
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly selective synthesis of D-amino acids from readily available L-amino acids by a one-pot biocatalytic stereoinversion cascade†
Danping Zhanga,
Xiaoran Jinga,
Wenli Zhanga,
Yao Nie *ac and
Yan Xu*ab
*ac and
Yan Xu*ab
aSchool of Biotechnology, Key Laboratory of Industrial Biotechnology, Ministry of Education, Jiangnan University, Wuxi 214122, China. E-mail: ynie@jiangnan.edu.cn; yxu@jiangnan.edu.cn
bState Key Laboratory of Food Science and Technology, Jiangnan University, Wuxi 214122, China
cSuqian Industrial Technology Research Institute of Jiangnan University, Suqian 223814, China
First published on 23rd September 2019
Abstract
D-Amino acids are key intermediates required for the synthesis of important pharmaceuticals. However, establishing a universal enzymatic method for the general synthesis of D-amino acids from cheap and readily available precursors with few by-products is challenging. In this study, we constructed and optimized a cascade enzymatic route involving L-amino acid deaminase and D-amino acid dehydrogenase for the biocatalytic stereoinversions of L-amino acids into D-amino acids. Using L-phenylalanine (L-Phe) as a model substrate, this artificial biocatalytic cascade stereoinversion route first deaminates L-Phe to phenylpyruvic acid (PPA) through catalysis involving recombinant Escherichia coli cells that express L-amino acid deaminase from Proteus mirabilis (PmLAAD), followed by stereoselective reductive amination with recombinant meso-diaminopimelate dehydrogenase from Symbiobacterium thermophilum (StDAPDH) to produce D-phenylalanine (D-Phe). By incorporating a formate dehydrogenase-based NADPH-recycling system, D-Phe was obtained in quantitative yield with an enantiomeric excess greater than 99%. In addition, the cascade reaction system was also used to stereoinvert a variety of aromatic and aliphatic L-amino acids to the corresponding D-amino acids by combining the PmLAAD whole-cell biocatalyst with the StDAPDH variant. Hence, this method represents a concise and efficient route for the asymmetric synthesis of D-amino acids from the corresponding L-amino acids.
Introduction
D-Amino acids, as chiral directing auxiliaries and chiral synthons in organic synthesis, play important roles in the production of pharmaceuticals and fine chemicals.1,2 Current important applications of D-amino acids include their use as key components in β-lactam antibiotics, fertility drugs, anticoagulants, and pesticides.1,3–5 D-Arylalanines, such as D-phenylalanine (D-Phe), are useful intermediates for the production of pharmaceuticals, including β-lactam antibiotics, small peptide hormones, and pesticides.4 The production of D-Phe is of great interest since pharmaceuticals that include analgesics, antistress agents, antidiabetics (e.g., nateglinide), and anticoagulants are synthesized from D-arylalanines.6–8Many methods for the synthesis of D-amino acids and their derivatives have been developed. In general, two fundamentally different routes exist, namely those that involve chemical methods and those that are biocatalytic in nature.9–12 Chemical methods generally synthesize D-amino acids through the chiral resolution of racemic D,L-amino acids or by asymmetric protocols from chiral or prochiral starting materials. High costs and low yields due to D-amino acid racemization are the major disadvantages of chemical methods.10,12 However, the development of biocatalysis methods has significantly progressed in the past decade. With enzymes as biocatalysts, D-amino acids can be produced under mild reaction conditions with high enantioselectivities, conversions, and space-time yields.12,13 Enzymatic approaches for the synthesis of D-amino acids can be divided into the following five types: (1) processes involving D-hydantoinase and D-carbamoylase,4,14 (2) the D-amino acid aminotransferase-promoted transfer of the amino group of a D-amino acid to an α-keto acid,15 (3) hydrolysis of an N-acyl-D-amino acid by N-acyl-D-amino acid amidohydrolase,16,17 (4) kinetic resolution of a racemic mixture by L-amino acid oxidase,4 and (5) the asymmetric reductive amination of an α-keto acid by D-amino acid dehydrogenase.4,12 Liu et al. used D-carbamoylase from Arthrobacter crystallopoietes coupled with D-hydantoinase from Agrobacterium tumefaciens for the enantioselective resolution of L-indolylmethylhydantoin into D-tryptophan in 99.4% yield and greater than 99% enantiomeric excess (ee).14 D-Amino acid aminotransferase from Bacillus subtilis WB600 was used to produce D-Phe at a final product concentration of 1.72 g L−1,18 while Isobe et al. used L-amino acid oxidase from Rhodococcus sp. AIU Z-35-1 to produce D-citrulline, D-glutamine, D-homoserine, and D-arginine.19 Although these methods were used to synthesize a variety of D-amino acids, they usually require specific substrates that are generally expensive and commercially unavailable.1,4,20
Cascade biocatalysis has been developed in recent years as a promising method for the efficient synthesis of pharmaceutical intermediates and fine chemicals.21–24 Cascade biocatalysis comprises multiple biocatalytic reactions in a single reaction vessel without the isolation of any intermediate, and starting from cheap and available substrates.21,23,25 Because L-amino acids are mostly available through fermentation from inexpensive and renewable natural sources, multi-enzyme cascades for the biocatalytic stereoinversions of L-amino acids into D-amino acids represent economically effective and environmentally benign methods.8,26–28
Herein, based on the reaction route involving L-amino acid deaminase and D-amino acid dehydrogenase,8 we constructed and optimized the biocatalytic cascade route for the synthesis of D-amino acids. The reaction route is assembled using two modules, namely an oxidative deamination module and a reductive amination module. L-Amino acid deaminase from Proteus mirabilis (PmLAAD) and meso-diaminopimelate dehydrogenases (DAPDHs) from various microorganisms were cloned and expressed separately in recombinant Escherichia coli BL21(DE3) and used in the biocatalytic cascade route. After characterization of the enzymes involved, recombinant E. coli cells that express PmLAAD and a suitable DAPDH were used in the cascade route for the synthesis of D-amino acids from L-amino acids (Scheme 1). As a model reaction, this cascade route efficiently and completely transformed L-phenylalanine (L-Phe) into D-Phe with high optical purity. In particular, the artificial biocatalytic cascade also transforms a variety of aromatic and aliphatic L-amino acids into the corresponding D-amino acids.
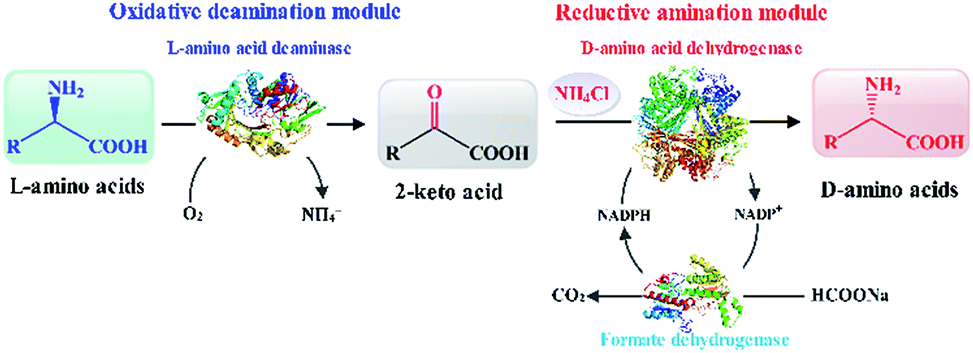 | ||
| Scheme 1 Depicting the one-pot biocatalytic cascade route for the synthesis of enantiomerically pure D-amino acids by L-amino-acid stereoinversion. | ||
Results and discussion
Constructing biocatalytic reaction route for stereoinversion of L-amino acids
As shown in Scheme 1, an L-amino acid is first converted into the corresponding 2-keto acid in the oxidative deamination module, after which the 2-keto acid is asymmetrically converted into the D-amino acid in the reductive amination module. This stereoinverting biocatalysis-based cascade reaction route was designed with the following basic principles in mind: (1) the reaction conditions in both reaction modules need to be compatible;21,29 (2) the reductive amination module must contain only irreversible reactions in order to direct the balance of the entire reaction and accumulate the final product; and (3) the 2-keto acid intermediate must not accumulate and no other intermediates can be generated.30 According to the above principles, L-amino acid deaminase (LAAD), which catalyzes the stereospecific oxidative deamination of L-amino acids to the corresponding 2-keto acids and ammonia, is better suited for the construction of the oxidative deamination module, compared to L-amino acid oxidase or L-amino acid dehydrogenase.31,32 D-Amino acid dehydrogenase (DAADH) is the desired biocatalyst for use in the reductive amination module, since it directly catalyzes the asymmetric reductive amination of an α-keto acid to the corresponding D-amino acid.4,10 Although LAAD and DAADH can potentially to be used to construct our artificial multi-enzyme cascade, coordinating these two enzyme-catalyzed reactions involving necessary cofactor-recycling system is challenging.12,21,33–35Oxidative deamination of L-Phe to phenylpyruvic acid
In order to construct the catalytic multi-enzyme cascade system, we used L-amino acid deaminase from Proteus mirabilis (PmLAAD) in the oxidative deamination module owing to its high activity toward L-Phe.36 During the oxidative deamination reaction, PmLAAD catalyzes the deamination of L-Phe to the corresponding α-imino acid with oxygen as the co-substrate, after which spontaneous hydrolysis produces phenylpyruvic acid (PPA) (Scheme 2).37–39 Because PmLAAD is a membrane-bound protein,40 different forms of the biocatalyst were prepared in order to investigate their effects on the yield of PPA from L-Phe. As shown in Table S1,† the yield of PPA was higher when the biocatalyst was in whole-cell form compared to other biocatalyst types. Therefore, the whole-cell biocatalyst was adopted as the PmLAAD that catalyzes the oxidative deamination reaction.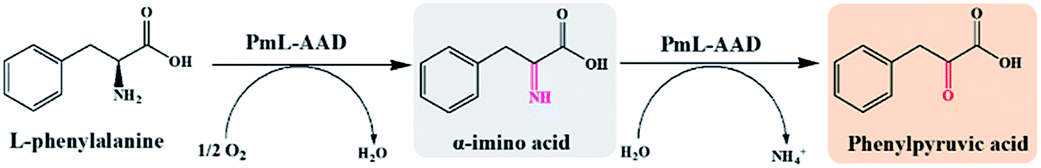 | ||
| Scheme 2 PmLAAD-catalyzed oxidative deamination of L-Phe. | ||
In addition, the reaction conditions for the catalytic PmLAAD whole-cell oxidative deamination were optimized. As shown in Fig. S1,† the PmLAAD whole-cell biocatalyst was more active in Tris–HCl buffer (50 mM, pH 9.0) at 45 °C and the optimum biocatalyst concentration was found to be 80 mg mL−1. Under the optimized reaction conditions, the catalytic activities of PmLAAD whole cells toward L-Phe, D-Phe, and PPA, were further evaluated, with the concentrations of L-Phe, D-Phe, and PPA determined for each reaction (Fig. 1). Only PPA was produced after 30 min when 50 mM L-Phe was used as the substrate. On the other hand, the concentration of D-Phe was unchanged and no PPA or L-Phe was detected during the reaction process when D-Phe was used as the substrate. In a similar manner, PPA was also not a substrate in this system. Clearly, the PmLAAD whole-cell biocatalyst was only active towards L-Phe and the expected PPA was the only product formed in the oxidative-deamination reaction process, which is consistent with requirements of the designed stereoinverting cascade reaction route.
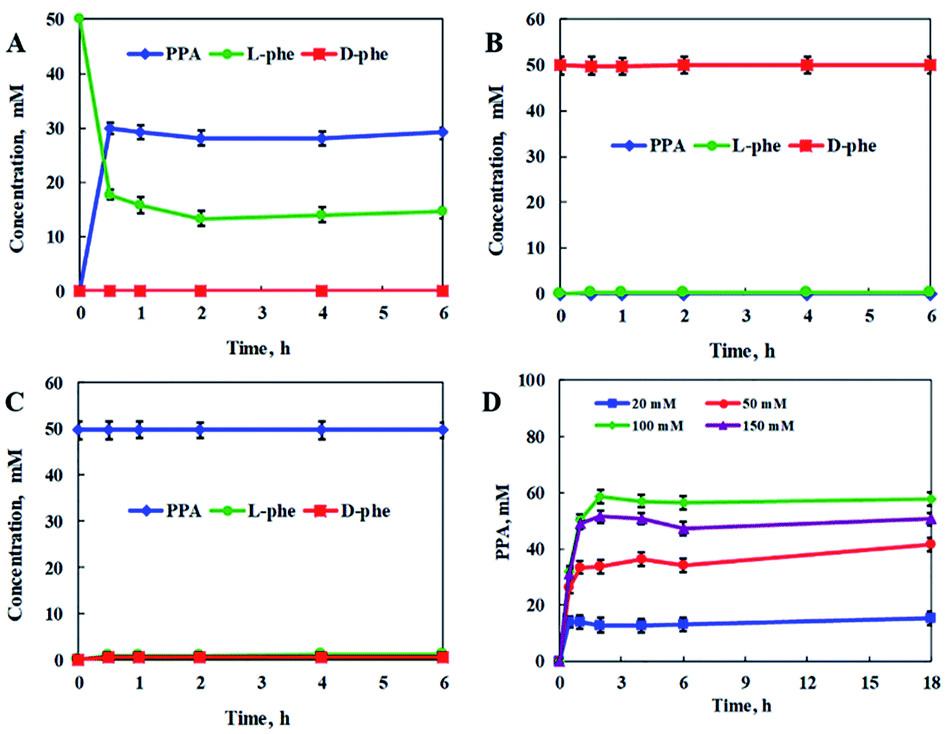 | ||
| Fig. 1 Catalytic PmLAAD whole-cell oxidative deamination processes with: (A) L-Phe, (B) D-Phe, and (C) PPA as substrates. (D) PPA formation at different concentrations of L-Phe. Reactions were carried out in Tris–HCl buffer (50 mM, pH 9.0) at 45 °C and a PmLAAD whole-cell concentration of 80 mg mL−1. The values were averaged from triplicate measurements and the standard deviations are indicated as error bars. | ||
We also investigated the effect of substrate concentration on the catalytic PmLAAD whole-cell oxidative deamination process. L-Phe was rapidly converted in the early stages of the reaction, after which its concentration plateaued. A PPA yield of 58.7% was obtained at an L-Phe concentration of 100 mM, while higher concentrations of L-Phe led to lower PPA yields. Clearly, PmLAAD whole cells mainly catalyze the oxidative deamination of L-Phe to PPA, which is favorable for use in the biocatalytic stereoinverting cascade system.
Reductive amination of PPA to D-Phe
Meso-diaminopimelate dehydrogenases (DAPDHs) that belong to the D-amino acid dehydrogenase family are candidates for the catalytic conversion of a 2-keto acid to the corresponding D-amino acid.4,41,42 DAPDHs catalyze the reductive amination of 2-keto acids using NADPH as the electron donor (Scheme 3). In this work, DAPDH from Ureibacillus thermosphaericus (UtDAPDH), DAPDH from Lysinibacillus fusiformis (LfDAPDH), and DAPDH from Symbiobacterium thermophilum (StDAPDH) and its H227V variant bearing a mutation at the substrate-binding pocket (StDAPDH/H227V) were employed to catalyze reductive amination.7,43,44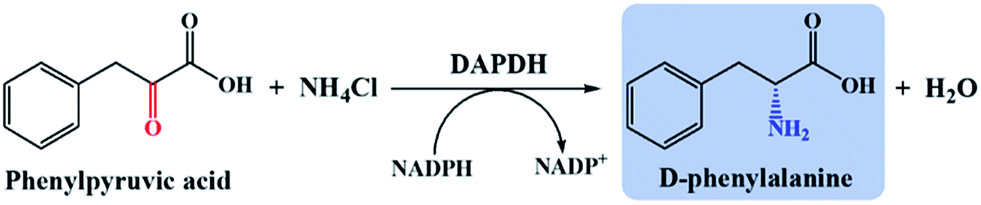 | ||
| Scheme 3 DAPDH-catalyzed phenylpyruvic acid reductive amination. | ||
As summarized in Table 1, the DAPDHs examined in this study were all active toward PPA, with the activity of StDAPDH/H227V (specific activity of 0.46 μmol min−1 mg−1) the highest among the four DAPDHs. Michaelis–Menten kinetic data for the DAPDHs towards PPA were also determined; Km and kcat values were calculated by non-linear fitting and are also summarized in Table 1. Although the affinities of StDAPDH and StDAPDH/H227V toward PPA as the substrate were similar, with comparable Km values, StDAPDH/H227V exhibited the highest reaction rate (kcat = 1.13 s−1) among the DAPDHs studied, which led to StDAPDH/H227V exhibiting a higher catalytic efficiency compared to the other DAPDHs. The variation at H227 in StDAPDH enlarges the substrate binding pocket allowing bulky substrates, such as PPA, easier access to the enzyme active site.7 Therefore, StDAPDH/H227V was selected for the construction of the reductive amination module.
| Enzyme | Specific activity (μmol min−1 mg−1) | Km (mM) | kcat (s−1) | kcat/Km (s−1 mM−1) |
|---|---|---|---|---|
| a Kinetic data were acquired in 50 mM Na2CO3/NaHCO3 buffer (pH 9.0) at 30 °C at NH4Cl and NADPH concentrations of 200 mM and 0.5 mM, respectively. The concentration of PPA varied between 2 and 40 mM. | ||||
| StDAPDH/H227V | 0.460 ± 0.030 | 12.48 ± 0.23 | 1.13 ± 0.06 | 0.091 ± 0.003 |
| StDAPDH | 0.014 ± 0.002 | 15.21 ± 0.31 | 0.13 ± 0.03 | 0.009 ± 0.002 |
| UtDAPDH | 0.042 ± 0.004 | 3.07 ± 0.18 | 0.18 ± 0.02 | 0.059 ± 0.006 |
| LfDAPDH | 0.035 ± 0.001 | 7.61 ± 0.14 | 0.14 ± 0.05 | 0.018 ± 0.004 |
To further confirm the feasibility of the stereoinverting multi-enzyme cascade constructed with the above-mentioned reaction route, the catalytic activities of StDAPDH/H227V toward PPA, L-Phe, and D-Phe were further evaluated, with the concentrations of PPA, L-Phe, and D-Phe determined in each case (Fig. 2). With PPA as the substrate, D-Phe was produced with an optical purity (ee) greater than 99% and a quantitative yield after 4 h. However, no reaction was observed with L-Phe or D-Phe as the substrate, nor was PPA produced, which indicates that the target product (D-Phe) cannot be converted into PPA, the reaction intermediate in the overall reaction process, and the reaction equilibrium in the cascade reaction system is directed toward the production of D-Phe.
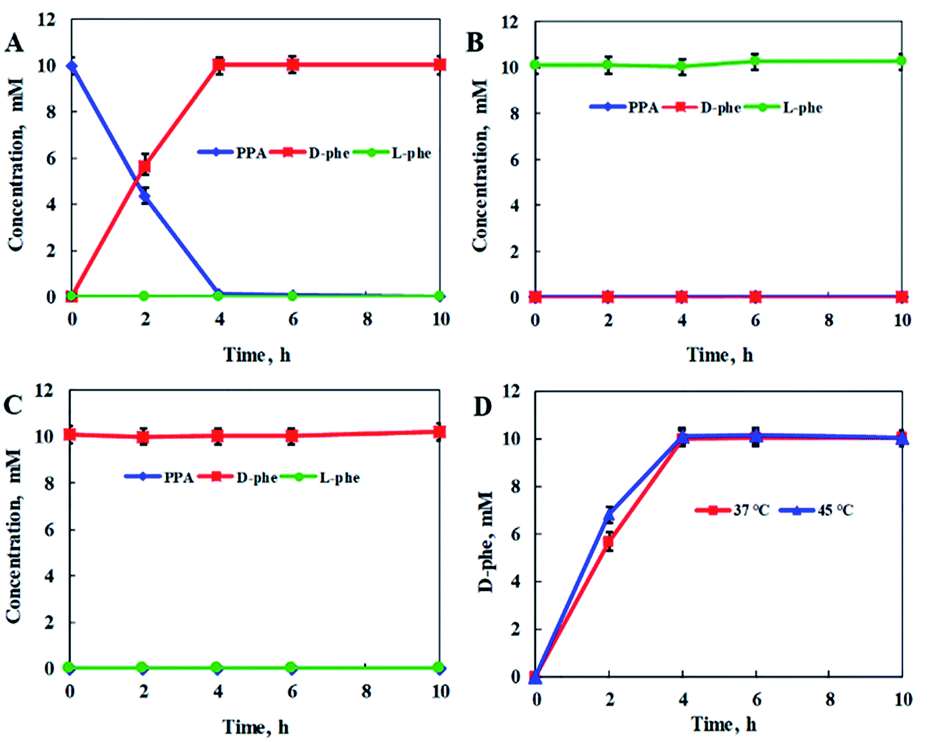 | ||
| Fig. 2 DAPDH-catalyzed reductive aminations with: (A) PPA, (B) L-Phe, (C) D-Phe as substrates. (D) D-Phe formation under different reaction temperatures. The reaction mixture contained StDAPDH/H227V (4 mg mL−1), NADPH (20 mM), NH4Cl (30 mM), and Tris–HCl buffer (50 mM, pH 9.0). Unless otherwise noted, the reaction temperature was 37 °C for the StDAPDH/H227V-catalyzed reactions. The values were averaged from triplicate measurements and the standard deviations are indicated as error bars. | ||
Considering that the optimum reaction temperature of the PmLAAD whole-cell biocatalyst is 45 °C, the influence of reaction temperature on the catalytic activity of StDAPDH/H227V was investigated in order to coordinate the reaction conditions of the two biocatalysts. Fig. 2D reveals that little difference in the StDAPDH/H227V-catalyzed reaction efficiency was observed at 45 °C compared to that at 37 °C, providing D-Phe in a quantitative yield after 4 h. Therefore, the optimum reaction temperature in the stereoinverting biocatalytic linear cascade was determined to be 45 °C for both the whole-cell oxidative deamination catalyzed by PmLAAD and the StDAPDH/H227V-catalyzed reductive amination.
Assembling the L-Phe to D-Phe stereoinverting cascade system
After analyzing the catalytic characteristics of PmLAAD whole-cells and StDAPDH/H227V in each reaction step, the two biocatalysts were used to construct the stereoinverting cascade setup. The theoretical ratio of the PmLAAD whole-cell biocatalyst to StDAPDH/H227V in the cascade-reaction system should be 1 U![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 U. Because the activities of the PmLAAD whole-cell biocatalyst and StDAPDH/H227V were 0.01 and 0.46 μmol min−1 mg−1, respectively, the theoretical concentration of the PmLAAD whole-cell biocatalyst and StDAPDH/H227V were required to be 100 and 2 mg mL−1, respectively. However, considering that the reductive amination module is critical for the production of D-Phe, the concentration of StDAPDH/H227V in the cascade reaction system was increased to 4 mg mL−1 to further improve the efficiency of the reductive amination module.
:1 U. Because the activities of the PmLAAD whole-cell biocatalyst and StDAPDH/H227V were 0.01 and 0.46 μmol min−1 mg−1, respectively, the theoretical concentration of the PmLAAD whole-cell biocatalyst and StDAPDH/H227V were required to be 100 and 2 mg mL−1, respectively. However, considering that the reductive amination module is critical for the production of D-Phe, the concentration of StDAPDH/H227V in the cascade reaction system was increased to 4 mg mL−1 to further improve the efficiency of the reductive amination module.
Due to the NADPH dependence of DAPDH catalyzing the reductive amination reaction, meanwhile, the cofactor-recycling system involving Burkholderia stabilis formate dehydrogenase (BsFDH) was applied to couple the StDAPDH/H227V-catalyzed reductive amination for further enhancement of efficiency of the one-pot biocatalytic stereoinverting cascade. The BsFDH exhibited the specific activity of 2.88 U mg−1 in Tris–HCl buffer (50 mM, pH 9.0) at 45 °C (Fig. S2†). By using BsFDH for NADPH regeneration, the yield of D-Phe was maintained at the same level as that with excessive loading of NADPH for the reductive amination module (10 mM PPA), even when only 1 mM NADP+ was added to initiate the cofactor regeneration (Fig. S3†). Then the PmLAAD whole-cell biocatalyst (100 mg mL−1), StDAPDH/H227V (4 mg mL−1), and BsFDH (0.35 mg mL−1, 1 U) were assembled to construct the one-pot stereoinverting cascade. Subsequently, the D-Phe was obtained in a yield of 76.2% by the addition of 30 mM L-Phe, 90 mM NH4Cl, 60 mM sodium formate, and 3 mM NADP+ to the 50 mM Tris–HCl (pH 9.0) reaction buffer during the cascade reaction at 45 °C (Fig. S4†).
The concentrations of the PmLAAD whole-cell biocatalyst, StDAPDH/H227V, and BsFDH in the cascade reaction system were next optimized in order to further reduce the amount of residual L-Phe substrate in the reaction system, which would lower the optical purity of the final D-Phe product. As shown in Table 2, when 40 mg mL−1 of the PmLAAD whole cells, 4.5–12 mg mL−1 StDAPDH/H227V, and 0.35–0.7 mg mL−1 BsFDH were added to the reaction system, 30 mM D-Phe was produced entirely from the L-Phe substrate and no PPA intermediate was accumulated. To a certain extent, increasing the amount of the catalyst for reductive amination and reducing the amount of the catalyst for oxidative deamination enhanced the catalytic efficiency of the one-pot stereoinverting cascade, leading to high yield and optical purity of the D-Phe. Consistent with the observation of PmLAAD whole-cell oxidative deamination process towards L-Phe, rapid accumulation of PPA in early stage of the reaction would inhibit continuous conversion of L-Phe, and the reductive amination reaction would be the rate-limiting step of the cascade process. Consequently, by optimizing the concentrations of the biocatalysts, enantiomerically pure D-Phe (>99% ee) was produced in quantitative yield with 40 mg mL−1 PmLAAD whole cells, 4.5 mg mL−1 StDAPDH/H227V, and 0.35 mg mL−1 BsFDH.
| PmLAAD (mg mL−1) | StDAPDH/H227V (mg mL−1) | BsFDH (mg mL−1) | L-Phe (mM) | D-Phe (mM) | Optical purity of D-Pheb (% ee) |
|---|---|---|---|---|---|
| a The reaction mixture consisted of 30 mM L-Phe, 3 mM NADP+, 90 mM NH4Cl, 60 mM sodium formate, and 50 mM Tris–HCl buffer (pH 9.0). The reactions were carried out at 45 °C and 220 rpm for 24 h.b Optical purity of D-Phe (% ee) was determined by HPLC after FDAA derivatization. | |||||
| 40 | 4.5 | 0.35 | 0 | 30.0 | >99 |
| 40 | 9 | 0.7 | 0 | 30.0 | >99 |
| 40 | 12 | 0.7 | 0 | 30.0 | >99 |
| 60 | 4.5 | 0.35 | 4.8 | 25.2 | 68 |
| 60 | 9 | 0.7 | 1.8 | 28.2 | 88 |
| 60 | 12 | 0.7 | 0 | 30.0 | >99 |
| 80 | 4.5 | 0.35 | 6.4 | 23.6 | 57 |
| 80 | 9 | 0.7 | 3.9 | 26.1 | 74 |
| 80 | 12 | 0.7 | 0 | 30.0 | 79.6 |
Using the optimum biocatalyst composition in the catalytic cascade system, the stereoinversion of L-Phe into D-Phe was tracked by monitoring the changes in the concentrations of L-Phe, D-Phe, and PPA. As shown in Fig. 3, quick consumption of L-Phe in the early stages of the cascade reaction was observed for the oxidative deamination reaction and simultaneously the PPA intermediate was detected in the cascade reaction system. Meanwhile, D-Phe was generated and subsequently accumulated from the PPA intermediate by the reductive amination reaction. For the increased substrate concentration up to 80 mM of L-Phe, the cascade reaction was almost complete after 6 h, and both the optical purity and the conversion of the product were close to 100%. Therefore, coordinating the two reaction modules of the oxidative deamination and the reductive amination by applying appropriate proportion of the corresponding catalysts to regulate the reaction rates in the cascade would be important to improve the substrate concentration and reaction efficiency. In an enlarged reaction system, optically pure D-Phe was produced with 91% isolation yield and >99.9% ee. The product was further confirmed to be D-Phe by MS, 1H-NMR, and 13C-NMR (Fig. S5–S7†). Consequently, we constructed a biocatalytic cascade system for the asymmetric synthesis of D-Phe through stereoinversion involving oxidative deamination and reductive amination.
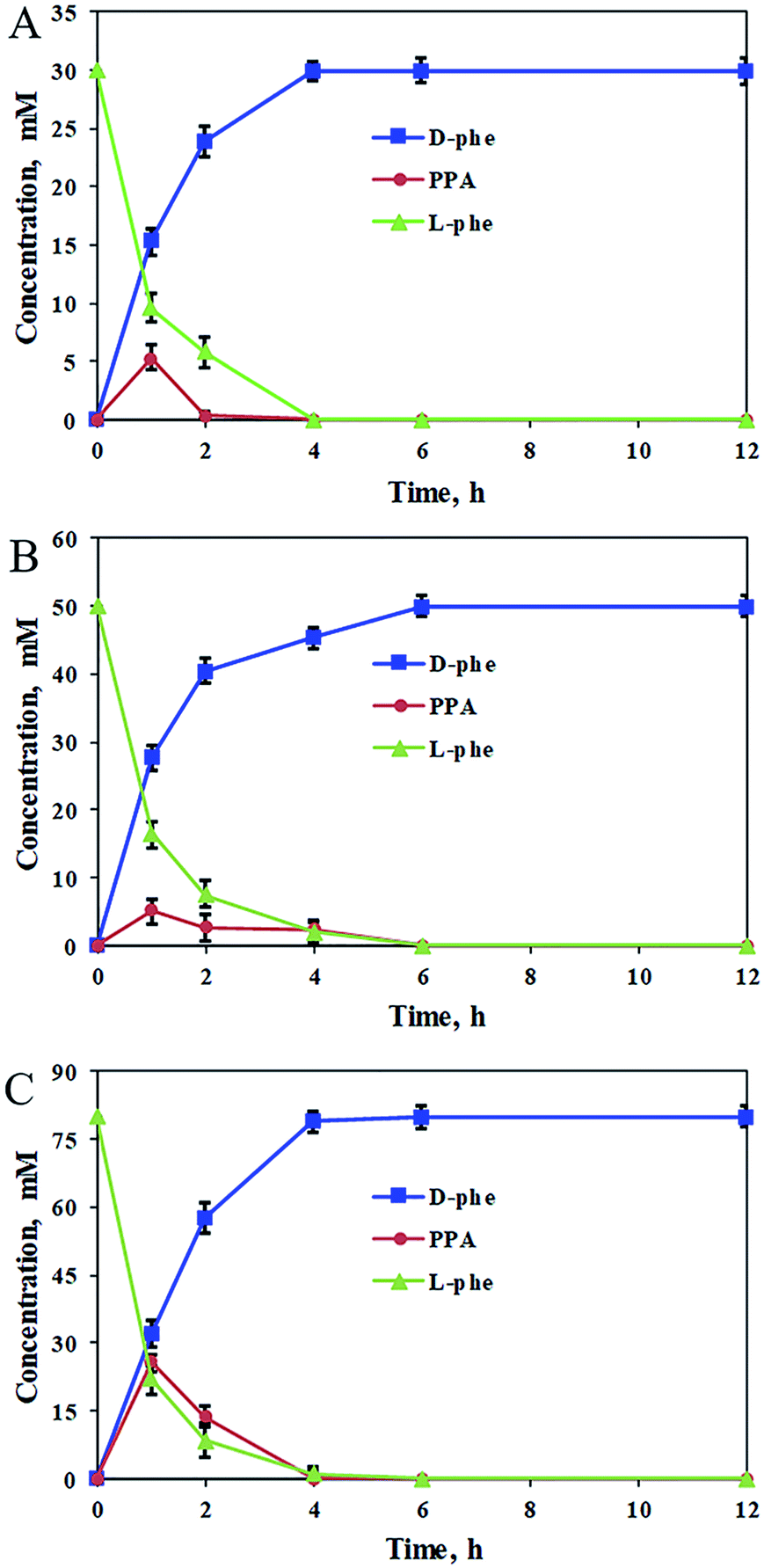 | ||
| Fig. 3 Profiles for the stereoinversion of L-Phe into D-Phe. (A) The cascade reaction system contained 30 mM L-Phe, 3 mM NADP+, 90 mM NH4Cl, and 60 mM sodium formate; (B) the cascade reaction system contained 50 mM L-Phe, 5 mM NADP+, 150 mM NH4Cl, and 100 mM sodium formate; (C) the cascade reaction system contained 80 mM L-Phe, 8 mM NADP+, 240 mM NH4Cl, and 160 mM sodium formate. All the reactions were carried out in 2 mL Tris–HCl buffer (50 mM, pH 9.0) comprising 40 mg mL−1 PmLAAD wet cells, 4.5 mg mL−1 StDAPDH/H227V, and 0.35 mg mL−1 BsFDH at 220 rpm and 45 °C for 12 h. The values were averaged from triplicate measurements and the standard deviations are indicated as error bars. | ||
Substrate scope of the biocatalytic cascade system
The applicability of the biocatalytic cascade system for the stereoinversion of L-Phe, and its construction strategy, were further evaluated for the asymmetric conversions of a variety of natural and noncanonical L-amino acids into the corresponding D-amino acids. As shown in Table 3, the L-amino acids examined were mostly converted into the desired D-amino acids. For substrates with bulky groups, such as L-Phe, L-homophenylalanine, 2-chloro-L-phenylalanine, and 3-chloro-L-phenylalanine, which are aromatic amino acids, the PmLAAD–StDAPDH/H227V cascade system produced the corresponding D-amino acids in high conversion (100%) and optical purity (>99% ee). In addition, the constructed PmLAAD–StDAPDH/H227V cascade system efficiently converted aliphatic amino acids, such as L-leucine, L-norvaline, and L-glutamic acid to the corresponding D-amino acids, with D-glutamic acid and D-norvaline even obtained in quantitative conversion and >99% ee. Therefore, the constructed biocatalytic cascade system is efficient and applicable to the asymmetric synthesis of D-amino acids through stereoinversion involving oxidative deamination and reductive amination.| Substrate | PmLAAD–StDAPDH/H227V | |
|---|---|---|
| Conversion (%) | Optical purityb (% ee) | |
| a Reaction conditions: 10 mM substrate, 1 mM NADP+, 20 mM sodium formate, and 30 mM NH4Cl were added to 50 mM Tris–HCl buffer (pH 9.0). The cascade biocatalysts were PmLAAD whole cells (40 mg mL−1), StDAPDH/H227V (4.5 mg mL−1), and BsFDH (0.35 mg mL−1). The reactions were carried out at 45 °C.b Optical purity (% ee) was determined by HPLC after FDAA derivatization. | ||
| L-Leucine | 70.1 | 68.3 |
| L-Glutamic acid | 100 | >99 |
| L-Norvaline | 75 | >99 |
| L-Tyrosine | 45.3 | >99 |
| L-Phenylalanine | 100 | >99 |
| L-Homophenylalanine | 100 | >99 |
| 2-Chloro-L-phenylalanine | 100 | >99 |
| 3-Chloro-L-phenylalanine | 100 | >99 |
| 4-Chloro-L-phenylalanine | 76.3 | 52.1 |
Conclusions
A versatile and highly efficient biocatalytic cascade system was constructed for the asymmetric synthesis of D-amino acids through the stereoinversions of L-amino acids using a combination of PmLAAD whole cells (oxidative deamination module), DAPDHs (reductive amination module), and BsFDH (cofactor regeneration). To understand the basic principles required for constructing the biocatalytic stereoinverting cascade, the catalytic characteristics of the oxidative deamination module and the reductive amination module were first analyzed. Using L-Phe as the model substrate, the catalytic activities of PmLAAD whole cells and the adopted DAPDHs toward L-Phe, D-Phe, and PPA were shown to guarantee the feasibility of the cascade stereoinversion process. The cascade reaction system was then constructed by assembling the two modules involving necessary cofactor recycling. By optimizing the concentrations of the involved biocatalysts and the reaction conditions such that both biocatalytic modules are compatible, L-Phe was quantitatively converted into D-Phe with greater than 99% ee using the one-pot cascade process. In particular, the cascade biocatalytic system also efficiently converted a variety of readily available and inexpensive aromatic and aliphatic L-amino acids into the corresponding D-amino acids. The developed stereoinverting catalytic system is a promising, sustainable, and cost-efficient alternative for the synthesis of a broad range of D-amino acids.Experimental
Materials
Enzymes, vectors, oligonucleotides, and other reagents for DNA cloning and amplification were obtained from the Takara-Bio Co., Japan and the Novagen Co., USA. The L-amino acids, 2-keto acids, and D-amino acids were purchased from the Sinopharm Chemical Reagent Co., Ltd. NADPH and NADP+ were purchased from Sigma-Aldrich (USA). Acetonitrile and methanol used for high performance liquid chromatography (HPLC) were of chromatographic grade (Honeywell Co., USA). All other chemicals were of analytical grade and commercially available.Cloning the gene-encoding PmLAAD, DAPDHs and BsFDH
The gene-encoding PmLAAD (GenBank accession no. EU669819.1), UtDAPDH (GenBank accession no. AB636161.1), LfDAPDH (GenBank accession no. WP_069481537.1), StDAPDH (GenBank accession no. BAD40410.1), StDAPDH/H227V, and BsFDH (GenBank accession no. ACF35003.1) were cloned and ligated into the pET-28a(+) vector. The pET-28a(+)-PmLAAD, pET-28a(+)-UtDAPDH, pET-28a(+)-LfDAPDH, pET-28a(+)-StDAPDH, pET-28a(+)-StDAPDH/H227V, and pET-28a(+)-BsFDH recombinant plasmids were transformed into E. coli BL21(DE3) cells.Enzyme expression and purification
Recombinant E. coli BL21(DE3) was propagated in 1 L of Luria–Bertani medium containing 100 mg mL−1 kanamycin at 37 °C. The culture was induced by the addition of isopropyl β-D-1-thiogalactopyranoside (IPTG) to a final concentration of 0.1 mM when the optical density (λ = 600 nm) was 0.6–0.8, after which it was incubated for an additional 16 h at 17 °C at 200 rpm. After centrifugation at 6000 × g and 4 °C for 20 min, the recombinant cells were washed with potassium phosphate buffer. The PmLAAD whole cells were cryopreserved at −20 °C for further experiments.To purify UtDAPDH, LfDAPDH, StDAPDH, StDAPDH/H227V, and BsFDH, the recombinant E. coli BL21(DE3) cells were resuspended in a lysis buffer containing 20 mM Tris–HCl and 150 mM NaCl (pH 7.5). Cell lysates were produced with a high-pressure homogenizer (APV, Germany) and then centrifuged at 12000 × g for 30 min at 4 °C to remove cell debris. Proteins were purified by Ni affinity chromatography with an Äkta Purifier 10 (GE Healthcare, USA). The gradient elution buffer for the Ni-NTA column was composed of 20 mM Tris–HCl (pH 7.5), 150 mM NaCl, and 0–500 mM imidazole. The imidazole in the protein solutions was removed using a PD-10 desalting column (GE Healthcare, USA). Protein concentrations were determined using a bicinchoninic acid assay kit (CWBIO, China). The resulting enzyme solutions were used for activity assays and catalytic reactions.
Activity assays and kinetics
PmLAAD whole cells were incubated in 50 mM Tris–HCl buffer (pH 9.0) comprising 100 mM L-Phe in a final volume of 5.0 mL at 37 °C. After reaction for 60 min, the mixture was centrifuged at 12000 × g for 5 min, after which a 15 μL aliquot of the supernatant was added to 1 mL of ferric chloride solution and incubation was continued for 2 min at room temperature. The concentration of PPA was determined by measuring the absorbance at 640 nm.45 One unit of PmLAAD-whole-cell catalytic activity is defined as 1 μmol of PPA generated per minute under the assay conditions.31 All data were averaged over three replicates and significant differences (p < 0.05) were recorded.
The activity assaying system for DAPDH-catalyzed reductive amination consisted of 20 mM 2-keto acid, 200 mM NH4Cl, 0.5 mM NADPH, and 50 mM Na2CO3–NaHCO3 buffer (pH 9.0) at a final volume of 200 μL.1,43 After incubation, reactions were performed in 96-well plates using a Spectramax M2e microplate reader (Molecular Devices, USA) and initiated by the addition of a 20 μL enzyme solution. The OD340 was monitored at 30 °C using a molar extinction coefficient of 6.22 mM−1 cm−1. One unit of enzyme activity is defined as 1 μmol NADPH consumed per minute under the assay conditions. All data were averaged over three replicates for each substrate and significant differences (p < 0.05) were determined.
Kinetic data for the DAPDHs toward PPA were determined by measuring the initial rates at various concentrations of the substrate. The reductive amination reactions were carried out with various concentrations of PPA, 200 mM NH4Cl, and 0.5 mM NADPH in 50 mM Na2CO3–NaHCO3 buffer (pH 9.0) at 30 °C. The concentration of PPA varied between 2 and 40 mM. The Michaelis–Menten kinetic parameters were obtained from a non-linear least-squares fit of the Michaelis–Menten equation to the experimental data and were further validated using double reciprocal Lineweaver–Burk plots. All data were averaged over three replicates for each substrate concentration and significant differences (p < 0.05) were determined.
The activity assaying system for BsFDH consisted of 50 mM sodium formate, 2 mM NADP+, and 50 mM Tris–HCl buffer (pH 7.5) at a final volume of 200 μL. After incubation, reactions were performed in 96-well plates using a Spectramax M2e microplate reader (Molecular Devices, USA) and initiated by the addition of a 20 μL enzyme solution. The OD340 was monitored at 30 °C using a molar extinction coefficient of 6.22 mM−1 cm−1. One unit of the enzyme was defined as the amount that catalyzed the formation of 1 μmol of NADPH per minute under the assay conditions. All data were averaged over three replicates for each substrate and significant differences (p < 0.05) were determined.
Single step reaction
To examine the catalytic activity of PmLAAD whole cells toward different components relevant to oxidative deamination, 50 mM L-Phe, 50 mM PPA, or 50 mM D-Phe substrate was separately added to a reaction mixture containing PmLAAD wet cells (80 mg mL−1) and Tris–HCl buffer (50 mM, pH 9.0). The oxidative deamination reactions were carried out at 45 °C for 6 h.To examine the catalytic activity of StDAPDH/H227V toward different components relevant to reductive amination, 10 mM L-Phe, 10 mM PPA, or 10 mM D-Phe substrate was separately added to a reaction mixture containing StDAPDH/H227V (4 mg mL−1), NADPH (20 mM), NH4Cl (30 mM), and Tris–HCl buffer (50 mM, pH 9.0). The reductive amination reactions were carried out at 37 °C for 10 h.
Combination of biocatalysts for cascade reaction
To construct the stereoinverting cascade, PmLAAD wet cells (40–80 mg mL−1) that catalyze oxidative deamination, StDAPDH/H227V (4.5–12 mg mL−1) that catalyze reductive amination, and BsFDH (0.35–0.7 mg mL−1) that catalyze NADPH regeneration were combined in the one-pot biocatalytic system. The reaction mixture was composed of 30 mM L-Phe, 3 mM NADP+, 90 mM NH4Cl, 60 mM sodium formate, and the three biocatalysts at various concentrations in 2 mL Tris–HCl buffer (50 mM, pH 9.0). Reactions were carried out at 220 rpm and 45 °C, and were terminated by the addition of an equal volume of 10% (w/v) trichloroacetic acid solution. The amount of PPA generated in the reaction process was determined using ferric chloride solution,45 and the amounts of L-Phe and D-Phe were determined by HPLC after derivation with 1-fluoro-2,4-dinitrophenyl-5-L-alanine amide (FDAA).Asymmetric synthesis of D-amino acids from L-amino acids
Various L-amino acids, including L-leucine, L-glutamic acid, L-tyrosine, L-norvaline, L-Phe, L-homophenylalanine, 2-chloro-L-phenylalanine, 3-chloro-L-phenylalanine, and 4-chloro-L-phenylalanine were used as substrates for the asymmetric synthesis of the corresponding D-amino acids using the cascade reaction system involving PmLAAD whole cells, StDAPDH/H227V, and BsFDH. The reaction mixture in 2 mL Tris–HCl buffer (50 mM, pH 9.0) contained 10 mM substrate, 1 mM NADP+, 30 mM NH4Cl, 20 mM sodium formate, and the combined PmLAAD whole-cell, StDAPDH/H227V, and BsFDH biocatalysts. The cascade biocatalysts were composed of PmLAAD wet cells (40 mg mL−1), StDAPDH/H227V (4.5 mg mL−1), and BsFDH (0.35 mg mL−1). The cascade reactions were carried out at 220 rpm and 45 °C. The reactions were terminated by adding equal volumes of 10% (w/v) trichloroacetic acid solution. The concentrations of the L-amino acids and D-amino acids were determined by HPLC following FDAA derivation.Determining the amount of PPA
The amount of PPA in the reaction process was determined using ferric chloride solution,37 which was prepared as follows: 0.1 M ferric chloride was dissolved in 6 mL dimethyl sulfoxide, followed by the addition of 4 mL deionized water and 200 μL acetic acid, after which the solution was cooled in an ice bath. To determine the amount of PPA, a 15 μL aliquot of a reaction sample was added to 1 mL of the ferric chloride solution, and the mixture was incubated at room temperature for 2 min. The amount of PPA was immediately determined by measuring the absorbance at 640 nm using the above-mentioned microplate reader.45Determining L- and D-amino acids
The FDAA reagent (Sigma-Aldrich, USA) was used to produce diastereomeric derivatives of the amino acids. A 10 μL sample of the amino acid, 8 μL of 1 M NaHCO3, and 40 μL of 1% (w/v) FDAA in acetone were mixed and heated for 1 h at 40 °C. When the sample was cooled to room temperature, 8 μL of 1 N HCl and 934 μL of 40% (v/v) aqueous acetonitrile were added to the mixture, after which it was vortexed and filtered (0.22 μm) for HPLC.46 Samples were analyzed using a Develosil ODS-UG-5 column (150 mm × 4.6 mm). The HPLC conditions for analyzing phenylalanine were: mobile phase A water (0.05% trifluoroacetic acid), mobile phase B acetonitrile (0.05% trifluoroacetic acid), with gradients from 20% B to 40% B over 40 min, and from 40% B to 50% B over 5 min at a flow rate of 1 mL min−1, detection at 340 nm, 40 °C, and injection volume of 20 μL (Fig. S8†). The HPLC conditions for analyzing other amino acids were: mobile phase A 5% acetonitrile (0.05% trifluoroacetic acid, 1% methanol), mobile phase B 60% acetonitrile (0.05% trifluoroacetic acid, 1% methanol), linear gradient from 0% B to 100% B over 45 min at a flow rate 1 mL min−1, injection volume 20 μL (Fig. S9–S16†).47Preparation, isolation, and characterization of D-Phe
In a 50 mL reaction system, 80 mM L-Phe, 40 mg mL−1 PmLAAD whole cells, 4.5 mg mL−1 StDAPDH/H227V, 0.35 mg mL−1 BsFDH, 8 mM NADP+, 240 mM NH4Cl, and 160 mM sodium formate were added in the reaction buffer (50 mM Tris–HCl, pH 9.0). Reactions were carried out at 220 rpm and 45 °C. After 6 h, pH of the reaction mixture was adjusted by diluted HCl and the precipitated enzymes were removed by centrifugation. The product was isolated by a cation-exchange resin (732) column and eluted by 6% NH3·H2O.20 The product was characterized by MS, 1H-NMR and 13C-NMR as follows (Fig. S5–S7†): 1H-NMR (400 MHz, D2O), δ/ppm: 3.11 (m, 1H), 3.28 (m, 1H), 3.98 (m, 1H), 7.38 (m, 5H); 13C-NMR (100 MHz, D2O), δ/ppm: 36.34, 56.03, 129.11, 129.37, 135.11, 173.89.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21676120, 31872891), the Program of Introducing Talents of Discipline to Universities (111-2-06), the High-End Foreign Experts Recruitment Program (G20190010083), the Program for Advanced Talents within Six Industries of Jiangsu Province (2015-NY-007), the National Program for Support of Top-notch Young Professionals, Postgraduate Research & Practice Innovation Program of Jiangsu Province (KYCX18_1794), the Fundamental Research Funds for the Central Universities (JUSRP51504), the Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions, Top-notch Academic Programs Project of Jiangsu Higher Education Institutions, the Jiangsu Province “Collaborative Innovation Center for Advanced Industrial Fermentation” Industry Development Program, the Program for the Key Laboratory of Enzymes of Suqian (M201803), and the National First-Class Discipline Program of Light Industry Technology and Engineering (LITE2018-09).Notes and references
- K. Vedha-Peters, M. Gunawardana, J. D. Rozzell and S. J. Novick, J. Am. Chem. Soc., 2006, 128, 10923–10929 CrossRef CAS PubMed.
- F. Paradisi, P. A. Conway, A. R. Maguire and P. C. Engel, Org. Biomol. Chem., 2008, 6, 3611–3615 RSC.
- P. Merviel, S. Najas, H. Campy, S. Floret and F. Brasseur, Minerva Ginecol., 2005, 57, 29–43 CAS.
- X. Z. Gao, Q. Y. Ma and H. L. Zhu, Appl. Microbiol. Biotechnol., 2015, 99, 3341–3349 CrossRef CAS PubMed.
- F.-R. Alexandre, D. P. Pantaleone, P. P. Taylor, I. G. Fotheringham, D. J. Ager and N. J. Turner, Tetrahedron Lett., 2002, 43, 707–710 CrossRef CAS.
- F. Khorsand, C. D. Murphy, A. J. Whitehead and P. C. Engel, Green Chem., 2017, 19, 503–510 RSC.
- X. Z. Gao, F. Huang, J. H. Feng, X. Chen, H. L. Zhang, Z. X. Wang, Q. Q. Wu and D. M. Zhu, Appl. Environ. Microbiol., 2013, 79, 5078–5081 CrossRef CAS PubMed.
- F. Parmeggiani, S. T. Ahmed, M. P. Thompson, N. J. Weise, J. L. Galman, D. Gahloth, M. S. Dunstan, D. Leys and N. J. Turner, Adv. Synth. Catal., 2016, 358, 3298–3306 CrossRef CAS.
- M. Breuer, K. Ditrich, T. Habicher, B. Hauer, M. Kesseler, R. Sturmer and T. Zelinski, Angew. Chem., Int. Ed., 2004, 43, 788–824 CrossRef CAS PubMed.
- C. Nájera and J. M. Sansano, Chem. Rev., 2007, 107, 4584–4671 CrossRef PubMed.
- Organic Synthesis Using Biocatalysis, ed. S. K. Au, J. Groover, B. D. Feske and A. S. Bommarius, Elsevier, 2016 Search PubMed.
- Y. P. Xue, C. H. Cao and Y. G. Zheng, Chem. Soc. Rev., 2018, 47, 1516–1561 RSC.
- P. J. Almhjell, C. E. Boville and F. H. Arnold, Chem. Soc. Rev., 2018, 47, 8980–8997 RSC.
- Y. F. Liu, G. C. Xu, R. Z. Han, J. J. Dong and Y. Ni, Bioresour. Technol., 2018, 249, 720–728 CrossRef CAS PubMed.
- E. S. Park and J. S. Shin, Adv. Synth. Catal., 2015, 356, 3505–3509 CrossRef.
- R. Morán-Ramallal, R. Liz and V. Gotor, Org. Lett., 2008, 10, 1935–1938 CrossRef PubMed.
- S. Baxter, S. Royer, G. Grogan, F. Brown, K. E. Holt-Tiffin, I. N. Taylor, I. G. Fotheringham and D. J. Campopiano, J. Am. Chem. Soc., 2012, 134, 19310–19313 CrossRef CAS PubMed.
- R. X. Liu, S. P. Liu, S. Cheng, L. Zhang, Z. Y. Ding, Z. H. Gu and G. Y. Shi, Appl. Biochem. Microbiol., 2015, 51, 695–703 CrossRef CAS.
- K. Isobe, H. Tamauchi, K. Fuhshuku, S. Nagasawa and Y. Asano, Enzyme Res., 2010, 2010, 1–6 CrossRef PubMed.
- X. Chen, Y. F. Cui, X. K. Cheng, J. H. Feng, Q. Q. Wu and D. M. Zhu, ChemistryOpen, 2017, 6, 534–540 CrossRef CAS PubMed.
- J. H. Schrittwieser, S. Velikogne, M. Hall and W. Kroutil, Chem. Rev., 2017, 118, 270–348 CrossRef PubMed.
- S. Wu, Y. Zhou, T. Wang, H. P. Too, D. I. Wang and Z. Li, Nat. Commun., 2016, 7, 11917–11930 CrossRef CAS PubMed.
- W. Song, J. H. Wang, J. Wu, J. Liu, X. L. Chen and L. M. Liu, Nat. Commun., 2018, 9, 1–9 CrossRef PubMed.
- J. A. Houwman, T. Knaus, M. Costa and F. G. Mutti, Green Chem., 2019, 21, 1–12 RSC.
- A. Al-Shameri, N. Borlinghaus, L. Weinmann, P. N. Scheller, B. M. Nestl and L. Lauterbach, Green Chem., 2019, 21, 1396–1400 RSC.
- Y. P. Xue, Y. G. Zheng, Z. Q. Liu, X. Liu, J. F. Huang and Y. C. Shen, ACS Catal., 2014, 4, 3051–3058 CrossRef CAS.
- F. Parmeggiani, S. L. Lovelock, N. J. Weise, S. T. Ahmed and N. J. Turner, Angew. Chem., Int. Ed., 2015, 54, 4608–4611 CrossRef CAS PubMed.
- C. J. W. Walton, F. Parmeggiani, J. E. B. Barber, J. L. McCann, N. J. Turner and R. A. Chica, ChemCatChem, 2018, 10, 470–474 CrossRef CAS.
- T. K. F. G. Mutti, N. S. Scrutton, M. Breuer and N. J. Turner, Science, 2015, 349, 1525–1529 CrossRef PubMed.
- S. P. France, L. J. Hepworth, N. J. Turner and S. L. Flitsch, ACS Catal., 2017, 7, 710–724 CrossRef CAS.
- L. Liu, G. S. Hossain, H. D. Shin, J. H. Li, G. C. Du and J. Chen, J. Biotechnol., 2013, 164, 97–104 CrossRef CAS PubMed.
- Y. Song, J. Li, H. D. Shin, G. Du, L. Liu and J. Chen, Sci. Rep., 2015, 5, 12614–12625 CrossRef CAS PubMed.
- M. B. Quin, K. K. Wallin, G. Zhang and C. Schmidt-Dannert, Org. Biomol. Chem., 2017, 15, 4260–4271 RSC.
- F. Aalbers and M. Fraaije, ChemBioChem, 2019, 20, 20–28 CrossRef CAS PubMed.
- J. M. Sperl and V. Sieber, ACS Catal., 2018, 8, 2385–2396 CrossRef CAS.
- J. O. Baek, J. W. Seo, O. Kwon, S. I. Seong, I. H. Kim and C. H. Kim, J. Basic Microbiol., 2011, 51, 129–135 CrossRef CAS PubMed.
- G. Molla, R. Melis and L. Pollegioni, Biotechnol. Adv., 2017, 35, 657–668 CrossRef CAS PubMed.
- E. Rosini, R. Melis, G. Molla, D. Tessaro and L. Pollegioni, Adv. Synth. Catal., 2017, 359, 3773–3781 CrossRef CAS.
- G. S. Hossain, H. D. Shin, J. H. Li, G. C. Du, J. Chen and L. Liu, RSC Adv., 2016, 6, 82676–82684 RSC.
- Y. Hou, G. S. Hossain, J. H. Li, H. D. Shin, G. C. Du and L. Liu, Appl. Microbiol. Biotechnol., 2016, 100, 2183–2191 CrossRef CAS PubMed.
- H. Akita, Y. Fujino, K. Doi and T. Ohshima, AMB Express, 2011, 1, 1–8 CrossRef PubMed.
- J. Hayashi, T. Seto, H. Akita, M. Watanabe, T. Hoshino, K. Yoneda, T. Ohshima and H. Sakuraba, Appl. Environ. Microbiol., 2017, 83, 1–13 CrossRef.
- X. Z. Gao, X. Chen, W. D. Liu, J. H. Feng, Q. Q. Wu, L. Hua and D. M. Zhu, Appl. Environ. Microbiol., 2012, 78, 8595–8600 CrossRef CAS PubMed.
- X. Z. Gao, Z. Zhang, Y. N. Zhang, Y. Li, H. Zhu, S. Wang and C. Li, Appl. Environ. Microbiol., 2017, 83, 476–486 Search PubMed.
- Y. Hou, G. S. Hossain, J. Li, H. D. Shin, L. Liu and G. Du, Appl. Microbiol. Biotechnol., 2015, 99, 8391–8402 CrossRef CAS PubMed.
- R. L. Hanson, B. L. Davis, S. L. Goldberg, R. M. Johnston, W. L. Parker, T. P. Tully, M. A. Montana and R. N. Patel, Org. Process Res. Dev., 2008, 12, 1119–1129 CrossRef CAS.
- R. Bhushan and H. Bruckner, Amino Acids, 2004, 27, 231–247 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra06301c |
| This journal is © The Royal Society of Chemistry 2019 |