentration. Inset: Stern–Volmer plot of [Q] versus Io/I. | ||
| Complex | K sv × 10−5, M−1 | k q × 10−10, M−1 s−1 | K F × 10−5, M−1 | n |
|---|---|---|---|---|
| bpqPtCl2 | 2.47 ± 0.03 | 4.97 ± 0.2 | 1.93 ± 0.02 | 0.98 ± 0.03 |
| dmbpqPtCl2 | 3.47 ± 0.11 | 6.98 ± 0.3 | 2.97 ± 0.07 | 0.98 ± 0.03 |
| bbqPtCl2 | 4.78 ± 0.19 | 9.61 ± 0.5 | 5.34 ± 0.10 | 1.01 ± 0.05 |
Results from Tables 3 and 4, clearly indicate that these Pt(II) complexes have substantial binding affinities to both CT-DNA and BSA. The binding abilities (from both UV-Vis and fluorescence studies) of the studied complexes with CT-DNA strongly suggest the groove binding mode, while the hydrophobic interactions are responsible for the strong interaction of BSA with the Pt(II) complexes. The bbqPtCl2 complex shows the greatest DNA and BSA binding affinity due to the extended π-conjugation on its ligand. Furthermore, all the complexes are coordinated with planer quinoxaline moiety which can associatively interact or insert into DNA/BSA. However, this moiety is almost perpendicular to the Pt(N^N)Cl2 plane, thereby reducing the extent of intercalation. The π-surface of the ligand and its hydrophobicity increase as the order of the binding abilities of the complex in the order bbqPtCl2 > dmbpqPtCl2 > bpqPtCl2. The rate of aqua substitution with S-donor nucleophiles follows the same order of reactivity, thus, the binding affinities are corroborated with the trend in the kinetic results.
2.12 Molecular docking with B-DNA
The dichloro Pt(II) complexes (bpq/dmbpq/bbqPtCl2) were docked onto B-DNA to obtain a theoretical insight into their surface molecular interactions, probable binding sites and preferred orientations with CT-DNA duplex. The minimum energy of the docked poses (Fig. 12 for bpq/dmbpq/bbqPtCl2) revealed that the Pt(II) complexes fitted into the curved contours of the B-DNA located in the minor grooves within the G–C (∼13.4 Å) bases sequence. The relative magnitude of the binding energy of complexes when docked to DNA was found to be −255.67, −273.23 and −284.34 kJ mol−1 for bpqPtCl2, dmbpqPtCl2 and bbqPtCl2, respectively. The computed E(lowest energy pose) values are consistent with the observed trend in binding strength from the absorption and fluorescence quenching titration studies (see Table 3). The complexes, bpqPtCl2 and bbqPtCl2 have the least and greatest binding affinities to B-DNA and the increasing binding order is bpqPtCl2 < dmbpqPtCl2 < bbqPtCl2. The hydrophobic contacts of the ligands of the bbqPtCl2 complexes also interact with non-polar functional groups within the minor groove of B-DNA to produce stable bbqPtCl2–DNA non-covalent complexes. The highest non-covalent binding affinity of bbqPtCl2 can facilitate its facile attack by the nucleophilic N7 atoms of guanine of DNA, forming 1,2 intrastrand adducts. As observed for the trend of the rate constants, the activity of Pt(II) complexes is expected to be higher for bbqPtCl2 due to extra and favorable non-covalent interactions at DNA. The metal complexes interact strongly with B-DNA base pairs possibly by grove binding. Complexes used in this study preferentially interact with groups in the minor groove according to a perfect fit-cooperative non-repulsive model. | ||
| Fig. 12 Computational docking poses, illustrating the non-covalent interactions of bpqPtCl2 (a), dmbpqPtCl2 (b) and bbqPtCl2 (c) with B-DNA duplex. | ||
2.13 In vitro cytotoxicity of the Pt(II) complexes on Vero and HepG2 cells
 | ||
| Fig. 13 Percentage of cell viability of human liver cancer cells: HepG2 cells when treated with 10, 20, 30, 40 and 50 μM of each of Pt(II) complex for 24 h. | ||
2.13.2.1 Acridine orange/ethidium bromide(AO/EB) staining. In order to study the morphological changes due to apoptosis caused by the Pt(II) complexes, fluorescence microscopic analysis was performed using the AO/EB differential staining technique. AO can pervade intact cell membranes and stain the nuclei green, whereas EB can only stain the nuclei of cells that have lost membrane integrity.45 Staining images are depicted in Fig. 14. From Fig. 14, it is inferred that the morphology of the untreated HepG2 cancer cells (control) remain intact and their stained images showed the characteristics green fluorescence associated with stained cells. Contrary to this, the cells treated with a fixed dose of 50 μM of each Pt(II) complex, clearly revealed significant morphological changes imaged as yellow coloured fluorescence. Thus, these complexes induce early apoptotic cell damage characterised by membrane blebbing. The stained orange colour fluorescence is certainly due to cells in their late apoptotic induced changes. The stains appear as dense spots because of the formation of highly condensed chromatin aggregates. The red colour fluorescence is due to cells that have been necrosised. Typical dead cells appear as bright red coloured fluorescence. Overall, the results indicate that necrosis of the HepG2 cells occurred at a significantly lower dose (50 μM) of the Pt(II) complexes. This is in good agreement with their reported high in vitro cytotoxicity by the MTT assay. This implies that the Pt(II) complexes prompted early apoptosis in HepG2 cancer cell line while they were non-toxic to the normal Vero cells.
 | ||
| Fig. 14 AO/EB and DAPI staining of the HepG2 cancer cell line; cells were treated with a fixed concentration of 50 μM of each Pt(II) complex (b–d) for 24 h and were compared with the control (a). | ||
2.13.2.2 4′,6-Diamidino-2-phenylindole (DAPI) staining. DAPI is a fluorescent dye that binds strongly to adenine-thymine regions in the DNA helix and passes through an intact cell membrane. Up on DAPI treatment, chromosomal DNA of apoptotic cells causes a positive DAPI staining image while non-apoptotic cells are non-responsive. Binding of DAPI to DNA produces a ∼20-fold enhancement in the fluorescence, apparently due to the displacement of water molecules from both DAPI and the minor groove.46 As a result, the apoptotic shrunken nuclei appear as bright blue-coloured fluorescent images owing to the condensed state of the nuclei.47 To further detect the changes in nuclear morphology of the cells induced by Pt(II) complexes in the HepG2 cancer cell line, the DAPI nucleic acid staining technique was performed. Fluorescence microscopy images of DAPI stained HepG2 cells observed 24 h after incubating them with and without 50 μM of each Pt(II) complex are shown in Fig. 14. While the structures of nuclei of the untreated control cells were intact, those treated with Pt(II) complexes showed bright fetches, an indication of the condensation of chromatin and possible nuclear fragmentations in the cancer cells. Thus, the results from MTT assay and fluorescence microscopy analysis (AO/EB and DAPI staining assays), show that inhibition of HepG2 cell occurred through both necrosis and apoptosis induction (early stages), casting these complexes as good potential therapeutics against the HepG2 cell line and other related cancer cell lines.
3 Conclusions
Seven member N^N-chelated dichloro and diaqua Pt(II) complexes were synthesized to understand the effect of the chelate ligands and their substituents on the reactivity and also their interactions with DNA and BSA. The substitution kinetics with S-donor nucleophiles proceeds through a simultaneous substitution of the aqua ligands from the complexes, followed by a forced dechelation of the ligand from the Pt(II) centre by the thiourea nucleophiles. The latter is induced by the strong trans influence of the coordinated thiourea ligands in the substituted intermediates. The rate of enhancement and decrease in pKa1/2 values for dmbpqPt(OH2)22+ when compared to bpqPt(OH2)22+ is predominantly due to trans σ-effect. The pKa1/2 values for bpqPt(OH2)22+ are lower than those for bpqPt(OH2)22+ and the latter reacts slightly more than the former due to the better π-back bonding of the bbq ligand. This leads to an increase in electrophilicity on the Pt(II), thus higher rates and the observed increased reactivity trend is bpqPt(OH2)22+ < dmbpqPt(OH2)22+ < bbqPt(OH2)22+. The substitution kinetics follows an associative mechanism, as supported by the large negative entropy of activation and small positive enthalpy of activation values. Isokinetic liner relationships between the activation parameters support a single reaction pathway for the complexes with all the nucleophiles. The seven-membered puckered chelate form by N,N bidentate affected electron charge transfer into the ligands. Electron donor groups reinforced σ-inductive effect towards the Pt atom while a fused phenyl ring synergistically promoted π-back bonding from metal centered d-orbitals of the Pt to π* MOs on the ligand.Binding data calculated from the UV-Visible absorption and EtBr-CT-DNA competitive fluorescence titrations of the Pt(II) complexes showed very good binding abilities of the studied complexes with CT-DNA via groove binding interactions. The increasing order of binding abilities of the complexes is bpqPtCl2 < dmbpqPtCl2 < bbqPtCl2. These binding abilities were validated by theoretical molecular docking studies, and trend in the stability of the docking pose. From the MTT assay data, it is concluded that all the three studied Pt(II) complexes exhibited higher cytotoxic potency towards HepG2 cell lines than the widely used clinical chemotherapeutic agent cisplatin. They also displayed high selectivity between tumor cells and normal Vero cell lines. Among the three, bbqPtCl2 complex has shown the least activity, which may be due to non-specific deactivation by S-donor biomolecules in the cytosol since it is the most reactive of the three. The AO/EB and DAPI staining images suggested that cell death was through necrosis and induction of apoptosis in its early stages.
4 Experimental
4.1 Materials and reagents
Synthetic manipulations were performed under an inert atmosphere of nitrogen. Solvents such as acetonitrile and diethyl ether were purchased from Sigma-Aldrich, distilled and dried by standard literature methods prior to use. All the reagents were obtained from Sigma-Aldrich or Merck and used without any further purification.4.2 Synthesis of ligands
The ligands 2,3-bis(2′pyridyl)-quinoxaline (bpq)48 and 2,3-bis(2′pyridyl)benzo[g]quinoxaline (bbq)49 were prepared according to a published procedure and the other ligand 6,7-dimethyl-2,3-di(2-pyridyl)quinoxaline (dmbpq) was purchased from Sigma-Aldrich. Electron spray time-of-flight mass spectral (ESI+ TOF MS), 1H NMR data and elemental analysis results are comparable to those in reported earlier.24 TOF MS and 1H NMR spectra are given in ESI Fig. S15, S16, S17 and S18,† respectively.4.3 Synthesis of dichloro platinum(II) complexes
The complexes (bpqPtCl2, dmbpqPtCl2 and bbqPtCl2) were prepared by a literature procedure50 with minor modifications, details are given in ESI.† The complexes were characterized by TOF MS, 1H NMR and elemental analysis. TOF MS and 1H NMR spectra are given in ESI Fig. S19–S21 and S22–S24,† respectively.4.4 Preparation of diaqua platinum(II) complexes
Owing to the low water solubility of the dichloro platinum(II) complexes (bpqPtCl2, dmbpqPtCl2 and bbqPtCl2), they were converted to their aqua analogs by removing chlorides using silver perchlorate, AgClO4 according to a literature procedure (also refer ESI†).51 The solution volumes were adjusted to a final volume of diaquated platinum complexes (bpqPt(OH2)22+/dmbpqPt(OH2)22+/bbqPt(OH2)22+) using 0.1 M HClO4 to guarantee that the complex exists as the aqua form during the kinetic measurements.4.5 pKa determination of the aqua complexes
Spectrophotometric measurements over pH changes (1–10) were performed to obtain pKa values of the aqua Pt(II) complexes. A Varian Cary 100 Bio UV-visible spectrophotometer was used to record changes in absorbance within the wavelength range 200–800 nm while a Jenway 4330 was used to measure pH of the solution. Details are given in ESI.†4.6 Instrumentation and physical measurements
A Waters ESI+ TOF Micro-mass LCT Premier Spectrometer operated in the positive ion mode was used to record low-resolution ESI+ mass spectra of the ligands and complexes. Mass spectra for synthesised complexes namely, bpqPtCl2, dmbpqPtCl2 and bbqPtCl2 are presented in ESI Fig. S13, S14 and S15,† respectively. Elementary compositions of the ligands and complexes were determined on a Thermo Scientific Flash 2000 analyser. 195Pt-NMR data were obtained from a Bruker Avance DPX 400 spectrometer at a frequency of 400 MHz using either 5 mm BBOZ probe or a 5 mm TBIZ probe at 30 °C. Kinetic and pKa measurements were recorded on a Varian Cary 100 Bio UV-Visible spectrophotometer coupled to a Varian Peltier temperature controller having an accuracy of ±0.05 °C. A Shimadzu UV-1800 UV-visible spectrophotometer fitted with CPS 240A was used for study the binding interactions of the Pt(II) complexes with CT-DNA and BSA. The temperature of the instrument was controlled to within ±0.1 °C by the Peltier effect. Emission spectra were recorded using a PerkinElmer LS 45 Fluorescence Spectrometer. The pH of the solutions was recorded on a Jenway 4330 conductivity & pH meter equipped with 4.5 μm glass electrode. The pH meter was calibrated at 25 °C using standard buffer solutions at pH values of 4.0, 7.0 and 10.0.4.7 X-ray crystallography
Single-crystal diffraction data for dmbpqPtCl2 was collected on a Bruker SMART APEX2 area detector diffractometer equipped with an Oxford Instruments Cryojet operating at a temperature of T = 100(2) K and an Incoatec microsource operating at 30 W power. The diffraction data were measured using ω and ϕ scans, using a MoKα (λ = 0.71073 Å) radiation beam at a crystal-to-detector distance of 50 mm. The maximum resolution that was achieved was θ = 27.499° (0.77 Å). The diffraction pattern was indexed, the total number of runs and images was based on the strategic calculation from the program COSMO.52 The unit cell was refined using SAINT (Bruker) on 9996 reflections, 38% of the observed reflections. The structure was solved with ShelXS-2013 (ref. 53) structure solution program and by using Olex2 (ref. 54) as the graphical interface. The structural model was refined with version 2016/6 of ShelXL55 using Least Squares minimisation. All non-hydrogen atoms were refined anisotropically. Hydrogen atom positions were calculated geometrically and refined using the riding model. The crystal structure and summary of the crystal data and refinement details are Table 4.4.8 Kinetic procedure
The kinetics of substitution of aqua ligands from the complexes bpqPt(OH2)22+, dmbpqPt(OH2)22+ and bbqPt(OH2)22+ by S-donor thiourea nucleophiles were monitored using a Varian Cary 100 Bio UV-visible spectrophotometer within the wavelength range of 200–800 nm. Details are provided in ESI.†4.9 Computational calculations and molecular dockings
Gaussian 09 program package56 was used to get insightful information about the low energy structures of complexes. The geometry optimizations, frequency calculations and molecular orbital generations were carried out by means of Density Functional Theoretical, DFT using the B3LYP functional method with the Los Alamos National Laboratory 2 Double-Zeta (LANL2DZ).57 The structural data of the complexes were computed in water solution, taking into account the solvolysis effects by the conductor polarizable continue model (CPC model).58 Natural bond orbital (NBO) atomic charges were performed using the Gaussian “Pop = NBO” to determine the spin densities and atomic charges of the complexes.59 All three diaqua platinum complexes were modeled as cationic species of formal charge +2.Molecular docking technique is a useful tool for study the metal complex-DNA interactions for the rational drug design, by probing the molecular interactions of their ligands with potential binding sites of the specific region of the nucleic acids. Thus, the Pt(II) complexes were docked onto B-DNA using the HEX8.0 software.60
4.10 DNA binding studies
4.11 Bovine serum albumin (BSA) binding studies
Quenching of fluorescence emission of BSA by the complexes (bpqPtCl2, dmbpqPtCl2 and bbqPtCl2) was performed in order to determine their binding constants on the protein. The concentration of BSA was measured spectrophotometrically using the Beer-Lambert formulation where a εmax value of 4.4 × 104 M−1 cm−1 is assumed at 278 nm (λmax).64 Stock concentration of BSA (10.4 μM) was prepared in 5.0 mM Tris–HCl/50 mM NaCl buffer at pH = 7.2. The emission spectra of equilibrated 30 μM of the buffered BSA solutions in the absence and presence of Pt(II) complexes of concentration ranging from 0 to 130 μM were acquired at room temperature. The emission changes were recorded in the λem range of 250 to 450 nm, with the excitation wavelength set at 278 nm for all binding interactions with the complexes. Details are provided in ESI.†4.12 In vitro anticancer activities
The three different Pt(II) complexes were tested for their in vitro cytotoxicity property using Vero and HepG2 cells using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay.65 Briefly, 100 μL of both Vero and HepG2 cells (1 × 104 cells/mL) were seeded in 96-well micro plates and the plates were incubated at 37 °C for 24 h in a humidified atmosphere of 5% CO2. After 24 hours of incubation, the cells were observed for 90% confluence using a phase contrast microscope. Then each cell type was treated separately with 100 μL of 10, 20, 30, 40 and 50 μM of each of the Pt(II) complex (bpqPtCl2, dmbpqPtCl2 and bbqPtCl2) and allowed to incubate at 37 °C in a humidified atmosphere of 5% CO2 for 24 h. A control blank, where no metal complexes were added was included along with the treated wells. At the end of the incubation, 20 μL of MTT solution (5 mg mL−1 in phosphate buffer saline, PBS) was added to each well and the plates were incubated for another 2 to 4 h. The excess medium in the wells was removed and the purple formazan crystals in each well were dissolved by adding 100 μL of dimethyl sulfoxide (DMSO). The contents of the plates were mixed well for 5 min and the absorbance of each well was measured at 560 nm using a micro titre plate reader. The percentage of cell viability was calculated using the formula, Cell viability (%) = (A560 of treated cells/A450 of control cells) × 100, where, A450 is absorbance at 450 nm of the control (cells treated with 1% DMSO) and A560 is absorbance of cells treated with different test Pt(II) complexes. The corresponding IC50 values were determined by nonlinear regression analysis of the % cell viability versus concentration data using OriginPro 9.1®.7 The viability of both Vero and HepG2 cells after treatment with the test Pt(II) complexes were compared to that observed in the control cells and the % of cell viability was calculated. The cells were treated with varying concentrations (10 to 50 μM) of the test complexes at an interval of 10 μM. Further, the concentration of each test complex which inhibits 50% of the cell growth (IC50 for a 24 h incubation period) value was determined and expressed in μM of concentration.
4.12.2.1 AO-EB staining assay. The morphological changes caused by the Pt(II) complexes in HepG2 cells were further investigated by using AO-EB staining procedure.66 This inferred whether the inhibition is due to apoptotic induction or non-specific necrosis.67 AO intercalates into the DNA of viable cells and gives a green fluorescence and thus the viable cells appear as a green nuclei while early apoptotic cells appear as condensed or fragmented nuclei. EB intercalates into the DNA of non-viable cells, giving bright orange nuclei of the dead cells after AO staining.68 Briefly, HepG2 cells were seeded in six well plates (4 × 105 cells per well) and incubated at 37 °C for 24 h in a humidified atmosphere of 5% CO2. The cells were treated with 50 μM of the respective Pt(II) complex and allowed to incubate for 24 h at 37 °C as described in the previous section. At the end of the incubation, the adhered cells were washed 3 times with 100 μL of PBS. 2 μL of EB and AO (1 mg mL−1 AO and 1 mg mL−1 EB in PBS) were added to each well and fluorescence was immediately observed under the florescence microscope. It is known that AO stained living cells are uniformly green while early apoptotic cells are densely stained green/yellow, late apoptotic cells are densely stained orange and necrotic cells are stained orange/red. The HepG2 cells were separately exposed for 24 h to the three different complexes at a concentration of 50 μM and visualized for their morphological changes after the AO–EB staining process.
4.12.2.2 DAPI staining assay. The DAPI staining assay was also performed to deduce if apoptotic induction of HepG2 nuclei had occurred after treating them with the Pt(II) complexes. DAPI dye is effective for fluorescence staining of fixed-cells and thus the quantitation of their DNA content. Briefly, the HepG2 cells were grown in 6 well plates as described earlier and the cells were treated with 50 μM of each of the three Pt(II) complexes separately. In the DAPI staining assay, the treated cells were fixed with 80% ethanol at room temperature for 30 min. The fixing medium was removed and the cells were washed with PBS (phosphate buffer saline, pH ∼ 7.2) at the end of 24 h incubation period. The cells were washed thrice with PBS and fixed using 3.7% (w/v) paraformaldehyde-phosphate buffer saline for 15 min at room temperature. The cells were then stained using DAPI (1 μg mL−1, dissolved in PBS buffer) and kept for 5 min in the dark at room temperature. The stained HepG2 cells from each treatment were mounted on a slide, and their images were observed under a fluorescent microscope with a green/blue filter after excitation at 350 nm and observing their emission at 460 nm. The stained cells (both in AO/EB and DAPI staining assay) visual images were captured for their morphological changes using FLoid® cell imaging station (Life Technologies).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from the University of KwaZulu-Natal, South Africa for a postdoctoral fellowship to Dr Rajesh Bellam.References
- B. H. Sørensen, P. Werth, I. H. Lambert and P. J. Bednarski, Metallomics, 2018, 10, 323–336 RSC.
- A. M. Mansour and O. R. Shehab, Dalton Trans., 2018, 47, 3459–3468 RSC.
- M. Gaber, H. A. El-Ghamry and S. K. Fathalla, Spectrochim. Acta, Part A, 2015, 139, 396–404 CrossRef CAS.
- J. Reedijk, Chem. Rev., 1999, 99, 2499–2510 CrossRef CAS.
- M. Ashby, Comments Inorg. Chem., 1990, 10, 297–313 CrossRef CAS.
- T. Soldatović, S. Jovanović, Ž. D. Bugarčić and R. van Eldik, Dalton Trans., 2012, 41, 876–884 RSC.
- OriginPro 9.1, OriginLab Corporation, One Roundhouse Plaza, suite 303, Northampton, MA 01060, USA, 1800-969-7720 Search PubMed.
- N. Summa, W. Schiessl, R. Puchta, N. van Eikema Hommes and R. van Eldik, Inorg. Chem., 2006, 45, 2948–2959 CrossRef CAS.
- S. Hochreuther, R. Puchta and R. van Eldik, Inorg. Chem., 2011, 50, 8984–8996 CrossRef CAS.
- A. Mambanda and D. Jaganyi, Dalton Trans., 2011, 40, 79–91 RSC.
- J. Bogojeski, Ž. D. Bugarčić, R. Puchta and R. van Eldik, Eur. J. Inorg. Chem., 2010, 2010, 5439–5445 CrossRef.
- W. K. Lo, G. S. Huff, D. Preston, D. A. McMorran, G. I. Giles, K. C. Gordon and J. D. Crowley, Inorg. Chem., 2015, 54, 6671–6673 CrossRef CAS.
- C. R. Arana and H. D. Abruna, Inorg. Chem., 1993, 32, 194–203 CrossRef CAS.
- S. Hochreuther, S. T. Nandibewoor, R. Puchta and R. van Eldik, Dalton Trans., 2012, 41, 512–522 RSC.
- G. V. Rao, R. Bellam and N. R. Anipindi, Transition Met. Chem., 2012, 37, 189–196 CrossRef CAS.
- R. Bellam and D. Jaganyi, Int. J. Chem. Kinet., 2017, 49, 182–196 CrossRef CAS.
- R. Bellam, N. R. Anipindi and D. Jaganyi, J. Mol. Liq., 2018, 258, 57–65 CrossRef CAS.
- A. Hofmann, D. Jaganyi, O. Q. Munro, G. Liehr and R. van Eldik, Inorg. Chem., 2003, 42, 1688–1700 CrossRef CAS.
- W. P. Asman and D. Jaganyi, Int. J. Chem. Kinet., 2017, 49, 545–561 CrossRef CAS.
- I. M. Wekesa and D. Jaganyi, Dalton Trans., 2014, 43, 2549–2558 RSC.
- S. J. S. Kerrison and P. J. Sadler, J. Chem. Soc., Chem. Commun., 1977, 861–863 RSC.
- B. M. Still, P. A. Kumar, J. R. Aldrich-Wright and W. S. Price, Chem. Soc. Rev., 2007, 36, 665–686 RSC.
- J. R. Priqueler, I. S. Butler and F. D. Rochon, Appl. Spectrosc. Rev., 2006, 41, 185–226 CrossRef CAS.
- R. Bellam, D. Jaganyi, A. Mambanda and R. Robinson, New J. Chem., 2018, 42, 12557–12569 RSC.
- C.-X. Sheng, S. Singh, A. Gambetta, T. Drori, M. Tong, S. Tretiak and Z. Vardeny, Sci. Rep., 2013, 3, 2653 CrossRef PubMed.
- P. Atkins and J. De Paula, Physical chemistry for the life sciences, Oxford University Press, USA, 2011 Search PubMed.
- M. Sirajuddin, S. Ali and A. Badshah, J. Photochem. Photobiol., B, 2013, 124, 1–19 CrossRef CAS.
- O. Novakova, H. Chen, O. Vrana, A. Rodger, P. J. Sadler and V. Brabec, Biochemistry, 2003, 42, 11544–11554 CrossRef CAS PubMed.
- K. Chichak, U. Jacquemard and N. R. Branda, Eur. J. Inorg. Chem., 2002, 2002, 357–368 CrossRef.
- T. Biver, Appl. Spectrosc. Rev., 2012, 47, 272–325 CrossRef CAS.
- F. J. Meyer-Almes and D. Porschke, Biochemistry, 1993, 32, 4246–4253 CrossRef CAS.
- J. C. Peberdy, J. Malina, S. Khalid, M. J. Hannon and A. Rodger, J. Inorg. Biochem., 2007, 101, 1937–1945 CrossRef CAS PubMed.
- M. Cory, D. D. McKee, J. Kagan, D. Henry and J. A. Miller, J. Am. Chem. Soc., 1985, 107, 2528–2536 CrossRef CAS.
- I. Mitra, S. Mukherjee, B. Misini, P. Das, S. Dasgupta, W. Linert and S. C. Moi, New J. Chem., 2018, 42, 2574–2589 RSC.
- S. Satyanarayana, J. C. Dabrowiak and J. B. Chaires, Biochemistry, 1993, 32, 2573–2584 CrossRef CAS.
- V. G. Vaidyanathan and B. U. Nair, J. Inorg. Biochem., 2003, 94, 121–126 CrossRef CAS.
- M. P. Barrett, C. G. Gemmell and C. J. Suckling, Pharmacol. Ther., 2013, 139, 12–23 CrossRef CAS.
- L. Lerman, J. Mol. Biol., 1961, 3, 18–30 CrossRef CAS.
- F. Dimiza, F. Perdih, V. Tangoulis, I. Turel, D. P. Kessissoglou and G. Psomas, J. Inorg. Biochem., 2011, 105, 476–489 CrossRef CAS.
- F. Samari, B. Hemmateenejad, M. Shamsipur, M. Rashidi and H. Samouei, Inorg. Chem., 2012, 51, 3454–3464 CrossRef CAS.
- X. Yu, R. Liu, F. Yang, D. Ji, X. Li, J. Chen, H. Huang and P. Yi, J. Mol. Struct., 2011, 985, 407–412 CrossRef CAS.
- U. Kragh-Hansen, F. Hellec, B. de Foresta, M. Le Maire and J. V. Møller, Biophys. J., 2001, 80, 2898–2911 CrossRef CAS.
- Z.-J. Cheng, H.-M. Zhao, Q.-Y. Xu and R. Liu, J. Pharm. Anal., 2013, 3, 257–269 CrossRef CAS.
- E. Ramachandran, D. Senthil Raja, N. P. Rath and K. Natarajan, Inorg. Chem., 2013, 52, 1504–1514 CrossRef CAS.
- R. Tokala, S. Thatikonda, U. S. Vanteddu, S. Sana, C. Godugu and N. Shankaraiah, ChemMedChem, 2018, 13, 1909–1922 CrossRef CAS.
- M. Barcellona, G. Cardiel and E. Gratton, Biochem. Biophys. Res. Commun., 1990, 170, 270–280 CrossRef CAS.
- R. Jadeja, K. Vyas, K. Upadhyay and R. Devkar, RSC Adv., 2017, 7, 17107–17116 RSC.
- H. A. Goodwin and F. Lions, J. Am. Chem. Soc., 1960, 82, 5013–5023 CrossRef CAS.
- H. A. Goodwin and F. Lions, J. Am. Chem. Soc., 1959, 81, 6415–6422 CrossRef CAS.
- J. Granifo, M. E. Vargas, M. a. T. Garland and R. Baggio, Inorg. Chim. Acta, 2000, 305, 143–150 CrossRef CAS.
- H. Ertürk, A. Hofmann, R. Puchta and R. van Eldik, Dalton Trans., 2007, 2295–2301 RSC.
- COSMO-V1.61, Software for the CCD Detector Systems for Determining Data Collection Parameters, Bruker axs, Madison, WI, 2009 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.1, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- E. D. Glendening and F. Weinhold, J. Comput. Chem., 1998, 19, 593–609 CrossRef CAS.
- G. Macindoe, L. Mavridis, V. Venkatraman, M.-D. Devignes and D. W. Ritchie, Nucleic Acids Res., 2010, 38, W445–W449 CrossRef CAS.
- O. Stern and M. Volmer, Phys. Z., 1919, 20, 183–188 CAS.
- C. Parker and W. Rees, Analyst, 1962, 87, 83–111 RSC.
- M. Puchalski, M. Morra and R. Von Wandruszka, Fresenius' J. Anal. Chem., 1991, 340, 341–344 CrossRef CAS.
- E. Alarcon, A. Aspee, M. Gonzalez-Bejar, A. Edwards, E. Lissi and J. Scaiano, Photochem. Photobiol. Sci., 2010, 9, 861–869 RSC.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS.
- W. C. Satterfield, N. K. Banada, A. Dunlap, K. S. Steimer, R. Kurrle and T. H. Finkel, Apoptosis, 1996, 1, 69–84 CrossRef.
- U. V. Mallavadhani, N. R. Vanga, M. K. Jeengar and V. Naidu, Eur. J. Med. Chem., 2014, 74, 398–404 CrossRef CAS.
- I. M. Ghobrial, T. E. Witzig and A. A. Adjei, Ca-Cancer J. Clin., 2005, 55, 178–194 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S6 shows UV-Vis spectral changes with pH, dependence plots of kobs on [Nu], Eyring plots, iso-kinetic plots, Fig. S7 and S8 shows ORTEP view and schematic packing diagram of the dmbpqPtCl2, respectively, Fig. S9 DFT-optimized structures, HOMO and LUMO maps for bpq/dmbpq/bbqPt(OH)22+ complexes, Fig. S10 and S11 shows absorption spectral titration with CT-DNA; inset: plot of [CT-DNA] vs. [DNA]/(εa − εf) and fluorescence emission spectra of EtBr bounded to CT-DNA in the presence bpqme/bbqPtCl2; inset: Stern–Volmer plot of [Q] vs. Io/I, respectively, Fig. S12 shows absorption spectra of BSA with and without each Pt(II) complex, Fig. S13 and S14 shows fluorescence emission spectra of BSA in the presence of bpqmePtCl2/bbqPtCl2; inset: Stern–Volmer plot of [Q] vs. Io/I and Scatchard plots, respectively, Fig. S15–S24 shows TOF-MS spectra and 1H NMR spectrum of the free ligands and the corresponding Pt(II) complexes. Table S1 summarise λmax in UV and visible regions for both chloro and aquated Pt(II) complexes, Tables S2 and S3† summarises chosen wavelengths for kinetic traces to get kobs values and the second order rate constants, k2 at 25, 45 and 55 °C for the reactions of complexes with S-donor nucleophiles, respectively, Table S4† summarises crystallographic data and structure refinement details for complex dmbpqPtCl2 Table S5† summarises DFT-calculated data for bpq/dmbpq/bbqPt(OH2)22+ complexes and the detailed synthetic procedures, preparation of diaqua platinum(II) complexes, pKa determination of the aqua complexes, preparation of complex and nucleophile solutions for kinetic analysis, kinetic procedure, molecular docking and DNA/BSA binding studies. CCDC 1857551. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra06488e |
| This journal is © The Royal Society of Chemistry 2019 |