
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceO-Methylation steps during strobilurin and bolineol biosynthesis†
Karen E.
Lebe
* and
Russell J.
Cox
 *
*
Institute for Organic Chemistry, BMWZ, Leibniz Universität Hannover, Schneiderberg 38, 30167 Hannover, Germany. E-mail: russell.cox@oci.uni-hannover.de
First published on 4th October 2019
Abstract
Strobilurins are potent antifungal polyketides produced by basidiomycete fungi. Two genes encoding O-methyltransferases (O-MeT) are present in the biosynthetic gene cluster of strobilurin A 1. In previous studies, the two O-MeT enzymes Str2 and Str3 were found to catalyse the final steps of the biosynthesis of 1. Here, we show by in vivo expression experiments, that O-methylation during strobilurin biosynthesis is regiospecific. O-MeT Str2 acts first and selectively catalyses the methylation of the carboxyl group of strobilurin and bolineol precursors. Str3 catalyses the subsequent methyl transfer to the enol group to form strobilurin A 1, but cannot methylate bolineol 4. Toxicity tests showed increasing antifungal activity of intermediates through the pathway and that bolineol 4 shows antifungal activity against A. oryzae NSAR1 with an MIC of 0.1 mg ml−1.
Introduction
Strobilurins and the closely related mucidins are polyketide natural products produced by various basidiomycete fungi.1 Basidiomycetes are increasingly recognised as sources of interesting bioactive compounds.2 Compounds such as strobilurin A 1 from the fungus Strobilurus tenacellus possess potent antifungal properties, and because of this they played an important role as lead structures during the creation of the β-methoxyacrylate class of agricultural fungicides.3 The key β-methoxyacrylate toxophore targets the Qo site of complex III of mitochondrial electron transport chain and prevents adenosine triphosphate (ATP) synthesis.4 The novel biochemical mode of action attracted e.g. Syngenta and BASF to develop potent compounds (e.g. azoxystrobin 2 and Kresoxim methyl 3) with increased photo-stability and selectivity, which are among the most widely used fungicides worldwide (Fig. 1).5 The strobilurin fungicides are estimated to make up 25% of the fungicide market and 6.7% of the total crop protection market (worth $3.4 billion in 2015).6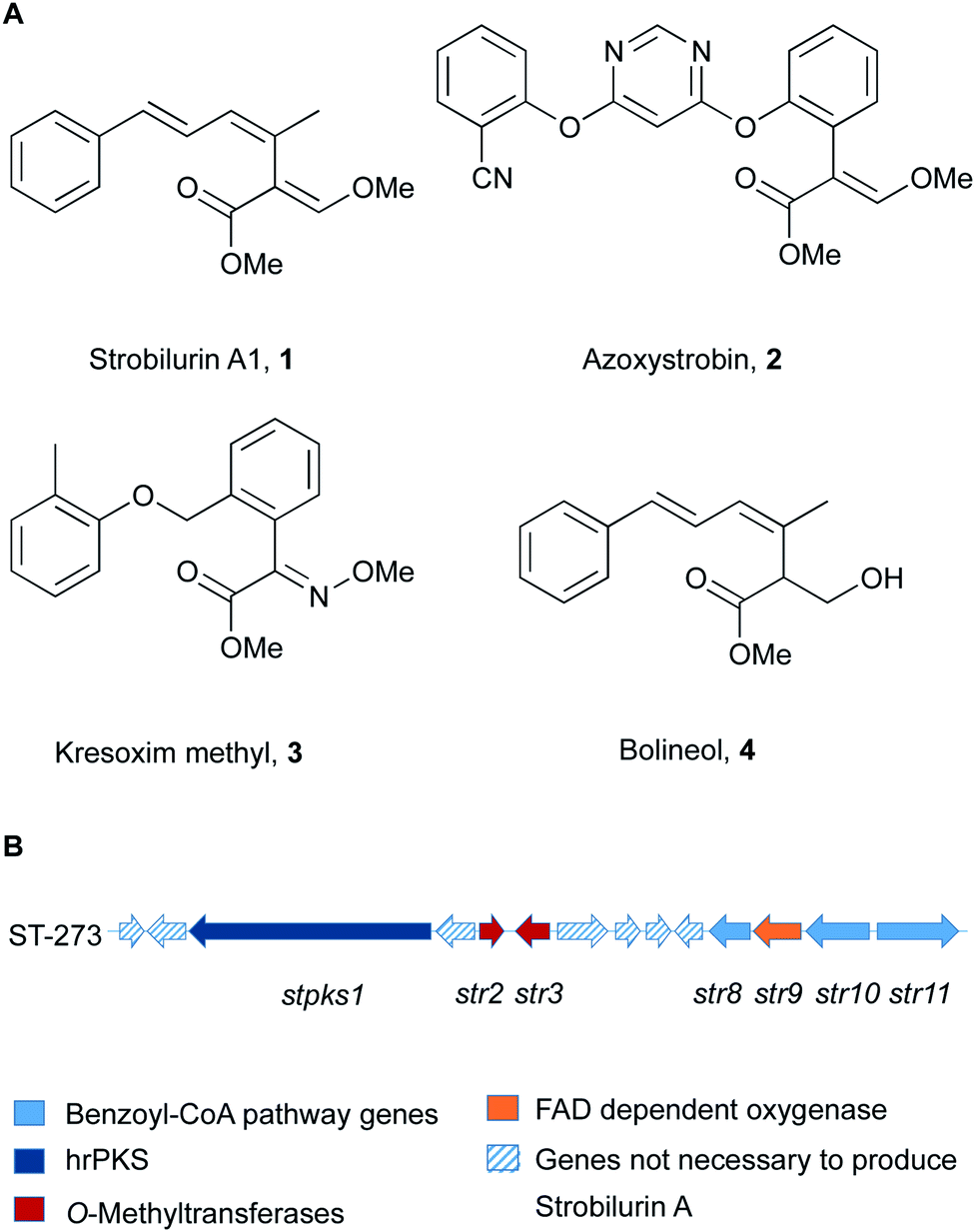 | ||
| Fig. 1 (A) Structures of key natural (1, 4) and synthetic (2, 3) strobilurins; (B) strobilurin A biosynthetic gene cluster (BGC). | ||
More than 20 different strobilurin natural products are known, with the β-methoxyacrylate toxophore as their common structural element. A very close relation of 1 is the formally reduced congener bolineol 4 which was isolated as a co-metabolite of strobilurins from Bolinea lutea, an apparent ascomeycete.7 The producing fungus was recently renamed as Strobilurus lutea as sequencing data revealed that it is a basidiomycete.6
Isotopic feeding experiments showed that strobilurin A 1 is a polyketide.8 The biosynthetic steps to strobilurin A 1 have been reported recently by our group (Scheme 1).6 Genome sequencing of S. tenacellus and S. lutea revealed that each organism harboured a biosynthetic gene cluster (BGC) responsible for the production of 1. A highly reducing polyketide synthase (PKS), encoded by stPKS1, uses benzoyl CoA 5, derived from phenylalanine, as a starter unit to produce the tetraketide 6.
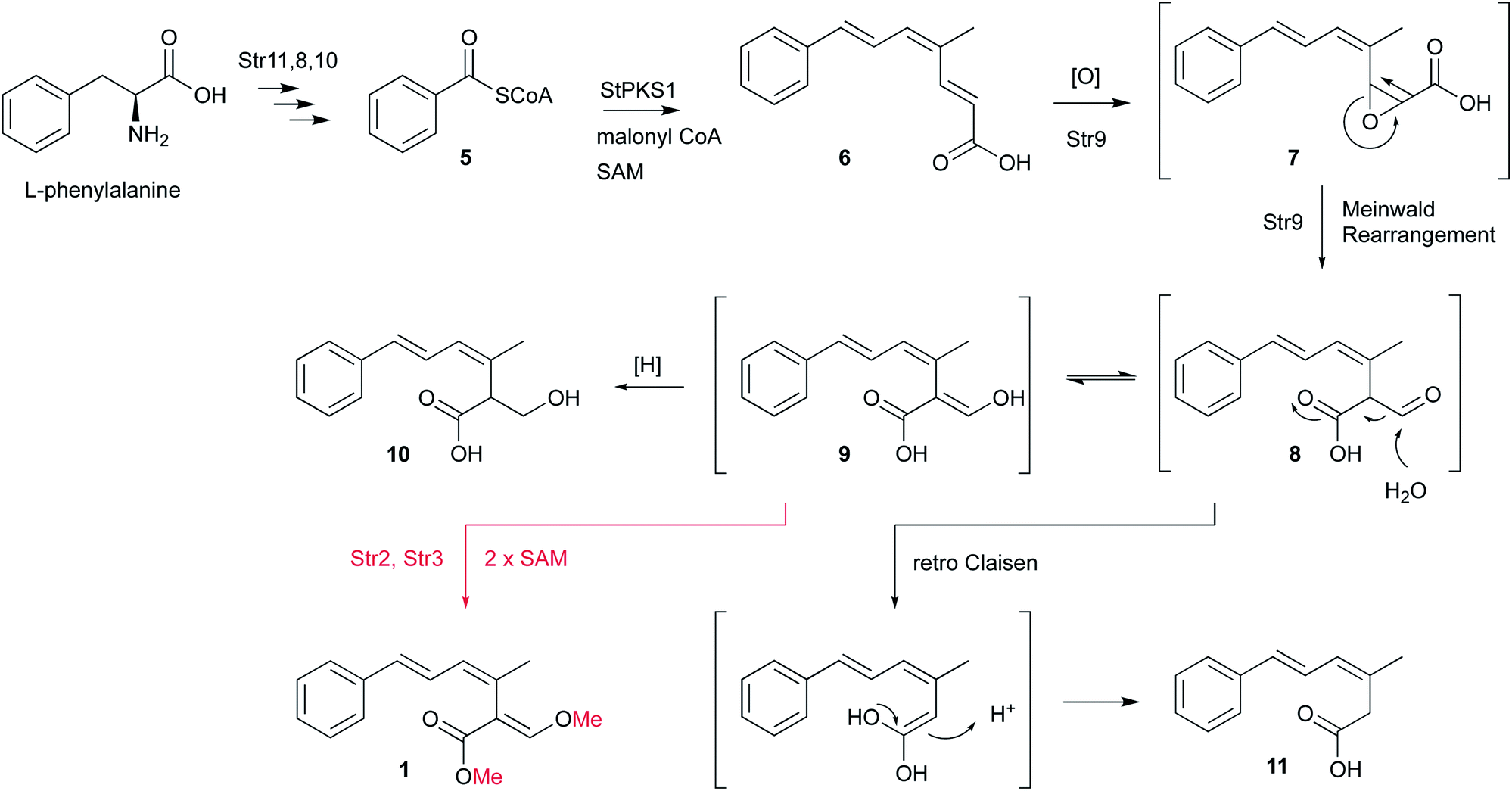 | ||
| Scheme 1 Biosynthetic steps to strobilurin A 1, compounds in brackets are proposed intermediates. | ||
The first enzyme free intermediate is prestrobilurin A 6, which is the substrate for an unusual Meinwald oxidative rearrangement to form the β-methoxyacrylate toxophore of 1. The rearrangement step is catalysed by the FAD dependent oxygenase Str9 as shown both in vivo and in vitro.6
In order to complete the biosynthesis to 1, two O-methyltransferases (O-MeT) Str2 and Str3 are required.6O-Methyltransferases catalyse the transfer of a methyl group from S-adenosyl-L-methionine to a hydroxyl or carboxyl group of the acceptor molecule to form methyl ether or methyl ester derivatives, respectively.9 Structure–activity studies of strobilurins revealed that the two O-methyl groups are crucial for antifungal activity and therefore all synthetic strobilurins possess two methoxy groups.5
During the investigation of the biosynthesis of 1 in the heterologous host A. oryzae, the likely aldehyde intermediate 8 was shown to be highly reactive in the absence of Str2 and Str3 and it undergoes rapid retro-Claisen reaction to give carboxylic acid 11. Intermediate 8 can also be reduced by Stl2, or by endogenous A. oryzae enzymes, to give 10. Bolineol 4 could be formed by methylation of 10 directly, or possibly by reduction of a methyl ester of 9.6 Inclusion of both str2 and str3 gives a mixture of 1 and 4. However, the detailed roles of the two O-MeT enzymes during the biosynthesis of strobilurin A 1 and bolineol 4 has remained unexplored. Here we show the roles of Str2 and Str3 in these processes.
Results
Two expression vectors for fungal transformation were constructed using yeast recombination. The vector pTYGS-niaD was used to clone either str2 or str3 (see ESI†). In combination with previously constructed plasmids pTYGS-arg-stPKS, pTYGS-ade-str11-str8-str10 and pTYGS-met-str9,6 all four plasmids were used to transform A. oryzae strain NSAR1, which harbours four auxotrophic lesions on its genome allowing the introduction of up to four DNA fragments in parallel.10–12Heterologous expression of str3
In order to investigate the function of Str3 in strobilurin A 1 and bolineol 4 biosynthesis, str2 was omitted from the expression system, by co-expressing str3 and the minimal gene set (Table 1, expt 3). Genetic analysis of the transformants showed incorporation of gene str3 in the genome of A. oryzae NSAR1 (Fig. S3†). A. oryzae strains expressing these genes produced 10 and 11 (Fig. 2Id). Compounds 10 and 11 were already found by expression of the minimal gene set (Fig. 2Ib and Table 1, expt 1). Both compounds were identified by comparing retention time and mass fragmentation to previously isolated standards, followed by isolation and NMR analysis.6| Expt | str11, str8, str10 | stpks1 | str9 | str3 | str2 | Products |
|---|---|---|---|---|---|---|
| 1 | ✓ | ✓ | ✓ | — | — | 10, 11 |
| 2 | ✓ | ✓ | ✓ | ✓ | ✓ | 1, 4, 10, 11 |
| 3 | ✓ | ✓ | ✓ | ✓ | — | 10, 11 |
| 4 | ✓ | ✓ | ✓ | — | ✓ | 4, 11, 13 |
 | ||
| Fig. 2 (I) LCMS evaporative light scattering (ELS) chromatograms of organic extracts of A. oryzae NSAR1 expression strains (arbitrary units). (a) untransformed A. oryzae NSAR1, * unrelated compounds; (b) A. oryzae NSAR1 + stPKS1 + str11 + str8 + str10 + str9; (c) A. oryzae NSAR1 + stPKS1 + str11 + str8 + str10 + str9 + str2 + str3, inset trace shows extracted ion chromatogram for m/z 259.1; (d) A. oryzae NSAR1 + stPKS1 + str11 + str8 + str10 + str9 + str3; (e) A. oryzae NSAR1 + stPKS1 + str11 + str8 + str10 + str9 + str2, inset trace shows DAD trace; (II) mass spectra in ES+ mode. (a) Expt 3 at 7.23 min; (b) expt 4 at 7.28 min; (c) expt 4 at 8.86 min. | ||
Compounds 11 and 4 coelute but detailed analysis of the mass spectrum (ES+ mode) of the peak eluting at 7.2 min showed that only the un-methylated compound 11 was detected by its distinctive [M − H2O + H]+ (185.2), [M + H]+ (203.2) and [M + Na]+ (225.1) ions (Fig. 2IIa). Bolineol 4 could not be detected under these conditions. As both 10 and 11 were lacking O-methylations, we reasoned that either Str3 is not functional or that it can only catalyse a second O-methylation step.
Interestingly, some of the transformants showed a slightly unhealthier phenotype than others when growing on DPY agar. After LCMS analysis of the extracts, the unhealthier phenotype was shown to correlate to a higher production of 10 (Fig. S4†). Growing A. oryzae NSAR1 on DPY agar plates containing various concentrations of 10, revealed a minimal inhibitory concentration (MIC) of 0.4 mg ml−1 for 10 against A. oryzae NSAR1 (Fig. S5†).
Heterologous expression of str2
The gene str3 was omitted from the expression system, by co-expressing str2 and the minimal gene set (Table 1, expt 4). This led to one major peak in the ELSD traces of the extracts of transformants (Fig. 2Ie). According to the retention time (RT = 7.3 min) and the mass spectrum, the major compounds produced were 11 and 4 (compared to synthetic standard). Bolineol 4 was detected to co-elute with 11 by its distinctive [M − H2O + H]+ (229.2) and [M + Na]+ (269.3) ions (Fig. 2IIb). A new minor compound 13 eluting at 8.7 min was also detected in the DAD trace of the producing fungus (Fig. 2Ie, inset trace). The amount produced was too little for effective purification and full structural analysis by NMR. Nevertheless, the mass spectrum correlates to an O-methylated 11: [M + H]+ (217.2) and [M + Na]+ (239.2) ions (Fig. 2IIc). Further support for this structure is given by its characteristic UV spectrum (maxima at 212 nm, 292 nm), which matches UV spectrum for compound 10 (Fig. S6†). In this experiment compounds 10 and 1 could not be detected.Toxicity of bolineol 4
Overexpression of str2 in the absence of str3 resulted in extremely unhealthy A. oryzae cultures on DPY agar. We reasoned that this is caused by bolineol production. In order to test this hypothesis, a toxicity test with 4 towards untransformed A. oryzae was conducted. Bolineol 4 could not be isolated from the expression host, due to co-elution with 11. Therefore, desmethylbolineol 10 was isolated from A. oryzae NSAR1 transformed culture (expt 1, 30 mg l−1 culture) and was smoothly converted to 4 by treatment with trimethylsilyldiazomethane.13 The agar toxicity test revealed an MIC of 0.1 mg ml−1 for 4 against A. oryzae NSAR1 (Fig. S7†).Discussion
Our results show that O-methylation during strobilurin biosynthesis is regioselective. The two O-MeT enzymes Str2 and Str3 share 66% amino acid identity but display different reactivity towards their substrates. Str2 selectively catalyses the transfer of a methyl group to the carboxyl group of 9 to give a methyl ester, whereas Str3 catalyses the methyl transfer to an enol, but only after O-methylation by Str2, and it appears unable to O-methylate bolineol 4.In nature, there are several examples of O-MeT enzymes which share a very similar amino acid sequence but show different substrate- or regio-selectivity. For example, two O-MeT enzymes from Clarkia breweri that share 83% identity: IEMT and CO-MET, which methylate eugenol/isoeugenol and caffeic acid/5-hydroxyferulic acid, respectively.14
According to plate toxicity tests with 4 and 10, O-methylation by Str2 caused four times higher toxicity toward A. oryzae. Similar experiments testing strobilurin A 1 fungicidal activity revealed a MIC of 25 μg ml−1 against a variety of plant pathogenic fungi growing on agar.15 Therefore, a second O-methylation leads to 16 times higher toxicity against fungi compared to the unmethylated compound 10, and four times higher compared to the single methylated 4. Thus the toxophore group of strobilurins is more potent in its methylated form. This result also suggests a possible order of events during the evolution of the strobilurin pathway (Scheme 2).
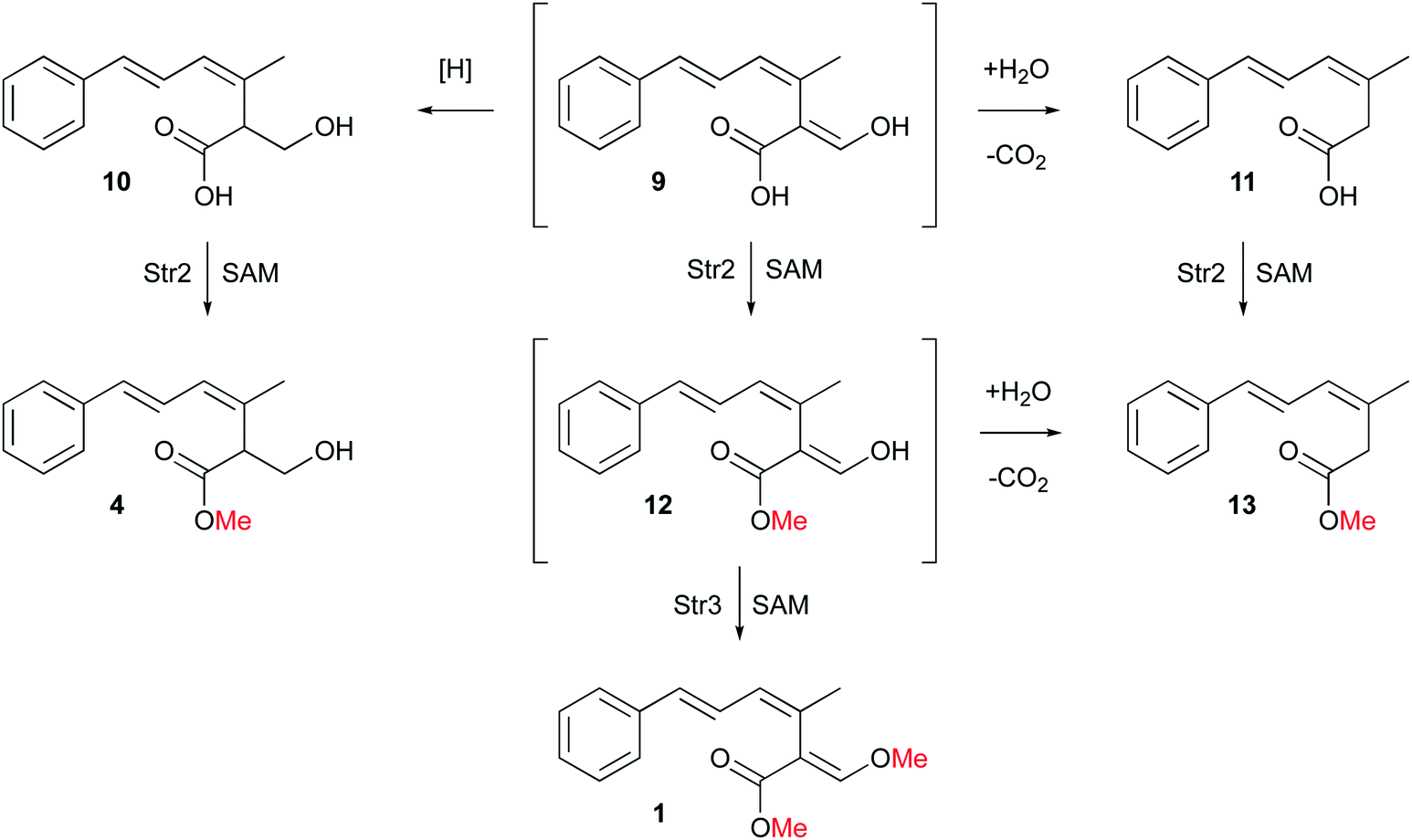 | ||
| Scheme 2 Detailed biosynthetic steps to strobilurin A 1 and bolineol 4. | ||
Initial production of the polyketide precursor 6 and its oxidative rearrangement by Str9 and reduction by Stl2 gives a weakly antifungal compound 10. Later gain by the pathway of the O-MeT Str2 then allowed the formation of the more potent bolineol 4. Finally, duplication of Str2 to give Str3 and its change in selectivity allowed the formation of the most potent compound 1. The formation of 1 does not require the activity of Stl2 and the presence of this enzyme in the pathway is now redundant. Its activity appears to be opposed by the oxidase Str4.6 Methylation of 12 by Str3 also stabilises the β-oxygenated acrylate toxophore and prevents retro-Claisen degradation to 13.
Similar observations are known during the biosynthesis of other natural products, such as Pterostilbene, which bears a higher antifungal activity due to its two methoxyl groups, than its desmethylated precursor resveratrol.16,17
Overall, our results show that O-methylation by Str2 and Str3 are regioselective. These steps complete the pathway to 1 which is now fully elucidated. However, 4 could be produced either by methylation of 10 or by reduction of 12, or a combination of routes. Likewise 13 could be produced by methylation of 11 or decarbonylation of 12. Further in vitro experiments will be required to differentiate these possibilities.
Experimental
Analytical and preparative methods
Analytical LCMS data was obtained using a Waters LCMS system comprising of a Waters 2767 autosampler, Waters 2545 pump, a Phenomenex Kinetex column (2.6 μm, C18, 100 Å, 4.6 × 100 mm) equipped with a Phenomenex Security Guard precolumn (Luna, C5, 300 Å) and a flow rate of 1 ml min−1. Detection was carried out by a diode array detector (Waters 2998) in the range 210 to 600 nm and an ELSD detector (Waters 2424) together with a mass spectrometer, Waters SQD-2 mass detector, operating simultaneously in ES+ and ES− modes between 150 and 1000 m/z. A solvent gradient was run over 15 min starting at 10% acetonitrile/90% HPLC grade water (0.05% formic acid) and ramping to 90% acetonitrile.Preparative LCMS was used to purify compounds from a raw extract or from a reaction mixture. This consisted of a Waters mass-directed autopurification system comprising of a Waters 2767 autosampler, Waters 2545 pump system, a Phenomenex Kinetex Axia column (5 μm, C18, 100 Å, 21.2 × 250 mm) equipped with a Phenomenex Security Guard precolumn (Luna, C5, 300 Å). A solvent gradient was run over 10 min or 15 min starting at 10% acetonitrile/90% HPLC grade water (0.05% formic acid) and ramping to 90% acetonitrile. The flow was set to 20 ml min−1 and the post-column flow was split (100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and the minority flow was made up with HPLC grade MeOH + 0.045% formic acid to 1 ml min−1 for simultaneous analysis by diode array detector (Waters 2998) in the range 210 to 600 nm and an ELSD detector (Waters 2424) together with a mass spectrometry, Waters SQD-2 mass detector, operating simultaneously in ES+ and ES− modes between 150 and 1000 m/z. Detected peaks were collected into glass test tubes. Combined tubes were evaporated (vacuum centrifuge).
:1) and the minority flow was made up with HPLC grade MeOH + 0.045% formic acid to 1 ml min−1 for simultaneous analysis by diode array detector (Waters 2998) in the range 210 to 600 nm and an ELSD detector (Waters 2424) together with a mass spectrometry, Waters SQD-2 mass detector, operating simultaneously in ES+ and ES− modes between 150 and 1000 m/z. Detected peaks were collected into glass test tubes. Combined tubes were evaporated (vacuum centrifuge).
NMR measurements were acquired on Bruker DRX 400, Bruker Avance 500 or Bruker Avance 600 MHz spectrometers (Institute for Organic Chemistry, Leibniz Universität Hannover). Chemical shifts are shown in parts per million (ppm) in comparison to the TMS (Tetramethylsilane) standard. Coupling constants J are quoted in Hz.
Strobilurin A 16
δ H (500 MHz, CDCl3) 7.43 (1H, s, H-11), 7.35 (2H, d, J = 8.3, H-8/8′), 7.30 (2H, d, J = 8.3, H-9/9′), 7.15 (1H, m, H-10), 6.62 (1H, dd, J = 15.6, J = 10.6, H-5), 6.49 (1H, d, J = 15.6, H-6), 6.26 (1H, brd, J = 10.6, H-4), 3.85 (3H, s, OMe), 3.74 (3H, s, OMe [ester]), 1.98 (3H, s, Me-12); δC (125 MHz, CDCl3) 168.0 (C-1), 159.0 (C-11), 138.0 (C-7), 131.5 (C-3), 131.3 (C-6), 129.9 (C-4), 128.6 (C-8/8′), 127.3 (C-10), 126.7 (C-5), 126.5 (C-9/9′), 111.0 (C-2), 62.1 (C–OMe), 51.8 (C–OMe [ester]), 23.8 (Me-12); HRMS (ES+): measured 281.1157, calc. for C16H18O3Na 281.1154.Synthesis of bolineol 47
(3Z,5E)-2-(Hydroxymethyl)-3-methyl-6-phenylhexa-3,5-dienoic acid 10 (0.011 g, 0.048 mmol, 1.0 eq.) was treated with TMS-CHN2 (0.016 g, 0.14 mmol, 3.0 eq., Sigma) in MeOH at RT for 16 h. The solvent was removed in vacuo and the product was purified using preparative LCMS. Synthetic and isolated bolineol were spectroscopically identical.δ H (400 MHz, CDCl3) 1.81 (m, 3H, H-11), 3.74 (3H, s, H-13), 3.68 (1H, dd, J = 11.1, 5.5, H-2), 3.99 (1H, dd, J = 8.7, 5.5, H-12), 4.10 (1H, dd, J = 11.1, 8.7, H-12), 6.22 (1H, dq, J = 11.1, 1.3, 4-H), 6.55 (1H, d, J = 15.4, H-6), 7.00 (1H, dd, J = 15.3, 11.0, H-5), 7.23 (1H, m, H-10), 7.32 (m, 2H, H-8/8′), 7.41 (2H, m, H-9/9′).
Desmethylbolineol 106
δ H (CDCl3, 400 MHz), 7.41 (2H, m, H-9/9′), 7.31 (2H, m, H-8/8′), 7.20 (1H, m, H-10), 7.02 (1H, dd, J = 15.3, J = 11.4, H-5), 6.56 (1H, d, J = 15.3, H-6), 6.24 (1H, d, J = 11.4, H-4), 4.08 (1H, m, H-2), 4.08 (1H, m, H-12a), 3.71 (1H, dd, J = 5.0, J = 11.0, H-12b); δC (CDCl3, 100 MHz) 179.9 (C-1), 137.3 (C-7), 133.5 (C-6), 131.1 (C-4), 130.9 (C-3), 128.8 (C-8/8′), 127.9 (C-10), 126.6 (C-9/9′), 123.6 (C-5), 61.9 (C-12), 49.9 (C-2), 21.3 (C-11); HRMS corresponding to C14H16O3 [M − H]− calc. 231.1021, measured 231.1021.(3Z,5E)-6-Phenyl-3-methylhexadienoic acid 116,18
δ H (400 MHz, CDCl3) 1.96 (s, 3H, H-11), 3.32, (s, 2H, H-2), 6.22 (d, J = 10.8 Hz, 1H, H-4), 6.53 (d, J = 15.4, 1H, H-6), 6.96 (dd, J = 10.9, J = 15.5, 1H, H-5), 7.22 (m, 1H, H-10), 7.31 (m, 2H, H-8/8′), 7.40 (m, 2H, H-9/9′); δC (100 MHz, CDCl3) 24.7 (C-11), 38.0 (C-2), 124.4 (C-5), 126.5 (C-9/9′), 127.6 (C-10), 128.7 (C-8/8′), 129.6 (C-4), 130.7 (C-3), 132.5 (C-6), 137.6 (C-7), 177.2 (C-1). HRMS corresponding to C13H15O2 [M + H]+ calc. 203.1072, measured 203.1071.Biological methods
Preparation of vectors expressing stpks1, str11, str8, str10 and str9 have been previously described.6 Preparations of vectors expressing str2 and str3 are described in the ESI.† Transformation, fermentation, molecular analyses and extraction procedures are also described in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
KEL thanks the Leibniz Universität Hannover for funding. DFG is thanked for the provision of LCMS and NMR equipment (INST 187/621-1, INST 187/686-1). RJC thanks Risa Nofiani for a standard sample of 1. The publication of this article was funded by the Open Access fund of Leibniz Universität Hannover.Notes and references
- T. Anke, F. Oberwinkler, W. Steglich and G. Schramm, J. Antibiot., 1977, 30, 806–810 CrossRef CAS.
- B. Sandargo, C. Chepkirui, T. Cheng, L. Chaverra-Muñoz, B. Thongbai, M. Stadler and S. Hüttel, Biotechnol. Adv., 2019, 37(6), 107344 CrossRef .
- H. Sauter, W. Steglich and T. Anke, Angew. Chem., Int. Ed., 1999, 38, 1329–1349 CrossRef CAS.
- W. Becker, G. Vonjagow, T. Anke and W. Steglich, FEBS Lett., 1981, 132, 329–333 CrossRef CAS.
- D. W. Bartlett, J. M. Clough, J. R. Godwin, A. A. Hall, M. Hamer and B. Parr-Dobrzanski, Pest Manage. Sci., 2002, 58, 649–662 CrossRef CAS PubMed.
- R. Nofiani, K. de Mattos-Shipley, K. E. Lebe, L.-C. Han, Z. Iqbal, A. M. Bailey, C. L. Willis, T. J. Simpson and R. J. Cox, Nat. Commun., 2018, 9, 3940 CrossRef.
- C. T. Bedford, D. Perry and R. K. Sharma, Nat. Prod. Res., 2008, 22, 1535–1539 CrossRef CAS PubMed.
- Z. Iqbal, L.-C. Han, A. Soares-Sello, R. Nofiani, G. Thormann, A. Zeeck, R. Cox, C. Willis and T. Simpson, Org. Biomol. Chem., 2018, 16, 5524–5532 RSC.
- H. L. Schubert, R. M. Blumenthal and X. Cheng, Trends Biochem. Sci., 2003, 28, 329–335 CrossRef CAS.
- F. J. Jin, J.-I. Maruyama, P. R. Juvvadi, M. Arioka and K. Kitamoto, FEMS Microbiol. Lett., 2004, 239, 79–85 CrossRef CAS.
- T. Fujii, H. Yamaoka, K. Gomi, K. Kitamoto and C. Kumagai, Biosci., Biotechnol., Biochem., 1995, 59, 1869–1874 CrossRef CAS.
- O. Yamada, S. Na Nan, T. Akao, M. Tominaga, H. Watanabe, T. Satoh, H. Enei and O. Akita, J. Biosci. Bioeng., 2003, 95, 82–88 CrossRef CAS.
- E. Kühnel, D. D. P. Laffan, G. C. Lloyd-Jones, T. M. del Campo, I. R. Shepperson and J. L. Slaughter, Angew. Chem., Int. Ed., 2007, 46, 7075–7078 CrossRef.
- J. Wang and E. Pichersky, Arch. Biochem. Biophys., 1999, 368, 172–180 CrossRef CAS.
- K. Beautement, J. M. Clough, J. Paul, J. de Fraine and C. R. A. Godfrey, Pestic. Sci., 1991, 31, 499–519 CrossRef CAS.
- M. Alessandro, S. Di Marco, F. Osti and A. Cesari, Phytopathol. Mediterr., 2000, 3, 357–365 Search PubMed.
- R. G. K. Pezet, O. Viret and H. Richter, Vitis, 2004, 43, 145–148 CAS.
- N. Y. Grigorieva, V. A. Popovsky, A. V. Stepanov and E. D. Lubuzh, Russ. Chem. Bull., 2010, 59, 2086–2093 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra06412e |
| This journal is © The Royal Society of Chemistry 2019 |