
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMicroencapsulation of eucalyptol in polyethylene glycol and polycaprolactone using particles from gas-saturated solutions
Jubril Olayinka Akolade *ab,
Mohammed Baloguna,
Andri Swanepoela,
Rasheed Bolaji Ibrahimc,
Abdullahi Ahmed Yusufd and
Philip Labuschagnea
*ab,
Mohammed Baloguna,
Andri Swanepoela,
Rasheed Bolaji Ibrahimc,
Abdullahi Ahmed Yusufd and
Philip Labuschagnea
aPolymers and Composites, Chemicals Cluster, CSIR, South Africa. E-mail: akoladejubril@yahoo.com; Tel: +27 718921863
bBiotechnology Advanced Research Centre, Sheda Science and Technology Complex, Nigeria. E-mail: jo.akolade@shestco.gov.ng; Tel: +234 7035722795
cDepartment of Biochemistry, Kwara State University, Molete, Nigeria
dDepartment of Zoology and Entomology, University of Pretoria, South Africa
First published on 23rd October 2019
Abstract
Eucalyptol is the natural cyclic ether which constitutes the bulk of terpenoids found in essential oils of Eucalyptus spp. and is used in aromatherapy for treatment of migraine, sinusitis, asthma and stress. It acts by inhibiting arachidonic acid metabolism and cytokine production. Chemical instability and volatility of eucalyptol restrict its therapeutic application and necessitate the need to develop an appropriate delivery system to achieve extended release and enhance its bioactivity. However, the synthesis method of the delivery system must be suitable to prevent loss or inactivation of the drug during processing. In this study, supercritical carbon dioxide (scCO2) was explored as an alternative solvent for encapsulation and co-precipitation of eucalyptol with polyethylene glycol (PEG) and/or polycaprolactone (PCL) using the particles from gas-saturated solution (PGSS) process. Polymers and eucalyptol were pre-mixed and then processed in a PGSS autoclave at 45 °C and 80 bar for 1 h. The mixture in scCO2 was micronized and characterized. The presence of eucalyptol in the precipitated particles was confirmed by infrared spectroscopy, gas chromatography and mass spectrometry. The weight ratios of PEG–PCL blends significantly influenced loading capacity and encapsulation efficiency with 77% of eucalyptol encapsulated in a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 composite blend of PEG–PCL. The particle size distribution of the PGSS-micronized particles ranged from 30 to 260 μm. ScCO2 assisted microencapsulation in PEG and PCL reduced loss of the volatile drug during a two-hour vaporization study and addition of PCL extended the mean release time in simulated physiological fluids. Free radical scavenging and lipoxygenase inhibitory activities of eucalyptol formulated in the PGSS-micronized particles was sustained. Findings from this study showed that the scCO2-assisted micronization can be used for encapsulation of volatile drugs in polymeric microparticles without affecting bioactivity of the drug.
:1 composite blend of PEG–PCL. The particle size distribution of the PGSS-micronized particles ranged from 30 to 260 μm. ScCO2 assisted microencapsulation in PEG and PCL reduced loss of the volatile drug during a two-hour vaporization study and addition of PCL extended the mean release time in simulated physiological fluids. Free radical scavenging and lipoxygenase inhibitory activities of eucalyptol formulated in the PGSS-micronized particles was sustained. Findings from this study showed that the scCO2-assisted micronization can be used for encapsulation of volatile drugs in polymeric microparticles without affecting bioactivity of the drug.
Introduction
Eucalyptol is the chemotypic compound native to essential oils of Eucalyptus spp. It is a monoterpene cyclic ether (1,3,3-trimethyl-2-oxabicyclo[2.2.2]octane), also commonly referred to as 1,8-cineole and can be found in appreciable quantities in other aromatic plants belonging to the family Myrtaceae and Lamiaceae.1,2 Eucalyptol is generally recognized as safe and approved for use as a flavourant, fragrance or additive in foods, cosmetics and drugs.3 Traditionally, the use of eucalyptol in otorhinolaryngology for the treatment of upper respiratory diseases like asthma and sinusitis dates back to the 19th century.4 In aromatherapy, it is used as a steam inhalation antitussive for the treatment of colds and is recommended as a demulcent for sore throats.5Clinically, the monoterpenoid is as an anti-inflammatory agent prescribed for control of mucus hypersecretion. Earlier in vitro studies by Juergens et al.6 reported for the first time inhibitory effects of eucalyptol on cytokine production and arachidonic acid metabolism in human blood monocytes. They further showed the modulatory effects of the monoterpenoid on inflammatory mediators in cultured human lymphocytes7 and the rationale for its use as a mucolytic agent in treatment of upper and lower airway diseases.8 Other preclinical screening of eucalyptol against microbial and viral infections,9,10 gastric ulcer and colitis,11,12 hepatic injury and oxidative stress,13,14 pain and pneumonia,15,16 cancer and tumours,17,18 cardiovascular and neurodegenerative diseases19,20 have also been attributed to its inherent ability to modulate redox and inflammation processes.
We have previously demonstrated that eucalyptol is the principal bioactive constituent that may be responsible for anti-hyperglycaemic properties of essential oil from Hoslundia opposita in chemically-induced diabetic rats.21 Other in vitro studies also showed inhibitory properties of eucalyptol against carbohydrate digesting enzymes.22,23 Despite the multiple pharmacological attributes of eucalyptol, its poor aqueous solubility, chemical instability and high volatility have restricted its oral administration for treatment of various metabolic diseases. When ingested, it is also easily metabolized by liver microsomal enzymes to its hydroxylated derivatives, 2α-hydroxy-1,8-cineole and 3α-hxdroxy-1,8-cineole.24 Thus, there is need to protect the drug from degradation while in transit to the target site of action.
Slow release of eucalyptol was achieved following encapsulation in a polymer blend prepared using solvent displacement method.25 Also, a self-microemulsifying drug delivery system loaded with eucalyptol significantly attenuated lipopolysaccharide-induced endothelial injury in mice, when compared to non-formulated drug at the same dose.26 The higher inhibitory effect of the drug on inflammatory markers was attributed to the improved bioavailability of the drug via encapsulation in the micro-drug delivery system. Encapsulation coupled with micronization in a polymeric matrix can prevent vaporization and protect against oxidative and enzymatic degradation of eucalyptol. An appropriate delivery system developed to encapsulate eucalyptol can improve its stability and solubility as well as sustain the drug release and enhance its bioactivity.
Conventional microencapsulation techniques such as solvent displacement and self-emulsifying systems require large volumes of organic solvents and post-treatment processes that are time consuming and of high energy demand to reduce residual solvent levels to safe limits. Supercritical carbon dioxide (scCO2) technology offers an alternative method for microencapsulation of essential oils that avoids the excessive use of organic solvents or post-processing treatment techniques. Essential oils have been encapsulated in liposomes and polymers using scCO2 techniques such as rapid expansion of supercritical solution (RESS) and particles from gas-saturated solution (PGSS) respectively.27–30
The RESS technique employs scCO2 as a solvent and uses organic co-solvents such as ethanol that can denature the bioactive with unwanted residual effect on the drug formulation. Unlike RESS that uses scCO2 as a solvent, the PGSS process exploits the ability of scCO2 to liquefy and plasticize a range of polymers such as polyethylene glycol (PEG) and polycaprolactone (PCL) at relatively high pressures and low temperatures by lowering the point at which the polymers melt. Generally, the PGSS process for drug delivery formulations involves saturation of a mixture of drug and polymer or blend of polymers with scCO2. This is followed by expansion of the gas-saturated solutions at ambient temperature accompanied by rapid pressure reduction through a nozzle into a spray chamber resulting in co-precipitation of the polymer and drug blend.31,32 This allows for volatile compounds such as eucalyptol to be incorporated into polymers at lower temperatures, reducing the risk of vaporization during processing.
Essential oils from different plants and individual terpene constituents such as limonene have been successfully encapsulated in octenyl succinic-anhydride modified starch, PCL and PEG using PGSS.29,30,33,34 Varona et al.30 showed that higher antibacterial activity was achieved for lavandin essential oil particles produced by PGSS-drying as compared to those produced by spray drying. The use of PCL alone as drug delivery carrier is limited due to its high hydrophobicity. Hence, PEG is widely used owing to its aqueous solubility. However, in order to establish controlled release of drugs during dissolution of PEG in physiological fluids, blending of the PEG with hydrophobic polymers such as PCL has been shown to impact positively on drug kinetics and efficacy.35,36 Thus, in this study, the suitability of PEG and PCL blends for micronization and encapsulation of the natural anti-inflammatory agent, eucalyptol in scCO2 as co-precipitated particles from gas saturated solution was investigated. We also evaluated bioactivities of the polymer–eucalyptol particles using free radical (DPPH) and lipoxygenase (LOX) inhibition assays.
Results and discussion
Chemical characterization
Polymers and eucalyptol were successfully processed, co-precipitated and micronized using supercritical fluid technology. FTIR spectroscopy established the presence of the eucalyptol in the PGSS particles (Fig. 1). A previous study reported that the C–O–C stretch was detected at 990 cm−1 in eucalyptol.37 In the present analysis the ether functional group was observed at 984 cm−1 for the PGSS-micronized particles encapsulated with eucalyptol (Fig. 1A). The absorption band of the PCL carbonyl, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O functional group observed at 1720 cm−1 for the empty particles shifted to 1723 cm−1 in the scCO2-processed ternary PEG–PCL–eucalyptol composites. Similarly there were shifts of the hydroxyl, C–O–H of PEG to 1102 cm−1 and the ether, C–O–C of eucalyptol to 985 cm−1 (Fig. 1B). The shifts in peaks are indicative of possible intermolecular interaction between components of the ternary composite. The presence of eucalyptol in PGSS-micronized particles was further confirmed using GC-MS (Fig. 2). The cyclic ether was detected at ∼13 min and the MS data aligned with that of the reference library for 1,8-cineole.
O functional group observed at 1720 cm−1 for the empty particles shifted to 1723 cm−1 in the scCO2-processed ternary PEG–PCL–eucalyptol composites. Similarly there were shifts of the hydroxyl, C–O–H of PEG to 1102 cm−1 and the ether, C–O–C of eucalyptol to 985 cm−1 (Fig. 1B). The shifts in peaks are indicative of possible intermolecular interaction between components of the ternary composite. The presence of eucalyptol in PGSS-micronized particles was further confirmed using GC-MS (Fig. 2). The cyclic ether was detected at ∼13 min and the MS data aligned with that of the reference library for 1,8-cineole.
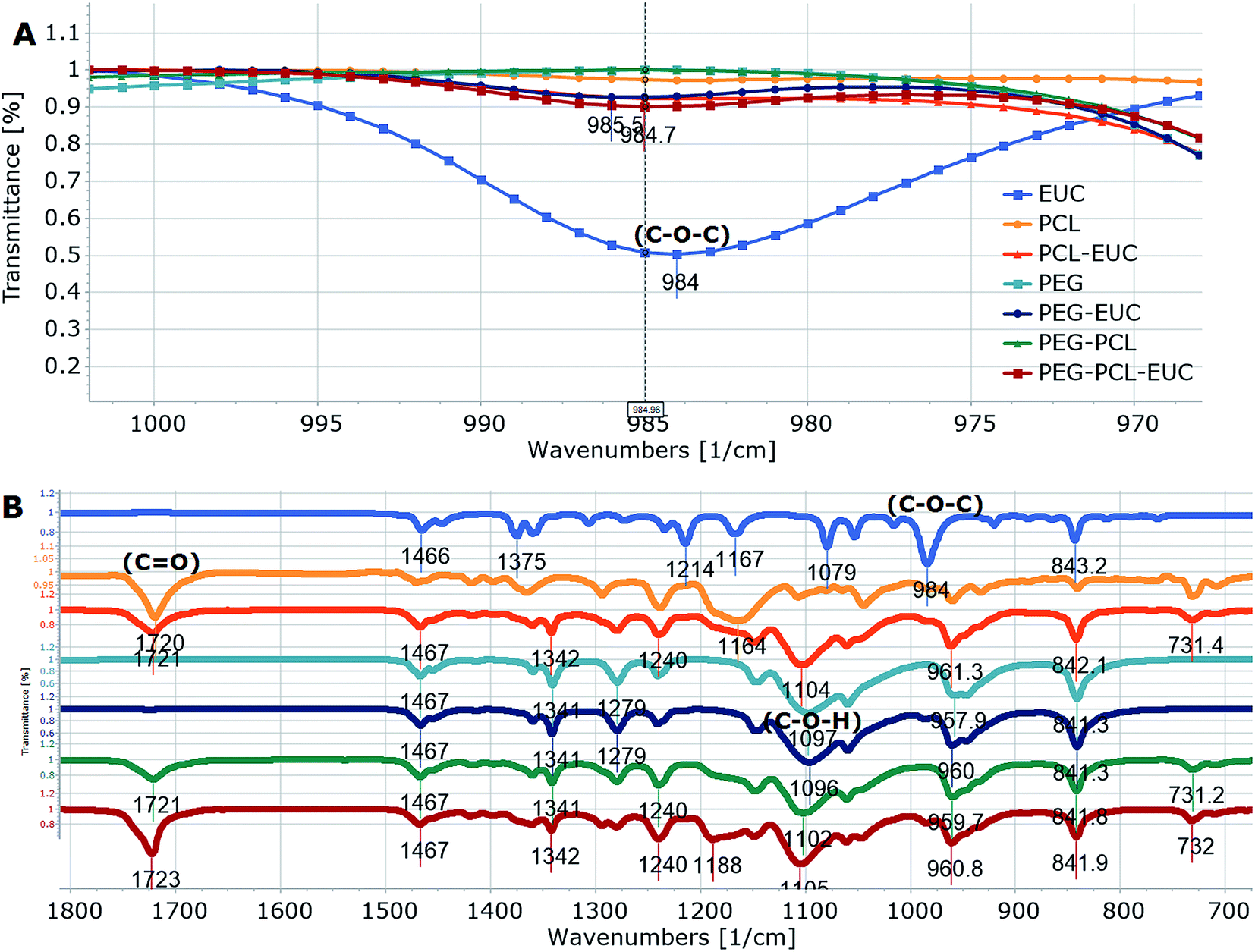 | ||
| Fig. 1 FTIR spectra of polyethylene glycol and polycaprolactone processed in supercritical carbon dioxide with or without eucalyptol (A = ether functional group occurring at around 1000 cm−1; B = different functional groups between 1800–700 cm−1). | ||
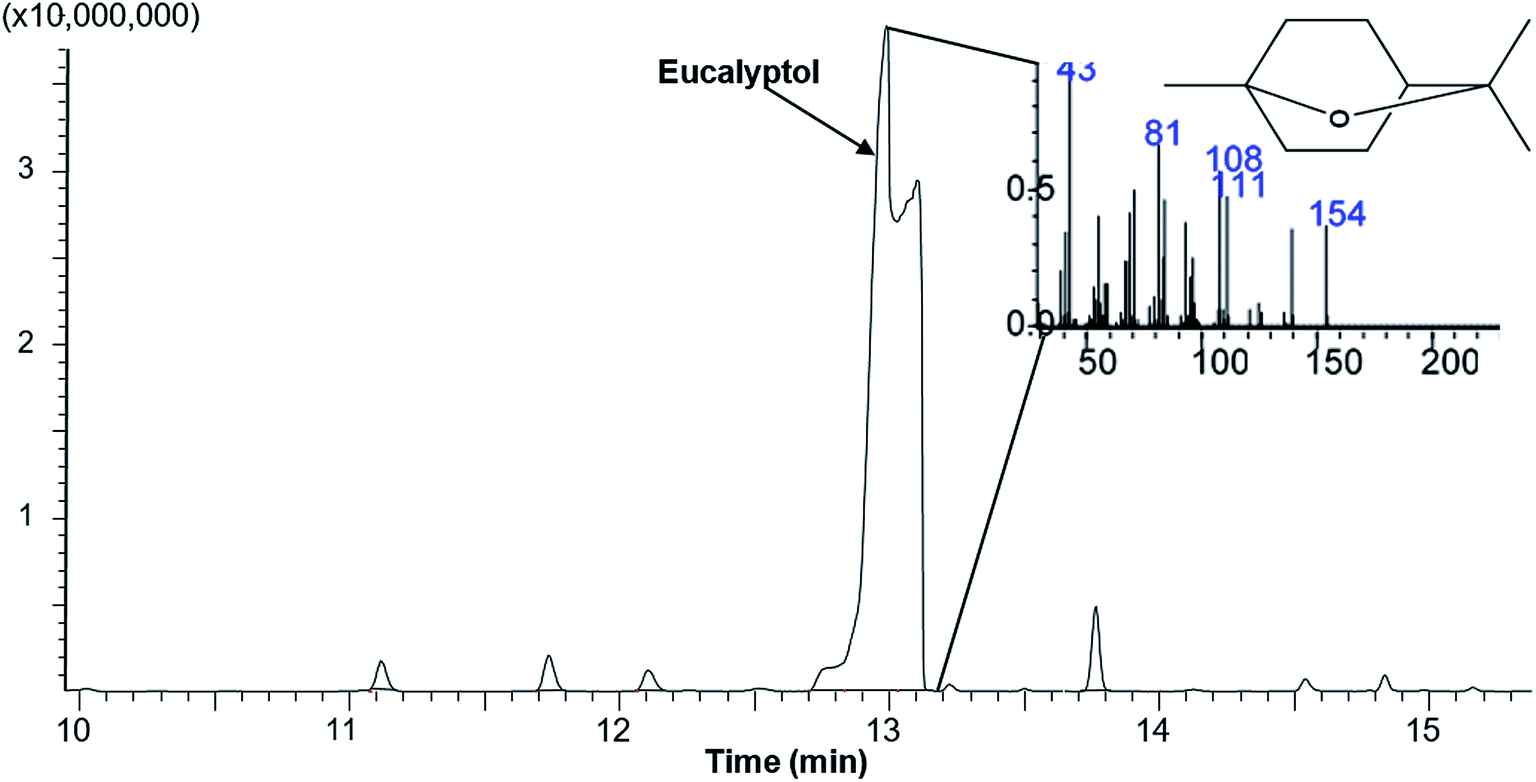 | ||
| Fig. 2 A representative total ion chromatogram and mass spectra showing presence of eucalyptol (retention time 12.95 min) in polymeric microparticles. | ||
Loading and encapsulation characteristics
Loading capacity and encapsulation efficiency of PEG and PCL of varying weight ratios following processing with eucalyptol in scCO2 are shown in Table 1. A 20% weight loading of the eucalyptol to the polymers was processed by PGSS but only 17.5% was detectable in PEG-encapsulated particles compared with 19% loading for the PCL micronized particles. Similar percentage yields have been reported in other studies using different terpene actives.30,38 In this study, encapsulation efficiency was significantly (p < 0.05) lower in PEG4000 (61%), when compared to PCL10000 (77%) or the polymeric blends (63–77%). Computed results showed that encapsulation efficiencies recorded in this study were either higher or within range of those previously reported in literature. Gîtin et al.39 showed that encapsulation efficiency of garlic volatile oil was between 26–49% and that of lavandin oil was reported by Varona et al.29 to be between 14–66%, when processed in PEG6000 and PEG9000, respectively. These significant variations in encapsulation efficiencies may be due to differences in the molecular weights of the polymers, PGSS processing conditions or analytical methods employed for determination of encapsulated volatile constituents. A previous study by Chen et al.40 showed higher content of ibuprofen in PEG6000 micronized particles, when compared to PEG2000. Similarly, a higher content of β-carotene was encapsulated in PCL10000 as against PCL6000 using the same PGSS processing conditions.38 Solution viscosity, which is a function of molecular weight has been shown to be a limiting factor that significantly influences polymer encapsulation efficiency.41| % | Loading capacity | Encapsulation efficiency |
|---|---|---|
| a Values are mean ± SEM of three determinations and those with different superscripts across a column are significantly different from each other (p < 0.05). | ||
| PCL (100) | 19.03 ± 0.14c | 76.78 ± 1.05c |
| PEG (50)–PCL (50) | 13.83 ± 1.06a | 63.42 ± 1.32a |
| PEG (60)–PCL (40) | 16.08 ± 0.12b | 70.62 ± 0.90b |
| PEG (70)–PCL (30) | 17.21 ± 0.10b,c | 72.88 ± 0.12b |
| PEG (80)–PCL (20) | 17.38 ± 0.04b,c | 77.01 ± 0.48c |
| PEG (90)–PCL (10) | 18.01 ± 0.03b,c | 77.36 ± 0.31c |
| PEG (100) | 17.47 ± 0.01b,c | 60.69 ± 0.52a |
The loading capacity and encapsulation efficiency of the polymeric blends decreased as weights ratios of PCL to PEG increased (Table 1). A drastic decrease in loading capacity to 14% and reduction in encapsulation efficiency by 13% was observed in the blend containing equal weight ratios of PCL and PEG. In contrast to this study, Thonggoom et al.42 reported that loading capacity and encapsulation efficiency increased with increasing block length of PCL when clove volatile oil was microencapsulated in diblock co-polymers of PCL and PEG. They attributed the concentration-dependent effect to stronger hydrophobic interactions and hydrogen bonding in the PEG-b-PCL. In another study, absolute encapsulation of eugenol in PCL using an emulsion-diffusion method was also attributed to the hydrophobicity of PCL.43 Previous studies in our laboratory also showed that co-processing of polymers in scCO2 facilitated hydrogen bond interactions between the –O–H group of PEG and CO group of polyvinyl pyrrolidone.31,44 Similarly, hydrogen bonding interactions may have occurred between PEG and PCL during processing in scCO2, which may be responsible for the higher incorporation of eucalyptol into the PEG–PCL when compared to the percentage in PEG only. However, the concentration-dependent decrease in loading capacity and encapsulation efficiency among the various weight ratios of PEG–PCL blends analysed in this study may likely be due to reduced intermolecular interactions when scCO2 is absorbed into the blends of PEG–PCL, thus restricting mobility and shielding of H-bond interactions between the co-polymers.44
Size, morphology and volatility
In order to achieve a greater degree of hydrophobicity and controlled release of eucalyptol from the PEG-based drug delivery carriers, the maximum weight fraction of PCL10000 (0.2) in blends of PEG–PCL that gave values not significantly different from the optimum loading capacity and encapsulation efficiency was selected for further characterization. Fig. 3 shows the vaporization profile of eucalyptol in the PGSS-micronized particles using thermal treatment at 37 °C. There was 96% loss of non-encapsulated eucalyptol oil within the 2 h experimental period, with a mean vaporization time of 58 min, whereas co-precipitation of drug with PEG4000, PEG–PCL and PCL10000 averaged about 40% loss, extending the mean vaporization time to 126, 133 and 154 min, respectively. Molecular weight, porosity and size of particles play a substantial role in the release of the volatile drug under thermal treatments. Hsieh et al.45 demonstrated controlled release of citronella volatile oil from chitosan microcapsules prepared in oil/water emulsion; slower release were observed under thermal treatments in particles with thicker cell wall membrane as well as those of larger sizes.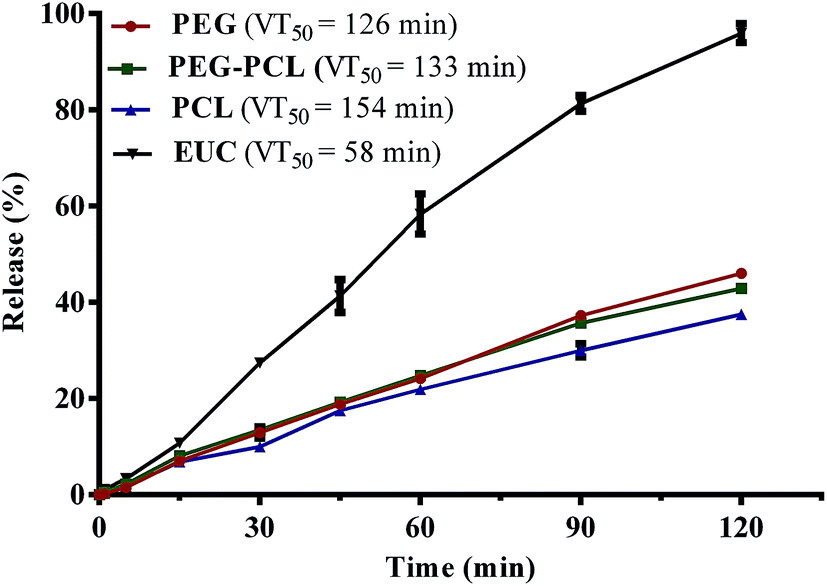 | ||
| Fig. 3 Loss of eucalyptol (EUC) at 37 °C from polyethylene glycol (PEG) and polycaprolactone (PCL) micronized particles produced using supercritical carbon dioxide (points on graph are mean ± SEM of three determinations; VT50 = mean vaporization time). | ||
In this study, processing of eucalyptol in scCO2 with polymeric composites and subsequent micronization yielded co-precipitated particles with a broad volume mean diameter of 70–260 μm using DLS technique and 30–45 μm in size from SEM extrapolation (Table 2). PEG–eucalyptol particles were smaller than those of PCL–eucalyptol or PEG–PCL–eucalyptol. Larger particles are more likely to have a smaller total specific area, therefore retarding vaporization of the eucalyptol. More so, microscopic observation of the polymeric composites revealed different degrees of porosity, further examination at higher magnification revealed the high porous nature of the PEG microparticles (Fig. 4). This physical characteristic may also contribute to the rate of vaporization of eucalyptol from PGSS-micronized particles during thermal treatment.
| Polymer–eucalyptol | Volume mean diameter (μm) | Particle size by SEM (μm) |
|---|---|---|
| a PCL = polycaprolactone; EUC = eucalyptol; PEG = polyethylene glycol. Values are mean ± SEM of three determinations and those with different superscripts across a column are significantly different from each other (p < 0.05). | ||
| PCL–EUC | 176.33 ± 1.00b | 42.54 ± 3.73 |
| PEG–PCL–EUC | 261.93 ± 7.81c | 35.81 ± 1.49 |
| PEG–EUC | 72.46 ± 3.45a | 34.10 ± 2.08 |
 | ||
| Fig. 4 Size and morphology of polyethylene glycol (PEG) and polycaprolactone (PCL) microparticles co-precipitated with eucalyptol (EUC) using supercritical carbon dioxide (A = ×1000 and B = ×6000). | ||
Thermal and storage properties
Alterations were observed in melting point and fusion enthalpy of polymers processed with or without eucalyptol (Fig. 5). The melting point of PGSS processed PEG4000 was higher and the heat of fusion was twice that of PCL10000 (Fig. 5; solid lines). The high crystalline nature of PEG4000 was also corroborated during preliminary view cells experiments, where complete melting of PEG4000 in carbon dioxide at 80 bar was observed at 48 °C, PCL melted at 40 °C, and 47 °C for the 4:1 PEG–PCL blend. Inclusion of eucalyptol into these polymers resulted in broadening of the melting peaks as evident from the DSC thermograms (Fig. 5; dash lines). This suggested some disruption in the crystal structures and consequently a higher degree of crystallinity heterogeneity. Since eucalyptol is a volatile liquid, the thermo-desorption peak was detected at 80.88 °C. This desorption peak of eucalyptol was not detected in the thermogram of polymer–eucalyptol microparticles during either the first or second heat cycles. However, the onset melting temperature and heat of fusion were lower in polymer–eucalyptol microparticles when compared to the empty polymer, suggesting that the presence of the eucalyptol in the polymeric matrix disrupted the polymer crystalline structure. In comparison with other studies, the PEG–PCL micronized particles showed a bimodal melting peak, indicating that its broad endotherm was contributed by both crystalline domains of PEG and PCL. Similar thermogram profiles have been reported in triblock PEG/PCL co-polymers synthesized by using ring opening polymerization46 and PEG/PLA co-polymers produced with scCO2.47 In the ternary composites of PEG–PCL–eucalyptol, there was a significant decrease in the heat of fusion (−7.5%) when compared to the PEG co-polymer processed without the drug, although only a slight change (−1 °C) occurred in the melting temperature (Table 3). In the homopolymer of PEG the change in enthalpy was only −4.5% when eucalyptol was incorporated using the PGSS process.
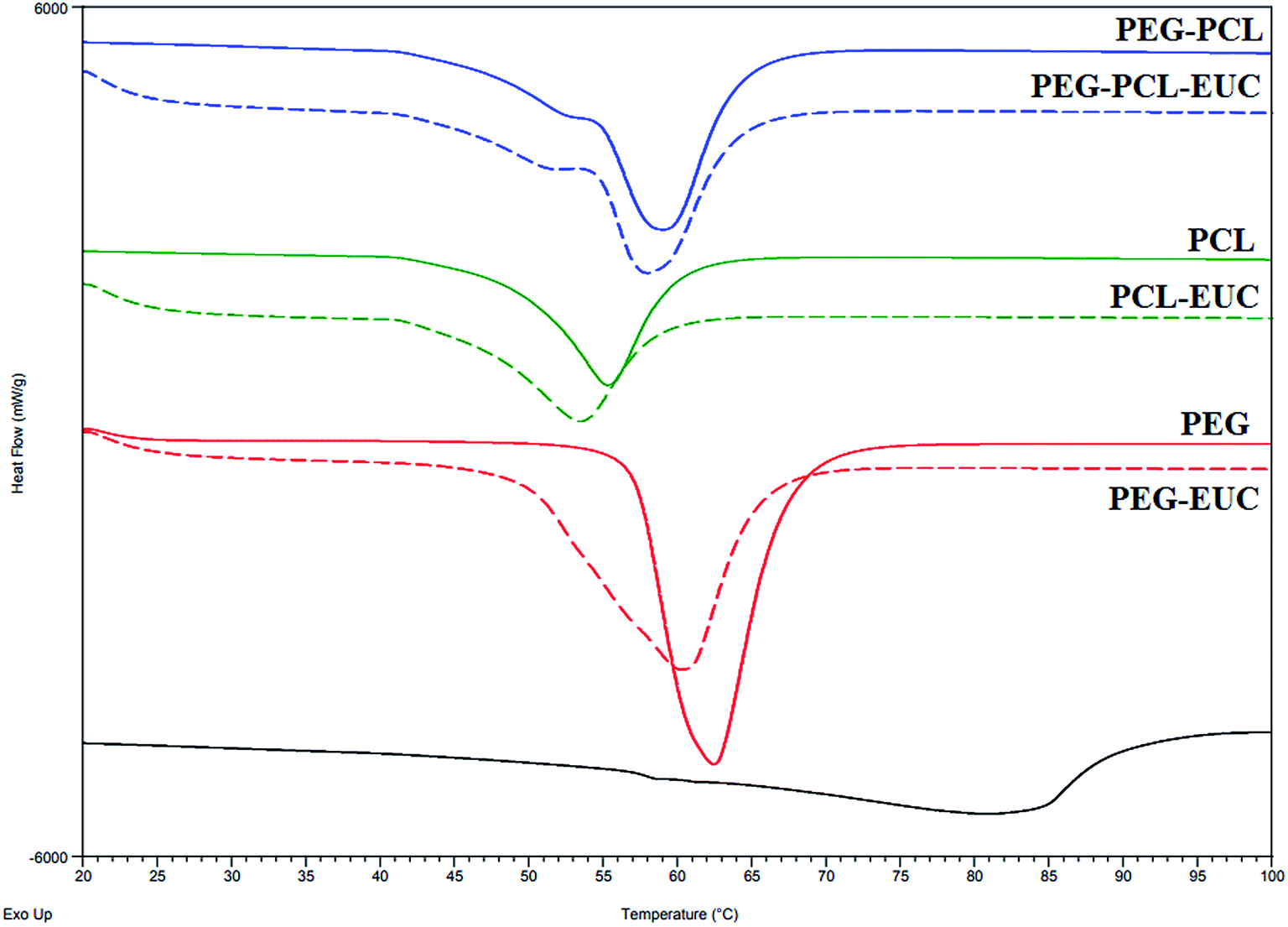 | ||
| Fig. 5 Thermograms (first heat cycle) of polyethylene glycol (PEG) and polycaprolactone (PCL) microparticles formulated with or without eucalyptol (EUC) using supercritical carbon dioxide. | ||
| Melting temperature °C | Fusion enthalpy, J g−1 | |||
|---|---|---|---|---|
| Processed in scCO2 without eucalyptol | Processed in scCO2 with eucalyptol | Processed in scCO2 without eucalyptol | Processed in scCO2 with eucalyptol | |
| a scCO2 = supercritical carbon dioxide. | ||||
| PCL | 55.30 | 53.46 | 82.48 | 75.88 |
| PEG–PCL | 59.08 | 57.97 | 140.10 | 129.52 |
| PEG | 62.42 | 60.23 | 178.30 | 170.30 |
The weight fraction of crystalline PEG (wcrPEG) processed with PCL with or without eucalyptol was calculated as fraction of the heat of fusion of the PEG–PCL blend to the weight fraction (wPEG) value of the heat of fusion of PEG homopolymer.44 The wPEG of the polymer composite for this study was 0.8. Thus, calculated crystallinity of PEG4000 after blending with PCL10000 in supercritical fluid was 0.98, with a further reduction to 0.91 on incorporation of eucalyptol. This reduction suggests high levels of inter-dispersion of the drug into the matrix of the polymeric blends. The degree of crystallinity of microparticulate drug delivery carriers is very crucial to sustained entrapment of the volatile drug during storage and controlled release of drug in physiological fluids following administration.33
The drug loading capacity of the PGSS-micronized particles as influenced by storage for four months either in the refrigerator (4 °C) or in the humidity chamber (30 °C; relative humidity of 70%) is shown in Fig. 6. Overall, loss of the volatile drug was significantly lower in refrigerated samples (p < 0.05), than those stored in the humidity chamber. After four months of refrigeration, only 3.5% of eucalyptol was lost in PEG microparticles, whereas 14% and 21% losses were recorded in PEG–PCL and PCL microparticles, respectively. Correspondingly, analyses of the samples stored in the humidity chamber revealed aggravated losses of 21%, 29% and 35% of eucalyptol from PEG, PEG–PCL and PCL co-precipitated particles. The highly crystalline nature of formulations containing PEG may be attributed to the significantly (p < 0.05) reduced loss of oil during storage. There could also be some degree of H-bond interactions between PEG and eucalyptol as evident from the FTIR study (Fig. 1). The reduction in PEG or PEG–PCL heat of fusion following PGSS processing with eucalyptol could also suggest some crystalline reinforcement action of H-bond interactions between PEG and eucalyptol (Table 3).
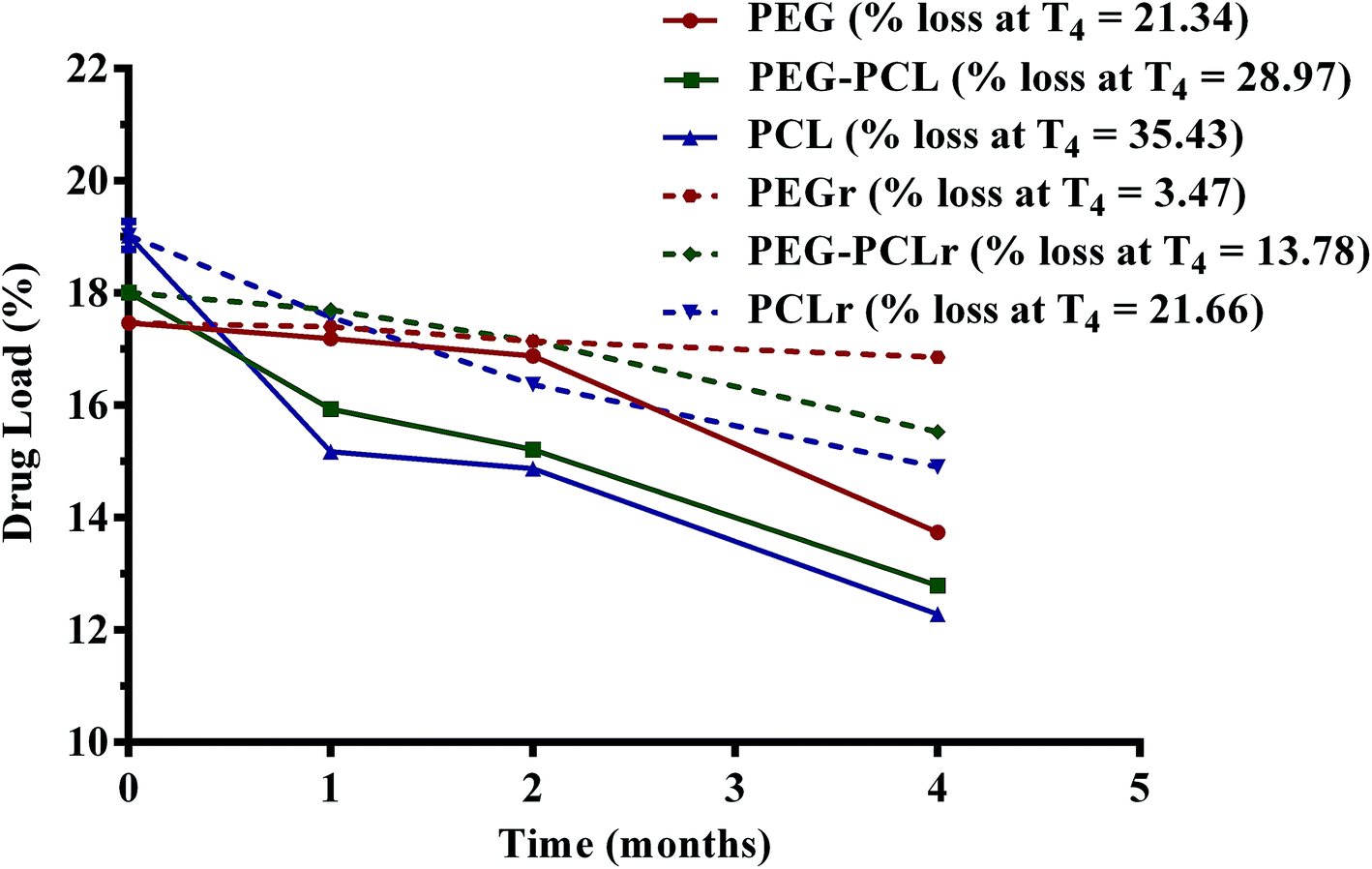 | ||
| Fig. 6 Influence of storage conditions on loading capacity of polyethylene glycol (PEG) and polycaprolactone (PCL) microparticles formulated with eucalyptol using supercritical carbon dioxide (*solid lines = storage at 30 °C and 70% relative humidity; dash lines = refrigeration at 4 °C; T4 = at 4 months; points on graph are mean ± SEM of three determinations). | ||
Release profiles
Cumulative release profiles of eucalyptol in simulated physiological fluids are shown in Fig. 7. A burst release of superficial eucalyptol from the PGSS-micronized particles was observed within 30 min of dispersion and mild agitation at 100 rpm in SGF. Release of 5.64%, 7.91% and 15.21% of eucalyptol from PCL, PEG–PCL and PEG respectively were quantified after a 2 h dissolution experiment in SGF. In SIF, cumulative release efficiency of the drug from PEG tripled after 8 h and the percentage release (49%) was almost twice that of PEG–PCL composite (27%). Only a marginal increase from 6 to 7% was recorded in PCL microparticles. Cumulatively, at the end of the 24 h experiment a quarter of the drug was released into the SPS solution from PCL microparticles, whereas 88% release efficiency was totalled for PEG micronized particles. Release of eucalyptol from the PEG–PCL co-polymer was retarded by 20% when compared to the PEG homopolymer, extending mean release time to 17 h as opposed to the 10 h computed for the PEG delivery system. The highly hydrophobic nature of PCL in aqueous solution can be attributed to the retarded release observed in formulations containing PCL. Thus, the incorporation of PCL into the PEG microparticles at weight ratios that gave optimum encapsulation efficiency resulted in an improved sustained release of eucalyptol. The PEG–PCL blend can be further optimized for controlled release of volatile drugs using various experimental design and approaches.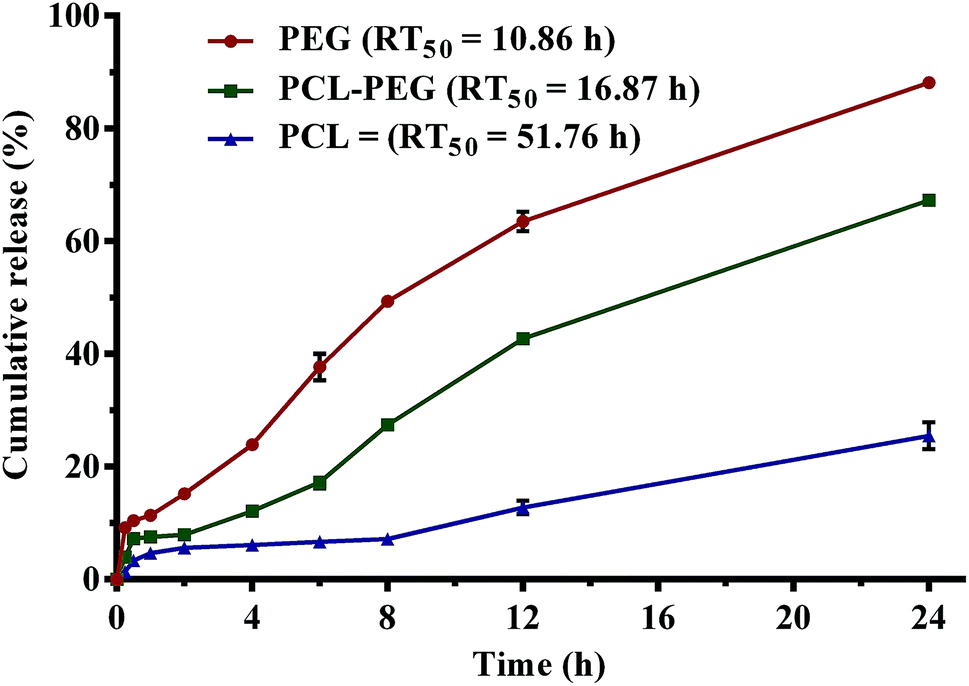 | ||
| Fig. 7 Cumulative release of eucalyptol from polyethylene glycol (PEG) and polycaprolactone (PCL) microparticles formulated using supercritical carbon dioxide in physiological fluids (points on graph are mean ± SEM of three determinations; RT50 = mean release time). | ||
Bioactivities
Finally, the influence of PGSS-micronized particles on the bioactivities of eucalyptol are shown in Fig. 8 and 9. DPPH assay is a simple, rapid and the most widely used method for in vitro evaluation of antioxidant property of compounds or extracts. It gives a violet colour that can be read and quantified in appropriate organic solvents using spectrophotometry. The degree of discoloration of DPPH solution when tested against eucalyptol or polymer–eucalyptol microparticles dissolved in DCM was computed as percentage radical scavenging activity (Fig. 8). The DPPH scavenging activity increased as the concentration of the drug increased, with mean radical scavenging concentration of 145 mg eucalyptol equivalent (Fig. 8). The polymeric microparticles; PEG, PCL and PEG–PCL formulated with 20% eucalyptol (∼200 mg), gave 40%, 42% and 49% radical scavenging activity, corresponding to approximately 85 mg, 95 mg and 135 mg eucalyptol equivalents, respectively. The calculated eucalyptol equivalent in the PEG–PCL microparticles was within range of the mean radical scavenging concentration of the drug. This data shows that the bioactivity of the eucalyptol was not affected following encapsulation into the various polymers using scCO2 PGSS process.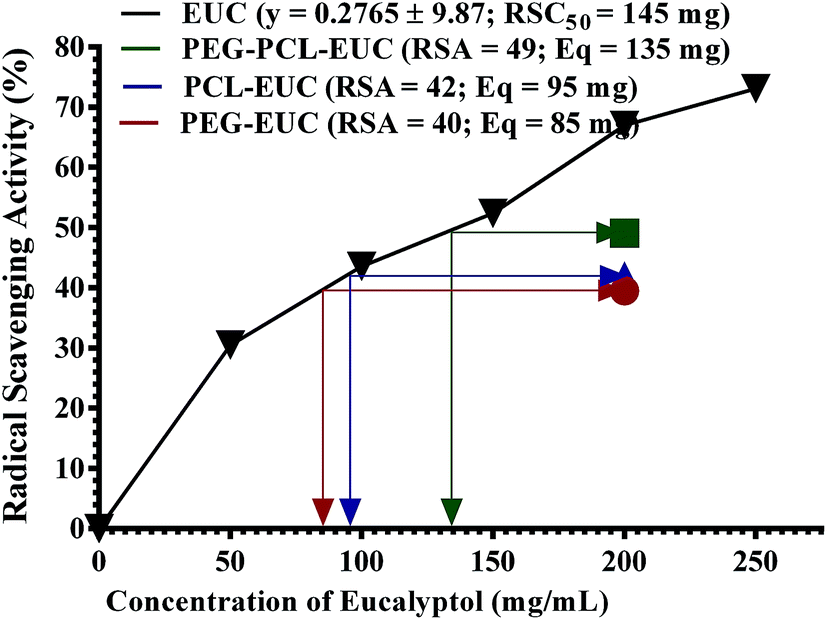 | ||
| Fig. 8 Free radical scavenging activity of polyethylene glycol (PEG) and polycaprolactone (PCL) microparticles formulated with eucalyptol (EUC) using supercritical carbon dioxide (RSA = radical scavenging activity; Eq = eucalyptol concentration equivalent; RSC50 = mean radical scavenging concentration). | ||
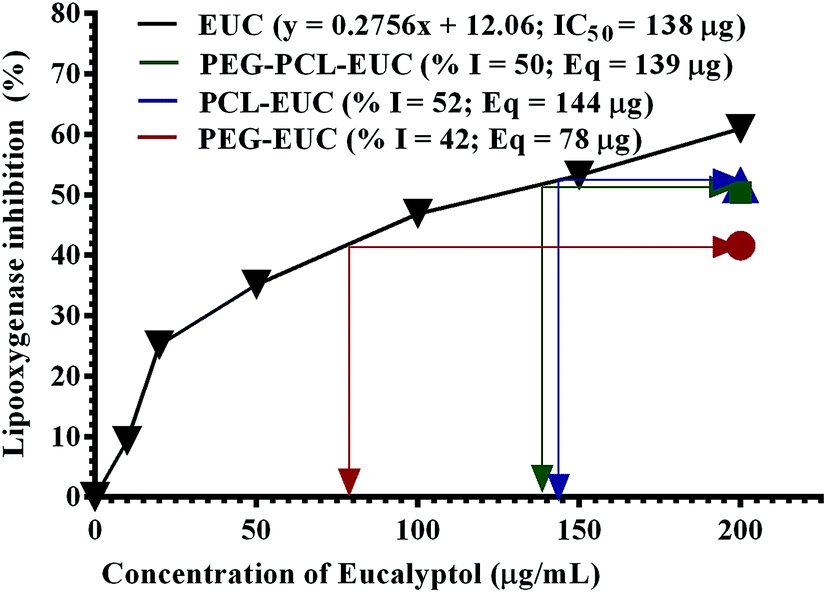 | ||
| Fig. 9 Lipoxygenase inhibitory activity of polyethylene glycol (PEG) and polycaprolactone (PCL) microparticles formulated with eucalyptol (EUC) using supercritical carbon dioxide (% I = percentage inhibition of lipoxygenase; Eq = eucalyptol concentration equivalent; RSC50 = mean radical scavenging concentration). | ||
LOX inhibition assay was also carried out to further substantiate the sustained activity of eucalyptol encapsulated in the PGSS-micronized particles. The inflammatory enzyme marker was inhibited by eucalyptol in a concentration-dependent manner with mean inhibitory concentration of 138 mg (Fig. 9). Eucalyptol is clinically approved as an anti-inflammatory agent and acts by inhibiting LOX, a key enzyme in leukotriene production.8 The polymeric microparticles; PEG, PEG–PCL and PCL formulated with 20% eucalyptol (∼200 mg) gave 42%, 50% and 52% LOX inhibition, corresponding to approximately 78 mg, 139 mg and 144 mg eucalyptol equivalents, respectively. Also, correlation analysis of response indices showed that the anti-inflammatory activity of polymer–eucalyptol particles are strongly linked to the encapsulation efficiency (r2 = 0.997), while the antioxidant activity was strongly correlated to the volume mean diameter (r2 = 0.925). These imply that the size and amount of entrapped oil in the polymer significantly influenced the bioactivity of the oil (Table 4).
| Loading capacity | Encapsulation efficiency | Volume mean diameter | Particle size | |
|---|---|---|---|---|
| DPPH | −0.388 | 0.664 | 0.925 | −0.140 |
| LOX | 0.502 | 0.997 | 0.860 | 0.706 |
Experimental
Materials
PEG (4000 g mol−1), PCL (10000 g mol−1), eucalyptol (density = 0.921 g mL−1), dichloromethane (DCM), acetonitrile, linoleic acid, lipoxygenase V from glycine max and 2,2-diphenyl-1-picrylhydrazyl (DPPH) were purchased from Sigma Aldrich (St. Louis, USA). Carbon dioxide (99.99% purity) was purchased from Air Products (Bryanston, South Africa). All other chemicals and reagents used were of analytical grade and purchased from reputable suppliers.
Processing in scCO2 by PGSS
Supercritical fluid apparatus for PGSS was set-up as previously described by Labuschagne et al.31 Processing in scCO2 was carried out in a Separex pilot-scale reactor (Separex Equipment, Champigneulles, France). Blends of liquefiable PEG were mixed with PCL of varying weight ratios (0.1–0.5). At different times, PEG (24 g), PCL (24 g) and PEG–PCL blends (24 g) were transferred into the PGSS autoclave (0.5 L capacity) which was pre-heated with electrical heaters to 45 °C and fitted with a mechanically driven stirrer. For polymer-drug processing, eucalyptol (6 mL; 20% w/w loading) was added to the polymer and the autoclave was sealed. CO2 gas was drawn from a standard commercial gas cylinder, pumped through a chamber, pre-set to the same temperature of 45 °C as the autoclave. The autoclave pressure was then set to 80 bar. The polymer–eucalyptol mixture was allowed to liquefy for 30 min and further stirred for 30 min at 150 rpm. The liquefied product was sprayed and micronized through a 500 μm capillary with a length of 5 mm, into a 10 L expansion chamber at atmospheric pressure. The PGSS-micronized particles were then collected for characterization and further studies.Infrared spectroscopy
Infrared spectra of particles were obtained as previously described by Labuschagne et al.31 using Attenuated Total Reflection-Fourier Transform Infrared (ATR-FTIR) spectroscopy (Spectrum 100 FTIR spectrometer PerkinElmer Inc., Norwalk, CT, USA). Wavenumbers ranged from 4000 cm−1 to 650 cm−1, using four scans with a resolution of 4 cm−1. Analyses of results were performed using Spectragryph-optical spectroscopy software.48Gas chromatography and mass spectrometry
GC-MS (QP2010SE, Shimadzu, Kyoto, Japan) was used to further confirm the presence of eucalyptol in the polymeric composites. The particles were washed thrice in hexane to remove non-encapsulated eucalyptol, the polymer–oil composites were then dissolved in water and/or acetone. The eucalyptol was recovered and reconstituted appropriately in hexane prior to GC analysis. GC-MS analysis was carried out by injecting 1 μL of sample at 250 °C with a split ratio of 10:1 onto an InertCap 5MS/NP capillary (30 m × 0.25 mm × 0.25 μm; GL Sciences, Tokyo, Japan). The ion source was operated at 200 °C and the oven temperature was programmed from 50 °C holding for 8 min and increased at 8 °C min−1 to 130 °C, then at to 200 °C at 5 °C min−1 with a final ramp to 280 °C at 15 °C min−1, and a hold for 4 min. Helium was used as carrier gas (flow rate 1.0 mL min−1 and velocity of 32 cm s−1). Mass spectra were recorded between 50 to 6000 m/z in the electron impact (EI) ionisation mode at 70 eV with a scan speed of 2500. Compounds were identified by comparing the obtained mass spectra with those from published commercial libraries NIST11 and Wiley (10th edition).
Thermal analysis
Differential scanning calorimetry (DSC Q2000, TA Instruments, New Castle, DE, USA) was used for thermal analysis of the samples. Samples (2–3 mg) were weighed in aluminium pans. A heat–cool–heat temperature programming protocol was adopted. The temperature ranged from 20 to 250 °C with a heating rate of 10 °C min−1 in a nitrogen atmosphere (flow rate = 10 mL min−1). Repeat runs on all samples were obtained to ensure reproducibility.Size and morphology
Volume-averaged mean diameters (VMD) of co-precipitated particles were measured using a Malvern Mastersizer 2000, multiple-angle, laser light scattering analyser (Malvern Instruments, Worcestershire, UK). Particles were dispersed in water and sonicated before measurement at 25 °C. Morphology was examined at varying magnification (×1000–10000) using a Zeiss Gemini Ultra Plus FEG SEM (Oberkochen, Germany). The particles were prepared by on carbon tape placed on an aluminium stub and coated with two layers of carbon using a sputter coater. Particle sizes were computed from multiple images of each sample using freely available imaging software (ImageJ, NIH, USA).
Loading capacity
Loading capacity was computed by determining indirectly the amount of oil in PGSS-micronized particles using gravimetric method as described by Yang and Ciftci49 with slight modifications. Briefly, 500 mg of particles in triplicates were placed in an oven and heated at 150 °C overnight for 12 h, instead of 30 min as reported by the authors. The empty particles were similarly analysed as blank. Loading capacity was determined by computing the amount of oil in particles (initial weight – final weight of samples) as a percentage of the PGSS-micronized particles.Encapsulation efficiency
For determination of encapsulation efficiency, superficial non-encapsulated eucalyptol in the co-precipitated particles (500 mg) was removed by washing thrice with hexane. The particles were then dissolved in DCM (5 mL). The sample was stirred vigorously for 6 h. The concentration of eucalyptol was quantified using UV-vis spectrophotometry (UV5, Mettler Toledo, Columbus, OH, USA). Standard solutions of eucalyptol ranging from 0.05–500 mg mL−1 were prepared in the extraction medium and the absorbance was read at its characteristic wavelength (λ = 285 nm) against the blank. A standard calibration curve for eucalyptol was computed and the extinction coefficient extrapolated (y = 0.0042x). Encapsulation efficiency was determined by computing the amount of recovered eucalyptol as a percentage of the total amount of the drug used for formulation.Vaporization study
Vaporization profiles of the volatile eucalyptol from the PGSS-micronized particles were determined using a digital moisture analyser (Radwag LLC, Miami, FL, USA) at elevated thermal treatment of 37 °C for 2 h. The empty particles were also analysed as blank. Decrease in weight was recorded and amount of evaporated eucalyptol was calculated at different time intervals. Mean vaporization time (VT50) was determined using linear regression analysis.Release profiles
Simulated gastric (SGF; 0.01 M HCl; pH 1.2), intestinal (SIF; 0.01 M phosphate buffer; pH 6.9) and physiological saline (SPS; 0.01 M phosphate buffer; 0.89% NaCl; pH 7.4) solutions were prepared in deionized water. Polymer–eucalyptol particles (500 mg) were dispersed in different 50 mL tubes containing the simulated fluids supplemented with 10% hexane. Cumulative release was carried out simultaneously in SGF for 2 h, followed by SIF to make up 8 h and then with SPS until 24 h. The mixtures were stirred at 100 rpm and 37 °C. At different time intervals (0.25, 0.5, 1, 2, 4, 8, 12, 24 h), the stirring was stopped, the mixture allowed to separate on standing, and an aliquot (50 μL) of the non-aqueous phase containing the released eucalyptol was sampled and quantified using spectrophotometry. Release efficiency (RE) of eucalyptol from the particles into the simulated fluids was calculated from the amount of the drug released into the fluid at a particular time (t) as a percentage of the total amount of the drug in the particles.50 Mean release time (RT50) was derived by subjecting the time course release data to linear regression analysis.Storage stability test
The PGSS-micronized particles were sealed in polypropylene containers, and then placed either in the humidity chamber which had been pre-set to 30 °C and 70% relative humidity or the refrigerator pre-set at 4 °C. The samples were monitored periodically using loading capacity as response index after 1, 2 and 4 months of storage.In vitro bioevaluation
Radical scavenging activity (RSA) was determined according to the method previously described with slight modifications.51 DPPH radical solution (0.1 mM) was prepared in DCM rather than methanol. Eucalyptol (50–250 mg) or polymer–eucalyptol particles (1000 mg) were dissolved separately in 5 mL of DCM. The solution (0.5 mL) was incubated in the dark for 30 min with 1.5 mL of DPPH solution. The absorbance (Δ) was read at 517 nm and the percentage RSA was computed.| RSA (%) = [1 − (ΔDPPH − Δsample)] × 100 |
Mean activity (RSA50) of eucalyptol was also determined and the eucalyptol equivalents of the PGSS-micronized particles were extrapolated.
Lipoxygenase inhibitory assay was carried out as previously reported with slight modifications.52 Stock solution of eucalyptol and polymer–eucalyptol particles were prepared in acetonitrile. Phosphate buffer (pH 6.3; 0.1 M) containing 100 μM linoleic acid was also prepared using distilled water. The reaction was initiated with the addition of 5-lipoxygenase diluted with phosphate buffer to 100 U. The increase in absorbance at 234 nm was recorded for 10 min. The percentage inhibition of LOX activity was computed by comparison with the acetonitrile control. The mean LOX inhibitory activity (IC50) of eucalyptol was determined and the eucalyptol equivalents of the PGSS-micronized particles were extrapolated.
Statistical analysis
Statistical evaluation of data was performed using Graph Pad Prism version 6. Data are expressed as mean of at least three replicates ± standard error of mean and statistically analyzed using one-way ANOVA followed by Turkey's posthoc test. Values were considered statistically significant at p < 0.05 (confidence level = 95%).Conclusions
We have shown here that scCO2 can be employed as an alternative solvent to aid microencapsulation of volatile drugs in polymeric composites. The PGSS process facilitated interaction between PEG and PCL and incorporation of eucalyptol into the PEG–PCL co-polymer. Blending of PEG with PCL at weight ratios of 4:1 improved loading capacity and encapsulation efficiency, extended mean release time in physiological buffers and reduced loss during storage. The bioactivity of the drug was maintained following encapsulation in PEG–PCL microparticles.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research work was funded by National Research Fund, South Africa (Grant no: 110789) under The World Academy of Sciences (TWAS/NRF) postdoctoral programme. The CSIR – Polymers and Composites Unit, Pretoria South Africa is duly acknowledged for supporting and hosting the first author for the fellowship.References
- C. J. Silva, L. C. Barbosa, A. J. Demuner, R. M. Montanari, A. L. Pinheiro, I. Dias and N. J. Andrade, Quím. Nova, 2010, 33, 104–108 CrossRef CAS.
- A. Giatropoulos, A. Kimbaris, A. Michaelakis, D. P. Papachristos, M. G. Polissiou and N. Emmanouel, Parasitol. Res., 2018, 117, 1953–1964 CrossRef.
- M. De Vincenzi, M. Silano, A. De Vincenzi, F. Maialetti and B. Scazzocchio, Fitoterapia, 2002, 73, 269–275 CrossRef CAS.
- N. Fox, Arch. Otolaryngol., 1927, 6, 112–122 CrossRef.
- W. H. Organization, Cough and cold remedies for the treatment of acute respiratory infections in young children, World Health Organization, Geneva, 2001 Search PubMed.
- U. R. Juergens, M. Stöber and H. Vetter, Eur. J. Med.Res., 1998, 3, 508–510 CAS.
- U. R. Juergens, T. Engelen, K. Racké, M. Stöber, A. Gillissen and H. Vetter, Pulm. Pharmacol. Ther., 2004, 17, 281–287 CrossRef CAS.
- U. Juergens, Drug Res., 2014, 64, 638–646 CrossRef CAS.
- K. Sato, S. Krist and G. Buchbauer, Flavour Fragrance J., 2007, 22, 435–437 CrossRef CAS.
- Z. Yang, N. Wu, Y. Fu, G. Yang, W. Wang, Y. Zu and T. Efferth, J. Biomol. Struct. Dyn., 2010, 28, 323–330 CrossRef CAS PubMed.
- F. Santos, R. Silva, A. Campos, R. De Araujo, R. L. Júnior and V. Rao, Food Chem. Toxicol., 2004, 42, 579–584 CrossRef CAS.
- G. F. R. Caldas, A. R. da Silva Oliveira, A. V. Araújo, S. S. L. Lafayette, G. S. Albuquerque, J. da Costa Silva-Neto, J. H. Costa-Silva, F. Ferreira, J. G. M. da Costa and A. G. Wanderley, PLoS One, 2015, 10, e0134558 CrossRef PubMed.
- F. A. Santos, R. M. Silva, A. R. Tomé, V. S. Rao, M. M. Pompeu, M. J. Teixeira, L. A. De Freitas and V. L. De Souza, J. Pharm. Pharmacol., 2001, 53, 505–511 CrossRef CAS.
- O. Ciftci, I. Ozdemir, S. Tanyildizi, S. Yildiz and H. Oguzturk, Toxicol. Ind. Health, 2011, 27, 447–453 CrossRef CAS PubMed.
- M. Takaishi, F. Fujita, K. Uchida, S. Yamamoto, M. Sawada, C. Hatai, M. Shimizu and M. Tominaga, Mol. Pain, 2012, 8, 86 CrossRef CAS.
- Y. Li, Y. Lai, Y. Wang, N. Liu, F. Zhang and P. Xu, Inflammation, 2016, 39, 1582–1593 CrossRef CAS.
- S. Murata, R. Shiragami, C. Kosugi, T. Tezuka, M. Yamazaki, A. Hirano, Y. Yoshimura, M. Suzuki, K. Shuto and N. Ohkohchi, Oncol. Rep., 2013, 30, 2647–2652 CrossRef CAS.
- J. Lee, S. J. Ha, J. Park, Y. H. Kim, N. H. Lee, Y. E. Kim, Y. Kim, K.-M. Song and S. K. Jung, Oncotarget, 2017, 8, 105995–106008 Search PubMed.
- S. Lahlou, A. F. Figueiredo, P. J. C. Magalhães and J. H. Leal-Cardoso, Can. J. Physiol. Pharmacol., 2002, 80, 1125–1131 CrossRef CAS PubMed.
- A. Khan, K. Vaibhav, H. Javed, R. Tabassum, M. E. Ahmed, M. M. Khan, M. B. Khan, P. Shrivastava, F. Islam and M. S. Siddiqui, Neurochem. Res., 2014, 39, 344–352 CrossRef CAS.
- J. O. Akolade, L. A. Usman, O. E. Okereke and N. O. Muhammad, J. Med. Food, 2014, 17, 1122–1128 CrossRef.
- S. S. Basak and F. Candan, J. Iran. Chem. Soc., 2010, 7, 216–226 CrossRef CAS.
- L. Jelenkovic, V. S. Jovanovic, I. Palic, V. Mitic and M. Radulovic, Trop. J. Pharm. Res., 2014, 13, 1421–1428 CrossRef CAS.
- M. Duisken, F. Sandner, B. Blömeke and J. Hollender, Biochim. Biophys. Acta, Gen. Subj., 2005, 1722, 304–311 CrossRef CAS PubMed.
- A. Sansukcharearnpon, S. Wanichwecharungruang, N. Leepipatpaiboon, T. Kerdcharoen and S. Arayachukeat, Int. J. Pharm., 2010, 391, 267–273 CrossRef CAS PubMed.
- F. Jiang, G. Wu, W. Li, J. Yang, J. Yan, Y. Wang, W. Yao, X. Zhou, Z. He and L. Wu, Eur. J. Pharm. Sci., 2019, 127, 14–23 CrossRef CAS.
- Z. Wen, B. Liu, Z. Zheng, X. You, Y. Pu and Q. Li, Chem. Eng. Res. Des., 2010, 88, 1102–1107 CrossRef CAS.
- Z. Wen, X. You, L. Jiang, B. Liu, Z. Zheng, Y. Pu and B. Cheng, Flavour Fragrance J., 2011, 26, 27–33 CrossRef CAS.
- S. Varona, S. Kareth, Á. Martín and M. J. Cocero, J. Supercrit. Fluids, 2010, 54, 369–377 CrossRef CAS.
- S. Varona, Á. Martín, M. Cocero and C. M. Duarte, Chem. Eng. Technol., 2013, 36, 1187–1192 CrossRef CAS.
- P. W. Labuschagne, S. G. Kazarian and R. E. Sadiku, J. Supercrit. Fluids, 2011, 57, 190–197 CrossRef CAS.
- P. W. Labuschagne, B. Naicker and L. Kalombo, Int. J. Pharm., 2016, 499, 205–216 CrossRef CAS.
- L. Machado, V. Pelegati and A. Oliveira, J. Supercrit. Fluids, 2016, 107, 260–269 CrossRef CAS.
- J. Ndayishimiye and B. S. Chun, J. Ind. Eng. Chem., 2018, 63, 201–207 CrossRef CAS.
- F. Ahmed and D. E. Discher, J. Controlled Release, 2004, 96, 37–53 CrossRef CAS.
- H. Xin, X. Sha, X. Jiang, W. Zhang, L. Chen and X. Fang, Biomaterials, 2012, 33, 8167–8176 CrossRef CAS.
- P. Raveendran, D. Zimmermann, T. Häber and M. Suhm, Phys. Chem. Chem. Phys., 2000, 2, 3555–3563 RSC.
- E. de Paz, Á. Martín, C. M. Duarte and M. J. Cocero, Powder Technol., 2012, 217, 77–83 CrossRef CAS.
- L. Gîtin, S. Varona and M. J. C. Alonso, Innovative Rom. Food Biotechnol., 2011, 9, 60 Search PubMed.
- W. Chen, X. Hu, Y. Hong, Y. Su, H. Wang and J. Li, Powder Technol., 2013, 245, 241–250 CrossRef CAS.
- A. B. Lokhande, S. Mishra, R. D. Kulkarni and J. B. Naik, J. Pharm. Res., 2013, 7, 414–420 CAS.
- O. Thonggoom, N. Punrattanasin, N. Srisawang, N. Promawan and R. Thonggoom, J. Microencapsulation, 2016, 33, 239–248 CrossRef CAS.
- M.-J. Choi, A. Soottitantawat, O. Nuchuchua, S.-G. Min and U. Ruktanonchai, Food Res. Int., 2009, 42, 148–156 CrossRef CAS.
- P. W. Labuschagne, M. J. John and R. E. Sadiku, J. Supercrit. Fluids, 2010, 54, 81–88 CrossRef CAS.
- W.-C. Hsieh, C.-P. Chang and Y.-L. Gao, Colloids Surf., B, 2006, 53, 209–214 CrossRef CAS.
- L. Piao, Z. Dai, M. Deng, X. Chen and X. Jing, Polymer, 2003, 44, 2025–2031 CrossRef CAS.
- D. R. Perinelli, M. Cespi, G. Bonacucina, A. Naylor, M. Whitaker, J. K. Lam, S. M. Howdle, L. Casettari and G. Palmieri, Curr. Drug Delivery, 2016, 13, 673–681 CrossRef CAS.
- F. Menges, Version, 2017, 1, 2016–2017 Search PubMed.
- J. Yang and O. N. Ciftci, Food Res. Int., 2016, 87, 83–91 CrossRef CAS PubMed.
- J. Varshosaz, N. Tavakoli, M. Minayian and N. Rahdari, AAPS PharmSciTech, 2009, 10, 158–165 CrossRef CAS.
- J. O. Akolade, H. O. B. Oloyede, M. O. Salawu, A. O. Amuzat, A. I. Ganiyu and P. C. Onyenekwe, J. Drug Delivery Sci. Technol., 2018, 45, 11–19 CrossRef CAS.
- Y. Frum and A. Viljoen, Skin Pharmacol. Physiol., 2006, 19, 329–335 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |