
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePalygorskite-anchored Pd complexes catalyze the coupling reactions of pyrimidin-2-yl sulfonates†
Huiying
Zhan
*a,
Rongrong
Zhou
a,
Xudong
Chen
a,
Quanlu
Yang
*a,
Hongyan
Jiang
b,
Qiong
Su
b,
Yanbin
Wang
b,
Jia
Li
b,
Lan
Wu
b and
Shang
Wu
 *b
*b
aCollege of Chemical Engineering, Lanzhou University of Arts and Science, Beimiantan 400, Lanzhou, Gansu 730000, People's Republic of China. E-mail: 674585005@qq.com; yangquanlu2002@163.com
bKey Laboratory for Utility of Environment-Friendly Composite Materials and Biomass in Universities of Gansu Province, College of Chemical Engineering, Northwest Minzu University, Lanzhou, Gansu 730030, People's Republic of China
First published on 26th September 2019
Abstract
In this work, an anchored Pd complex (PGS–APTES–Pd(OAc)2) was prepared via simple and green steps from the natural clay mineral palygorskite and was well characterized by XPS, XRD, IR, SEM, and EDX. This complex was further utilized as a fine catalyst for the C–C/C–N coupling reactions of pyrimidin-2-yl sulfonates. Subsequently, the cyclic utilization test indicated the high stability and sustainability of this PGS–APTES–Pd(OAc)2 catalyst, and no activation was required in the recycling process, providing an applicable and reusable catalyst in organic synthesis.
Introduction
The synthesis of heterocyclic compounds has been investigated in many studies for numerous times due to the important structural units of many medical and other bioactive molecules. Among them, molecules containing 3,4-dihydropyrimidin-2(1H)-one (DHPM) scaffolds are widely found in natural products and compounds with unique pharmaceutical activities (Fig. 1). For example, compounds A and B have been shown to be potent calcium blockers,1 and compounds C and D have been proven to possess calcium modulatory activities.2 In addition, other DHPM derivatives can be used as mitotic kinesin inhibitors and hepatitis B virus replication inhibitors.3 Therefore, the investigation of the functionalization and transformation of DHPM derivatives is of great value and significance.4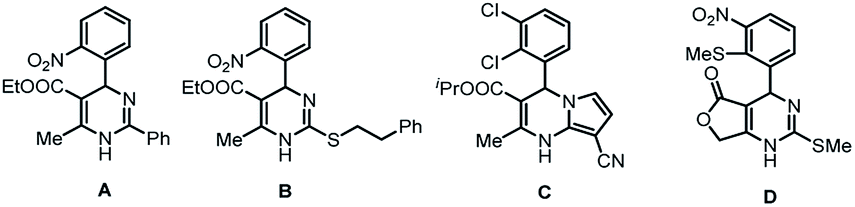 | ||
| Fig. 1 DHPMs with biological activities. | ||
Palladium-catalyzed C–C coupling reactions, with the advantages of high activity and concise reaction conditions, are widely applied in synthetic chemistry.5 They have also been used in the modification of DHPMs with considerable progress reported in recent years.6 However, these methods generally face the problem of the consumption of the precious metal catalyst in palladium homogeneous catalysis. Also, the catalyst is difficult to be recovered from the reaction system. This problem may cause serious loss of palladium and metal contamination of the products. Notably, in routine chemical and drug syntheses, the content of residual metals in the products is strictly monitored, and staining of palladium black from the reaction would invalidate the pharmaceutical products.7 Above all, these deficiencies limit their wide applications in industries. In order to overcome these shortcomings of the homogeneous Pd catalytic reactions, the development of supported palladium catalysts has become a research hotspot in the field of synthetic chemistry and has developed rapidly in recent years.8 The supported heterogeneous catalyst can be recycled via simple operations as the catalyst can be separated and recovered after the completion of the reaction.
Clay minerals, in view of their large specific surface area, rich pore structure and excellent ion exchange properties, have been adopted as excellent catalyst carriers.9 Among the previously reported materials, palygorskite (PGS), benefiting from its crystalline morphology, depositional mode and crystal structure, preserves high internal and external surface areas. It is a natural, non-toxic, cheap and easily available clay mineral. A Lewis acidification center and an alkalization center can be formed on the inner surface of palygorskite clay after treatment, which makes the palygorskite crystal not only meet the microporous and surface characteristics required for the heterogeneous catalytic reaction, but also have the capabilities of the catalytic cracking of molecular sieves.10 In 2010, Lei et al.10c reported the oxidation of alcohols using palygorskite as the catalyst and water as the solvent. Inspired by this work, we supposed that palygorskite may not only serve as a catalyst itself, but it might also be used as a good catalyst carrier. Herein, we reported the preparation of a palygorskite-anchored Pd complex (PGS–APTES–Pd(OAc)2), which was used as the catalyst for DHPMs in C–C coupling with arylboronic acids and terminal alkynes and C–N coupling with arylamines. In addition, the catalytic performance and recyclability of PGS–APTES–Pd(OAc)2 were investigated.
Results and discussion
Synthesis and characterization of the catalyst
Palygorskite was pretreated with dilute HCl10a and then, the activated PGS (1.0 g) was added to 150 mL xylene under ultrasonication for 1 h. A (3-aminopropyl)triethoxysilane (APTES) ethanol aqueous solution (EtOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O = 9:1) was hydrolyzed for 5 min; then, we added this to the PGS dispersion under vigorous stirring at 80 °C. The PGS–APTES product was obtained by filtration, washed with anhydrous ethanol, and then dried in vacuum at 50 °C. Finally, the product was crushed, sifted out through 200 mesh sieves, and extracted with ethanol.
:H2O = 9:1) was hydrolyzed for 5 min; then, we added this to the PGS dispersion under vigorous stirring at 80 °C. The PGS–APTES product was obtained by filtration, washed with anhydrous ethanol, and then dried in vacuum at 50 °C. Finally, the product was crushed, sifted out through 200 mesh sieves, and extracted with ethanol.
Then, 1 g PGS–APTES and 0.2 g Pd (OAc)2 were added into 20 mL deionized water, and the mixture was stirred at room temperature for 3 h. When the reaction was complete, the liquid was discarded by centrifugation, and the solid was washed with deionized water (3 × 20 mL) and acetone (3 × 20 mL) and dried in vacuum at 40 °C for 4 h; the gray powder obtained was PGS–APTES–Pd(OAc)2.
ICP-AES analysis revealed that the content of Pd in the PGS–APTES–Pd(OAc)2 catalyst was 0.463 wt% (0.0435 mmol g−1).
IR spectra were obtained for PGS, PGS–APTES and PGS–APTES–Pd(OAc)2 (Fig. 2). In the FTIR spectrum shown in Fig. 2(a), the absorption peaks appearing at 3614 cm−1 and 3542 cm−1 are attributed to the telescopic vibration of the –OH group (Al, Fe, and Mg) of PGS; also, the absorption peaks at 3442 cm−1 and 1641 cm−1 are ascribed to the telescopic vibration and the bending vibration of the adsorbent H2O. The absorption peaks at 1031 cm−1 and 779 cm−1 corresponded to the Si–O bond flexural vibration and the Si–O–Si bond telescopic vibration. The absorption peak at 1438 cm−1 was ascribed to Lewis acid. For PGS–APTES, a weaker N–H bending (in-plane) vibration absorption peak was found at 1562 cm−1. The absorption peaks at 2933 and 2877 cm−1 corresponded to the asymmetric and symmetric stretching vibrations of methylene. The absorption peak at 1438 cm−1 disappeared, and the C–H bending (in-plane) vibration absorption peak appeared at 1562 cm−1. These changes in the above-mentioned spectrum indicated that APTES was successfully grafted onto the surface of PGS. In Fig. 2(c), compared with the observations for PGS–APTES, the absorption bands at 1778 cm−1 and 1490 cm−1 are attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching and –CH3 antisymmetric stretching vibrations of Pd(OAc)2, respectively. It was revealed that coordination was formed between Pd(OAc)2 and PGS–APTES.
O stretching and –CH3 antisymmetric stretching vibrations of Pd(OAc)2, respectively. It was revealed that coordination was formed between Pd(OAc)2 and PGS–APTES.
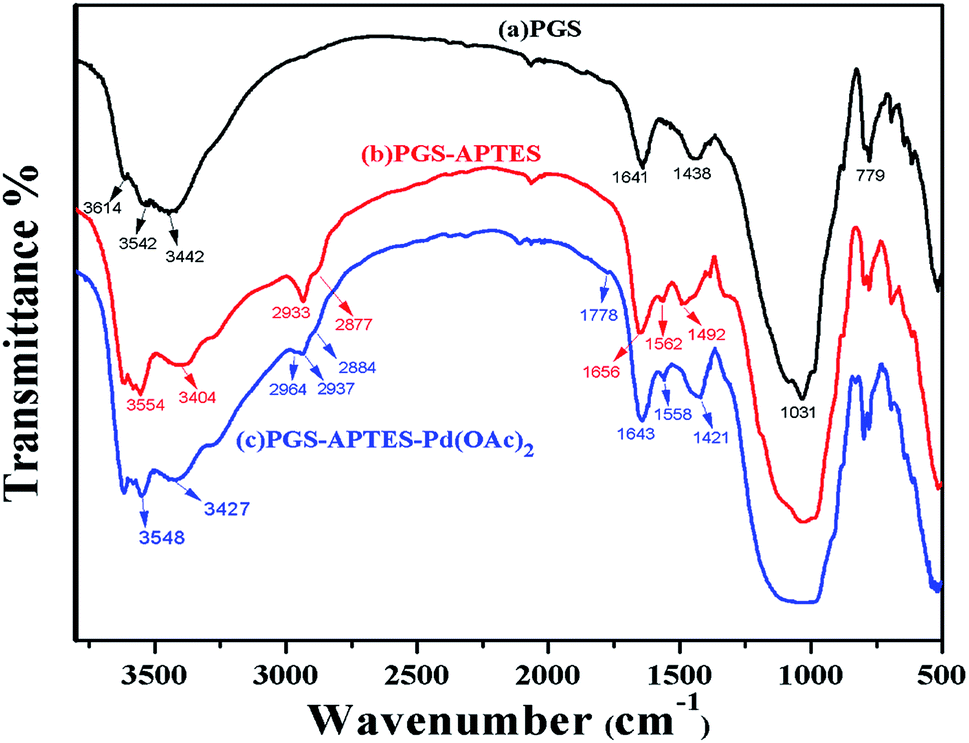 | ||
| Fig. 2 IR spectra of (a) PGS, (b) PGS–APTES and (c) PGS–APTES–Pd(OAc)2. | ||
From the XRD spectrum (Fig. 3), it is revealed that PGS, PGS–APTES and PGS–APTES–Pd(OAc)2 show the same diffraction peaks at 2θ = 8.3°, 21.3°, 26.7°, 36.8°, and 60.3°, indicating that the fibrous palygorskite is not likely to be layered montmorillonite. This ordered structure in palygorskite is difficult to change. In addition, the relatively low loading of Pd also caused no obvious changes in the characteristic diffraction peaks.
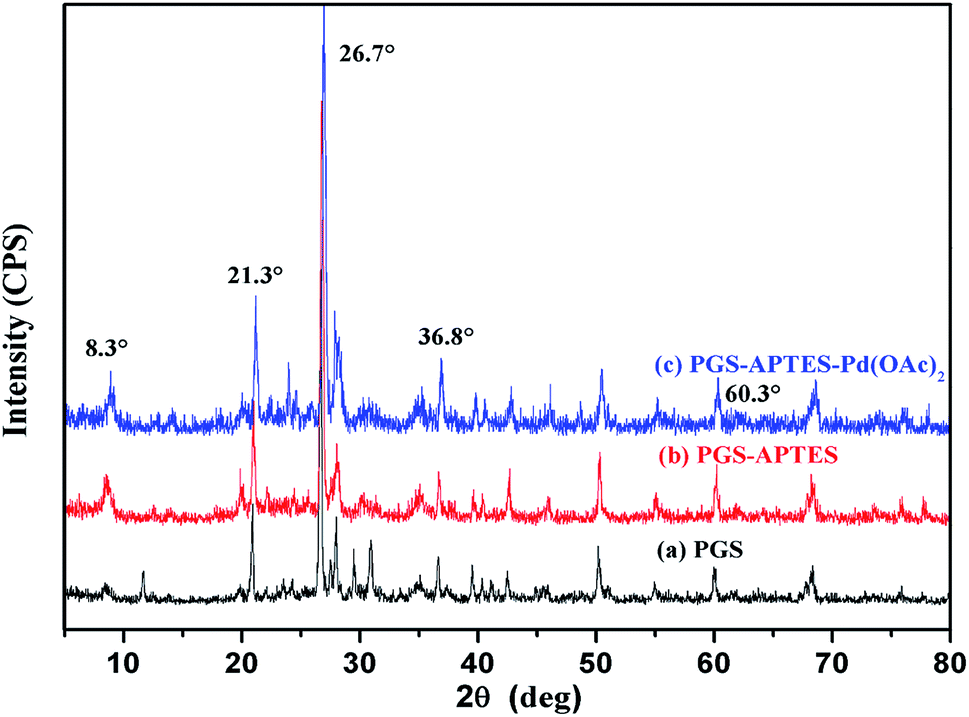 | ||
| Fig. 3 XRD spectrum of (a) PGS, (b) PGS–APTES and (c) PGS–APTES–Pd(OAc)2. | ||
The thermogravimetric analysis (Tg) of PGS, PGS–ATPES and PGS–ATPES–Pd is shown in Fig. 4. At 100 °C, there was a slight increase in the loss of the mass of PGS–APTES and PGS–APTES–Pd(OAc)2 compared with that for PGS, which may be because the water molecules of PGS were substituted by APTES molecules.11 In addition, at a temperature around 100 °C to 600 °C, the loss of the mass of PGS–APTES and PGS–APTES–Pd(OAc)2 further increased, which was regarded as the decomposition of the surfactant APTES molecules.11 Consequently, APTES was successfully supported on PGS.
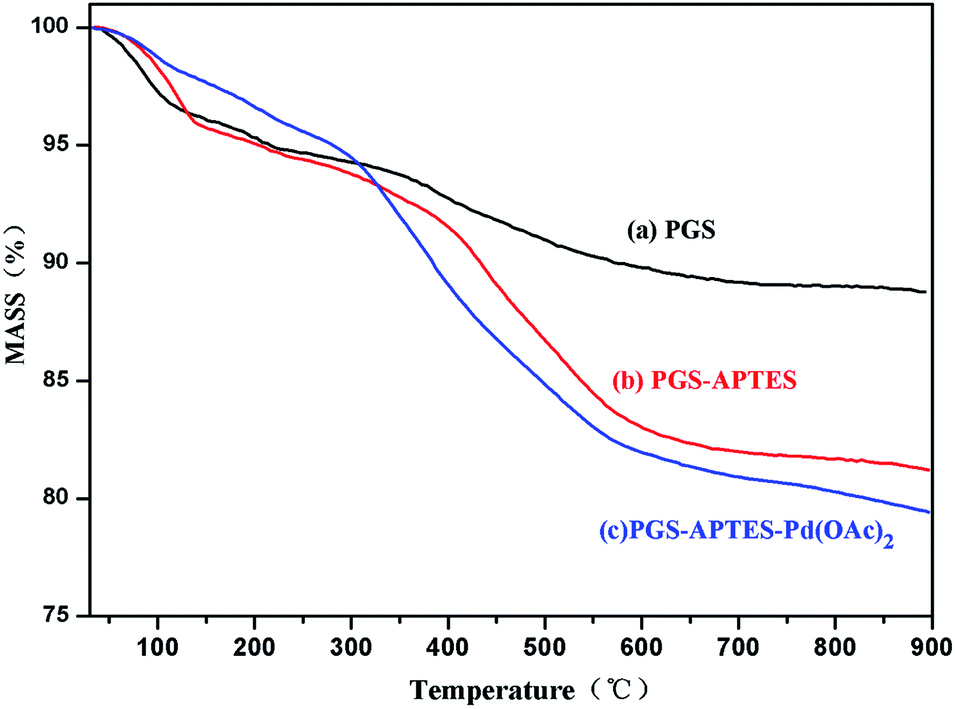 | ||
| Fig. 4 TG spectrum of (a) PGS, (b) PGS–APTES and (c) PGS–APTES–Pd(OAc)2. | ||
The SEM images of stony palygorskite, PGS–APTES and PGS–APTES–Pd(OAc)2 are shown in Fig. 5. The structure of a palygorskite-like typical rod can be clearly seen in Fig. 5(a), with a diameter of 50 nm and a length of 500–1000 nm. When grafted onto APTES (Fig. 5(b)), the rod structure did not change significantly, and the arrangement became loose and had more pores. After Pd(OAc)2 was loaded, no reunion of Pd particles was found (Fig. 5(c)). The corresponding EDX and EDS spectra are shown in Fig. 5(d–h), and it was demonstrated that N and Pd did exist in uniform distribution though the APTES and Pd loading was relatively less.
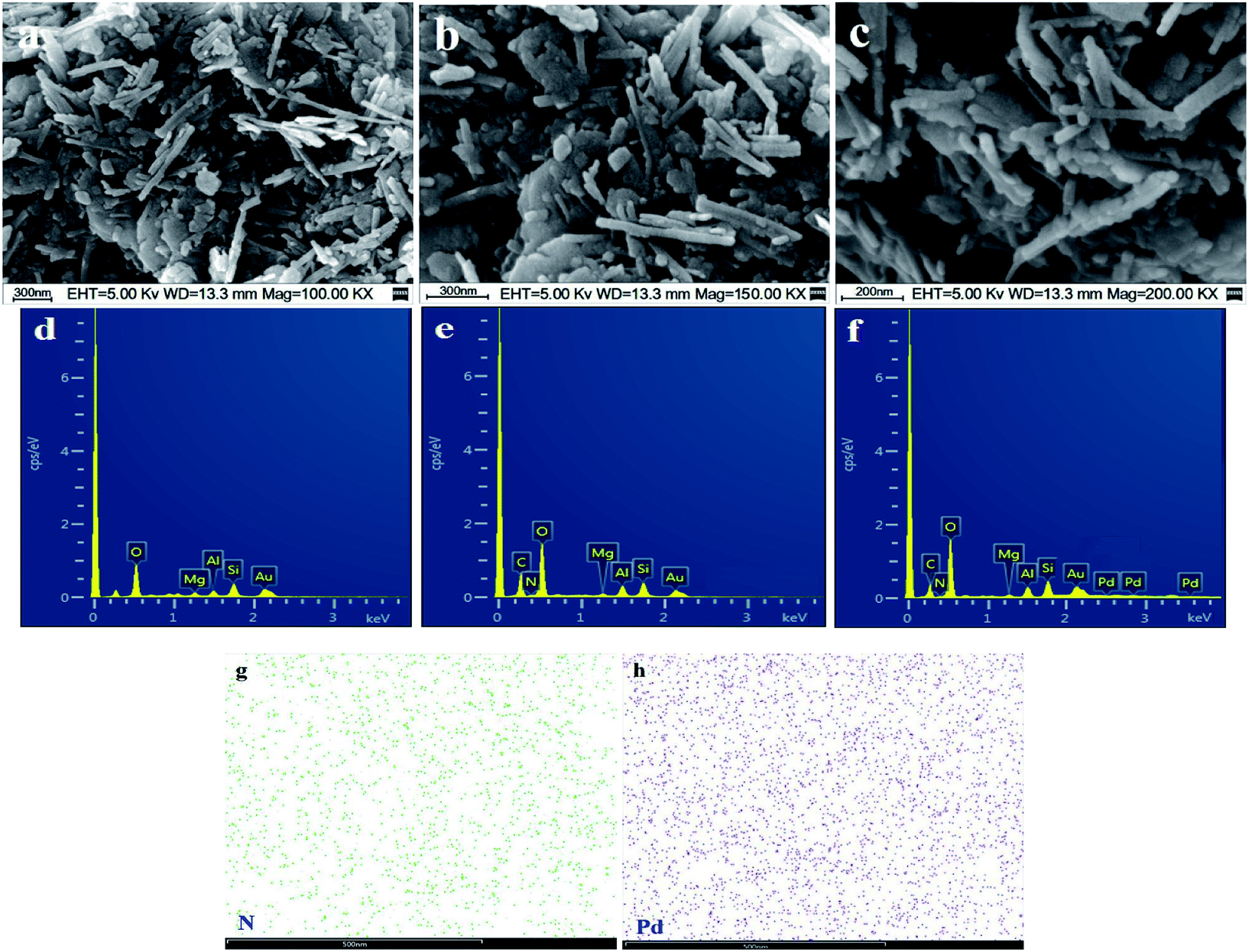 | ||
| Fig. 5 SEM and EDX images of (a and d) PGS, (b and e) PGS–APTES, and (c and f) PGS–APTES–Pd(OAc)2; (g and h) the corresponding EDS mapping of N and Pd. | ||
Fig. 6 shows the TEM images of PGS, PGS–APTES and PGS–APTES–Pd(OAc)2. The rod-like shape of palygorskite can be seen clearly, and the Pd particles are distributed on the surface of PGS. Obviously, the interactions between Pd and N at the surface of PGS resulted in the good dispersion of Pd particles at a high magnification (Fig. 6(d)).
 | ||
| Fig. 6 TEM images of (a) PGS, (b) PGS–APTES, and (c) PGS–APTES–Pd(OAc)2. (d) The TEM image of the catalyst at high magnification. | ||
In order to further determine whether Pd(OAc)2 is loaded onto the palygorskite surface in the form of adsorption or coordination bonds and the valence state of Pd before and after the loading process, the catalysts were characterized by XPS (Fig. 7). The N 1s binding energies of PGS–APTES and PGS–APTES–Pd(OAc)2 were 399.18 eV and 399.88 eV, respectively, increasing by 0.70 eV. In Fig. 7(c), the Pd 3d peaks consist of two components with binding energies of 343.78 eV (Pd 3d3/2) and 338.48 eV (Pd 3d5/2) for Pd(OAc)2 and 343.28 eV (Pd 3d3/2) and 337.98 eV (Pd 3d5/2) for PGS–APTES–Pd(OAc)2, respectively, which were reduced by 0.50 eV. In addition, this originated from Pd 3d3/2 and Pd 3d5/2 of Pd(II). Therefore, a coordination bond might be formed between Pd(OAc)2 and PGS–APTES.
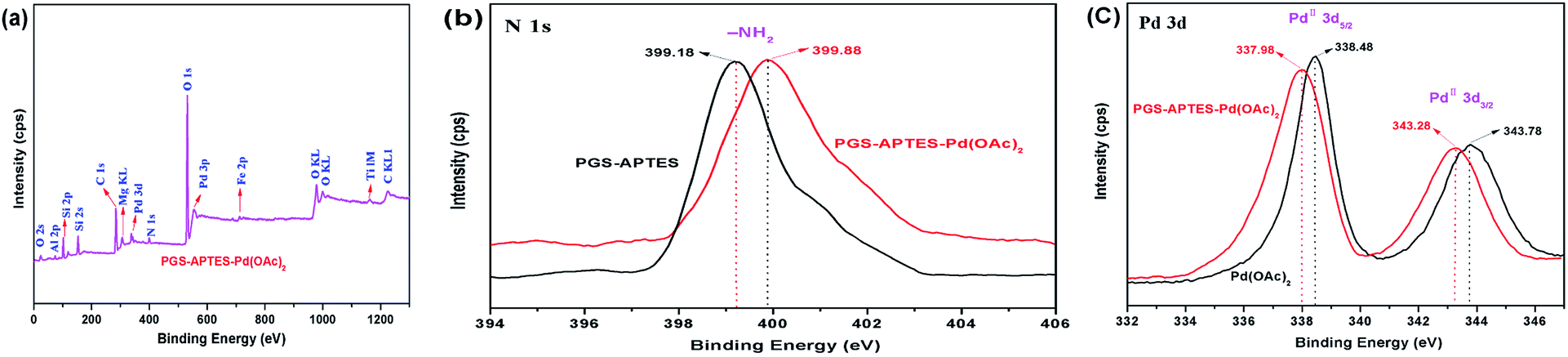 | ||
| Fig. 7 The XPS spectra of the catalyst: (a) full spectra, (b) N 1s, (c) Pd 3d. | ||
PGS–APTES–Pd(OAc)2–catalyzed Suzuki coupling reaction of pyrimidin-2-yl sulfonates
As the catalyst was well characterized, PGS–APTES–Pd(OAc)2 was used in the coupling reactions of pyrimidin-2-yl sulfonates. Ethyl 4-methyl-6-phenyl-2-(tosyloxy)pyrimidine-5-carboxylate (1a) and phenylboric acid (2a) were used as the substrates for the investigation of the reaction conditions of this PGS–APTES–Pd(OAc)2 catalytic system. The effects of the solvent, catalyst dosage and additives were screened (Table S1, in ESI†).First, 1,4-dioxane was used as the solvent, in which the ligand and base effects were studied (Table S1,† entries 1–7). As a result, DPE-Phos and X-Phos exhibited similar activities, but they both showed slightly lower activity than PPh3. The activity of the base for TBAB, NaOAc, Cs2CO3, K2CO3 and K3PO4 increased in turn. Therefore, when PPh3 was used as the ligand and K3PO4 was used as the base, the product could be obtained in a higher yield of 88% (Table S1,† entry 3).
Subsequently, the effect of the solvent was screened. 1,4-Dioxane can provide better conversion (Table S1,† entries 8 and 9). With regard to green chemistry, we tested other solvents, but they did not afford higher yields. In addition, the amount of catalysts was also investigated. We found out that with the increase in catalyst loading, the yield of 3a increased significantly. When the amount of the catalyst was 20 mg, the yield reached 88% (Table S1,† entries 3, 10–12). Similarly, the activities of the homogeneous catalysts Pd(OAc)2 and PGS–APTES–Pd(OAc)2 were compared using the same amount of Pd. The results indicated that the catalytic efficiency of Pd in PGS–APTES–Pd(OAc)2 was higher (Table S1,† entries 3 and 16) (Scheme 1).
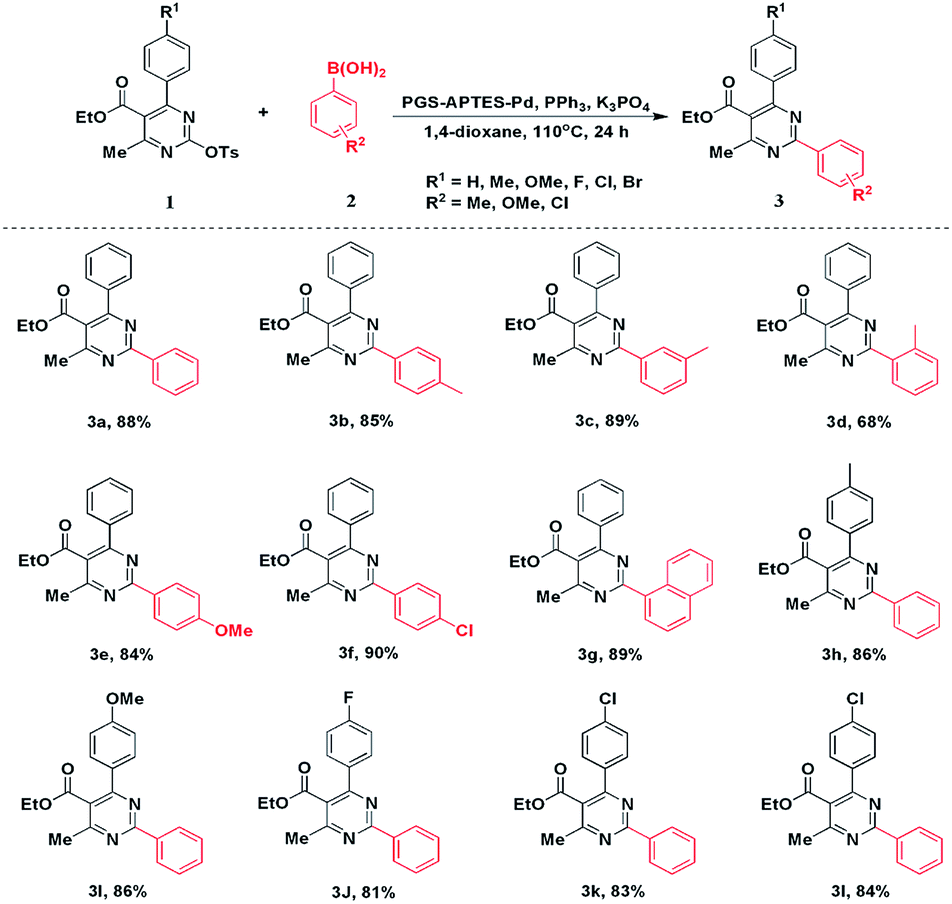 | ||
| Scheme 1 Scope of Suzuki reaction. | ||
Under optimized conditions, a series of arylboronic acids were reacted with 1a. Phenylboronic acids with ortho-, meta-, and para-methyl groups were satisfactorily tolerated, giving the corresponding products 3b–3d in good to moderate yields. In addition, the steric hindrance of coupling partners had slight influence on this coupling reaction. Other functional groups such as MeO- and Cl- could deliver the products 3e and 3f, respectively, in good yields. In addition, naphthalen-1-ylboronic acid could also afford the product 3g in 85% yield. Next, we explored the scope of DHPM-sulfonates with phenylboronic acid (2a). Compared with 1a, the DHPM-sulfonates with different functional groups such as Me-, MeO-, F-, Cl-, and Br- could be well tolerated, giving the corresponding products in good yields. These results revealed the applicable method of this PGS–APTES–Pd(OAc)2 catalytic system.
PGS–APTES–Pd(OAc)2-catalyzed Sonogashira coupling reaction of pyrimidin-2-yl sulfonates
Similarly, the prepared PGS–APTES–Pd(OAc)2 was used as the catalyst in the Sonogashira coupling reactions of pyrimidin-2-yl sulfonates with terminal alkynes. Compound 1a and phenylacetylene (4a) were used as the substrates to optimize the reaction conditions.Table S2 (in ESI†) shows that the Cu(II) salt has no activity (Table S2,† entry 4). The activity of the inorganic Cu(I) salt is very low (Table S2,† entry 1 and 2). We therefore tested the effect of ((thiophene-2-carbonyl)oxy)copper (CuTC), and it could deliver the corresponding product 5a in a moderate yield of 59% when X-phos was used as the ligand (Table S2,† entry 3). We therefore screened other ligands with CuTC as the co-catalyst. When DPE-Phos was used as the ligand instead of X-phos, the yield of the product increased significantly (Table S2,† entry 6). However, when PPh3 was used as the ligand, it could only afford a yield of 22% (Table S2,† entry 5). Further investigations indicated that 1,4-dioxane was the best solvent in this reaction (Table S2,† entries 6–8). In addition, the amount of the catalyst was also explored. The results showed that the reaction could be completed to the maximum extent when the amount of the catalyst was 20 mg (Table S2,† entries 6, 9–10). The optimized reaction time of the system showed that the best reaction time of the system was 48 h (Table S2,† entries 11–13). Similarly, we carried out control experiments on the system, and the results indicated that the catalyst and the ligand were indispensable (Table S2,† entries 14–15). From the above-described experiments, we concluded the optimized reaction conditions: PGS–APTES–Pd(OAc)2 (20 mg, Pd 0.463 wt%), CuTC (10 mol%), K3PO4 (2.0 equiv.), DPE-Phos (6 mol%), 1,4-dioxane (5 mL), with the temperature of 110 °C and 48 hours reaction time.
Under the optimized reaction conditions, the substituted DHPM-sulfonates (1) and terminal alkynes (4) were used to investigate the substrate scope (Scheme 2). Both aryl and alkyl terminal alkynes were compatible with the reaction system, and the corresponding products could be produced in moderate to good yields. For the substituted DHPM-sulfonates, the electronic effect of the substituents was more obvious (please compare 5e, 5f, and 5g).
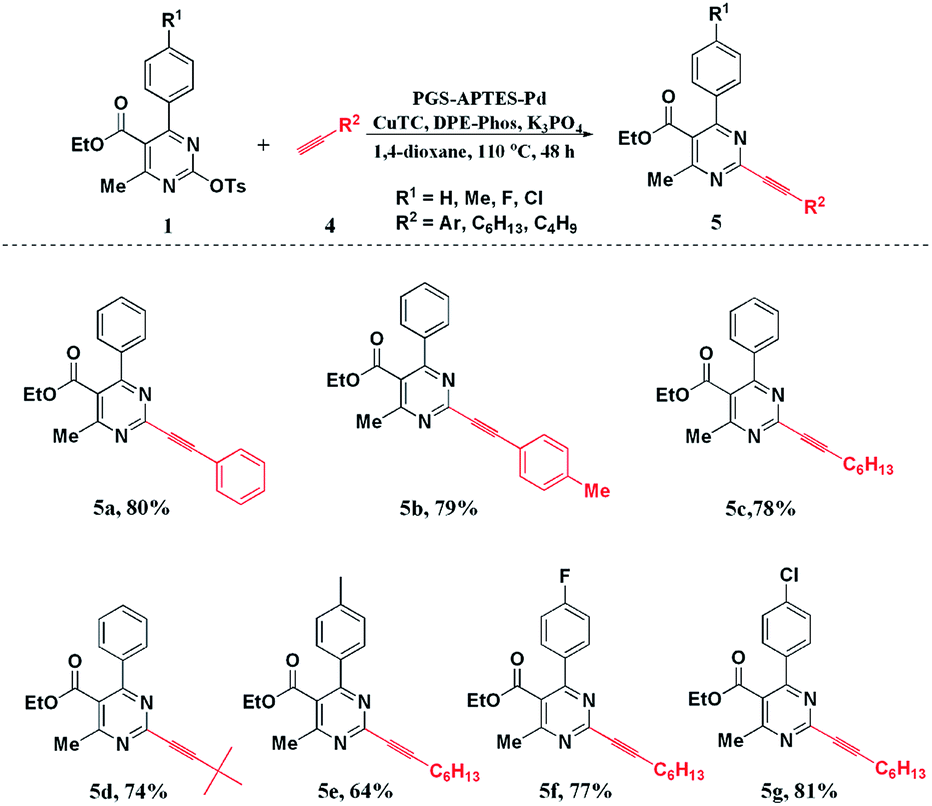 | ||
| Scheme 2 Scope of Sonogashira reaction. | ||
PGS–APTES–Pd(OAc)2-catalyzed C–N coupling reaction of pyrimidin-2-yl sulfonates
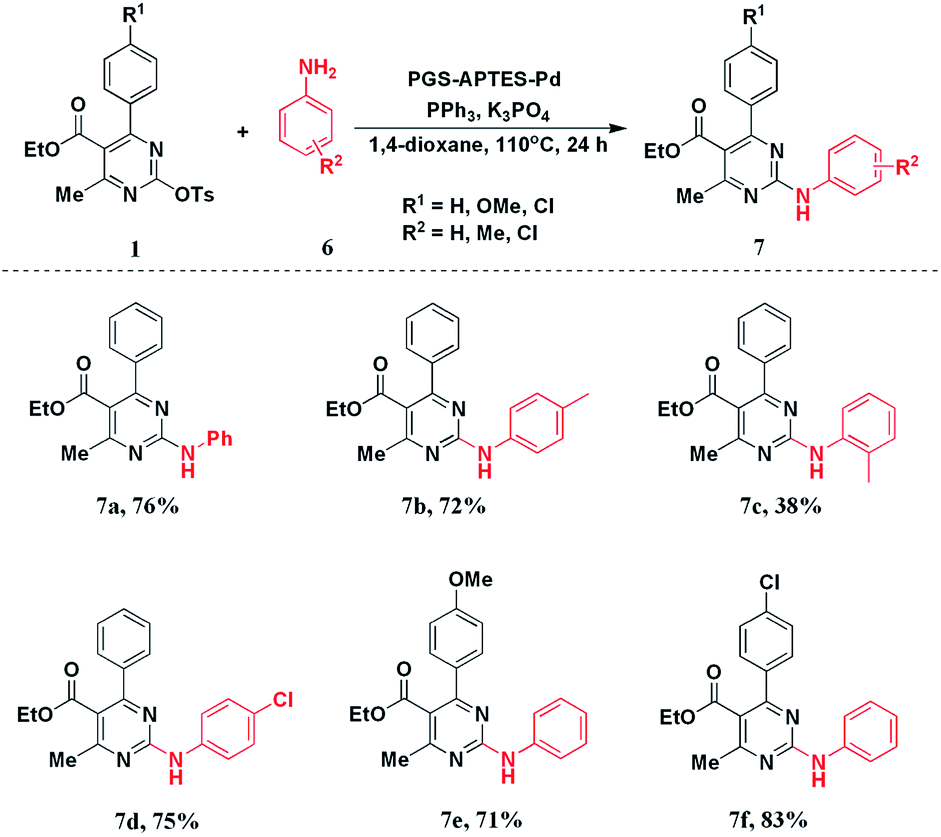
After realizing the C–C coupling reaction catalyzed by PGS–APTES–Pd(OAc)2, we applied the catalytic system to the C–N coupling reaction of DHPM-sulfonates with phenylamine derivatives. The results are listed in Table S3.†After screening different reaction conditions, we found out that when the amount of the catalyst was 20 mg and the solvent was 1,4-dioxane with K3PO4 as the base and PPh3 as the ligand, this reaction could afford the C–N coupling product in a good yield of 76%.
Based on the optimization conditions, we explored the scope of the DHPM-sulfonates with different substituted anilines. As a result, when different anilines were tested, the C–N coupling products could be obtained in a high yield regardless of an electron-withdrawing substituent (–Cl) or an electron-donating substituent (–Me). However, the steric hindrance effect of anilines was more obvious as o-methyl aniline only delivered 7c in 38% yield. For the substituted DHPM-sulfonates, the electronic effect of the substituents on the yield was negligible. Different functional groups could be well tolerated in good yields (Scheme 3).
 | ||
| Scheme 3 Scope of the C–N coupling reaction. | ||
Recycling of the catalyst
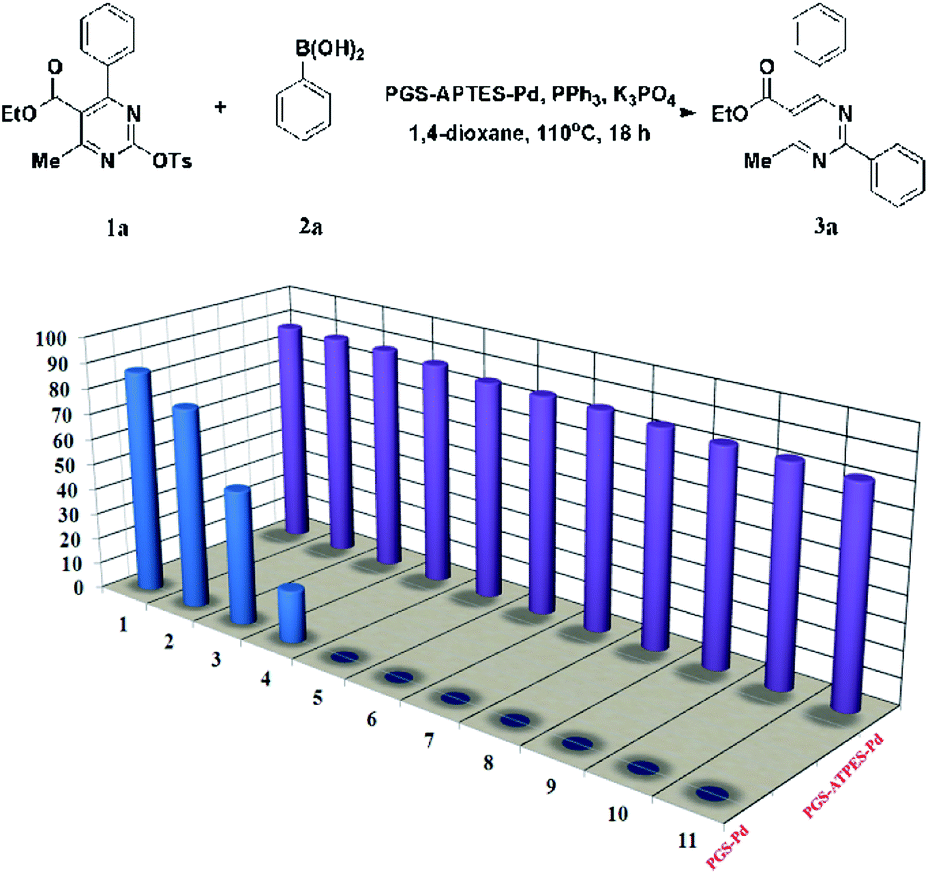
The lifetime of a heterogeneous catalyst plays a key role in the catalytic reaction for practical applications, which is superior to a homogenous one. The reusability of the PGS–APTES–Pd(OAc)2 catalyst for the Suzuki coupling reaction of DHPM-sulfonates 1a and phenylboronic acid 2a was also investigated. After each reaction, the mixture was cooled to room temperature, and the solid was separated by centrifugation, washed, dried, and then used again in the next run. The Pd content of the catalyst after each reaction was determined.In this test, PGS–APTES–Pd(OAc)2 and PGS–Pd (prepared by the impregnation method) were used as the catalysts (equivalent Pd). It can be seen from Fig. 8 that PGS–APTES–Pd(OAc)2 has good reusability, and the yield of the product for PGS–APTES–Pd(OAc)2 has no obvious downward trend after eleven cycles of the catalyst. However, the activity of PGS–Pd in the initial use was similar to that of PGS–APTES–Pd(OAc)2 and in the third cycle, its activity declined significantly. The results further indicated that the PGS–APTES–Pd(OAc)2 catalyst was stable, and the Pd content was almost unchanged (0.463–0.431%) after 11 cycles of use by ICP-AES. The cumulative TON value of the catalyst was 2216.
 | ||
| Fig. 8 Recycling of the catalyst in the Suzuki reaction. | ||
The content of Pd in the reaction solution was determined to be 76 ppb by ICP-MS analysis. In order to verify whether free Pd or PGS–APTES–Pd(OAc)2 in the reaction system plays a real catalytic role, we also carried out a “hot filtration test” on the reaction system.
Under the optimum conditions, 1a and 2a were used as the substrates and after 3 hours of reaction, the catalyst was separated from the reaction solution. At this time, the conversion of substrates was determined to be only 28%. After that, the filtrate was stirred for 24 hours under the standard conditions, but no reaction was detected. This result indicated a key catalytic role of PGS–APTES–Pd(OAc)2 against the trace amount of free Pd in the solution.
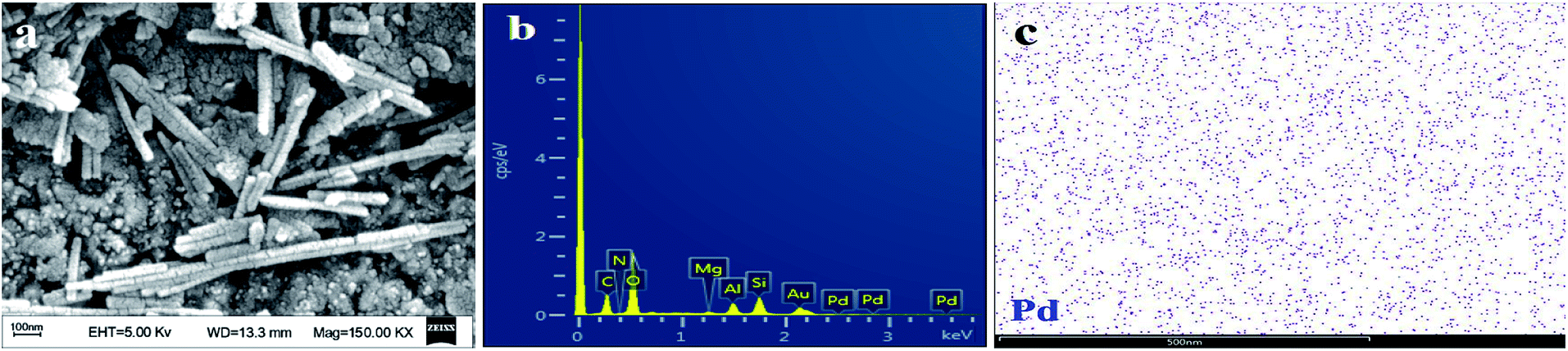
In addition, the morphology of the catalyst after the reaction was characterized (Fig. 9). Compared with the catalyst before the reaction, the palygorskite-supported catalyst was still in rod shape (Fig. 9(a)). After the reaction, there was no obvious accumulation of Pd particles on the surface, but the surface was uneven, which may be due to the corrosion of palygorskite by the reaction system. In addition, the EDX and EDS spectra (Fig. 9(b and c)) show that Pd is still uniformly loaded on the catalyst surface. Therefore, the catalyst has a good recycling performance.
 | ||
| Fig. 9 (a) SEM image of the reused catalyst; (b) the corresponding EDX spectrum and (c) EDS mapping of the reused catalyst. | ||
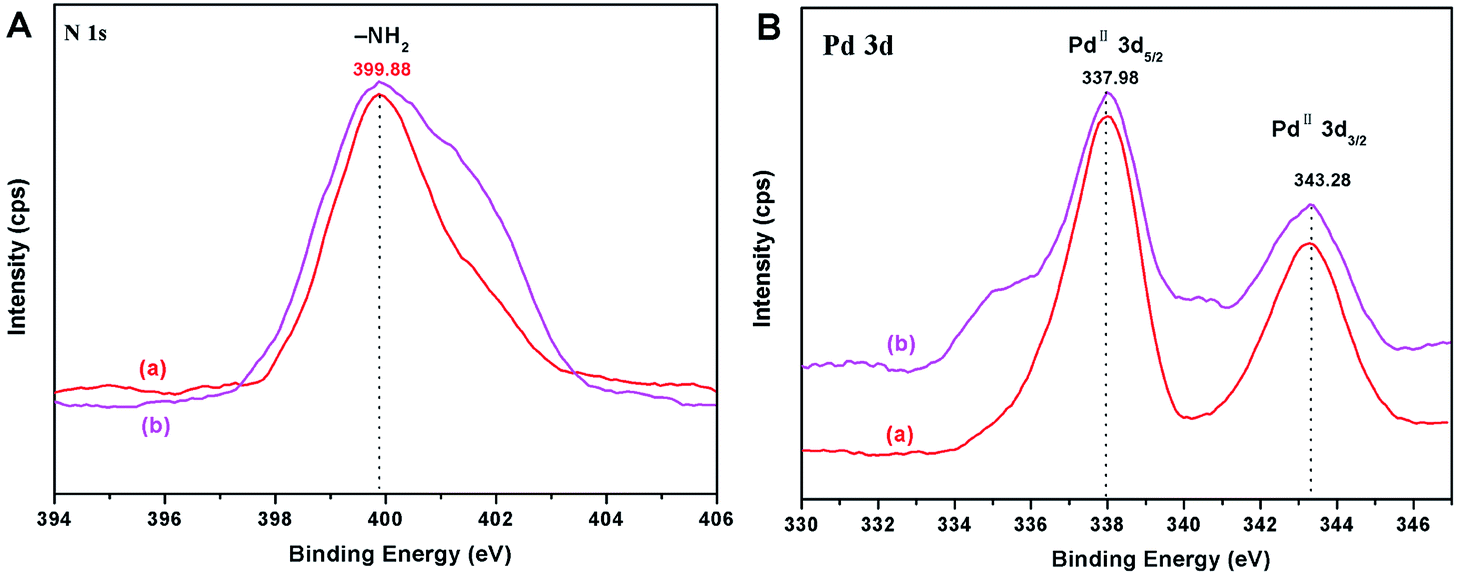
In order to further testify the stability of the catalyst, the catalyst was characterized by XPS after the reaction. As shown in Fig. 10, the binding energies of N 1s and Pd 3d have no significant change before and after the Suzuki reaction (Fig. 10). The XPS results indicated that the oxidation state of Pd was PdII both in the fresh and in the reused catalyst. This provided direct evidence for the high recyclability and stability of the PGS–APTES–Pd(OAc)2 catalyst.
 | ||
| Fig. 10 XPS spectra of (A) fresh and (B) reused catalyst. | ||
Proposed mechanism for the Suzuki reaction
Based on previous literature6 and our above-mentioned studies, a proposed mechanism for the Suzuki-type coupling reaction is depicted in Fig. 11. This PGS–APTES–Pd(OAc)2 catalytic process was most likely to undergo a cycle between Pd(0) and Pd(II) species via oxidative addition with 1a and transmetallation with arylboronic acid, followed by fast reductive elimination to afford the C–C coupling products. The substituents of PGS–APTES may probably have a steric effect to facilitate the reductive elimination step, which in turn enhanced the Suzuki coupling reaction better than that observed for the ordinary Pd catalysts. Pd(0) would be oxidized to Pd(II) after the reaction during the after-treatment by oxygen in air, and the catalyst was recovered.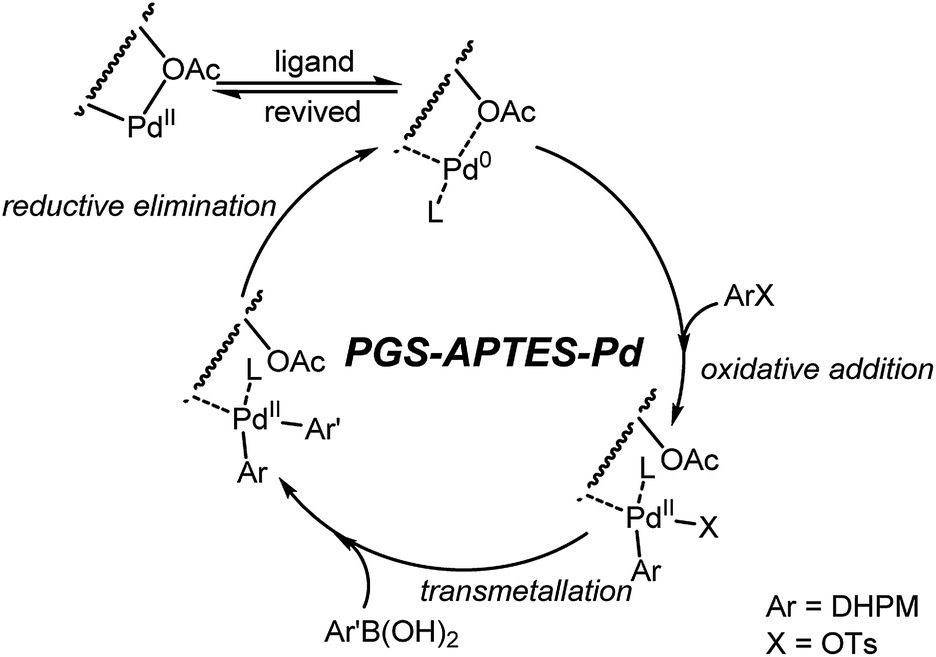 | ||
| Fig. 11 Proposed mechanism of the Suzuki coupling reaction. | ||
Conclusions
In conclusion, a practical and easily available PGS–APTES–Pd(OAc)2 catalyst was prepared using simple procedures. The catalyst could be applied in the C–C and C–N coupling reactions of pyrimidin-2-yl sulfonates with arylboronic acids, terminal alkynes and anilines smoothly, giving the corresponding products in moderate to good yields. Obviously, the stability and reusability of PGS–APTES–Pd(OAc)2 were superior to those of the PGS–Pd catalyst (prepared by the impregnation method) in the recycling test. This catalytic system will promote the application of the PAL materials in catalysis with the expanding of the palladium chemistry.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support from the National Natural Science Foundation of China (No. 21962017, 21464013, 51563022, 31760608 and 21467027), the Research Funds for the Colleges and Universities of Gansu Province (No. 2014A-130), the Fundamental Research Funds for the Central Universities (31920160010, 31920190016), and the Northwest Minzu University’s Double First-class and Characteristic Development Guide Special Funds-Chemistry Key Disciplines in Gansu Province (No. 11080316).References
- C. O. Kappe, Eur. J. Med. Chem., 2000, 35, 1043 CrossRef CAS.
- K. S. Atwal and S. Moreland, Bioorg. Med. Chem. Lett., 1991, 1, 291 CrossRef CAS.
- (a) Z. Q. Niu, Q. Peng and Z. B. Zhuang, Chem.–Eur. J., 2012, 1, 89813 Search PubMed; (b) R. A. Hughes and C. J. Moody, Angew. Chem., Int. Ed., 2007, 46, 7930 CrossRef CAS.
- For reviews on the DHPMs, see: (a) C. O. Kappe, Tetrahedron, 1993, 49, 6937 CrossRef CAS; (b) C. O. Kappe, Acc. Chem. Res., 2000, 33, 879 CrossRef CAS; (c) C. O. Kappe and A. Stadler, Org. React., 2004, 63, 1 CAS; (d) D. Dallinger, A. Stadler and C. O. Kappe, Pure Appl. Chem., 2004, 76, 1017 CAS; (e) L. Z. Gong, X. H. Chen and X. Y. Xu, Chem.–Eur. J., 2007, 13, 8920 CrossRef CAS; (f) M. A. Kolosov and V. D. Orlov, Mol. Diversity, 2009, 13, 5 CrossRef CAS; (g) Z. J. Quan, Z. Zhang, Y. X. Da and X. C. Wang, Chin. J. Org. Chem., 2009, 29, 876 CAS.
- (a) J. He, M. Wasa, K. S. L. Chan, Q. Shao and J.-Q. Yu, Chem. Rev., 2017, 117, 8754 CrossRef CAS; (b) J. Wang and G. Dong, Chem. Rev., 2019, 119, 7478 CrossRef CAS; (c) P. Ruiz-Castillo and S. L. Buchwald, Chem. Rev., 2016, 116, 12564 CrossRef CAS; (d) Á. Molnár, Chem. Rev., 2011, 111, 2251 CrossRef; (e) M. E. Trusova, M. Rodriguez-Zubiri, K. V. Kutonova, N. Jung, S. Bräse, F.-X. Felpin and P. S. Postnikov, Org. Chem. Front., 2018, 5, 41 RSC; (f) L. Liang, H. Y. Niu, M. S. Xie, G. R. Qu and H. M. Guo, Org. Chem. Front., 2018, 5, 3148 RSC; (g) L. Jin, W. Wei, N. Sun, B. Hu, Z. Shen and X. Hu, Org. Chem. Front., 2018, 5, 2484 RSC; (h) A. S. Kumari, S. Layek and D. D. Pathak, New J. Chem., 2017, 41, 5595 RSC.
- (a) Z. J. Quan, W. H. Hu, X. D. Jia, Z. Zhang, Y. X. Da and X. C. Wang, Adv. Synth. Catal., 2012, 354, 2939 CrossRef CAS; (b) Z. J. Quan, Y. Lv, Z. J. Wang, Z. Zhang, Y. X. Da and X. C. Wang, Tetrahedron Lett., 2013, 54, 1884 CrossRef CAS; (c) Z. J. Quan, W. H. Hu, Z. Zhang, X. D. Jia, Y. X. Da and X. C. Wang, Adv. Synth. Catal., 2013, 355, 891 CrossRef CAS; (d) Z. J. Quan, Y. Lv, F. Q. Jing, X. D. Jia, C. D. Huo and X. C. Wang, Adv. Synth. Catal., 2014, 356, 325 CrossRef CAS; (e) Z. Yan, Z. Quan, Y. Da, Z. Zhang and X.-C. Wang, Chem. Commun., 2014, 50, 13555 RSC; (f) N. H. T. Phan, H. Kim, H. Shin, H. Lee and J. Sohn, Org. Lett., 2016, 18, 5154 CrossRef; (g) H. Mathur, M. S. K. Zai, P. Khandelwal, N. Kumari, V. P. Verma and D. K. Yadav, Chem. Heterocycl. Compd., 2018, 54, 375 CrossRef CAS.
- (a) C. Y. Yi and R. M. Hua, Catal. Commun., 2006, 7, 377 CrossRef CAS; (b) J. Michalik, M. Narayana and L. Kevan, J. Phys. Chem., 1985, 89, 4553 CrossRef CAS; (c) W. M. H. Sachtler, F. A. P. Cavalcanti and Z. Zhang, Catal. Lett., 1991, 9, 261 CrossRef CAS; (d) V. Arun, N. Sridevi, P. P. Robinson, S. M. Manju and K. K. Yusuff, J. Mol. Catal. A: Chem., 2009, 4, 191 CrossRef.
- (a) B. J. Borah and D. K. Dutta, J. Mol. Catal. A: Chem., 2013, 366, 202 CrossRef CAS; (b) M. Fang, G. Fan and F. Li, Catal. Lett., 2014, 144, 142 CrossRef CAS; (c) L. Xue, B. Jia, L. Tang, X. F. Ji, M. Y. Huang and Y. Y. Jiang, Polym. Adv. Technol., 2004, 15, 346 CrossRef CAS; (d) M. Ranjbar-Mohammadi, S. Hajir Bahrami and M. Arami, J. Appl. Polym. Sci., 2013, 129, 707 CrossRef CAS; (e) S. Wu, H. C. Ma, X. J. Jia, Y. M. Zhong and Z. Q. Lei, Tetrahedron, 2011, 67, 250 CrossRef CAS; (f) H. C. Ma, W. Cao, Z. K. Bao and Z. Q. Lei, Catal. Sci. Technol., 2012, 2, 2291 RSC; (g) P. Cotugno, M. Casiello, A. Nacci, P. Mastrorilli and A. Monopoli, J. Organomet. Chem., 2014, 752, 1 CrossRef CAS.
- (a) A. Indra, C. S. Gopinath, S. Bhaduri and G. K. Lahiri, Catal. Sci. Technol., 2013, 3, 1625 RSC; (b) M. Massaro, S. Riela, G. Cavallaro, M. Gruttadauria, S. Milioto, R. Noto and G. Lazzara, J. Org. Chem., 2014, 749, 410 CrossRef CAS; (c) R. Arundhathi, D. Damodara, K. V. Mohan, M. L. Kantam and P. R. Likhar, Adv. Synth. Catal., 2013, 355, 751 CrossRef CAS; (d) W. Xu, H. M. Sun, B. Yu, G. F. Zhang, W. F. Zhang and Z. W. Gao, ACS Appl. Mater. Interfaces, 2014, 6, 20261 CrossRef CAS; (e) J. Olszówka, R. Karcz, B. Napruszewska, E. Biela′nska, R. Dula, M. Krzan, M. Nattich-Rak, R. P. Socha, A. Klimek, K. Bahranowski and E. M. Serwicka, Appl. Catal., A, 2016, 509, 52 CrossRef.
- (a) Z. W. Yang, X. Zhao, T. J. Li, W. L. Chen, Q. X. Kang, X. Q. Xu, X. X. Liang, Y. Feng, H. H. Duan and Z. Q. Lei, Catal. Commun., 2015, 65, 34 CrossRef CAS; (b) F. Wang, J. Zhang, C. Liu and J. H. Liu, Appl. Clay Sci., 2015, 105–106, 150 CrossRef CAS; (c) S. Wu, H. C. Ma and Z. Q. Lei, Synlett, 2010, 18, 2818 Search PubMed.
- (a) R. Zhu, Q. Chen, Q. Zhou, Y. Xi, J. Zhu and H. He, Appl. Clay Sci., 2016, 123, 239 CrossRef CAS; (b) H. Khalaf, O. Bouras and V. Perrichon, Microporous Mater., 1997, 8, 141 CrossRef CAS; (c) L. Zhou, H. Chen, X. Jiang, F. Lu, Y. Zhou, W. Yin and X. Ji, J. Colloid Interface Sci., 2009, 332, 16 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra06035a |
| This journal is © The Royal Society of Chemistry 2019 |