
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCu2−xSe nanoparticles (Cu2−xSe NPs) mediated neurotoxicity via oxidative stress damage in PC-12 cells and BALB/c mice†
Faning Leng‡
,
Yali Liu‡,
Guobing Li,
Wenjing Lai,
Qian Zhang,
Wuyi Liu,
Changpeng Hu,
Pantong Li,
Fangfang Sheng,
Jingbin Huang* and
Rong Zhang *
*
Department of Pharmacology, The Second Affiliated Hospital of Army Medical University, Chongqing, 400037, China. E-mail: xqpharmacylab@126.com; hjb20091364@126.com
First published on 11th November 2019
Abstract
Cu2−xSe nanoparticles (Cu2−xSe NPs) are widely used for optical diagnostic imaging and photothermal therapy due to their strong near-infrared (NIR) optical absorption. With the continuous expansion of applications using Cu2−xSe NPs, their biosafety has received increasing attention in recent years. Cu2−xSe NPs can enter the brain by crossing the blood–brain barrier, but the neurotoxicity of NPs remains unclear. The present investigation provides direct evidence that the toxicity of Cu2−xSe NPs can be specifically exploited to kill rat pheochromocytoma PC-12 cells (a cell line used as an in vitro model for brain neuron research) in dose- and time-dependent manners. These cytotoxicity events were accompanied by mitochondrial damage, adenosine triphosphate (ATP) depletion, production of oxidizing species (including reactive oxygen species (ROS), malondialdehyde (MDA) and hydrogen peroxide (H2O2)), as well as reductions in antioxidant defense systems (glutathione (GSH) and superoxide dismutase (SOD)). Moreover, our in vivo study also confirmed that Cu2−xSe NPs markedly induced neurotoxicity and oxidative stress damage in the striatum and hippocampal tissues of BALB/c mice. These findings suggest that Cu2−xSe NPs induce neurotoxicity in PC-12 cells and BALB/c mice via oxidative stress damage, which provides useful information for understanding the neurotoxicity of Cu2−xSe NPs.
1. Introduction
Nanoparticles (NPs), which range from 1 to 100 nm in size, have been widely used in the cosmetics, food additives, and biomedical industries, as well as in environmental technologies.1–4 Despite their widespread application in many fields, the impact of nanoparticles on human health is still unknown. Numerous studies have shown that nanoparticles can be absorbed and found in many organs, including the brain, liver, lung, spleen, and kidneys, after animals were exposed through oral administration, intraperitoneal injection or intravenous injection.5–12 Therefore, the biosafety of nanoparticles should be investigated. Cu2−xSe nanoparticles (Cu2−xSe NPs), important members of the family of copper chalcogenide nanomaterials, have attracted attention in cancer therapy because of their strong near-infrared (NIR) absorption.13–17 They have been widely used as photosensitizers for both photothermal therapy (PTT)18–21 and photodynamic therapy (PDT).22 Furthermore, our previous research suggested that Cu2−xSe NPs may potentially inhibit autophagy and combining Cu2−xSe NPs with traditional chemotherapeutic drugs could provide a fresh therapeutic strategy for treating cancer.23Copper chalcogenide nanomaterials are widely used in biological fields and their toxicity has received widespread attention. Li et al. compared the cytotoxicity of copper sulfide nanoparticles with gold nanoparticles in in vitro conditions.24 The results revealed that CuS was cytotoxic to human HEK 293 cells. Wei Feng et al. evaluated the cytotoxicity of copper sulfide in four types of cell lines.25 More recently, Guo et al. conducted an in vivo toxicity study of PEGylated CuS and gold nanoparticles in mice.26 Recent studies have shown that copper ions could enter the murine brain by crossing the blood–brain barrier.27,28 Moreover, it has also been reported that uncomplexed copper ions are harmful to humans and are especially involved in the progression of Alzheimer's29,30 and Wilson's disease.31–33 However, the safety of Cu2−xSe NPs in humans remains unclear. Thus, there is an urgent need for a comprehensive understanding and assessment of Cu2−xSe NPs.
Studies have revealed that oxidative stress and mitochondrial abnormalities play key roles in numerous neuropsychiatric disorders. For example, TiO2 NPs were shown to induce reactive oxygen species (ROS) generation and disturb the mitochondrial energy production in brain microglia.34,35 Quantum dots can trigger oxidative stress through multiple interactions, causing the overexpression of intracellular ROS.36 In addition, more studies have shown that when copper oxide nanoparticles (CuO NPs) enter human hepatocarcinoma cells, the Bax/Bcl-2 ratio increased and the mitochondrial membrane potential decreased.37 Therefore, whether Cu2−xSe NPs can induce toxic side effects on the nervous system, as well as the mechanism of neurotoxicity in mitochondria, need to be studied further.
In this report, in vitro and in vivo studies demonstrated the neurotoxicity of Cu2−xSe NPs using PC-12 cells and BALB/c mice (Fig. 1). The results showed that exposure of PC-12 cells to Cu2−xSe NPs resulted in a significant decrease in cell viability and an increase in apoptosis in dose-dependent manners. Cu2−xSe NPs induced apparent morphological changes and mitochondrial dysfunction in PC-12 cells, which were associated with intracellular ROS production. After exposure to Cu2−xSe NPs, pathologic changes, TdT-mediated dUTP nick-end labeling (TUNEL)-positive cells, cleaved caspase-3 immunoreactivity, as well as significant oxidative stress in the striatum and hippocampus of BALB/c mice, were observed. These findings suggest that Cu2−xSe NPs induced ROS-related mitochondrial apoptosis in PC-12 cells and BALB/c mice. Our study provides useful information for understanding the neurotoxicity of Cu2−xSe NPs.
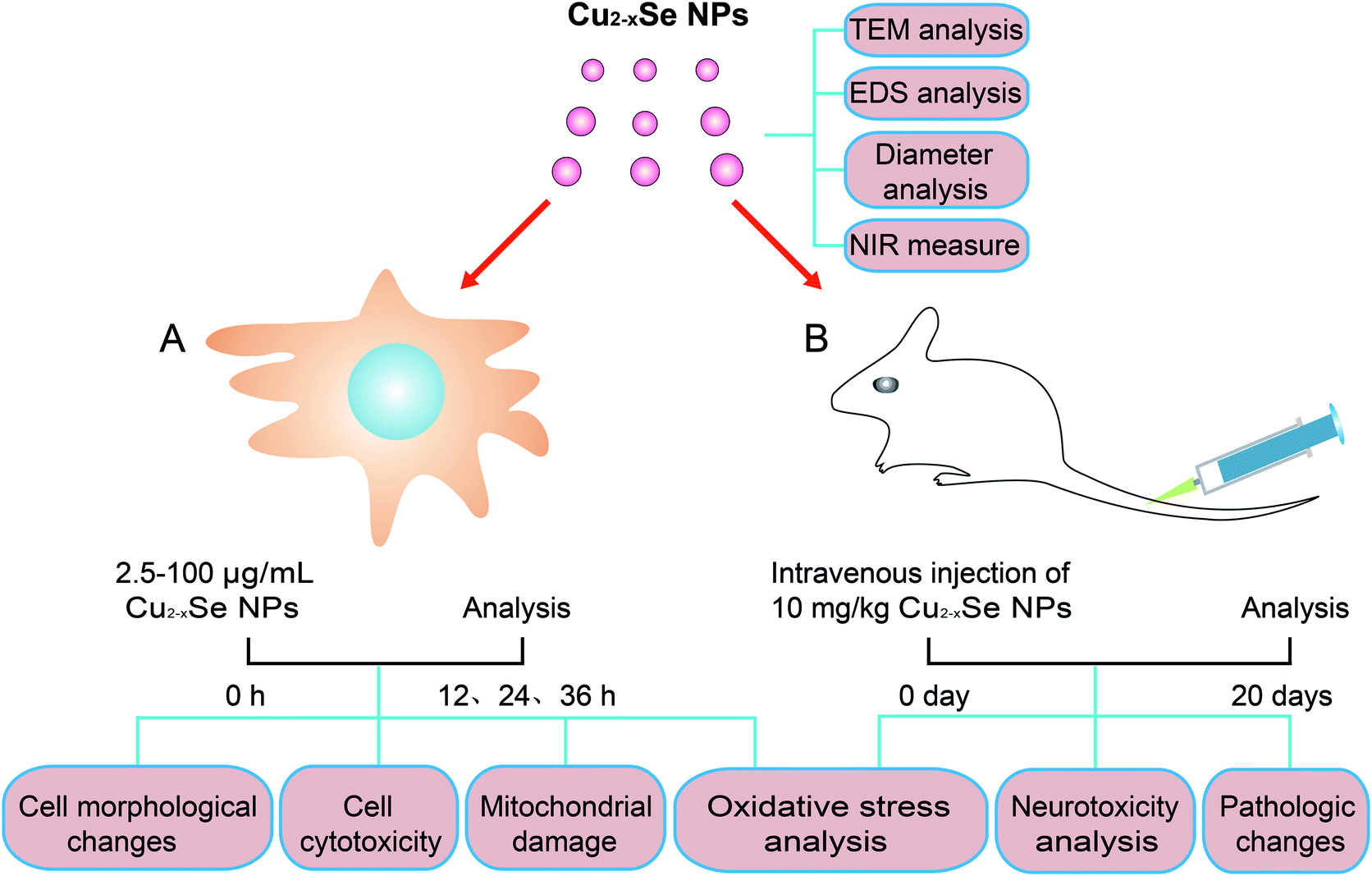 | ||
| Fig. 1 Schematic diagram of the experimental model for evaluating the neurotoxicity of Cu2−xSe NPs in vitro and in vivo. After the preparation and characterization of Cu2−xSe NPs, in vitro analysis was performed using PC-12 cells. The PC-12 cells were incubated with 2.5 to 100 μg mL−1 of Cu2−xSe NPs for 12, 24, or 36 h. Cell morphology, cell cytotoxicity, mitochondrial damage and oxidative stress were analyzed. In vivo analysis was performed as follow: BALB/c mice were intravenously injected with Cu2−xSe NPs (10 mg kg−1) every two days for 20 days for (i.e., 10 times) and pathologic changes, neurotoxicity, and oxidative stress in the striatums and hippocampal tissues were investigated. | ||
2. Materials and methods
2.1 Materials
Anhydrous copper sulfate (CuSO4) and selenium dioxide (SeO2) were purchased from Aladdin Chemistry Co., Ltd (Shanghai, China). Polyvinylpyrrolidone powder (PVP, Mw = 55000) was supplied by Sigma-Aldrich Co. (St. Louis, MO, USA). Ascorbic acid (AA) was obtained from Dingguo Changsheng Biotechnology Co., Ltd (Beijing, China).2.2 Preparation and characterization of Cu2−xSe NPs
The PVP-stabilized Cu2−xSe NPs were prepared as follows.17 In a round-bottomed flask, 30.0 mg of PVP was dissolved in 10 mL of distilled water, then 0.2 mL AA (0.2 g mL−1) and 0.5 mL SeO2 (0.25 M) were added to the PVP solution. After 10 min, 0.5 mL of CuSO4 (0.5 M) mixed with 0.3 mL AA (0.2 g mL−1) was poured into the flask under vigorous stirring. The mixture solution was placed in a 40 °C oil bath for 6 h to generate a Cu2−xSe NPs suspension solution, then centrifuged for 15 min at 8000 rpm, washed three times, and resuspended in distilled water. Transmission electron microscopy (TEM, JEM-2100, Japan) was used to observe the structure of Cu2−xSe NPs, energy-dispersive X-ray (EDS, JEM-2100) was used to analyze the composition of the Cu2−xSe NPs. The average hydrodynamic radius and surface charge of the Cu2−xSe NPs in distilled water and physiological saline were analyzed using a Malvern laser particle size analyzer (NanoZS90, Malvern, UK) daily for a week. The NIR absorption was measured via a spectrophotometer (CARY5000, USA). Finally, the Cu2−xSe NPs products were stored at 4 °C.2.3 Cell culture
The PC-12 cell line was obtained from the Cell Bank of Type Culture Collection of the Chinese Academy of Sciences in Shanghai. The cells were cultured in high-glucose Dulbecco's modified Eagle medium (DMEM, catalog number 12491, Invitrogen) supplemented with 10% fetal bovine serum (FBS, catalog number 10100, Gibco) in an incubator at 37 °C and 5% CO2.2.4 Cell cytotoxicity assay
Cell cytotoxicity was evaluated using the standard 3-(3,4-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Briefly, the cells were plated in 96-well plates at a density of 1 × 104 cells per well and were allowed to attach for 24 h. Then, the cells were treated with Cu2−xSe NPs (0, 2.5, 5, 10, 20, 40, 60, 80, and 100 μg mL−1) at 37 °C for 12, 24 and 36 hours. Then, 20 μL of MTT stock solution (5 mg mL−1, Sigma) was added to each well and the plate was incubated for 4 h at 37 °C. Then, the supernatant was carefully aspirated and the formazan crystals in the cells were dissolved with 150 μL dimethyl sulfoxide (Eastman Kodak, Columbus, GA, USA). The absorbance of the solution was recorded at 492 nm with a microplate reader (Varioskan Flash, USA). Each experiment was repeated three times and the results are expressed as a percentage of the untreated controls, which was set to 100%.2.5 Apoptosis determination
The cells were grown with different concentrations of Cu2−xSe nanoparticles. After 24 h of treatment, the cells were trypsinized, collected, and washed with phosphate-buffered saline (PBS). The apoptotic cells were measured with an annexin V/propidium iodide (PI) apoptosis detection kit (catalog number 556547, BD Biosciences) and analyzed by flow cytometry (FACScan, BD Biosciences). Then, quadrant analyses were performed and the stained cells were designated as apoptotic and the unstained cells were designated as live using annexin V-FITC and/or PI.2.6 Transmission electronic microscopy
For the TEM analysis, after 24 h incubation with Cu2−xSe NPs (100 μg mL−1), the PC-12 cells were harvested and washed with PBS, fixed with fresh glutaraldehyde (2.5%) 4 °C overnight, then fixed with osmium tetroxide (2%) for 2 h, dehydrated through a graded series of ethanol, and embedded in Epon. The samples were then sectioned using an ultramicrotome (Leica EM UC7) and stained with uranyl acetate and lead citrate. Finally, the ultrastructure of the PC-12 cells was observed using a transmission electron microscope (Hitachi H-7500, Japan) at 60 kV.2.7 Mitochondrial transmembrane potential assay
Diminished mitochondrial membrane potential (ΔΨm) was analyzed using the rhodamine-123 kit (catalog number C2007, Beyotime) according to the manufacturer's protocol. Briefly, after 24 h exposure to 50 and 100 μg mL−1 Cu2−xSe NP, the PC-12 cells were collected and washed twice with PBS. Then, the cells were dispersed in PBS and stained with rhodamine-123 (1 μmol L−1) in serum-free medium for 30 min at 37 °C in the dark. After that, the medium was removed and fresh medium was added for detection. The fluorescence intensity was subsequently monitored using a fluorescence microplate reader (Thermo Varioskan™ LUX) with excitation and emission settings of 507 and 529 nm, respectively. The results are expressed as a percentage of the control, which was set to 100%.2.8 ATP luminescence assay
An ATP Determination kit (catalog number S0026, Beyotime) was used to measure the ATP levels. The PC-12 cells were first lysed and centrifuged at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000×g for 5 min, then the cells were incubated with ATP detection working solution according to the manufacturer's instructions. The luminescence was measured by a microplate reader (Varioskan Flash). The final results are expressed as the percentage of the readings compared to untreated controls.
000×g for 5 min, then the cells were incubated with ATP detection working solution according to the manufacturer's instructions. The luminescence was measured by a microplate reader (Varioskan Flash). The final results are expressed as the percentage of the readings compared to untreated controls.
2.9 Intracellular ROS measurement and oxidative damage
Intracellular ROS was measured by the fluorescent probe 2′,7′-dichlorofluorescein diacetate (DCF-DA, catalog number C6827, Thermo Fisher). In cells, DCFH-DA reacts with ROS to form dichlorofluorescein (DCF), which is highly fluorescent. Briefly, PC-12 cells were seeded in 6-well plates at a density of 5.0 × 105 cells per well. After a 24 h exposure to Cu2−xSe NPs at concentrations of 50 and 100 μg mL−1, the cells were incubated with DCFH-DA at 37 °C for 30 min in 2 mL of working solution (10 mM DCFH-DA stock solution in methanol diluted 1000-fold to 10 μM with DMEM without serum). The fluorescence was then analyzed in a flow cytometer (FACScan) at an excitation wavelength of 488 nm and an emission wavelength of 525 nm.Kits were used to detect the levels of hydrogen peroxide (H2O2, catalog number S0038, Beyotime), malondialdehyde (MDA, catalog number S0131, Beyotime), glutathione (GSH, catalog number S0053, Beyotime) and superoxide dismutase (SOD, catalog number S0101, Beyotime) respectively. Briefly, PC-12 cells were seeded in 6-well plates at a density of 5.0 × 105 cells per well and different concentrations of Cu2−xSe NPs (50 and 100 μg mL−1) were added. After a 24 h treatment, the intracellular levels of H2O2, GSH, SOD, and MDA were measured by using H2O2, GSH, SOD, and MDA determination kits, respectively.
2.10 Animal treatment
BALB/c mice (25 g) were purchased from Vital River Laboratories (VRL, Beijing, China). All experimental procedures were conducted according to the ethical standards and protocols approved by the Army Medical University Institutional Animal Care and Use Committee, Chongqing, China. All mice were housed in groups under controlled environmental conditions throughout the entire process (temperature 23 ± 0.5 °C, humidity 50 ± 5%). Rodent diet and water were provided ad libitum.The animals were randomly divided into two groups, the control and Cu2−xSe NPs groups (n = 5 in each group). The Cu2−xSe NPs were distributed in physiological saline. Mice in the Cu2−xSe NPs group were intravenously injected with Cu2−xSe NPs (10 mg kg−1), and mice in the control group were injected with an equal volume of physiological saline, every two days during 20 days, i.e., 10 times. At 20 days, the animals were euthanized. The striatum and hippocampal tissues were harvested and fixed in 4% paraformaldehyde and the paraffin-embedded tissues were sectioned and analyzed by hematoxylin and eosin (H&E) staining, TUNEL and immunohistochemical analysis were performed as previously described.38,39 The striatum and hippocampal tissues were also lysed and the levels of H2O2, MDA, SOD and GSH were measured by using commercial kits according to the manufacturers' instructions as described above.
2.11 TUNEL assay
Apoptotic cells in the striatum and hippocampal tissues were detected using an In Situ Cell Death Detection kit (Roche, Mannheim, Germany) according to the manufacturer's instructions. After deparaffinization and permeabilization, the tissue sections were treated with proteinase K for 15 min, followed by incubation with the TUNEL reaction mixture that includes terminal deoxynucleotidyl transferase (TdT) and fluorescein-dUTP at 37 °C for 1 h. After washing three times with PBS, the sections were stained with Converter-POD which includes an anti-fluorescein antibody conjugated to horseradish peroxidase (POD) for 30 minutes. After three additional washes in PBS, the sections were stained with 0.05% DAB and examined under a light microscope.2.12 Histological and immunohistochemical evaluation
At the end of the treatment period, the striatum and hippocampal tissues from representative mice were sectioned, embedded in paraffin, and stained with H&E for histopathologic assessment. For immunohistochemical analysis, 4 μm-thick tissue sections were dewaxed and rehydrated in xylene and graded alcohols. Endogenous peroxidase activity was quenched by staining with Tris-buffered saline and Tween 20 (TBS-T) containing 3% hydrogen peroxide. Antigen retrieval was performed by incubating with 0.01 M citrate buffer at pH 6.0 for 20 min in a 95 °C oil bath. Following that, the sections were incubated with 10% goat serum for 1 h and stained with primary antibodies against cleaved-caspase-3 and Ezrin, then washed three times with PBS, stained with biotinylated secondary antibody for 1 h, followed by treatment with streptavidin–peroxidase complex for another 1 h. After washing three times in PBS, the slides were exposed to diaminobenzidine working solution. Finally, the slides were counterstained with DAPI.2.13 Statistical analysis
All data are expressed as the mean ± SD from at least three separate experiments. Statistical comparisons were performed using one-way analysis of variance (ANOVA) followed by Tuckey and Dunnett tests. *P < 0.05, **P < 0.01 and ***P < 0.001 were considered statistically significant.3. Results
3.1 Preparation and characterization of monodispersed Cu2−xSe NPs
To synthesize the Cu2−xSe NPs, we used 55 kDa PVP to prevent the nanoparticles from aggregating. The morphology of the prepared Cu2−xSe NPs was examined by TEM. As shown in Fig. 2A, the nanoparticles were spherical, uniform, highly-dispersed particles. The mean diameter of the Cu2−xSe NPs was measured daily for a week. No changes were found, indicating high dispersion and stability of the NPs (Fig. 2B and S1†). The hydrodynamic diameter of the particles was found to be 142.9 ± 9.5 nm and the ζ-potential was −19.3 ± 2.6 mV in distilled water and 151.4 ± 7.5 nm and −20.5 ± 2.4 mV in physiological saline, respectively. Absorption in the NIR range was analyzed from 650 to 1350 nm and the highest absorption of Cu2−xSe NPs was found at 1070 nm (Fig. 2C). Moreover, energy-dispersive X-ray spectroscopy (EDS) showed that the atomic ratio of the Cu2−xSe NPs was 1.3 (x = 0.7), explaining the serious copper deficiency in the Cu2−xSe NPs domains (Fig. 2D).40,41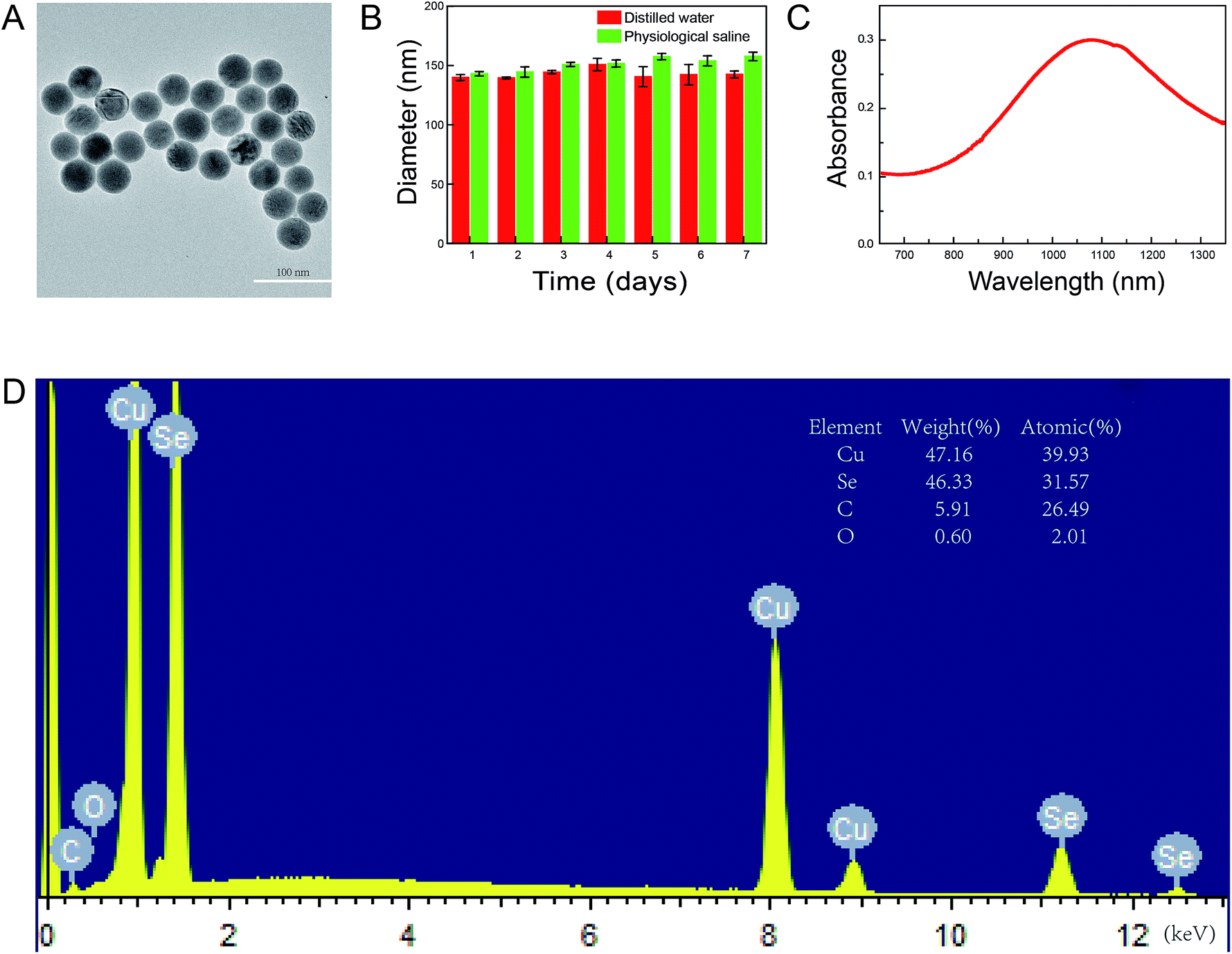 | ||
| Fig. 2 Characterization of the Cu2−xSe NPs. (A) TEM analysis of the Cu2−xSe NPs. Scale bars: 100 nm. (B) The hydrodynamic changes in particle radii in distilled water and physiological saline every day during one week by dynamic light scattering (DLS). (C) The absorption spectra in near-infrared. (D) EDS spectrum of the as-synthesized Cu2−xSe NPs. | ||
3.2 Cu2−xSe NPs decrease cell viability and induce apoptosis in PC-12 cells
PC-12 cell are a cell line that is derived from a rat adrenal medulla pheochromocytoma with typical characteristics of neurons and is widely used as a model for neuronal research in vitro. We first evaluated the effects of Cu2−xSe NPs on the cell morphology of PC-12 cells. As shown in Fig. 3A, the exposure of PC-12 cells to 50 and 100 μg mL−1 of Cu2−xSe NPs for 24 h resulted in an increase in the number of round and neurite-deficient cells compared to the control group, which exhibited stretched neurites in the periphery. Next, an MTT assay was used to investigate the cytotoxicity of the Cu2−xSe NPs. Our results revealed that PC-12 cells exposed to different concentrations of Cu2−xSe NPs (0, 2.5, 5, 10, 20, 40, 60, 80, and 100 μg mL−1) for 12, 24 or 36 h resulted in a significant decrease in cell viability in dose- and time-dependent manners (Fig. 3B). To quantitatively analyze the apoptotic rate of PC-12 cells treated with Cu2−xSe NPs, annexin V-FITC/PI staining and flow cytometry were employed. Exposure of the PC-12 cells to 50 or 100 μg mL−1 Cu2−xSe NPs for 24 h markedly increased the percentage of apoptotic cells in a dose-dependent manner (Fig. 3C). Together, these findings indicate that the Cu2−xSe NPs decreased cell viability and induced apoptosis in PC-12 cells.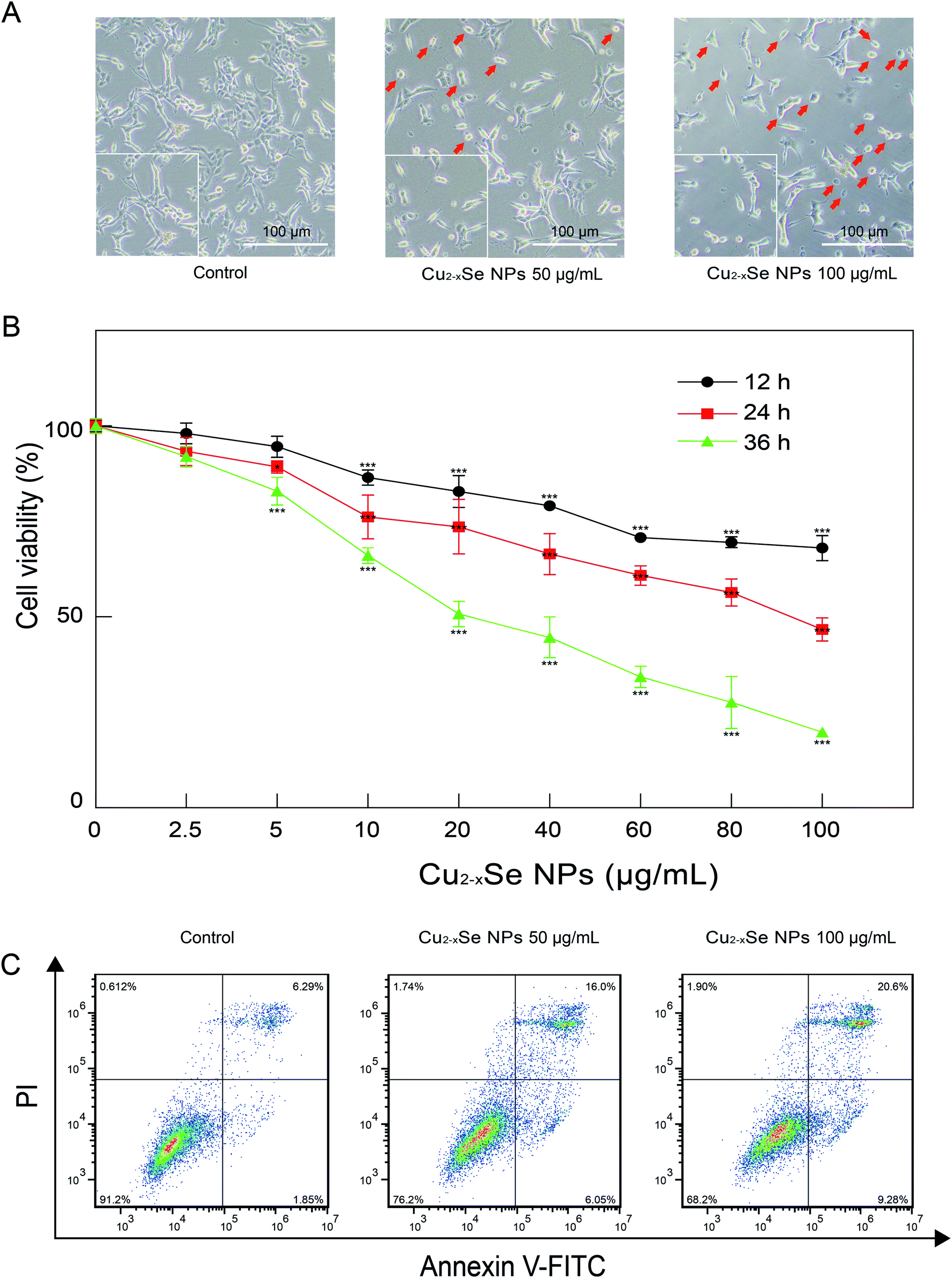 | ||
| Fig. 3 The toxic effects of Cu2−xSe NPs on PC-12 cells. (A) Morphological characterization of PC-12 cells treated with or without Cu2−xSe NPs (50, 100 μg mL−1) for 24 h. Scale bars: 100 μm. (B) Cell viabilities of PC-12 cells treated with different concentrations of Cu2−xSe NPs for (12, 24, and 36 h) detected by MTT assays. (C) PC-12 cells were treated with 50 and 100 μg mL−1 Cu2−xSe NPs for 24 h, followed by staining with annexin V-FITC/PI and analysis by flow cytometry. The data are expressed as the mean ± SD (n = 3), *P < 0.05, **P < 0.01, ***P < 0.001 compared to control. | ||
3.3 Cu2−xSe NPs induced mitochondrial damage, loss of MMP, ATP depletion, and oxidative stress damage in PC-12 cells
To determine the mechanism of Cu2−xSe NPs-induced apoptosis in PC-12 cells, TEM was used to observe ultrastructural changes in the PC-12 cells. Our TEM studies revealed that after treatment with Cu2−xSe NPs (100 μg mL−1) for 24 h, mitochondrial swelling appeared in the Cu2−xSe NPs-treated cells (Fig. 4A), indicating that Cu2−xSe NPs were taken up into the cells and induced mitochondrial apoptosis. Next, we examined the effects of Cu2−xSe NPs on the mitochondrial membrane potential in PC-12 cells. The results showed that exposure to 50 and 100 μg mL−1 Cu2−xSe NPs markedly decreased the fluorescence intensity of rhodamine-123 and increased mitochondrial injury (loss of ΔΨm) in dose-dependent manners (Fig. 4B). These results suggest that Cu2−xSe NPs induced a collapse of mitochondrial membrane potential (MMP) in PC-12 cells. Mitochondria are important organelles for cellular energy ATP production and mitochondrial dysfunction leads to ATP depletion.42–44 To further confirm the previous results, we next investigated the effects of Cu2−xSe NPs on intracellular ATP levels in PC-12 cells. As shown in Fig. 4C, treatment with Cu2−xSe NPs (50 or 100 μg mL−1) for 24 h resulted in a significant decrease in ATP levels in a dose-dependent manner. These results suggest that Cu2−xSe NPs induced mitochondrial damage in PC-12 cells.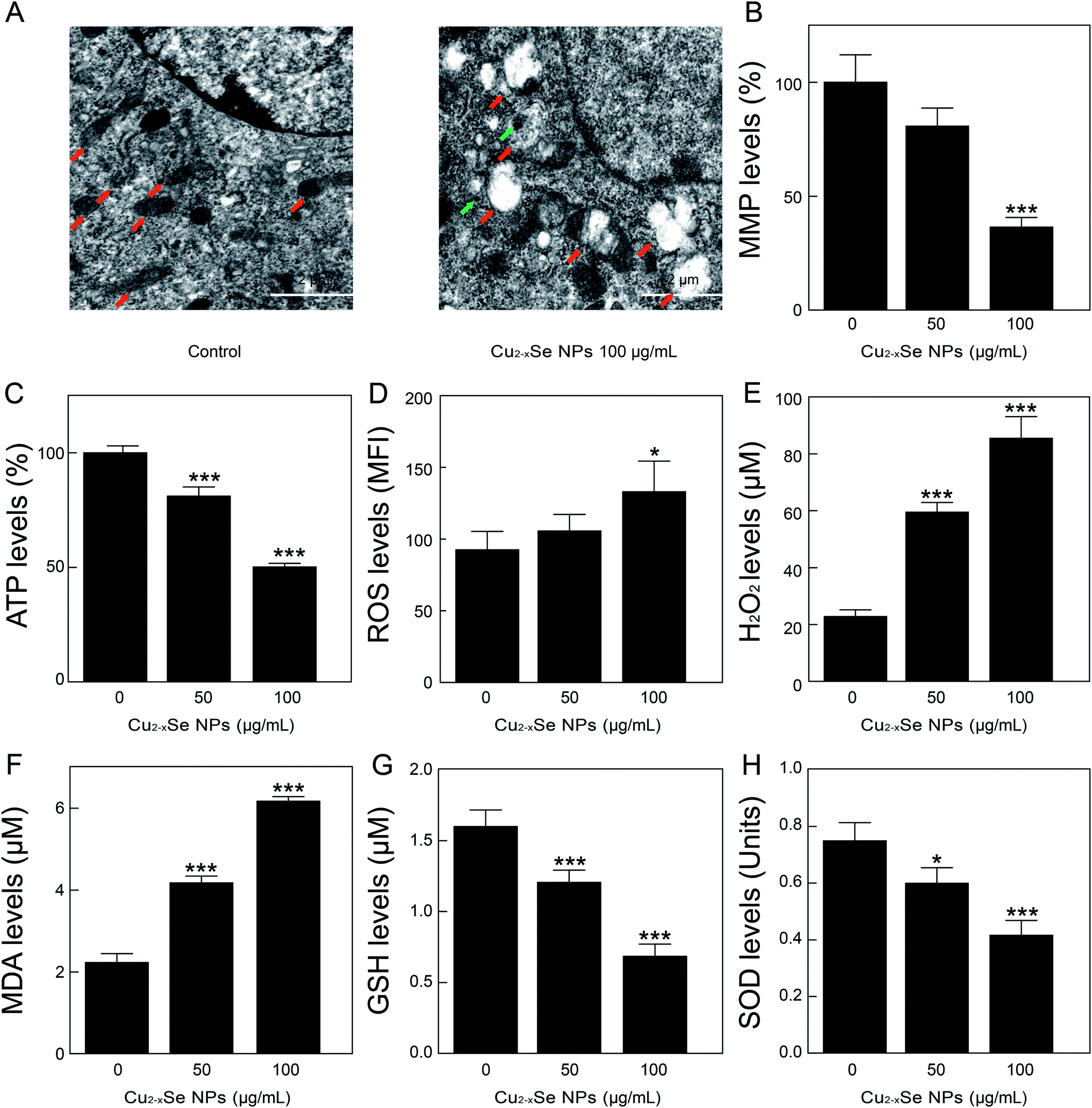 | ||
| Fig. 4 Cu2−xSe NPs induce mitochondrial damage and oxidative stress in PC-12 cells. (A) Representative TEM images depicting the mitochondrial ultrastructure of PC-12 cells treated with or without Cu2−xSe NPs (100 μg mL−1) for 24 h. Red arrow: mitochondrion. Green arrow: Cu2−xSe NPs. Scale bars: 2 μm. (B) PC-12 cells were treated with 50 and 100 μg mL−1 Cu2−xSe for 24 h. The mitochondrial membrane potential (MMP) was measured by rhodamine-123 staining and analyzed by a fluorescence microplate reader. (C) After the PC-12 cells were treated with 50 and 100 μg mL−1 Cu2−xSe for 24 h, cellular ATP concentration was measured using an ATP Determination kit. (D–H) Levels of ROS, H2O2, MDA, GSH, and SOD in PC-12 cells after treatment with or without Cu2−xSe NPs (50 or 100 μg mL−1) for 24 h. The data are expressed as the mean ± SD (n = 3), *P < 0.05, **P < 0.01, ***P < 0.001 compared to control. | ||
Studies had shown that oxidative stress is a key mechanism of NP-induced mitochondrial dysfunction.45,46 Additionally, oxidative stress was associated with increased production of oxidizing species, including ROS, H2O2, and MDA, and with a significant decrease in the effectiveness of antioxidant defenses systems, like SOD and GSH. Mitochondria are the major source of oxidative stress and produce superoxide anions during the process of electron transfer. Our results showed that the treatment of PC-12 cells with Cu2−xSe NPs resulted in a significant increase in the production of ROS, H2O2, and MDA in dose-dependent manners and a marked decrease in the levels of GSH and SOD in PC-12 cells (Fig. 4D–H). Taken together, these findings indicated that Cu2−xSe NPs induced mitochondrial dysfunction and oxidative stress in PC-12 cells.
3.4 Cu2−xSe NPs induce neurotoxicity via oxidative stress damage in vivo
To determine whether our in vitro findings could be replicated in vivo, BALB/c mice were used to investigate the molecular mechanisms in vivo. The striatum and hippocampus are crucial structures of the brain. The striatum plays essential roles in the modulation of movement pathways and the hippocampus is associated with learning and long-term memory and hippocampal dysfunction is closely associated with the risk of developing Parkinson's and Alzheimer's diseases.5,36,47–49 Next, we investigated whether exposure to Cu2−xSe NPs could cause striatum and hippocampal toxicity, including changes in tissue pathology, apoptosis, and oxidative injury. BALB/c mice were intravenously injected with Cu2−xSe NPs or physiological saline every two days for a total of 10 times over 20 days. The mice were euthanized at the termination of the experiment and the striatum and hippocampus were sectioned and stained with H&E to examine the tissue pathology and TUNEL analysis and immunohistochemical staining of cleaved caspase-3 were performed to analyze apoptosis in the cells. H&E staining of the Cu2−xSe NPs treatment group exhibited significant changes in pathology, with signs of necrosis, infiltration of inflammatory cells, and fibrosis, compared to BALB/c mice intravenously injected with physiological saline (Fig. 5 and 6). Additionally, Cu2−xSe NPs treatment dramatically increased the numbers of TUNEL-positive cells and cleaved caspase-3 immunoreactivity, both indicative of apoptosis, in the striatum and hippocampal tissues (Fig. 5 and 6). Next, all striatum and hippocampal tissues of each group were lysed and the levels of oxidative-stress-related biomarkers (H2O2, MDA, SOD, GSH) were measured. Consistent with our in vitro results, H2O2 and MDA levels were increased in BALB/c mice treated with Cu2−xSe NPs (10 mg kg−1) for 20 days and SOD and GSH activities were significantly decreased compared to the control group (Fig. 7A–D). Such findings suggest that Cu2−xSe NPs-mediated neurotoxicity in vivo is associated with damage from oxidative stress.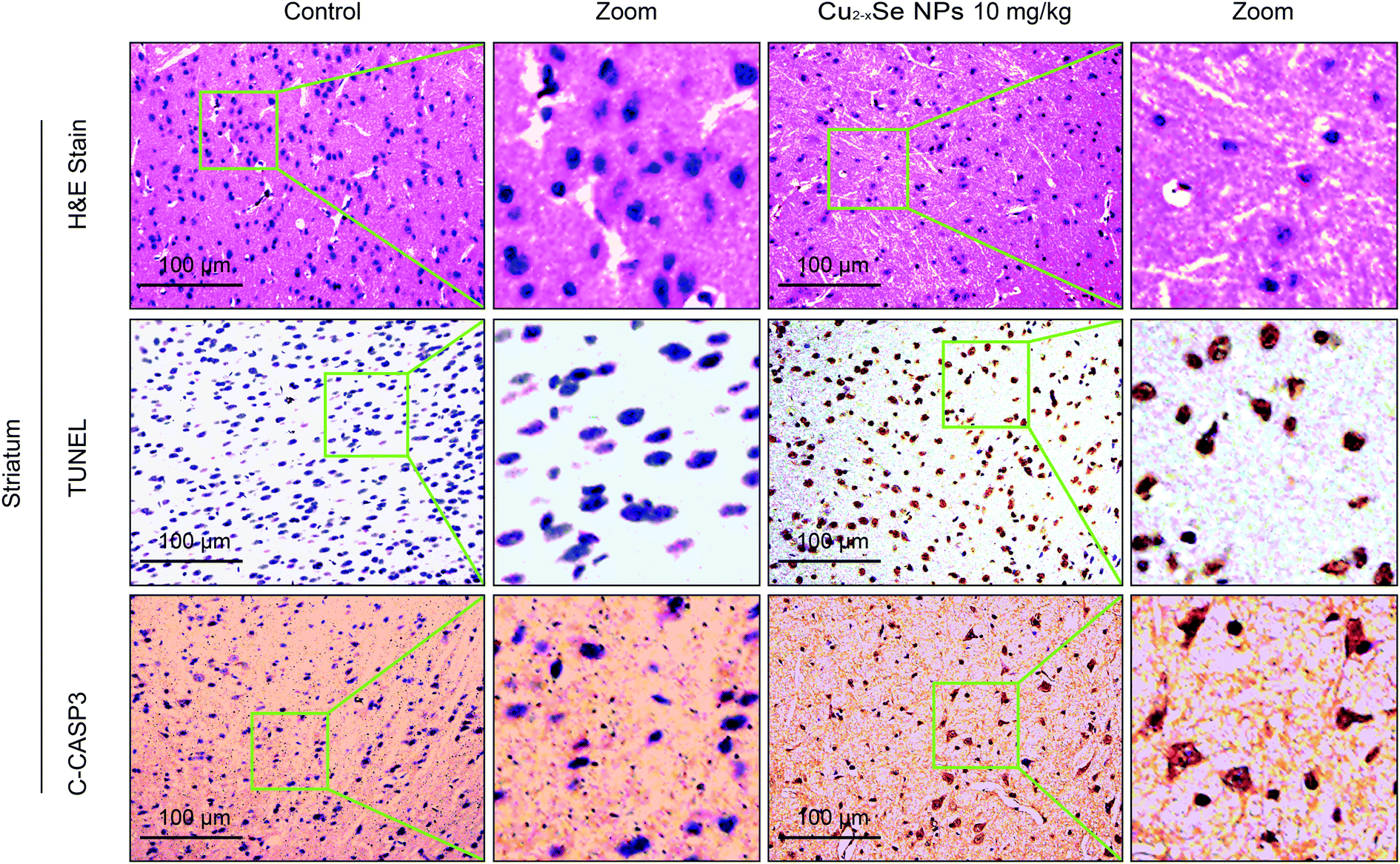 | ||
| Fig. 5 Cu2−xSe NPs induce neurotoxicity in vivo. The mice were euthanized after 20 days of intravenous injections every two days with physiological saline or 10 mg kg−1 Cu2−xSe NPs. The striatum tissues were sectioned and subjected to H&E staining, TUNEL assay, and immunohistochemical staining for cleaved caspase-3 and observed under a light microscope at 20× magnification (TCS SP5). Scale bars: 100 μm. | ||
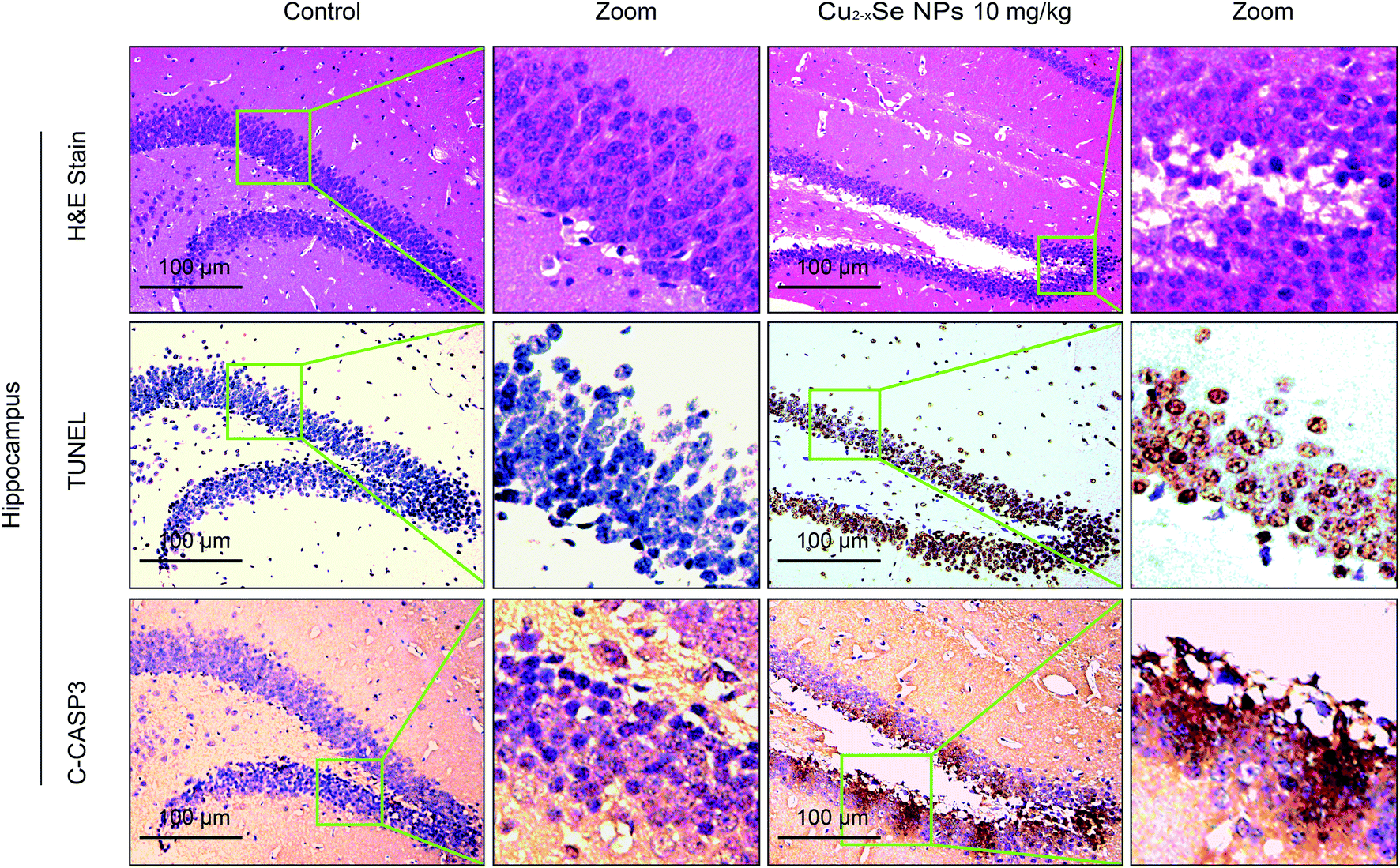 | ||
| Fig. 6 Cu2−xSe NPs induce neurotoxicity in vivo. The mice were euthanized after 20 days of intravenous injections every two days with physiological saline or 10 mg kg−1 Cu2−xSe NPs. The hippocampal tissues were sectioned and subjected to H&E staining, TUNEL assay, and immunohistochemical staining for cleaved caspase-3 and observed under a light microscope at 20× magnification (TCS SP5). Scale bars: 100 μm. | ||
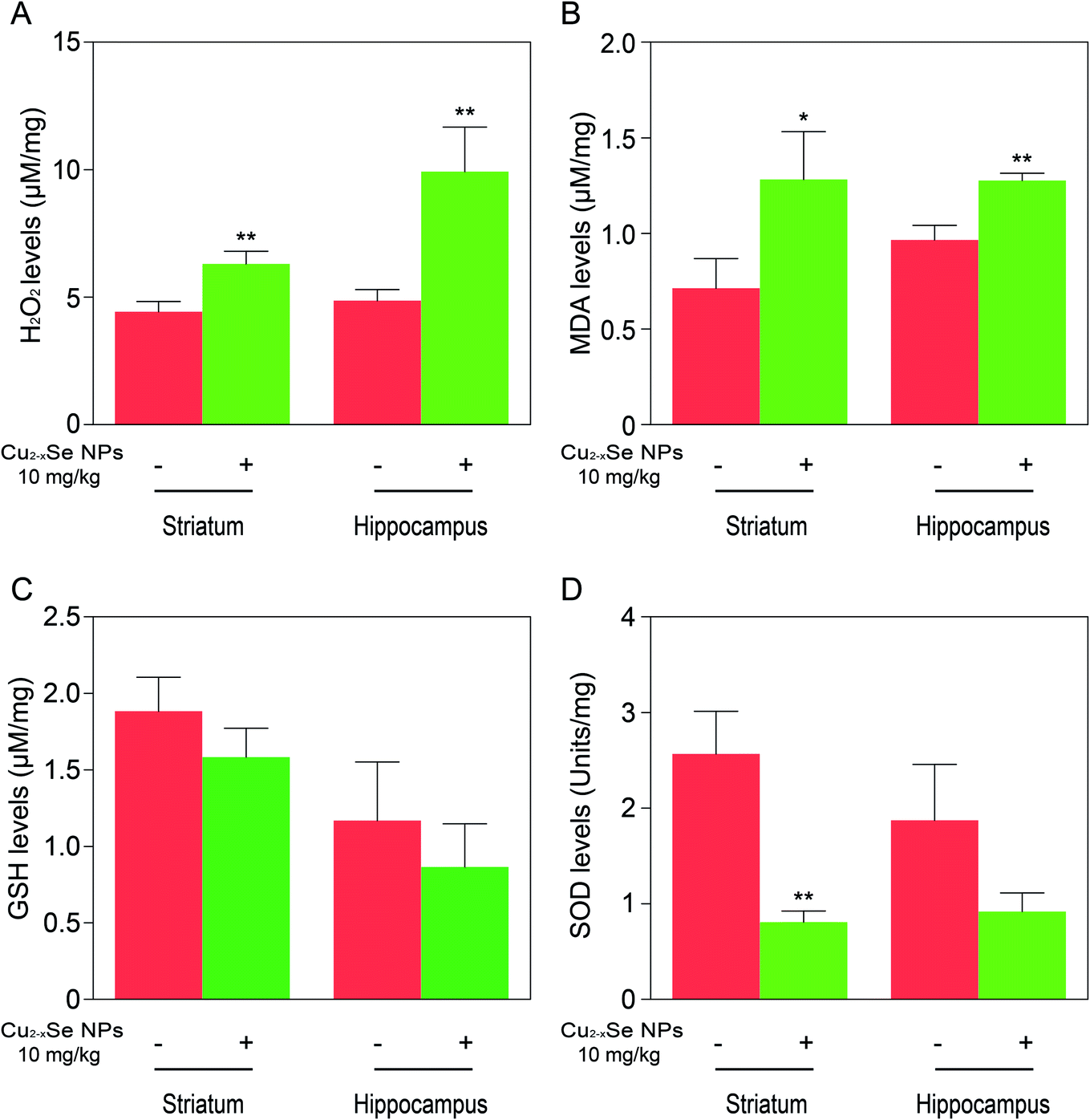 | ||
| Fig. 7 Cu2−xSe NPs induce oxidative stress damage in vivo. (A–D) Levels of H2O2, MDA, GSH, and SOD in the striatum and hippocampal tissues of BALB/c mice after treatment with or without Cu2−xSe NPs (10 mg kg−1) for 20 days. The data are expressed as the mean ± SD (n = 3), *P < 0.05, **P < 0.01, ***P < 0.001 compared to control. | ||
4. Discussion
Although nanomaterials have unique physicochemical properties, the toxicity of nanomaterials has received widespread attention. As an important member of copper chalcogenide nanomaterials, Cu2−xSe NPs have attracted much attention in cancer therapy due to its strong NIR absorption and photothermal effects. However, whether Cu2−xSe NPs have biological toxicity and how this might further affect other biological functions has not been reported. In the present investigation, we first used MTT assays and flow cytometry technology to confirm that Cu2−xSe NPs induced dose-dependent cell death and apoptosis in PC-12 cells. Our in vivo studies also revealed that Cu2−xSe NPs induced marked neurotoxicity in the striatum and hippocampal tissues of BALB/c mice, evidenced by pathologic changes, significantly increased numbers of TUNEL-positive cells, and increased cleaved caspase-3 immunoreactivity. Taken together, our findings suggested that Cu2−xSe NPs induced neurotoxicity in vitro and in vivo.Numerous studies have implicated mitochondrial dysfunction and oxidative stress damage in the neurotoxicity of metallic NPs.50–53 Chen et al. reported that nano-alumina particles could target mitochondria, causing changes in mitochondrial membrane potential and cellular oxidation levels.54 High doses of nano-silica resulted in damage to mitochondria and inhibition of mitochondrial biosynthesis.53 Meena et al. intravenously injected rats with TiO2 NPs. The results showed increased Ti concentrations in the brain, accompanied by reductions in SOD and GSH-Px, as well as excessive ROS and MDA production.55 Tian et al. concluded that ZnO NPs increased the MDA content and inhibited the activities of SOD and GSH-Px in the mouse brain after intraperitoneal injections.56 The antimicrobial activity of CuO NPs has been reported to act via the production of ROS57 and increasing oxidative stress was a key mechanism in PC-12 apoptosis induced by nano-Cu.58 Meanwhile, our innovation lies in confirming the neurotoxicity of Cu2−xSe NPs related to the generation of ROS in vitro and in vivo.
Consistent with these reports, our study demonstrated that the treatment of PC-12 cells with Cu2−xSe NPs (100 μg mL−1) for 24 h caused mitochondrial swelling, decreases in ATP production, and changes in mitochondrial membrane potential. After the cells were treated with Cu2−xSe NPs (100 μg mL−1) for 24 h, the levels of ROS, MDA and H2O2 were increased, whereas the GSH and SOD activities which were negatively correlated with oxidative stress, were significantly decreased. Similar oxidative stress effects were also observed in the striatum and hippocampus of Cu2−xSe NPs-treated BALB/c mice.
All of these results suggest that Cu2−xSe NPs caused neurotoxicity related to mitochondrial abnormalities and oxidative stress, as shown in Fig. 8. Cu2−xSe NPs penetrated the cell membrane and accumulated in the mitochondria, decreasing mitochondrial energy production and increasing mitochondrial oxidative damage associated with the pathogenesis of neurotoxicity. Since Cu2−xSe NPs are involved in the production of ROS, they may impair mitochondrial bioenergetics and generate oxidative stress.
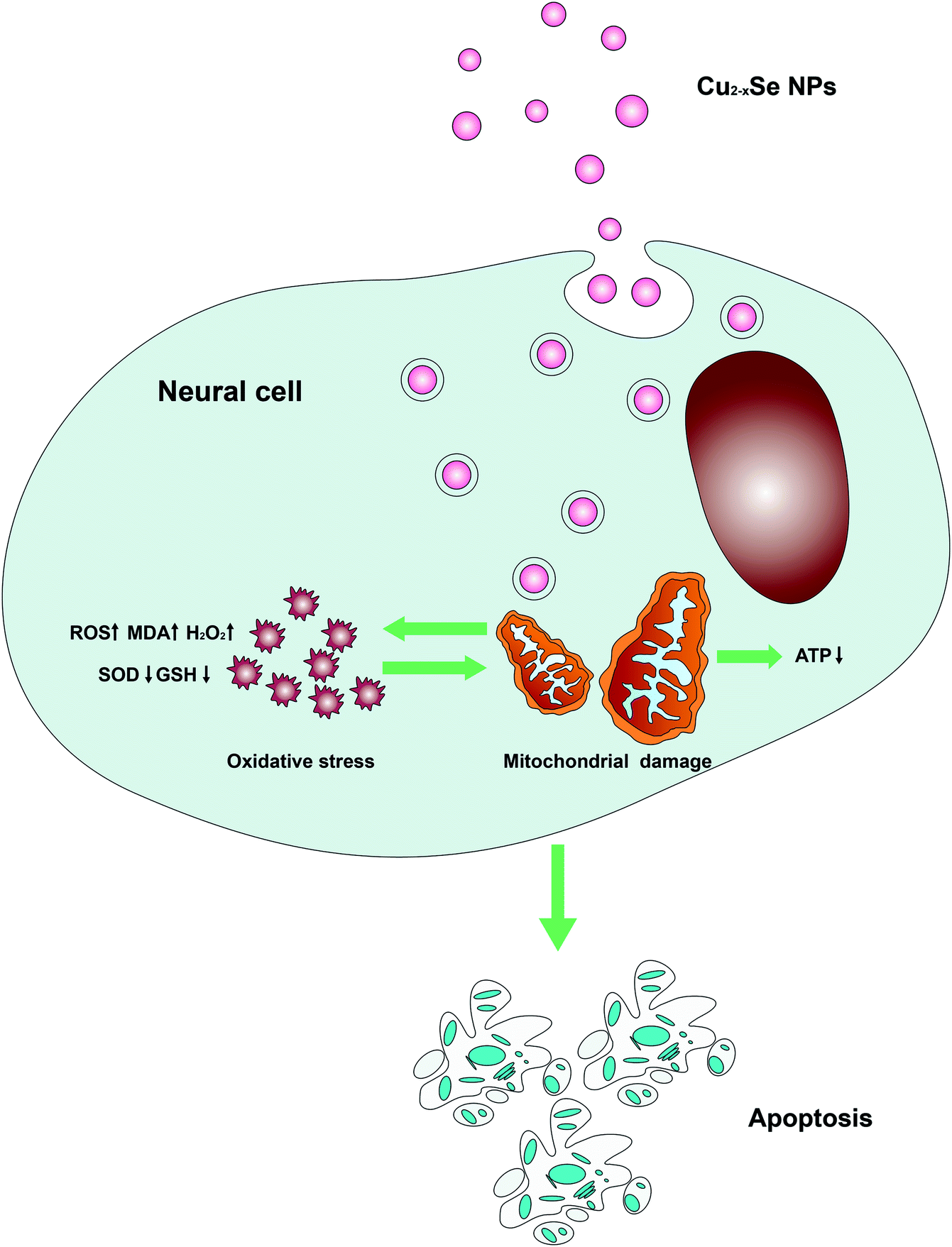 | ||
| Fig. 8 Model of Cu2−xSe NPs-induced apoptosis in PC-12 cells. Cu2−xSe NPs penetrated the cell membranes and accumulated in the mitochondria, decreased mitochondrial energy production, and increased mitochondrial oxidative damage involved in the pathogenesis of PC-12 cells apoptosis and neurotoxicity. | ||
It is worth mentioning that the biosafety of nanomaterials is closely related to their physicochemical characteristics, such as size, shape, charge and surface modifications.59–61 In future work, different shapes, sizes and surface modifications of Cu2−xSe NPs will be prepared and the in-depth mechanism of neurotoxicity will be studied.
5. Conclusions
In summary, our study investigated the neurotoxicological effects of Cu2−xSe NPs. The results demonstrated that Cu2−xSe NPs induced dose- and time-dependent cell death and apoptosis in PC-12 cells. Mitochondrial dysfunction and oxidative stress are believed to be the main molecular mechanisms underlying the neurotoxicity of Cu2−xSe NPs. These mechanisms were also confirmed in vivo. In the present work, we demonstrated the neurotoxicological effects of Cu2−xSe NPs and the potential mechanism is illustrated in Fig. 8. Our study provided useful information for understanding the neurotoxicity and safe application of Cu2−xSe NPs.Ethical statement
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Army Medical University and approved by the Animal Ethics Committee of Laboratory Animal Welfare and Ethics Committee of Army Medical University.Conflicts of interest
The authors report no conflicts of interest related to this work.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 31600806) and the Youth Talent Project of Army Medical University (Grant no. 2017R025).References
- D. Y. Joh, J. Kinder, L. H. Herman, S. Y. Ju, M. A. Segal, J. N. Johnson, G. K. Chan and J. Park, Nat. Nanotechnol., 2011, 6, 51–56 CrossRef CAS.
- F. Tang, L. Li and D. Chen, Adv. Mater., 2012, 24, 1504–1534 CrossRef CAS.
- S. Laurent, D. Forge, M. Port, A. Roch, C. Robic, L. Vander Elst and R. N. Muller, Chem. Rev., 2008, 108, 2064–2110 CrossRef CAS.
- S. Lanone and J. Boczkowski, Curr. Mol. Med., 2006, 6, 651–663 CrossRef CAS.
- X. Feng, A. Chen, Y. Zhang, J. Wang, L. Shao and L. Wei, Int. J. Nanomed., 2015, 10, 4321–4340 CAS.
- C. Medina, M. J. Santos-Martinez, A. Radomski, O. I. Corrigan and M. W. Radomski, Br. J. Pharmacol., 2007, 150, 552–558 CrossRef CAS.
- Y. Han, T. Wang, H. Liu, S. Zhang, H. Zhang, M. Li, Q. Sun and Z. Li, Nanoscale, 2019, 11, 11819–11829 RSC.
- M. L. Schipper, G. Iyer, A. L. Koh, Z. Cheng, Y. Ebenstein, A. Aharoni, S. Keren, L. A. Bentolila, J. Li, J. Rao, X. Chen, U. Banin, A. M. Wu, R. Sinclair, S. Weiss and S. S. Gambhir, Small, 2009, 5, 126–134 CrossRef CAS.
- J. V. Jokerst, T. Lobovkina, R. N. Zare and S. S. Gambhir, Nanomedicine, 2011, 6, 715–728 CrossRef CAS.
- J. You, J. Zhou, M. Zhou, Y. Liu, J. D. Robertson, D. Liang, C. Van Pelt and C. Li, Part. Fibre Toxicol., 2014, 11, 26–39 CrossRef.
- Z. Liu, W. Cai, L. He, N. Nakayama, K. Chen, X. Sun, X. Chen and H. Dai, Nat. Nanotechnol., 2007, 2, 47–52 CrossRef CAS.
- N. R. Jacobsen, P. Moller, P. A. Clausen, A. T. Saber, C. Micheletti, K. A. Jensen, H. Wallin and U. Vogel, Basic Clin. Pharmacol. Toxicol., 2017, 121, 30–43 CrossRef CAS.
- Y. Zhao, H. Pan, Y. Lou, X. Qiu, J. Zhu and C. Burda, J. Am. Chem. Soc., 2009, 131, 4253–4261 CrossRef CAS PubMed.
- J. M. Luther, P. K. Jain, T. Ewers and A. P. Alivisatos, Nat. Mater., 2011, 10, 361–366 CrossRef CAS.
- D. Dorfs, T. Hartling, K. Miszta, N. C. Bigall, M. R. Kim, A. Genovese, A. Falqui, M. Povia and L. Manna, J. Am. Chem. Soc., 2011, 133, 11175–11180 CrossRef CAS PubMed.
- S. Q. Lie, D. M. Wang, M. X. Gao and C. Z. Huang, Nanoscale, 2014, 6, 10289–10296 RSC.
- T. Yang, H. Y. Zou and C. Z. Huang, ACS Appl. Mater. Interfaces, 2015, 7, 15447–15457 CrossRef CAS PubMed.
- B. Li, Q. Wang, R. Zou, X. J. Liu, K. B. Xu, W. Y. Li and J. Q. Hu, Nanoscale, 2014, 6, 3274–3282 RSC.
- Q. Tian, F. Jiang, R. Zou, Q. Liu, Z. Chen, M. Zhu, S. Yang, J. Wang, J. Wang and J. Hu, ACS Nano, 2011, 5, 9761–9771 CrossRef CAS.
- C. M. Hessel, V. P. Pattani, M. Rasch, M. G. Panthani, B. Koo, J. W. Tunnell and B. A. Korgel, Nano Lett., 2011, 11, 2560–2566 CrossRef CAS.
- M. Zhou, J. Li, S. Liang, A. K. Sood, D. Liang and C. Li, ACS Nano, 2015, 9, 7085–7096 CrossRef CAS.
- S. Zhang, C. Sun, J. Zeng, Q. Sun, G. Wang, Y. Wang, Y. Wu, S. Dou, M. Gao and Z. Li, Adv. Mater., 2016, 28, 8927–8936 CrossRef CAS.
- Y. Liu, W. Liu, J. Huang, W. Lai, F. Leng, C. Hu, Q. Zhang, M. Zhou, Q. Tang, F. Sheng, G. Li and R. Zhang, Nanomedicine, 2019, 14, 1307–1321 CrossRef CAS.
- Y. Li, W. Lu, Q. Huang, M. Huang, C. Li and W. Chen, Nanomedicine, 2010, 5, 1161–1171 CrossRef CAS.
- W. Feng, W. Nie, Y. Cheng, X. Zhou, L. Chen, K. Qiu, Z. Chen, M. Zhu and C. He, Nanomedicine, 2015, 11, 901–912 CrossRef CAS.
- L. Guo, I. Panderi, D. D. Yan, K. Szulak, Y. Li, Y. T. Chen, H. Ma, D. B. Niesen, N. Seeram, A. Ahmed, B. Yan, D. Pantazatos and W. Lu, ACS Nano, 2013, 7, 8780–8793 CrossRef CAS.
- Z. Yang, Z. W. Liu, R. P. Allaker, P. Reip, J. Oxford, Z. Ahmad and G. Ren, J. R. Soc., Interface, 2010, 7, 411–422 CrossRef.
- Y. Liu, M. Nguyen, A. Robert and B. Meunier, Acc. Chem. Res., 2019, 52, 2026–2035 CrossRef CAS.
- S. Miranda, C. Opazo, L. F. Larrondo, F. J. Muñoz, F. Ruiz, F. Leighton and N. C. Inestrosa, Prog. Neurobiol., 2000, 62, 633–648 CrossRef CAS.
- W. Cerpa, L. Varela-Nallar, A. E. Reyes, A. N. Minniti and N. C. Inestrosa, Mol. Aspects Med., 2005, 26, 405–420 CrossRef CAS.
- L. Zhang, R. Bai, Y. Liu, L. Meng, B. Li, L. Wang, L. Xu, L. Le Guyader and C. Chen, Nanotoxicology, 2012, 6, 562–575 CrossRef CAS.
- I. L. Yurkova, J. Arnhold, G. Fitzl and D. Huster, Chem. Phys. Lipids, 2011, 164, 393–400 CrossRef CAS.
- D. Strausak, J. F. Mercer, H. H. Dieter, W. Stremmel and G. Multhaup, Brain Res. Bull., 2001, 55, 175–185 CrossRef CAS.
- J. Chen, X. Dong, Y. Xin and M. Zhao, Aquat. Toxicol., 2011, 101, 493–499 CrossRef CAS.
- R. Hu, L. Zheng, T. Zhang, G. Gao, Y. Cui, Z. Cheng, J. Cheng, M. Hong, M. Tang and F. Hong, J. Hazard. Mater., 2011, 191, 32–40 CrossRef CAS.
- J. Wu, T. Ding and J. Sun, NeuroToxicology, 2013, 34, 243–253 CrossRef CAS PubMed.
- M. A. Siddiqui, H. A. Alhadlaq, J. Ahmad, A. A. Al-Khedhairy, J. Musarrat and M. Ahamed, PLoS One, 2013, 8, e69534 CrossRef CAS.
- G. B. Li, T. Zhou, L. Liu, J. Chen, Z. Zhao, Y. Peng, P. Li and N. Gao, Blood Canc. J., 2013, 3, e108 CrossRef CAS.
- G. B. Li, R. Q. Fu, H. M. Shen, J. Zhou, X. Y. Hu, Y. X. Liu, Y. N. Li, H. W. Zhang, X. Liu, Y. H. Zhang, C. Huang, R. Zhang and N. Gao, Oncotarget, 2017, 8, 10359–10374 Search PubMed.
- K. Miszta, G. Gariano, R. Brescia, S. Marras, F. De Donato, S. Ghosh, L. De Trizio and L. Manna, J. Am. Chem. Soc., 2015, 137, 12195–12198 CrossRef CAS.
- S. Goel, F. Chen and W. Cai, Small, 2014, 10, 631–645 CrossRef CAS.
- M. Brandon, P. Baldi and D. C. Wallace, Oncogene, 2006, 25, 4647–4662 CrossRef CAS.
- J. Singleterry, A. Sreedhar and Y. Zhao, Mitochondrion, 2014, 17, 50–55 CrossRef CAS.
- T. van Zutphen, J. Ciapaite, V. W. Bloks, C. Ackereley, A. Gerding, A. Jurdzinski, R. A. de Moraes, L. Zhang, J. C. Wolters, R. Bischoff, R. J. Wanders, S. M. Houten, D. Bronte-Tinkew, T. Shatseva, G. F. Lewis, A. K. Groen, D. J. Reijngoud, B. M. Bakker, J. W. Jonker, P. K. Kim and R. H. Bandsma, J. Hepatol., 2016, 65, 1198–1208 CrossRef CAS.
- L. Shi, B. Hernandez and M. Selke, J. Am. Chem. Soc., 2006, 128, 6278–6279 CrossRef CAS.
- A. Nel, T. Xia, L. Madler and N. Li, Science, 2006, 311, 622–627 CrossRef CAS.
- D. Arneson, G. Zhang, Z. Ying, Y. Zhuang, H. R. Byun, I. S. Ahn, F. Gomez-Pinilla and X. Yang, J. Nanopart. Res., 2018, 17, 3894 Search PubMed.
- P. Huot, M. Levesque and A. Parent, Brain, 2007, 130, 222–232 CrossRef.
- M. P. Laakso, M. Lehtovirta, K. Partanen, P. J. Riekkinen and H. Soininen, Biol. Psychiatry, 2000, 47, 557–561 CrossRef CAS.
- V. Kumar and K. D. Gill, NeuroToxicology, 2014, 41, 154–166 CrossRef CAS.
- M. D. Prokić, S. S. Borković-Mitić, I. I. Krizmanić, J. J. Mutić, J. Đ. Trifković, J. P. Gavrić, S. G. Despotović, B. R. Gavrilović, T. B. Radovanović, S. Z. Pavlović and Z. S. Saičić, Ecotoxicology, 2016, 25, 1531–1542 CrossRef.
- K. Jomova, D. Vondrakova, M. Lawson and M. Valko, Mol. Cell. Biochem., 2010, 345, 91–104 CrossRef CAS.
- B. Song, Y. Zhang, J. Liu, X. Feng, T. Zhou and L. Shao, Nanoscale Res. Lett., 2016, 11, 291 CrossRef.
- Y. Chen and S. B. Gibson, Autophagy, 2008, 4, 246–248 CrossRef CAS.
- M. Ramovatar, K. Sumit and R. Paulraj, J. Nanopart. Res., 2015, 17, 49–62 CrossRef.
- L. Tian, B. Lin, L. Wu, K. Li, H. Liu, J. Yan, X. Liu and Z. Xi, Sci. Rep., 2015, 5, 16117 CrossRef CAS.
- G. Applerot, J. Lellouche, A. Lipovsky, Y. Nitzan, R. Lubart, A. Gedanken and E. Banin, Small, 2012, 8, 3326–3337 CrossRef CAS.
- P. Xu, J. Xu, S. Liu, G. Ren and Z. Yanget, J. Nanopart. Res., 2012, 14, 906–914 CrossRef.
- P. N. Navya and H. K. Daima, Nano Convergence, 2016, 3, 1–14 CrossRef CAS.
- M. M. Ugru, S. Sheshadri, D. Jain, H. Madhyastha, R. Madhyastha, M. Maruyama, P. N. Navya and H. K. Daima, Colloids Surf., A, 2018, 3, 1–27 CrossRef.
- K. J. Sourabh Monnappa, N. Firdose, G. Madhu Shree, K. Nath and P. N. Navya, Int. J. Nanotechnol., 2017, 14, 816–832 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra06245a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |