DOI:
10.1039/C9RA06024C
(Paper)
RSC Adv., 2019,
9, 31517-31526
Anti-tumor effect of volatile oil from Houttuynia cordata Thunb. on HepG2 cells and HepG2 tumor-bearing mice†
Received
3rd August 2019
, Accepted 23rd September 2019
First published on 3rd October 2019
Abstract
The aim of this paper is to study the anti-tumor mechanism of volatile oil from Houttuynia cordata Thunb. (sodium new houttuyfonate, SNH). In vitro, SNH exhibited a concentration-dependent cytotoxic effect against four human cancer lines (HepG2, A2780, MCF-7, SKOV-3). SNH treatment with different concentrations induced HepG2 cells to exhibit varying degrees of morphological changes in apoptotic features, such as round shape, cell shrinkage and formation of apoptotic body. It was observed that SNH caused the decrease in Bcl-2 mRNA expression and triggered the apoptosis of HepG2 cells. Wound healing assay and RT-PCR results showed that the decrease in the expression level of MMP9 and VEGF was observed in HepG2 cells after treatment with SNH for 48 h, suggesting that the extracellular matrix pathway degradation was involved in the HepG2 cells migration. Moreover, we got an insight into the binding mode of SNH into the MMP9 active site through 3D pharmacophore models. Docking study and molecular dynamics (MD) simulation analysis sheds light on that SNH was completely embedded into the MMP9 active site and formed hydrogen bonds with key catalytic residues of MMP9, including Ala191, His190, Ala189 and Glu227. The prediction of SNH binding interaction energies in the MMP9 was almost in good agreement with the original inhibitor EN140. In vivo experiments, both SNH and cyclophosphamide significantly reduced tumor weights and their tumor inhibitory rates were 50.78% and 82.61% respectively. This study demonstrated that SNH was an apoptosis inducer in HepG2 cells. SNH has four possible functions, that it could induce apoptosis by mitochondria pathway in HepG2 cells, inhibit the tumor growth, regulate Bcl-2 family mRNA expression and effectively subdue migration of hepatocellular carcinoma cells by decreasing the expression of MMP9 and VEGF. Therefore, SNH might be a potential candidate drug for the treatment of hepatocellular carcinoma, which could provide a reference for further clinical research.
Introduction
Cancer is a major public health problem in China and many other parts of the world.1,2 One in four deaths in China are due to cancer, and its morbidity and mortality are very high and increase year by year.3 Therapeutic methods for cancer have been long developing.4,5 However, due to the characteristics of recurrence and metastasis of cancer, the prognosis of cancer treatment is limited.6 The majority of cancer-related deaths are caused by distant metastases from the primary tumor site.7 Treatment failure mainly arises from cancer cell proliferation, invasion and metastasis, which ultimately lead to patient mortality. Many studies have shown that the destruction of the extracellular matrix (ECM) was a key link in the invasion and metastasis of cancer.8–10 Matrix metalloproteins (MMPs), which are involved in the destruction of extracellular matrices, are the main enzymes, especially MMP9, which has been found to be directly associated with the invasion.11 In addition, the ability of tumor metastasis is also associated with the decrease in apoptosis. Apoptosis is a mechanism by which cells undergo death to control cell proliferation or in response to DNA damage.12 Tumor production is related to the inhibition of apoptosis, and it is currently believed that the triggering of apoptosis is the result of cascaded gene expression. There are many genes involved in this process, such as, Caspase family, Bcl-2 family and so on, which regulate apoptosis as a potential inhibitor of apoptosis.13 Thus, inhibiting expression of MMP9 and anti-apoptotic genes may be critical in the treatment of malignant tumors.
Phytochemicals are considered as potential alternative or supplementary anticancer agents mainly because of their high effectiveness and low adverse reactions.14–17Houttuynia cordata Thunb. (HCT) is a member of the Saururaceae family and a well-known edible and medicinal plant distributed widely in China and it has complex medicinal ingredients, mainly including volatile oil, alkaloids, flavonoids, polysaccharides, organic acids and others.18 In previous study, the beneficial pharmacological effect of HCT crude extract was attributed to its anti-adipogenic and anti-obesity,19,20 anti-inflammatory,21,22 antimicrobial,23 antiviral24 and antitumor properties.25–27 Among these bioactivities, antitumor effect was considered as the most important function. The previous publication reported that HCT induced apoptotic cell death in human primary colorectal cancer cells through a mitochondria-dependent signalling pathway.28 In addition, previous articles have reported that polysaccharides in HCT may inhibit the proliferation of A549 lung cancer cells through cell cycle arrest and apoptosis,25 and alkaloid in HCT can inhibit H460 growth and induce apoptosis.26 Furthermore, quercetin, one of the main flavonoids in HCT, inhibits the growth of human melanoma cells27 and K562 human leukemic cells through inducing programmed cell death.29 These antitumor and anticancer effects are generally linked to inhibiting cell proliferation or inducing apoptosis. The positive findings of Houttuynia-related researches have instigated our interest in the potential antitumor effects of the volatile oil from HCT.
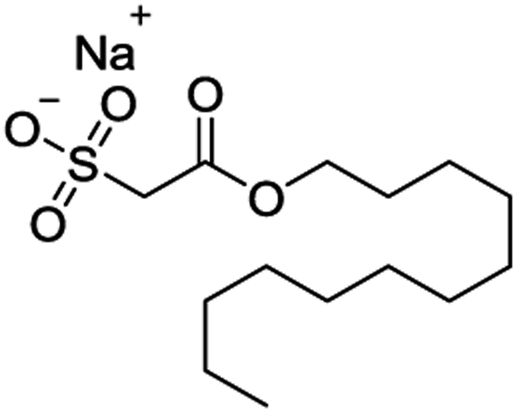
The volatile oil, the extract of HCT leaves,30 in which aldehydes (dodecanoyl acetaldehyde, lauric aldehyde) are one kind of the natural products and the major active compounds.31,32 Because of the unstable nature of aldehydes which is likely inactivated by polymerization after extracting, it must be made to a steady form by reforming the structure and keep its activity.33 Sodium new houttuyfonate (SNH, C14H27O5SNa, MW 330.41) is the addition compound of sodium bisulfite and dodecanoyl acetaldehyde of volatile oil, which has stable properties and the same effect as volatile oil of HCT. The structure of SNH is shown in Table 1. Several reports have shown antibacterial,34 antiviral35 and anti-inflammatory36,37 activities of SNH. However, in vitro and in vivo antitumor effects and the underlying mechanism of SNH have been little investigated. Thus, in this study, the effect of SNH on the apoptosis and migration pathways would be revealed. The current study estimated the apoptosis-induced and migration pathway of SNH in human cancer cells and explored the further antitumor effects in C57BL/6J tumor-bearing mice.
Table 1 Cytotoxic activities of SNH against tumor cell lines
| Compound |
Structure |
IC50a (μg mL−1) |
| HepG2 |
A2780 |
SKOV-3 |
MCF-7 |
|
IC50: concentration of the tested compound that inhibits 50% of cell growth. The IC50 was calculated by IBM SPSS Statistics software. All data are presented as means ± standard deviation of three independent experiment.
|
| SNH |
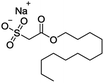
|
436.54 ± 25.89 |
354.61 ± 12.23 |
313.87 ± 19.35 |
147.59 ± 6.83 |
Results and discussion
Cytotoxicity and apoptosis assay
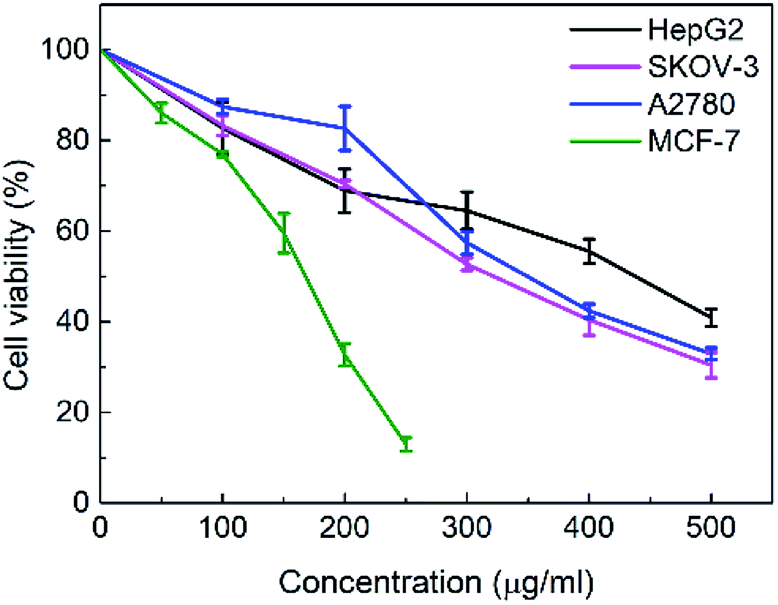
The in vitro cytotoxicities of the SNH against four human tumor cell lines HepG2 (hepatoma), MCF-7 (breast cancer), SKOV-3 (ovarian cancer) and A2780 (ovarian cancer) were evaluated by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay. The IC50 values are presented in Table 1. The results shown that SNH exhibited marked cytotoxicity against all the tested cancer cell lines, suggesting that the SNH cytotoxic effect against cancer cells lines could be broad spectrum. However, SNH is sensitive to different cancer cells (Fig. 1). Actually, SNH gave the best inhibitory results on MCF-7 cell line (IC50 = 147.59 μg mL−1). And SNH shown similar cytotoxicity to A2780 cells and SKOV-3 cells, probably because both cells are human ovarian cancer cells.
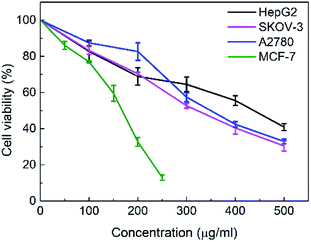 |
| | Fig. 1 Cytotoxic effect of SNH against four different human tumor cell lines after 48 h treatment at 37 °C. Cell viability percentage of hepatoma HepG2 cell line (black), breast cancer MCF-7 cell line (green), ovarian cancer SKOV-3 cell line (pink) and ovarian cancer A2780 cell line (blue) can be observed. Dada are shown as mean ± SD of 3 parallel wells. Statistically significant differences were found between the cytotoxic effects exhibited by the SNH in the HepG2 cell line at 200 μg mL−1 (*) and 300, 400, 500 μg mL−1 (**). (Independent-samples t test, *P < 0.05, **P < 0.01 versus the control group). | |
To gain insight into the cellular mechanism of SNH, the HepG2 cell line was selected for the further mechanism studies by DNA ladder, annexin V-FITC/PI staining, Hoechst 33258 staining, wound healing assay and real time RT-PCR analysis. When HepG2 cells were treated with SNH at different concentrations for 48 h, the cell changes were not obvious and inhibition rates of cell growth were significantly increased in every SNH-treated groups except 100 μg mL−1 group. Some cells in the 200 μg mL−1 SNH and 300 μg mL−1 SNH experimental groups exhibited rounding, shrinkage, and the formation of vesicles and apoptotic bodies was observed in 500 μg mL−1 SNH treated groups, which were hallmarks of apoptotic cells (Fig. 2). These changes were further examined by Hoechst 33258 staining. HepG2 cells of the untreated group were stained homogeneously, while some SNH-treated cells displayed significant changes in cell morphology, such as chromatin condensation and nuclear fragmentation. The SNH's cytotoxic effect was concentration-dependent in the 100–1000 μg mL−1 range, the IC50 being 436.54 μg mL−1. To further confirm whether SNH could induce apoptosis in HepG2 cells, DNA laddering is often used to judge if the decrease in cell viability is due to apoptosis rather than necrosis. To explore the mechanism by which SNH caused cytotoxicity in cultured HepG2 cells, we performed DNA laddering detection. As shown in (Fig. 3), the appearance of typical DNA ladder band of SNH-treated HepG2 cells revealed DNA fragmentation characteristic of apoptotic cells, suggesting that the cytotoxic effect of the SNH is concomitant with apoptotic events.
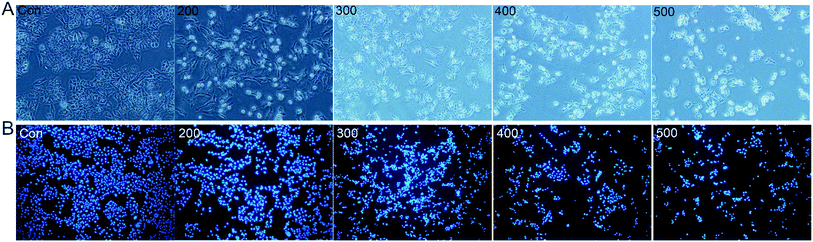 |
| | Fig. 2 SNH induces apoptosis in HepG2 cells. HepG2 cells were treated with SNH (0, 200, 300, 400 and 500 μg mL−1) for 48 h. (A) Morphological changes of HepG2 cells were observed with a phase contrast microscope (200× magnification). (B) The nuclear morphology changes of HepG2 cells were detected by Hoechst 33258 staining (100× magnification). | |
 |
| | Fig. 3 HepG2 cells were treated with different concentrations of SNH (0, 100, 200, 300, 400 and 500 μg mL−1) for 48 h, and DNA fragments was collected and 20 μL of final sample was loaded in all lanes. Electrophoresis was performed on 1.2% agarose gel. Data shown are representative of three separate experiments. | |
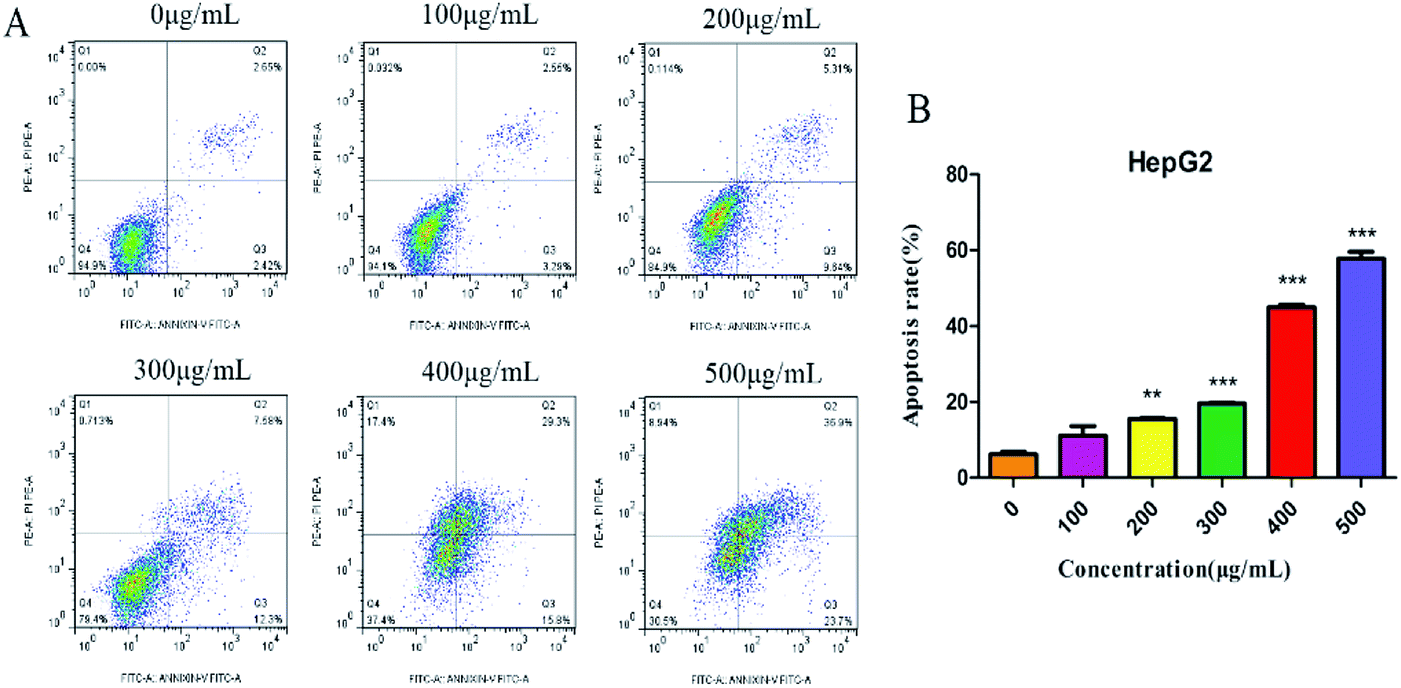
Annexin V-FITC/PI double-staining assay was further used to detect the apoptotic cells. SNH-incubated HepG2 cells were stained with annexin V (AV) and propidium iodide (PI). The apoptotic cells were quantified by flow cytometry. As shown in (Fig. 4), apoptotic cell (early apoptotic cells and late apoptotic cells) ratios increased in a dose-dependent manner from 5.84% to 60.6%. Thus, SNH could inhibit HepG2 cells growth through inducing cell apoptosis. Sun et al.38 investigated the antitumor effects of volatile oil from HCT on Ehrlich ascites cancer in mice and found that the highest inhibition rate of cancer cells mitosis was up to 45.7%. Furthermore, Wang et al.39 revealed that the volatile oil of HCT significantly inhibited the proliferation of lung cancer A549 cells. Volatile oil from HCT also significantly inhibited the proliferation and induced the apoptosis of Raji cell by blocking G1/M transformation of cells.40 Our results were consistent with these findings that SNH can promote apoptosis of cancer cells. These findings prompted us to further investigate the apoptotic process after treatment of cells with SNH.
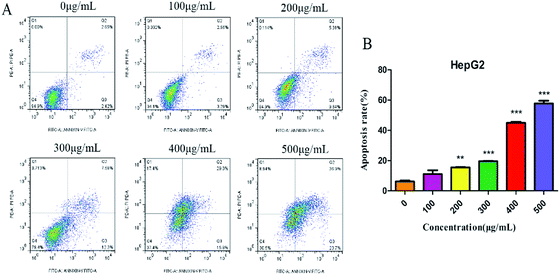 |
| | Fig. 4 HepG2 cells were treated with SNH (0, 100, 200, 300, 400 and 500 μg mL−1) for 48 h, and apoptosis was measured by flow cytometry after staining with annexin V/PI, (independent-samples t test, **P < 0.01, ***P < 0.001 versus the control group). | |
Effect of SNH on migration of HepG2 cells
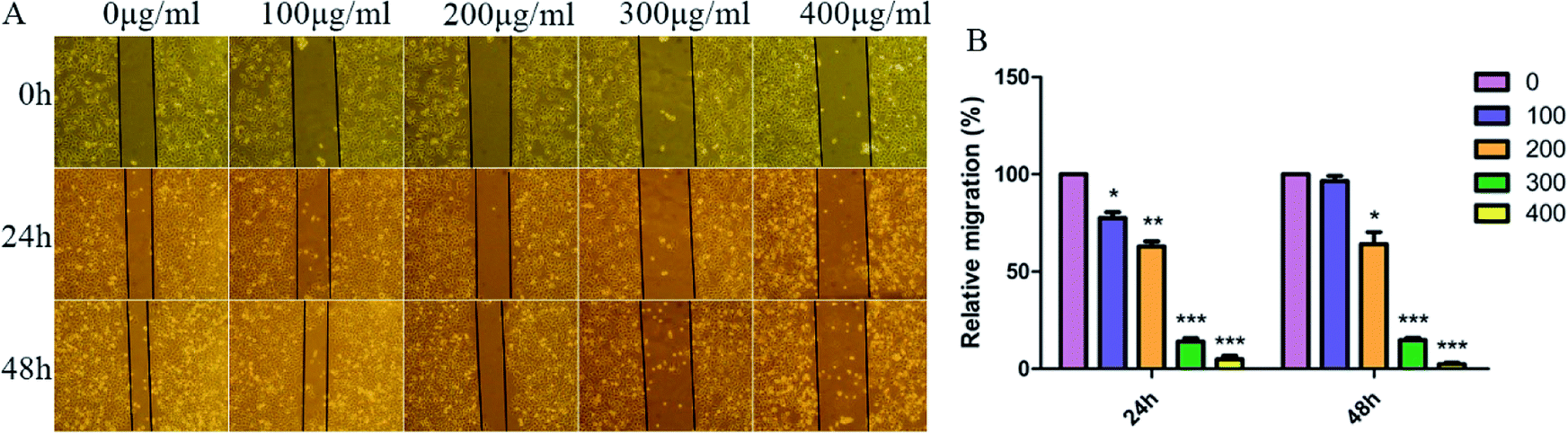
As we all know, cell–cell adhesion and interactions with extracellular matrices and basement membrane components regulate many physiological and pathological processes such as tissue development and tumor invasion.41,42 To test whether SNH plays an important role in regulation of cell migration in HepG2 cells, a wound-healing assay was performed. Confluent monolayers of HepG2 cells were scratched and treated with SNH (100, 200, 300 and 400 μg mL−1) for 48 h. Photographs were taken at different time points (0, 24, 48 h) after the trauma to observe changes in migration, and then the area of cell migration to the wound area was measured (Fig. 5A). SNH treated groups had a much slower wound healing rate compared to the control group. And SNH dose-dependently decreased the capacity of wound healing. It can be seen from (Fig. 5B) that at low concentrations, the mobility increases with the increase in time. In previous study, ethanol extract of HCT had certain inhibitory effect on HMC-1 cell movement,43 and the water extract of HCT suppressed the migration and proliferation of rat aortic smooth muscle cells.44 Our results are in agreement with those findings. Moreover, MMP9 is one of gelatinases which can degrade fibrous collagen unconditionally to move cancer cells. It not only creates a pathway for disintegrating the extracellular matrix, but also rearranges the ECM, thereby facilitating the metastasis of tumor cells.45 Therefore, MMP9 was considered prognostic factors in various types of cancer as well as potential targets to control invasive and metastatic cancers, which is agreement with the results by Agata et al. and Zoi et al.11,46
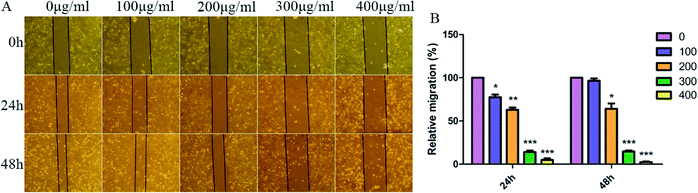 |
| | Fig. 5 SNH inhibited HepG2 cells migration in wound healing. HepG2 cells were treated with various concentrations of SNH (0, 100, 200, 300, and 400 μg mL−1) for 48 h. (A) Representative images (100× magnification) show cells migrating into wounded area in an in vitro scratch wound healing assay. (B) Quantification of the endothelial wound repair. The distance of cell migration to the wound area was measured in HepG2 cells after wounding. 0 μg mL−1 treatment was used as the standardized control for quantification. Values are means ± SD from three independent experiments. (Independent-samples t test, *P < 0.05; **P < 0.01; ***P < 0.001 versus the control group). | |
Docking assay and molecular dynamics simulation
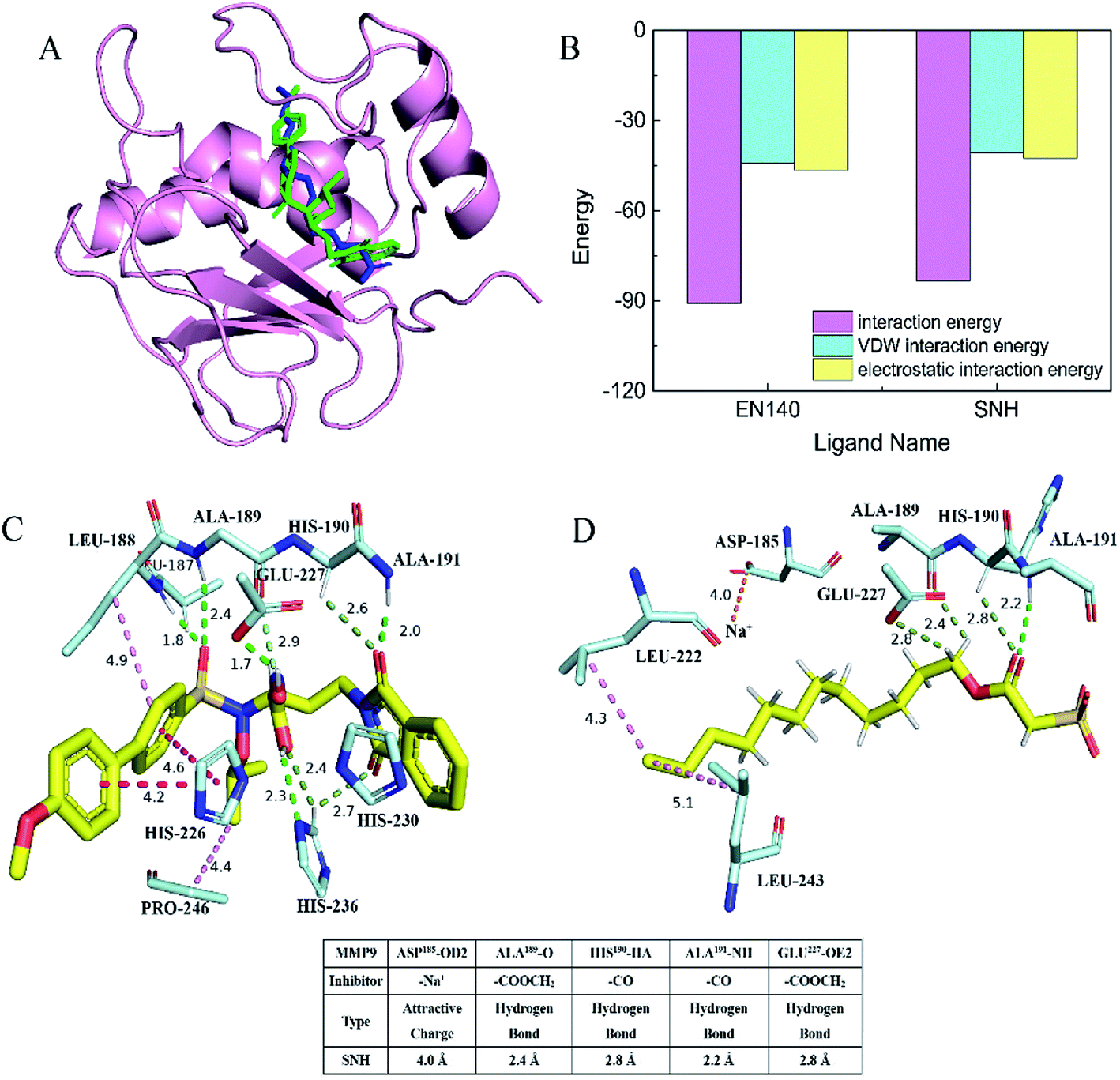
In order to determine whether SNH inhibits MMP9 protein, thereby affecting HepG2 cell migration, docking assay and molecular dynamics simulation were performed to get an insight into the binding mode of SNH into the MMP9 active site. The SNH molecule was precisely docked into active pocket of MMP9 in DS software. As shown in (Fig. 6A), the docking results highlighted that the binding orientation of the both ligands into MMP9 was almost identical.47 The interaction between the carbonyl oxygen atoms (O20) made for SNH with the protein backbone are at 2.8 and 2.2 Å from chiral hydrogen atom of His190 and the amino of Ala191. Two hydrogen bonds are formed between the backbone of the SNH and the protein main chain (backbone carbonyl oxygen atoms of Ala189, carboxyl oxygen atom of Glu227), which are short strong hydrogen bonds. Similar hydrogen bonds are formed in the original inhibitor EN140 (EN140 ligand) in complex with the MMP9. Only a single attractive charge interaction (5.0 Å) is observed between Na+ of SNH and the carbonyl oxygen atoms of Asp185. Metal ions play an important role in protein mechanisms. Then, with the aim of verifying the stability of the SNH–MMP9 interactions, the MMP9 complexes were subjected to molecular dynamic simulations. The SNH conformation with higher score was submitted to the ‘Calculate Binding Energies module’ of DS for binding energy calculation. The binding energy between SNH molecular and MMP9 was −172.7831 kcal mol−1, as shown in Table 2. Furthermore, it can be seen from (Fig. 6B), the total energies of the interaction between the EN140 ligand and the SNH ligand into MMP9 are about −90.6301 kcal mol−1 and −83.2091 kcal mol−1, respectively. Table S1† represents the numerical data of the contributions of important residues to the ligand binding. Shown in the histogram, the electrostatic interaction energy significantly larger than VDW energy, which indicates that electrostatic interaction is the main driving force for the formation of complexes.
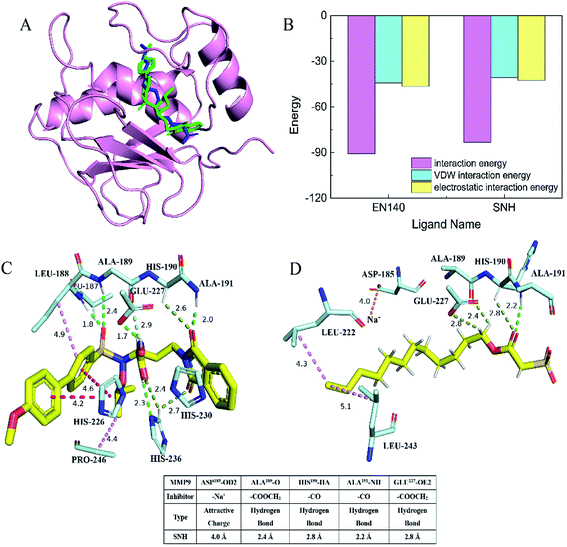 |
| | Fig. 6 Schematic representation of the crystal structures of the MMP9 catalytic domain bound to inhibitors SNH ligand and EN140 ligand. (A) Stereo diagrams of the MMP9 active site. Superposition of the SNH ligand and EN140 ligand active sites. The SNH ligand structure is coloured blue; the EN140 ligand structure is coloured green. (B) Total interaction energy statistics of SNH ligand and EN140 ligand complex surface on the MMP9. (C) A representation of the interaction bonds between the MMP9 catalytic domain and the EN140 ligand inhibitor. These results were analyzed and visualized using PyMOL (http://www.pymol.org). (D) Close-up of the MMP9 and SNH ligand complex. Four short hydrogen bonds are formed between Ala189, His190, Ala19, Glu227 and SNH ligand. Attractive charge is orange, conventional hydrogen bond is green, carbon hydrogen bond is light green and hydrophobic bond is shown in pink. | |
Table 2 Interaction and total binding energy of SNH and MMP9
| Interaction |
Donor atom |
Acceptor atom |
Chemistry bond |
Binding energy |
CDOCKER energy |
| SNH–MMP9 |
SNH:Na+ |
Asp185:OD2 |
Attractive charge |
−172.7831 kcal mol−1 |
54.9668 |
| Ala191:HN |
SNH:O20 |
Conventional hydrogen bond |
| His190:HA |
SNH:O20 |
Carbon hydrogen bond |
| SNH:H24 |
Ala189:O |
Carbon hydrogen bond |
| SNH:H25 |
Glu227:OE2 |
Carbon hydrogen bond |
| SNH:C19 |
LEU222:CG |
Alkyl |
| SNH:C19 |
LEU243:CG |
Alkyl |
From this virtual analysis, SNH is considered a highly active compound and has an inhibitory effect on MMP9 protein. The pharmacophore model used in this paper is a fast and effective tool to identify new ligand for specific target binding site. It is more reliable that the pharmacophore model uses molecular topological similarity and functional similarity of groups to analyze new ligands based on pharmacophore binding. Moreover, SNH confirmed its activity in vitro, which was verify the credibility of the docking result. Therefore, we hypothesized that SNH may inhibit the expression of MMP9 protein and thus reduce the migration of cancer cells.
Apoptosis and migration-related mRNA expression
The above results showed that SNH-induced apoptosis and migration were closely related to the mitochondrial pathway and extracellular matrix, respectively. It is well known that the Bcl-2 family is identified as an essential gene in controlling the mitochondrial pathway.48 Bcl-2, an important anti-apoptotic gene, acts on the mitochondria to change the redox state of mitochondrial mercapto, control its membrane potential and regulate the permeability of mitochondrial membrane to some apoptotic protein precursors, resulting in the release of certain cellular components, such as cytochrome c and Apaf-1 etc., which execute the apoptosis process.49 In addition, VEGF50 is a particularly interesting gene, and its decline is now recognized as a hallmark of anti-cancer. It plays important roles in neovascularization and tumor growth and metastasis of hepatocellular carcinoma.
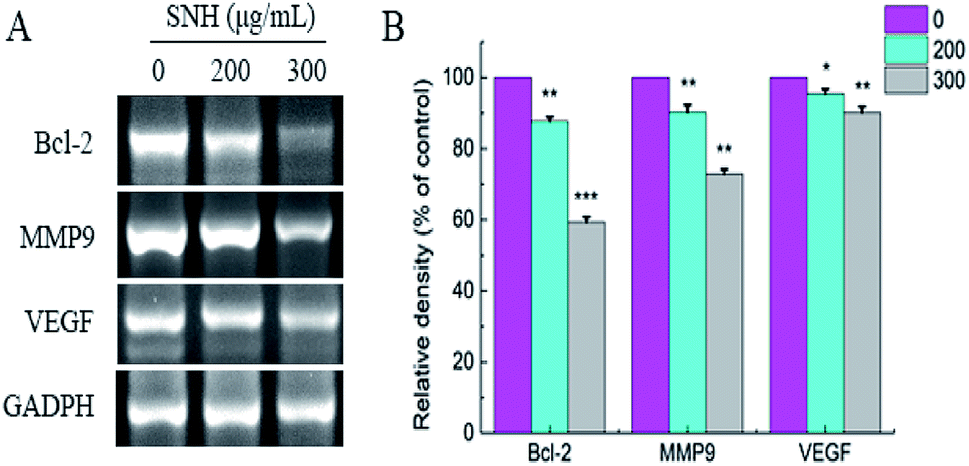
To confirm whether such a mechanism is involved in apoptosis and migration induced by SNH, the expressions of Bcl-2, VEGF, and MMP9 were determined by RT-PCR analysis. As shown in (Fig. 7), RT-PCR revealed a SNH-induced decrease in MMP9, Bcl-2 and VEGF expression levels in the HepG2 cells with a dose-dependent manner. Kim51et al. reported that HCT disturbed the expression of Bcl-2 family proteins (Bax, Bcl-2, and Bcl-xL) and induced the apoptosis in human HepG2 hepatocellular carcinoma cells. Previous study shown that the activation of NF-κB was also intensely inhibited by volatile oil of HCT33,46,52 NF-kappa B which plays a central role in tumor metastasis and angiogenesis53,54 regulated the transcription of many cytokines. It has been reported that there was a high correlation between NF-kappa B and VGF expression.55 HCC metastasis can be achieved by activating NF-κB-dependent MMP9 expression.56 Therefore, we deduced that the inhibitory effect of SNH on VEGF and MMP9 expression may occur partly through suppression of the level of NF-kappa B in HepG2 cells. Our findings indicated that SNH contributed to HepG2 cells apoptosis and metastasis by reducing the expression of MMP9, VEGF and Bcl-2.
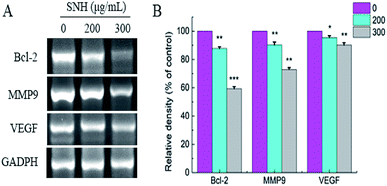 |
| | Fig. 7 Effect of SNH on the expression of apoptosis and migration-related mRNA in HepG2 cells assayed RT-PCR analysis. GADPH was employed as loading control, (independent-samples t test, *P < 0.05; **P < 0.01; ***P < 0.001 versus the control group). | |
Antitumor effects of SNH in vivo
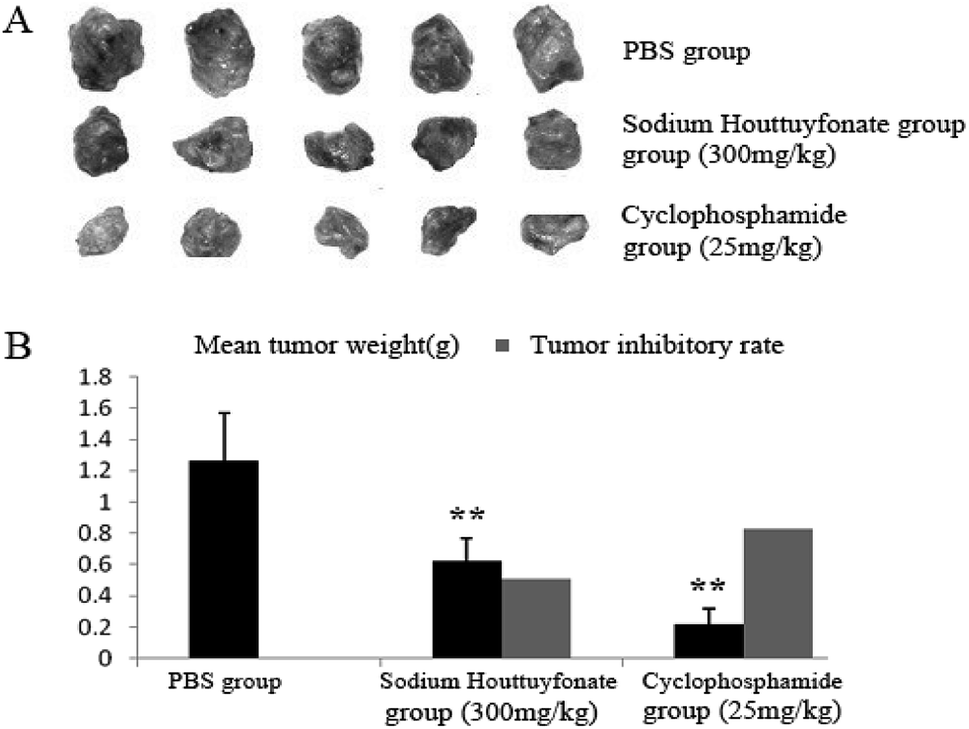
In this study, the therapeutic efficacy of SNH was evaluated via the tumor weight and tumor inhibitory rate of C57BL/6J tumor-bearing mice. SNH and cyclophosphamide could significantly inhibit the growth of tumor transplanted in mice. Compared with control group, SNH and cyclophosphamide could significantly inhibit the growth of tumor transplanted in mice. And tumor inhibitory rates of SNH group and cyclophosphamide group were 50.78% and 82.61% respectively. All the data were shown in (Fig. 8). Moreover, tumor growth was frequently accompanied by an immunosuppression regardless of tumor location and aetiology.57 Further studies is necessary to demonstrate the effect of SNH on immune organ, and the serum immunity-related cytokines in tumor-bearing mice were investigated to analyse the potential mechanism of their antitumor activity.
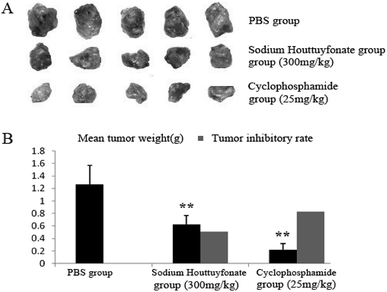 |
| | Fig. 8 Effect of SNH on tumors from HepG2 bearing mice. (A) Showed tumor tissues of each group. (B) Displayed mean tumor weights and tumor inhibitory rates. Data were presented as mean ± SD. (Independent-samples t test, **P < 0.01 versus the PBS group). | |
Experimental
Cell lines, chemicals and biochemicals
Sodium new houttuyfonate (SNH) was purchased from Xi'an Kailai Biological Engineering Co., Ltd. (Xi'an, China). We used high performance liquid chromatography to quantitatively analyze SNH with a purity of 98% (Fig. S1†). HepG2 cells were obtained from ATCC. Dulbecco's Modified Eagle Medium (DMEM) was purchased from Gibco (USA). Fetal bovine serum (FBS), MTT, penicillin and streptomycin all were purchased from Sigma Chemical Co. (St. Louis, MO, USA). Annexin V-FITC/PI apoptosis detection kit, Apoptosis-Hoechst staining kit and DNA ladder extraction kit were from Shanghai Beyotime Biological Technology Co. Ltd. (Shanghai China). The primer of GADPH, MMP9, VEGF and Bcl-2 were synthesized by Shanghai Sangon Biological Engineering Technology and Service Company. One step RT-PCR kit was bought from Qiagen (Germany). All other reagents were of the highest grade commercially available.
Cell culture and treatments
HepG2 cells were cultured in DMEM medium supplemented with 5% FBS, 100 units per mL penicillin and 0.1 mg mL−1 streptomycin in a humidified incubator at 37 °C and 5% CO2. HepG2 cells were subcultured at about 90% confluence, and fresh media were replaced every 2 days. For experiments, cells were grown under standard conditions and allowed to adhere overnight. Thereafter, HepG2 cells were incubated in DMEM supplemented with 0, 100, 200, 300, 400, 500 μg mL−1 of SNH respectively. The incubation time is 48 h for all indexes.
In vitro cytotoxic activity
Cytotoxic activities of SNH against HepG2, A2780, SKOV-3 and MCF-7 cell lines were evaluated by the MTT method. Briefly, the tumor cells from logarithmically growing cultures were seeded into 96-well plates at a concentration of 5000 cells per well. After cell adherence, the culture medium was removed and fresh medium containing various concentrations of SNH was added. They were incubated at 37 °C, 5% CO2 for 48 h. Three replicated wells were applied for per concentration. After incubation, the cells were treated with 20 μL of 5 mg mL−1 MTT and further cultured at 37 °C for 4 h. Subsequently, the old medium was removed and 100 μL of DMSO was added to each well. The absorbance of the 96-well plate was measured at 490 nm on a Microplate Reader Bio-Rad 550 after the crystals were fully dissolved.
DNA laddering detection
HepG2 cells were treated with different concentrations of SNH solutions for 48 h following to collect culture medium and adherent cells. DNA of each group was collected according to the DNA ladder kit. The DNA samples were electrophoresed on a 1.2% agarose gel containing 2 μL/50 mL GoodView nucleic acid dyes. The gel was examined and photographed by an ultraviolet gel documentation system (Bio-Rad, USA).
Hoechst 33258 staining
HepG2 cells grown in 6-well plates were treated with SNH for 48 h. After stimulating cell apoptosis, the culture medium containing compounds was removed. The cells were fixed for 10 minutes with 0.5 mL fixative, and then incubated in 1 mL nuclear fluorochrome Hoechst 33258 at room temperature for 5 minutes. Finally, an inverted fluorescence microscope was used to examine the chromatin condensation.
Annexin V-FITC/PI staining
The HepG2 cells (4 × 103 cells per well) were seeded in 96-well plates. After 24 h incubation, the culture medium was removed and the cells were treated with SNH for 48 h at 37 °C. Trypsinized cells with 1 mL trypsin–EDTA at 37 °C for 3 minutes until cells were detached completely. Then, 10% FBS–DMEM medium was added to terminate the trypsinization. The cells were transferred into an Eppendorf tube and centrifuged at 1500 rpm for 5 minutes. The supernatant was discarded washed with cold PBS and resuspended in 195 μL binding buffer, followed by addition of 5 μL FITC-labeled annexin V and 10 μL PI for 5 min at room temperature in darkness. Finally, each sample was analyzed with Flow cytometer (BD Biosciences). The gated HepG2 cells were then plotted for annexin V-FITC and PI in a 2-way dot plot to assess percentage of apoptotic cells. Apoptotic cells were defined as annexin V+/PI− and annexin V+/PI+ cells. Negative control: cells without treatment.
The wound-healing assay experiment
A monolayer scratch assay was used to observe the effect of SNH on the migratory ability of HepG2 cells. In brief, cells were counted and seeded in 6-well plates at a concentration of 3 × 105 mL−1. When the cells were grown to confluence, they were wounded by using a 10 mm diameter sterile pipette tip. The cells were gently washed three times with PBS, and then 2 mL of DMEM media containing various concentrations of SNH were added. The cells were incubated for 48 h. The wounded area was photographed at different times of 0, 24 and 48 h. The experiment was repeated 3 times. Gap area of the wound was measured using Photoshop software, and the data were normalized to the average of the control. When samples were compared with the control cells, differences were considered significant if P < 0.01.
Molecular docking
The X-ray crystal structure of complex MMP9 with an inhibitor (crystal structure of a hydroxamate based inhibitor EN140 in complex with the MMP9 catalytic domain) (PDB code: 4WZV) was downloaded from the RCSB Protein Data Bank (http://www.rcsb.org) with a resolution of 2.1 Å. Cleaning protein was prepared in ‘Prepare Protein module’. Removing the water and some other co-crystallized small molecules, the missing atoms residues, hydrogen atoms and side chains were building using the ‘Build and Edit Protein’ module. After the protein preparation, the binding site of the protein was defined based on volume occupied by the known ligand pose in an active site.58 Formation of sphere over the workspace of ligand indicates active site of the protein. The radius of the sphere was set according to the size of the SNH molecular (ligand). The original inhibitor EN140 (the structure of EN140 is shown in Fig. S2†) was removed from the complex as an active ligand. DS software was used to draw the structure of SNH, and the preparation of SNH (ligand) was performed using prepare ligands module. Potentials were assigned using CHARMm force field. Docking was performed to ensure the proper binding orientation and placement of each ligand and confirm the geometric fit of each ligand inside the active site. During the docking process, top 6 conformations were saved for SNH molecular ligand based on dock score value by CDOCKER methods.
Energy calculation
The interaction energies between the EN140 ligand and SNH ligand on MMP9 were calculated and compared by calculates the interaction energy module in simulation. The interaction energy is defined as the sum of the van der Waals and electrostatic energy. The interaction energy between sets of atoms across all conformations was calculated by using CHARMm.
To check the stability of the best hits of SNH conformation in MMP9, the binding energy of selected complexes was evaluated by the CHARMm implicit solvation models. The binding energy between a ligand (L) and a receptor (R) was calculated according to the following equation:
| Energybinding = Energycomplex − Energyligand − Energyreceptor |
Real time RT-PCR analysis
To test MMP9, VEGF and Bcl-2 expression in HepG2 cells treated with SNH, RT-PCR analysis was performed. HepG2 cells in six-cell plates treated with different concentrations of SNH for 48 h were harvested and each group included three replicates. Then, total RNA was extracted using Trizol reagent according to the manufacturer's instructions. After that, the extracted RNA was quantified by a spectrophotometer, NanoDrop 1000 (Bio-Rad, USA). RT-PCR was carried out by using one-step RT-PCR kit. Primers sequence of the studied genes was demonstrated in Table 3. After reversing transcription of mRNA at 50 °C for 30 min, thermocycling conditions were 94 °C for 2 min (polymerase activation), followed by 32 cycles of 94 °C for 10 s (denaturation), various annealing temperatures for 20 s, depending on the target gene (55 °C for GADPH; 55 °C for VEGF; 52 °C for Bcl-2, and 60 °C for MMP9), and by 72 °C for 40 s for elongation, and extending at 72 °C for 5 min. Normalization of data was performed using the housekeeping gene GADPH as an endogenous control. Real-time PCR run was done in three replicates for each sample.
Table 3 Primer sequences for reverse transcription-quantitative polymerase chain reaction
| Gene |
Forward primer (5′-3′) |
Reverse primer (5′-3′) |
| GADPH |
5′-AGAAGGCTGGGGCTCATTTG-3′ |
5′-AGGGGCCATCCACAGTCTC-3′ |
| VEGF |
5′-CGCAGCTACTGCCATCCAAT-3′ |
5′-CCCACAGGGATTTTCTTGTCTT-3′ |
| Bcl-2 |
5′-CACGCTGGGAGAACA-3′ |
5′-CTGGGAGGAGAAGATG-3′ |
| MMP9 |
5′-ACCTCGAACTTTAACAGCGACA-3′ |
5′-GATGCCATTCACGTCGTCCTTA-3′ |
HepG2 tumor-bearing mice tumor-bearing mouse model establishment and animal treatment
C57BL/6J mice (male, SPF, 7–8 W, and weighing 20 ± 2 g) were purchased from the Laboratory Animal Center of Nanjing Medical University. The mice were acclimatized in pathogen-free environment (22 ± 2 °C; 60% ± 5% humidity, and 12 h light/12 h dark cycle) for 7 days before the experiments. All experiments were carried out according to PR China legislation regarding the use and care of laboratory animals. All animals handling procedures were performed in strict accordance with the Guide for the Care and Use of Laboratory Animals (Ministry of Science and Technology of China, 2006). All experimental procedures were approved by the Ethical Committee for Laboratory Animals of Changzhou University.
The mice were given subcutaneous injection (1 × 107 HepG2 cells in 0.2 mL) at the right axillary to build up a HepG2 tumor-bearing model. After inoculation for 24 h, fifteen mice were randomly divided into three groups (n = 5) as follows: PBS group, SNH group and cyclophosphamide group. Then mice were daily gavaged with PBS or SNH (300 mg kg−1 d−1) or performed intraperitoneal injection with cyclophosphamide (25 mg kg−1 d−1) for 15 days. The dose of treatment group was chosen based on our pre-experiment, which shown no toxic effect on mice. After three days at the end of administration, all mice were sacrificed, and then the tumor tissues were dissected and weighed.
Tumor inhibitory rate was calculated according to the following formula: tumor inhibitory rate = [(average tumor weight of PBS negative control group − average tumor weight of experimental group)/average tumor weight of PBS negative control group] × 100%.
Conclusions
In this study, SNH was evaluated for antitumor activities in vitro and in vivo. SNH exhibited great cytotoxic activity against four human cancer lines. Action mechanism investigations shown that SNH could induce apoptosis by mitochondria pathway in HepG2 cells as well as inhibit the tumor growth. Furthermore, the decrease in MMP9 of MMPs family, the reduction in the expression level of VEGF and the variation of the ratio of migration in HepG2 cells suggested that metal matrix protease degradation pathway was involved in the SNH-mediated migration. Therefore, the findings demonstrated that SNH has a potential value in the treatment of cancer.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was partly supported by National Natural Science Foundation of China (Grant No. 21676029).
References
- W. Chen, R. Zheng, P. D. Baade, S. Zhang, H. Zeng, F. Bray, A. Jemal, X. Q. Yu and J. He, Ca-Cancer J. Clin., 2016, 66, 115–132 CrossRef.
- Y. T. N. Lee, J. Surg. Oncol., 2010, 17, 355–366 CrossRef.
- F. Wei, Y. Wu, L. Tang, F. Xiong, C. Guo, X. Li, M. Zhou, B. Xiang, X. Li and G. Li, Sci. China: Life Sci., 2017, 60, 117–121 Search PubMed.
- F. F. Moghadam, M. Bakhshandeh and H. Sahinbas, Journal of Cellular Immunotherapy, 2017, 3, 2–30 Search PubMed.
- C. Ogi and A. Aruga, Immunol. Lett., 2015, 164, 100–108 CrossRef CAS.
- X. J. Zhong, D. T. Li, X. L. Li, D. B. Mu and J. Y. Luo, Chin. J. Cancer, 2007, 26, 785–789 CAS.
- B. R. Lester and J. B. McCarthy, Cancer Metastasis Rev., 1992, 11, 31–44 CrossRef CAS.
- V. Azimian-Zavareh, G. Hossein, M. Ebrahimi and Z. Dehghani, Exp. Cell Res., 2018, 369, 90–104 CrossRef CAS.
- H. Li, H. Song, J. Luo, J. Liang, S. Zhao and R. Su, J. Exp. Clin. Cancer Res., 2012, 31, 39–50 CrossRef CAS.
- S. Pal, S. Moulik, A. Dutta and A. Chatterjee, Cancer Microenviron., 2014, 7, 71–78 CrossRef CAS.
- A. Jabłońska-Trypuć, M. Matejczyk and S. Rosochacki, J. Enzyme Inhib. Med. Chem., 2016, 23, 177–183 CrossRef.
- J. F. R. Kerr, A. H. Wyllie and A. R. Currie, Br. J. Cancer, 1972, 26, 239–257 CrossRef CAS.
- J. Y. Yuan and B. A. Yankner, Nature, 2000, 407, 802–809 CrossRef CAS.
- N. P. Gullett, A. R. M. R. Amin, S. Bayraktar, J. M. Pezzuto, D. M. Shin, F. R. Khuri, B. B. Aggarwal, Y. J. Surh and O. Kucuk, Semin. Oncol., 2010, 37, 258–281 CrossRef CAS.
- V. L. W. Go, D. A. Wong, M. S. Resnick and D. Heber, J. Nutr., 2001, 131, 179S–180S CrossRef CAS.
- J. M. Pezzuto, Biochem. Pharmacol., 1997, 53, 121–133 CrossRef CAS.
- M. Younas, C. Hano, N. Giglioli-Guivarc'H and B. H. Abbasi, RSC Adv., 2018, 8, 29714–29744 RSC.
- R. X. Hu, B. M. Xiao, Z. J. Tan, W. N. Zhao, B. Y. Xie, D. P. Xie and C. R. Wu, China Pharm., 2008, 17, 23–25 CAS.
- M. C. Lin, P. C. Hsu and M. C. Yin, Food Funct., 2013, 4, 322–327 RSC.
- L. C. Wang, T. M. Pan and T. Y. Tsai, J. Food Drug Anal., 2018, 26, 973–984 CrossRef CAS.
- J. Li and F. T. Zhao, Med. Recapitulate, 2014, 20, 306–307 Search PubMed.
- W. Li, P. Zhou, Y. Zhang and L. He, J. Ethnopharmacol., 2011, 133, 922–927 CrossRef.
- X. Q. Chen, Stud. Trace Elem. Health, 2007, 24, 17–18 CAS.
- K. Chiow, M. Phoon, T. Putti, B. K. Tan and V. T. Chow, Asian Pac. J. Trop. Med., 2016, 9, 1–7 CrossRef CAS.
- H. Kun, J. Can, C. Huanjun, W. Peipei, Y. Mei and D. Kan, Int. J. Biol. Macromol., 2018, 120, 228–296 Search PubMed.
- X. Xue, W. U. Huazhen, F. U. Tengfei, Y. Lan, Z. Luo, H. Zhou, J. Zhao and J. Meng, Mod. J. Integr. Tradit. Chin. West. Med., 2016, 25, 2972–2978 Search PubMed.
- M. Yanarojana, T. Nararatwanchai, S. Thairat and S. Tancharoen, Anticancer Res., 2017, 37, 6619–6628 CAS.
- K. C. Lai, Y. J. Chiu, Y. J. Tang, K. L. Lin, J. H. Chiang, Y. L. Jiang, H. F. Jen, Y. H. Kuo, S. Agamaya and J. G. Chung, Anticancer Res., 2010, 30, 3549–3556 Search PubMed.
- C. Wangchauy and S. Chanprasert, J. Chem. Pharm. Res., 2012, 4, 2590–2598 Search PubMed.
- D. Li, J. P. Liu, X. Han, Y. F. Wang, C. H. Wang, Z. Li and G. C. Wang, Chem. Nat. Compd., 2017, 53, 365–367 CrossRef CAS.
- J. M. Kang, I. H. Cha, Y. K. Lee and H. S. Ryu, J. Korean Soc. Food Sci. Nutr., 1997, 26, 214–221 CAS.
- Y. X. Wu, Heilongjiang Academy of Agricultural Sciences, 2014, 4, 105–106 Search PubMed.
- P. Pan, Y. J. Wang, L. Han, X. Liu, M. Zhao and Y. F. Yuan, J. Ethnopharmacol., 2010, 131, 203–209 CrossRef CAS.
- H. Lu, X. Wu, Y. Liang and J. Zhang, Chem. Pharm. Bull., 2006, 54, 936–940 CrossRef CAS.
- J. M. Pang, W. J. Dong, Y. H. Li, X. J. Xia, Z. H. Liu, H. Z. Hao, L. M. Jiang and Y. L. Liu, Molecules, 2017, 22, 1697–1704 CrossRef.
- J. Chen, W. Wang, C. Shi, X. Hou, J. Wan and J. Fang, Herb. Med., 2014, 1283–1288 CAS.
- W. Li, T. Fan, Y. Zhang, T. Fan, Z. Ping, X. Niu and L. He, Phytother. Res., 2013, 27, 1629–1639 CrossRef.
- M. M. Sun, J. Today Health, 2015, 14, 3 Search PubMed.
-
Y. F. Wang, Master's degree thesis, Zhengzhou University, 2016.
- Z. Zhang, N. Zhao, Y. Wang, M. Zou, Z. Zhao, X. Zhang and G. Wang, J. Zhengzhou Univ., 2016, 51, 474–477 Search PubMed.
- J. Behrens, U. Frixen, J. Schipper, M. Weidner and W. Birchmeier, Semin. Cell Biol., 1992, 3, 169–178 CrossRef CAS.
- F. Hyafil, C. Babinet and F. Jacob, Cell, 1981, 26, 447–454 CrossRef CAS.
- I. S. Kim, J. H. Kim, S. K. Jin, C. Y. Yun, D. H. Kim and J. S. Lee, J. Ethnopharmacol., 2007, 112, 90–95 CrossRef.
- J. H. Han, S. N. Park, M. S. Yoon and O. B. Choi, Korean J. Pharmacogn., 2011, 42, 182–186 Search PubMed.
- J. J. Yang, Z. M. Mao, Y. G. Wan, W. Wu, Y. R. Huang, G. Shi, W. B. Han and J. Yao, China J. Chin. Mater. Med., 2016, 41, 3805–3813 Search PubMed.
- Z. Piperigkou, D. Manou, K. Karamanou and A. D. Theocharis, Methods Mol. Biol., 2018, 1731, 325–348 CrossRef CAS.
- N. Elisa, C. Anna Rita, G. Cristina, B. Antonino, B. Barbara, C. Caterina, T. Tiziano, V. Laura, O. Elisabetta and N. Susanna, J. Med. Chem., 2016, 58, 7224–7240 Search PubMed.
- T. Moldoveanu, A. V. Follis, R. W. Kriwacki and D. R. Green, Trends Biochem. Sci., 2014, 39, 101–111 CrossRef CAS.
- J. Henry-Mowatt, C. Dive, J.-C. Martinou and D. James, Oncogene, 2004, 23, 2850–2860 CrossRef CAS.
- D. Abramovich, F. Parborell and M. Tesone, Biol. Reprod., 2006, 75, 434–441 CrossRef CAS.
- J. M. Kim, I. H. Hwang, I. S. Jang, K. Min, I. S. Bang, S. J. Park, Y. J. Chung, J. C. Joo and M. G. Lee, Integr. Cancer Ther., 2017, 16, 360–372 CrossRef.
- W. Wang, X. Hu, P. Shen, N. Zhang and Y. Fu, Microb. Pathog., 2017, 107, 12–16 CrossRef CAS.
- N. Yamanaka, N. Sasaki, A. Tasaki, H. Nakashima, M. Kubo, T. Morisaki, H. Noshiro, T. Yao, M. Tsuneyoshi and M. Tanaka, Anticancer Res., 2004, 24, 1071–1075 Search PubMed.
- Q. Zhu, X. Xu, X. Liu, J. Lin, Y. Kan, Y. Zhong, F. Liu and J. Xu, Res. Vet. Sci., 2015, 100, 245–251 CrossRef CAS.
- S. Huang, J. B. Robinson, A. Deguzman, C. D. Bucana and I. J. Fidler, Cancer Res., 2000, 60, 5334–5339 CAS.
- J. Zhang, D. L. Zhang, X. L. Jiao and Q. Dong, Eur. Rev. Med. Pharmacol. Sci., 2013, 17, 2372–2382 CAS.
- L. Zhang, X. Li, X. Xu and F. Zeng, Carbohydr. Res., 2005, 340, 1515–1521 CrossRef CAS.
- Y. Fu, Y. N. Sun, K. H. Yi, M. Q. Li, H. F. Cao, J. Z. Li and F. Ye, Molecules, 2017, 22, 959–971 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra06024c |
| ‡ Equivalent authors. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
Weiwei
Ji‡
a,
Hui
Zhong
a,
Luyao
Wang
a,
Xiaolin
Zhu
ab and
Jie
Zhu
ab
*ab,
Weiwei
Ji‡
a,
Hui
Zhong
a,
Luyao
Wang
a,
Xiaolin
Zhu
ab and
Jie
Zhu
ab