
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective oxidation of methacrolein to methacrylic acid over H4PMo11VO40/C3N4-SBA-15†
Lilong Zhou a,
Shanshan Zhanga,
Zhengjie Lia,
Jason Scottb,
Zhikun Zhanga,
Runjing Liu*a and
Jimmy Yun*ab
a,
Shanshan Zhanga,
Zhengjie Lia,
Jason Scottb,
Zhikun Zhanga,
Runjing Liu*a and
Jimmy Yun*ab
aCollege of Chemical and Pharmaceutical Engineering, Hebei University of Science and Technology, Shijiazhuang, Hebei province 050018, P. R. China. E-mail: liurj2002@163.com
bSchool of Chemical Engineering, The University of New South Wales, Sydney, NSW 2052, Australia. E-mail: jimmy.yun@unsw.edu.au
First published on 23rd October 2019
Abstract
Molybdovanadylphosphoric acid (HPMV) was supported on a carbon nitride-modified SBA-15 (CN-SBA-15) molecular sieve to enhance its catalytic performance for oxidation of methacrolein (MAL) to methacrylic acid (MAA). HPMV/CN-SBA exhibited increased catalytic activity (20%) and five times greater MAA selectivity (98.9%) compared to bulk HPMV. HPMV supported on CN-SBA-15 exhibited much better catalytic performance as compared to that on other supports, such as KIT-6, HY zeolite, TiO2, Al2O3, SiO2, CNTs, and NH3-modified CNTs. The supported HPMV was well characterized by FT-IR, XRD, SEM, N2 physical desorption, TG-DTA, NH3-TPD, CO2-TPD, XPS, and solid-state NMR. The CN minimized the interaction between the silica support and HPMV. HPMV was successfully separated from SBA-15, which was restricted by CN to increase stability and prevent interaction between the catalysts and support that would lead to decomposition of the catalysts during calcination and reaction. HPMV reacted with amino groups on the CN, which improved MAA selectivity and enhanced the thermal stability of the supported heteropoly acid (HPA) catalysts. This work identifies a new approach to preparing highly efficient and stable supported HPA catalysts for oxidation reactions.
Introduction
As an important intermediate chemical, methacrylic acid (MAA) is produced at a scale of 2.5–3 billion kilograms per year, and is used for producing methyl methacrylate, functional polymers, and coatings.1 Approximately half of the MAA is produced via the acetone cyanohydrin (ACH) process, which necessitates the use of toxic HCN and corrosive H2SO4, and generates significant amounts of NH4HSO4 as solid waste.2 The toxic chemistry and byproduct disposal issues associated with the ACH process have resulted in new factories being prohibited in many developed and developing countries. Oxidation of methacrolein (MAL) to MAA over heteropolyacid catalysts, such as molybdophosphoric or molybdovanadylphosphoric compounds (H4PMo11VO40, HPMV), is a compelling alternative for the green production of MAA.3,4 However, the low surface area of these heteropoly acid (HPA) materials remains a primary constraint in terms of catalytic efficiency.A suitable approach to address the low surface area issue is to load the HPA on a porous support,5 such as porous SiO2, zeolites, activated carbon, polymers, and metal oxides.6–16 Of these, silica is considered a good option due to its thermal stability under high-temperature reaction conditions in conjunction with its low cost. Legagneux et al. supported H4SiW12O40 and H3PW12O40 on porous SiO2 using maceration, and both HPAs reacted with the silica support to give [![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) SiOH2]xH4−xSiW12O40 and [SiOH2]3PW12O40, respectively, which led to partial decomposition of the Keggin structure upon heating.9 Kanno et al. supported HPMV on SiO2 for oxidation of MAL to MAA; however, the supported HPAV decomposed to MoO3 during the reaction.11 To surmount the decomposition problem, they instead loaded HPMV onto NH3-modified SiO2. The modified catalyst exhibited better catalytic performance with MAL conversion and MAA selectivity at 9% and 89%, respectively.12 However, the NH3-modification of SiO2 was performed at 900 °C, which is not suitable for scaling up of the reaction. Despite the improved performance, the supported catalysts continued to display poor thermal stability under the reaction conditions, and MAL conversion remained too low. It has been reported that the substandard thermal stability originates from the reaction between the H4PMo11VO40 catalyst and the silica support, whereby silicon atoms replace the central atom of the HPA structure.17
SiOH2]xH4−xSiW12O40 and [SiOH2]3PW12O40, respectively, which led to partial decomposition of the Keggin structure upon heating.9 Kanno et al. supported HPMV on SiO2 for oxidation of MAL to MAA; however, the supported HPAV decomposed to MoO3 during the reaction.11 To surmount the decomposition problem, they instead loaded HPMV onto NH3-modified SiO2. The modified catalyst exhibited better catalytic performance with MAL conversion and MAA selectivity at 9% and 89%, respectively.12 However, the NH3-modification of SiO2 was performed at 900 °C, which is not suitable for scaling up of the reaction. Despite the improved performance, the supported catalysts continued to display poor thermal stability under the reaction conditions, and MAL conversion remained too low. It has been reported that the substandard thermal stability originates from the reaction between the H4PMo11VO40 catalyst and the silica support, whereby silicon atoms replace the central atom of the HPA structure.17
Consequently, modifications to the silica surface so as to enhance thermal stability and improve the catalytic performance of the supported HPA catalyst are still required. One potential support modifier is carbon nitride (CN). Incorporating nitrogen atoms into a carbon nanostructure has been shown to enhance the conductive, mechanical, field-emission, and energy-storage properties of carbon materials.18–31 More importantly, the carbon nitride surface possesses high levels of amino groups,22 and adding NH3 to heteropolyacid catalysts has been previously reported to improve its performance for selective oxidation reactions.32 The potential then exists to use CN as a barrier to HPA degradation when it is supported on silica while concurrently promoting HPA catalytic performance via NH3 inclusion into the structure.
In the work presented here, the prospect of using CN as a favourable modifier for silica-supported HPA catalysts was examined. HPMV was loaded onto a CN-modified SBA-15 support and assessed as a catalyst for oxidizing MAL to MAA. The CN loading, synthesis conditions, and preparation time of the catalysts were optimized.
Results and discussion
Catalyst characteristics
Od), vs.(Mo–Ob–Mo), and vs.(Mo–Oc–Mo), respectively, and these are the characteristic vibration bands of the Keggin structure.34 The vibrational band at 1036 cm−1 can be assigned to vs.(V–O) in VO2+, which was formed from the migration of V atoms in the Keggin structure to the secondary structure during calcinations.37
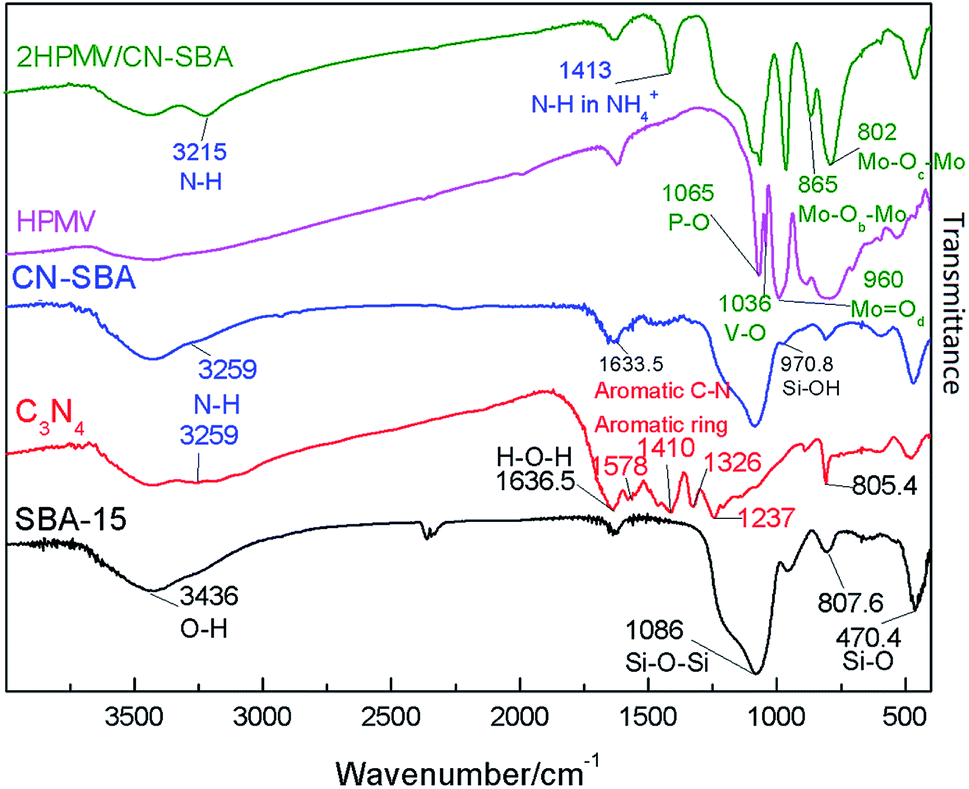 | ||
| Fig. 1 FT-IR spectra of SBA-15, CN, CN-SBA, calcined HPMV, and 2HPMV/CN-SBA. | ||
The four vibrational bands that are characteristic of a Keggin structure also emerged in the 2HPMV/CN-SBA spectrum. When compared with neat HPMV (Fig. S1†), the vs.(MoOd) of xHPMV/CN-SBA shows a shift from 961 to 960 cm−1, vs.(Mo–Ob–Mo) has shifted from 874 to 865 cm−1, and vs.(Mo–Oc–Mo) has shifted from 780 to 801.9 cm−1, while vs.(P–O) remains unchanged. The shifts suggest that there has been an interaction between the HPAV and amino groups on the CN-SBA support, and it also suggests that HPMV was successfully supported on CN-SBA. A new vibrational band emerged at 1413 cm−1, which can be assigned to vs.(N–H) in NH4+.39 The band indicates that HPMV reacted with the amino groups on the CN-SBA surface to form HPA ammonium salt.
Fig. S5–S9† shows the FTIR spectra of the HPMV, non-calcined catalysts, calcined catalysts, and used catalysts with different HPMV loadings, as well as catalysts prepared at different temperatures with different synthesis time. Apart from the V–O vibrational bands in VO2+ appearing in the calcined and used HPMV spectra, there is no obvious difference between the calcined HPMV spectrum and the non-calcined HPMV or used HPMV spectra (Fig. S1†). The spectra of the as-prepared HPMV/CN-SBA exhibited characteristic Keggin structure (1060, 960, 870, and 796 cm−1), silica (1086 and 465 cm−1), and CN (1460 and 1412 cm−1) vibrational bands, which also indicated that HPMV was successfully loaded onto CN-SBA (Fig. 1, S5–S9†).
After calcination, a new band emerged at 1036 cm−1 that was assigned to the V–O vibration band. Disappearance of the band at 1460 cm−1 may have occurred from the reaction between HPMV and amino groups during calcinations (Fig. S6†). After increasing the HPMV loading to n = 3 (75 wt%), the Mo–O vibrational band at 595 cm−1, belonging to MoO3, appears in the calcined and used catalyst spectra (Fig. S5–S7†). Appearance of the Mo–O vibration suggests that if excessive HPMV loading occurs, catalyst decomposition may be promoted. The preparation temperature and synthesis time have little influence on the structure of the supported catalysts (Fig. S8 and S9†) over the range considered.
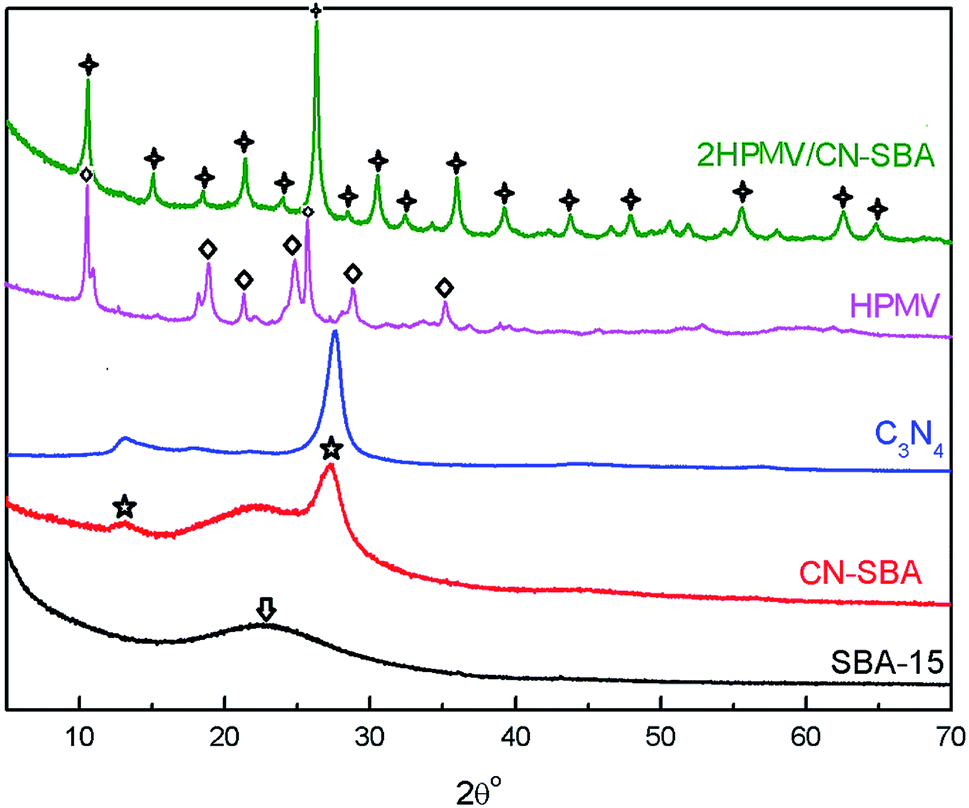 | ||
| Fig. 2 XRD patterns of SBA-15, CN-SBA, C3N4, calcined HPMV, and 2HPMV/CN-SBA. Quadrangular star = (NH4)xH4−xPMo11VO40; arrow = amorphous silica; ◇ = HPMV; ☆ = C3N4. | ||
Typical HPA patterns with a Keggin structure are a feature of the neat HPMV sample, which can be associated with a triclinic crystal phase.32 When HPMV was loaded on CN-SBA, two HPMV crystal forms are evident in the 2HPMV/CN-SBA patterns, cubic and triclinic. The overlaying crystal spectra potentially reflect (NH4)xH4−xPMo11VO40 (cubic crystal, 2θ = 10.6°, 15.0°, 19.3°, 21.4°, 26.3°, 30.5°, and 36.0°) and H4PMo11VO40 (triclinic crystal, 2θ = 9.0°, 9.3°, 18.5°, 24.8°, and 28.7°) (Fig. S10†).32,39 The spectrum depicting (NH4)xH4−xPMo11VO40 reveals that HPMV has reacted with amino groups on the CN-SBA surface. The diffraction patterns of C3N4 also can be observed at 27° for composite samples.
After calcining, the HPMV patterns have disappeared, with only (NH4)xH4−xPMo11VO40 remaining. This suggests that HPMV continued to react with amino groups during calcination. However, when the HPMV loading was deficient (e.g., 0.5HPMV/CN-SBA) or too high (4HPMV/CN-SBA, 5HPMV/CN-SBA), diffraction patterns representing MoO3 and V2O5 (25.6° and 27.3°, 20.5°, Fig. S10b†) appeared for the calcined supported catalysts. The MoO3 and V2O5 originate from degradation of the supported HPA during calcination. MoO3 diffraction patterns are also observed in the used 3HPMV/CN-SBA, 4HPMV/CN-SBA, and 5HPMV/CN-SBA patterns (Fig. S11†), which were generated from the decomposition of HPMV. It appears that if there is insufficient or excessive HPMV loaded onto the CN-SBA support, the thermal stability of HPA is diminished, and it undergoes partial decomposition. MoO3 diffraction patterns are also observed in the patterns of calcined 2HPMV/CN-SBA prepared at temperatures of 100 °C or greater and preparation times of 8 h or longer (Fig. S12 and S13†). The crystal diameters (Table S1†) calculated by the Scherrer formula show that the catalyst size decreased after being supported and increased with the increase in the loading amount. The findings indicate that an excessive preparation temperature and/or preparation time also lead to HPA decomposition.
| Catalysts | Specific surface area/m3 g−1 | Pore volume/cm3 g−1 | Average pore diameter/nm |
|---|---|---|---|
| HPMV | 3.3 | 0.014 | 17.8 |
| NH4PMV | 2.4 | 0.008 | 15.6 |
| SBA-15 | 1018 | 1.307 | 6.6 |
| CN-SBA | 499.4 | 1.019 | 7.7 |
| 0.5HPMV/CN-SBA | 165.6 | 0.40 | 8.2 |
| 1HPMV/CN-SBA | 175.2 | 0.32 | 7.8 |
| 2HPMV/CN-SBA | 118.0 | 0.20 | 8.1 |
| 3HPMV/CN-SBA | 66.7 | 0.11 | 7.3 |
| 4HPMV/CN-SBA | 66.4 | 0.086 | 5.7 |
| 5HPMV/CN-SBA | 50.4 | 0.089 | 5.9 |
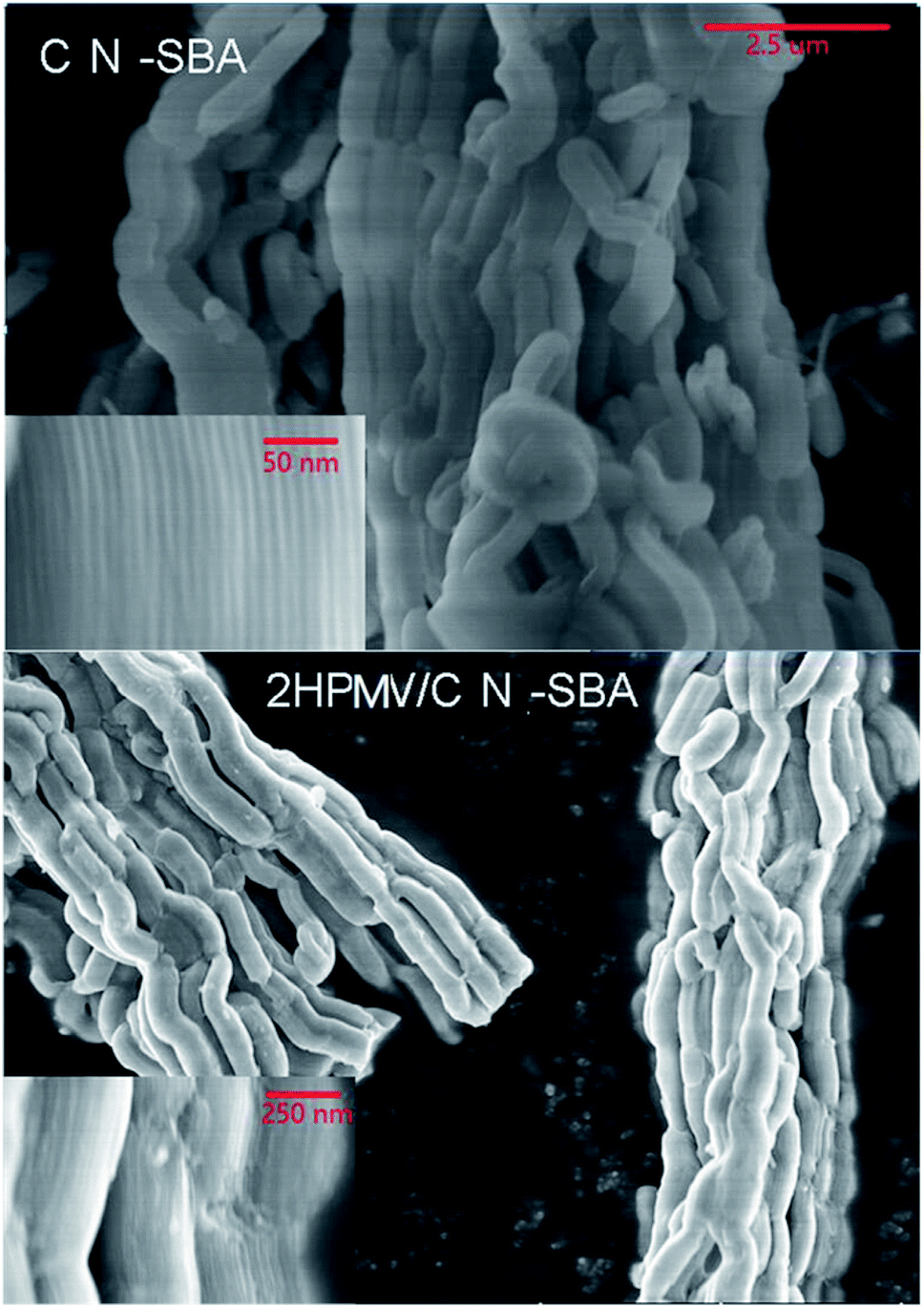 | ||
| Fig. 3 SEM images of CN-SBA and 2HPMV/CN-SBA. | ||
With increasing HPMV loading, the large plate per cubic particles firstly appear at the end, on the corners, and in the cracks of the support. The appearance of the plate per cubic originates from the HPMV initially entering the pores of the support and reacting with amino groups on the pore walls during preparation. With increasing HPMV loading, NHPMV crystals continued to accumulate in the pores, and eventually grew out of the pores to form large particles. Pore exits are absent at the end, corners, and cracks of the support, with the large particles consequently growing at these points. This may also account for the decrease in surface area with increased HPMV loading. Concurrently, HPMV may also react with amino groups on the outer surface of the support to form the plate-shaped particles. The BET results and scanning electron microscopy (SEM) images indicate that excessive HPMV loading decreases the surface area and is detrimental for the pore structure of the support.
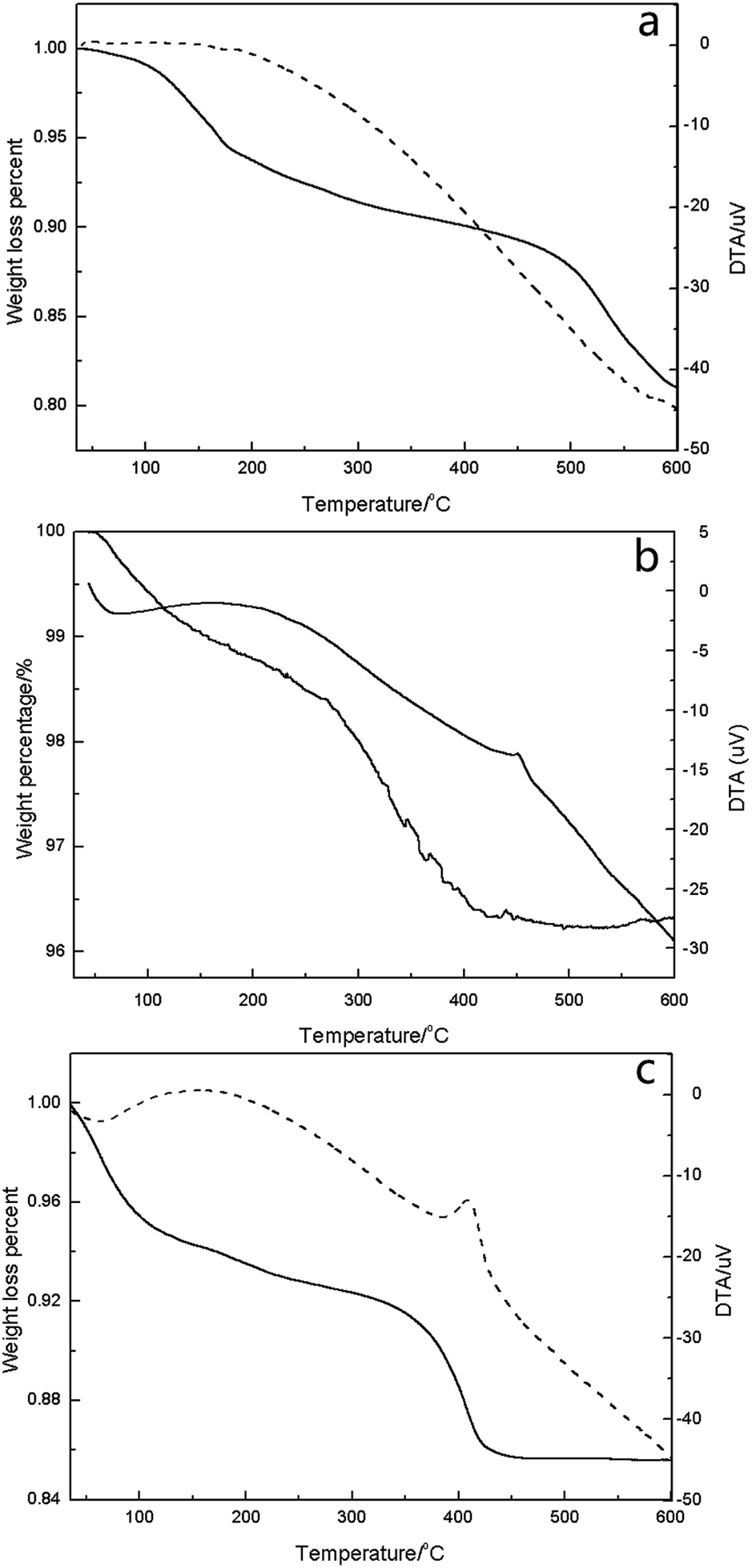 | ||
| Fig. 4 TG-DTA curves of CN-SBA, HPMV and 2HPMV/CN-SBA ((a) CN-SBA; (b). HPMV; (c) 2HPMV/CN-SBA). | ||
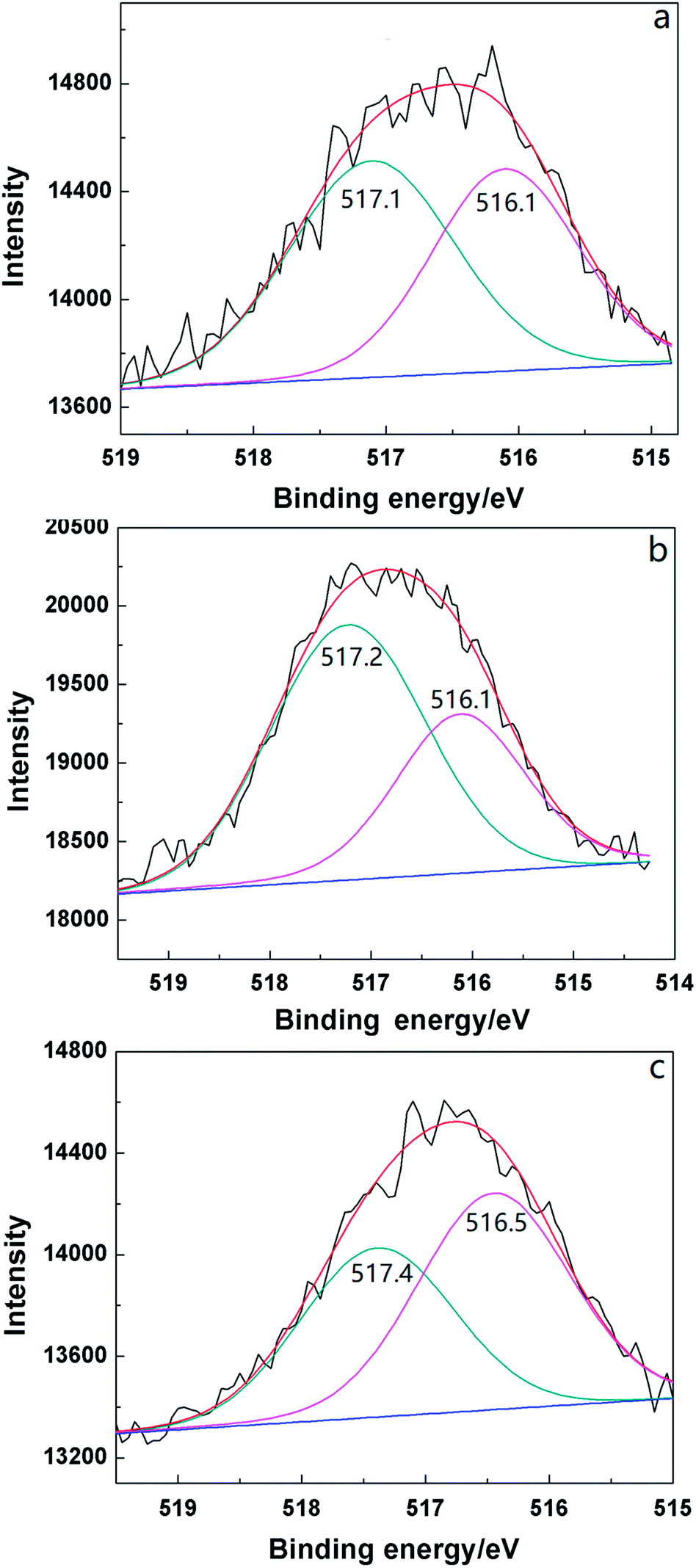 | ||
| Fig. 5 XPS spectra of V 2p in 2HPMV/CN-SBA (a) before and (b) after calcination, and (c) following the reaction. | ||
In the case of 2HPMV/CN-SBA (Fig. 5c), removal of the constitutional water had occurred at approximately 420 °C, which was 30 °C lower than bulk HPMV. Subsequently, it appears that the thermal stability of HPA can be weakened when it is loaded on a support. The decrease of thermal stability of supported HPMV may be caused by the decrease in the catalyst crystal size. The reaction temperature for oxidizing MAL to MAA is 280 to 350 °C,10–13,17,32,34,37,39–43 and therefore, the thermal stability of HPMV/CN-SBA is sufficient for application in this reaction. The temperature decreases with increasing HPMV loading, at which the supported catalysts completely decomposed (Fig. S21†). It is apparent that an increased HPMV presence lowers the thermal stability of the supported catalyst.
There were at least four different nitride species present within the carbon nitride (Fig. S24†). The peak at 400.6 eV can be attributed to amino groups and is analogous to nitride species on carbon nitride.24 The other three peaks at 400.0, 399.4, and 398.6 eV can be assigned to nitrogen species in the form of N–O, C–N, and N–H, respectively.25,27,28 These findings confirm the existence of carbon nitride and amino groups on the support surface.
Vanadyl and molybdenum species in the Keggin structure are the active species for oxidizing MAL to MAA.37 Two distinct vanadyl species were detected in the XPS profiles of the supported catalysts, V5+ at 517.1 eV and V4+ at 516.1 eV38 (Fig. 6). The ratio of V4+ in 2HPMV/CN-SBA (45.5%) was higher than that in HPMV/SBA (33.3%), with the binding energy of both vanadyl species also being lower for HPMV/SBA (Table S2†). These findings suggest that the presence of ammonia decreased the chemical states of the vanadyl species. After calcination, the ratio of V5+ increased from 54.5% to 64.3% due to V4+ oxidation by oxygen in the air flow. Following the reaction, the V5+ had decreased to 43.5% because it was reduced to V4+ by the MAL.
 | ||
| Fig. 6 NH3-TPD curves of neat HPMV, neat NH4PMV, and HPMV/CN-SBA after calcination. Catalysts were prepared at 80 °C with 2 h mixing and were calcined at 360 °C for 12 h. | ||
Two types of molybdenum species with binding energies of 232.8 and 231.6 eV are presented in Fig. S25,† which could be assigned to Mo6+ and Mo5+, respectively, in the Keggin structure.37 The Mo5+ was almost completely oxidized upon calcination, and then reappeared during the reaction. The binding energy of both molybdenum species in 3HPMV/CN-SBA before and after calcination was lower than that for HPMV/SBA. The binding energy change indicates that the ammonia reduced the chemical states of the supported heteropoly compounds, which was also demonstrated in earlier work.39 The low chemical states of the vanadyl and molybdenum species are beneficial for improving the selectivity toward MAA.
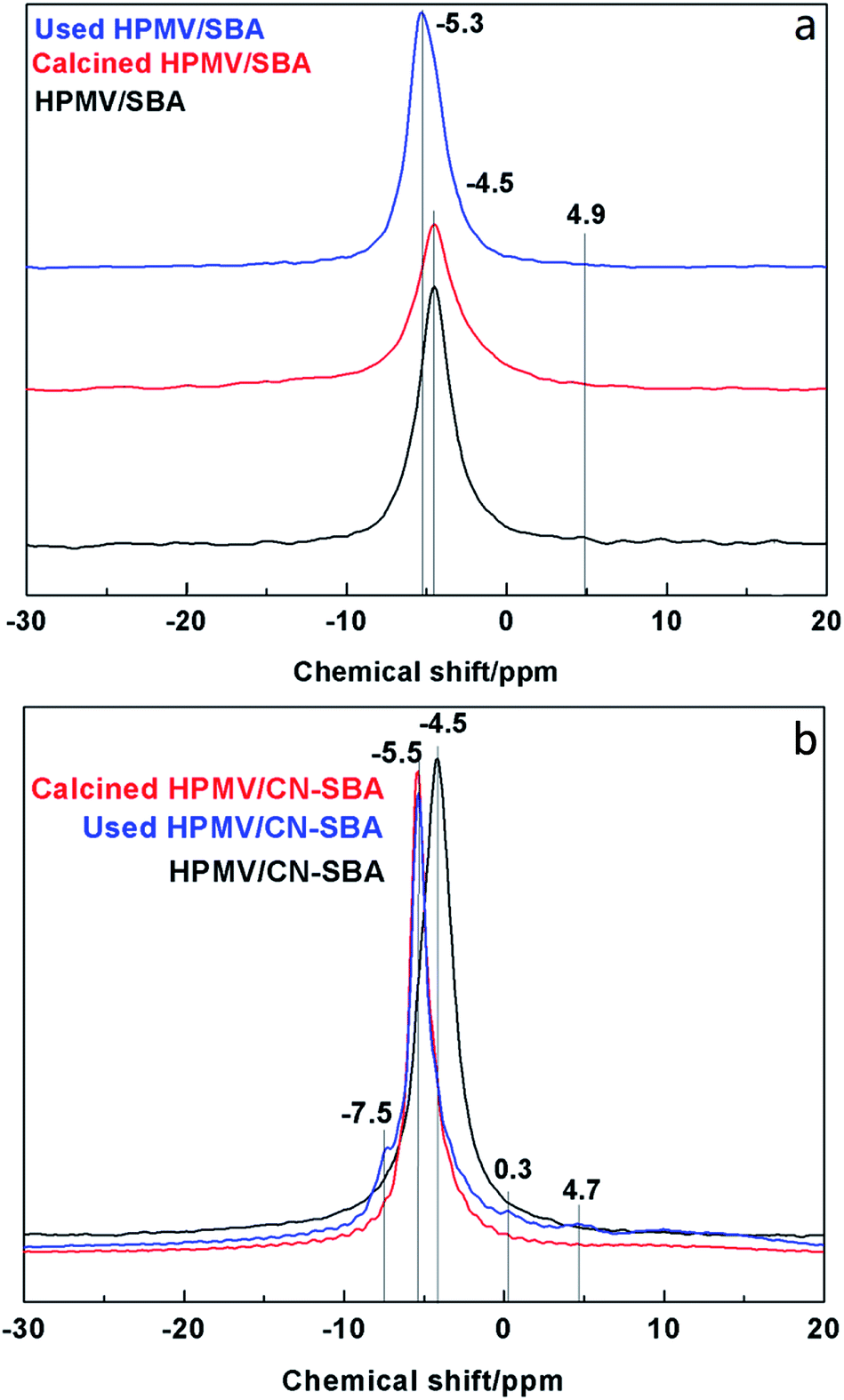 | ||
| Fig. 7 31P NMR spectra of (a) HPMV/SBA and (b) 2HPMV/CN-SBA before and after calcination and reaction. | ||
The HPMV/SBA and HPMV/CN-SBA 29Si NMR spectra are shown in Fig. 8. Chemical shifts at −110, −102.5, and −91 ppm can be attributed to the Q4, Q3, and Q2 silicon species in Si–(OSi)n(OH)4−n, respectively.38 Qn is the number of siloxane bonds linking the Si site to the silica framework. The T2 and T3 (Si–R, R is the organic groups) chemical shifts that should appear at −67 and −54 ppm are almost invisible. For CN-SBA, the chemical shifts of Q4, Q3, and Q2 move to −111, −104, and −90 ppm, respectively, and appear to arise from the influence of carbon nitride on the SBA-15 surface. Carbon nitride is rich in electrons that result in silicon species chemically shifting to high field, which also suggests that SBA-15 was successfully modified by carbon nitride. A new chemical shift at −99 ppm can be assigned to silica species interacting with amino groups. When HPMV is added to the CN-SBA support, the chemical shift of silicon species in Si-(OSi)n(OH)4−n results in movement to −109.5 and −102 ppm, which was the same as that for HPMV/SBA. Disappearance of the chemical shift at −99 ppm and weakening of the chemical shift at −90 ppm following calcination may arise from a reaction between HPMV and the amino groups on carbon nitride that causes a decrease in the electron cloud density and breaks the interaction between silica and amino groups. The 29Si NMR results demonstrate that carbon nitride strongly combined with SBA-15, and HPMV remains segregated from SBA-15.
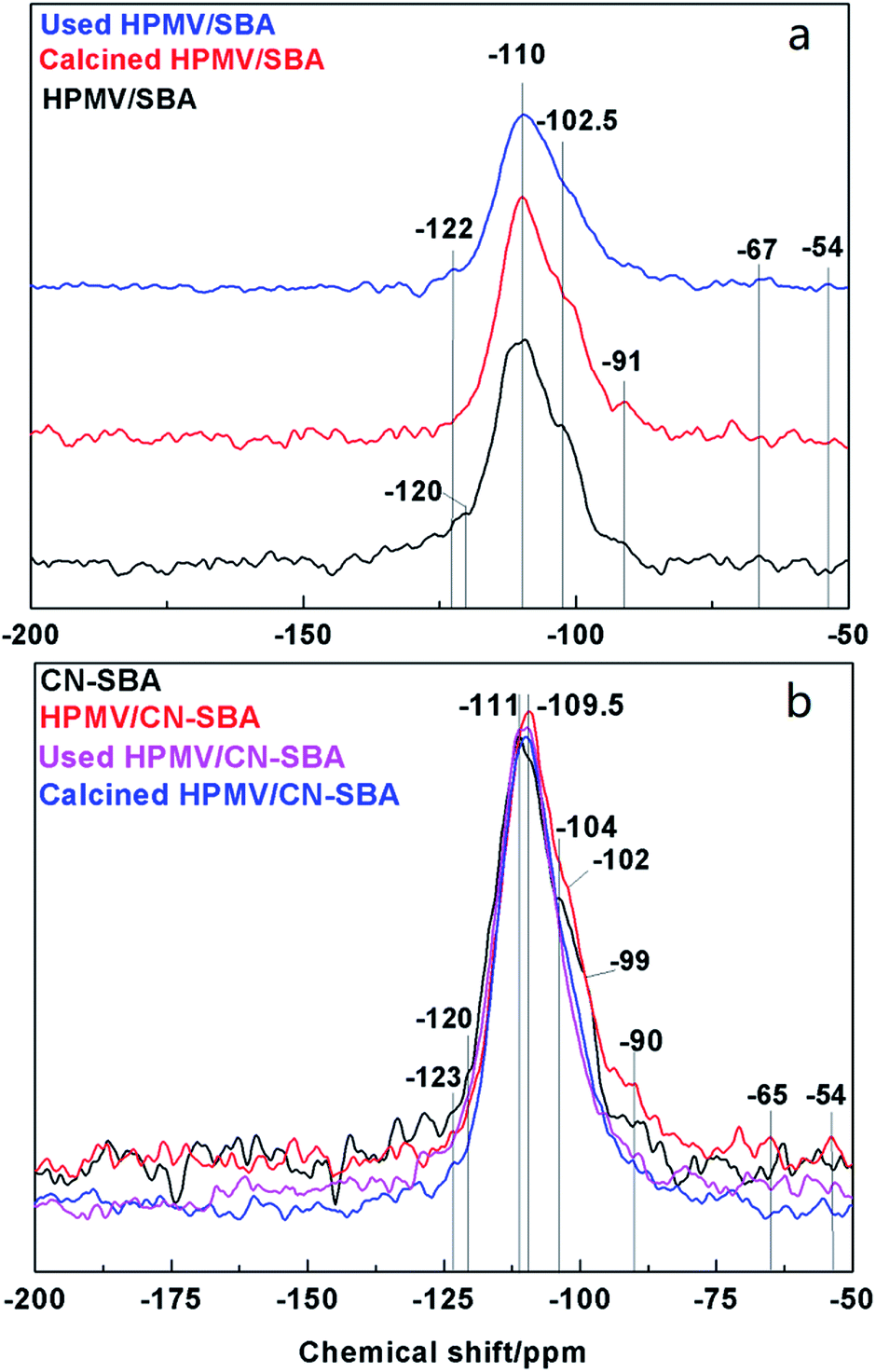 | ||
| Fig. 8 29Si NMR spectra of HPMV/SBA and 2HPMV/CN-SBA before and after calcination and post-reaction. | ||
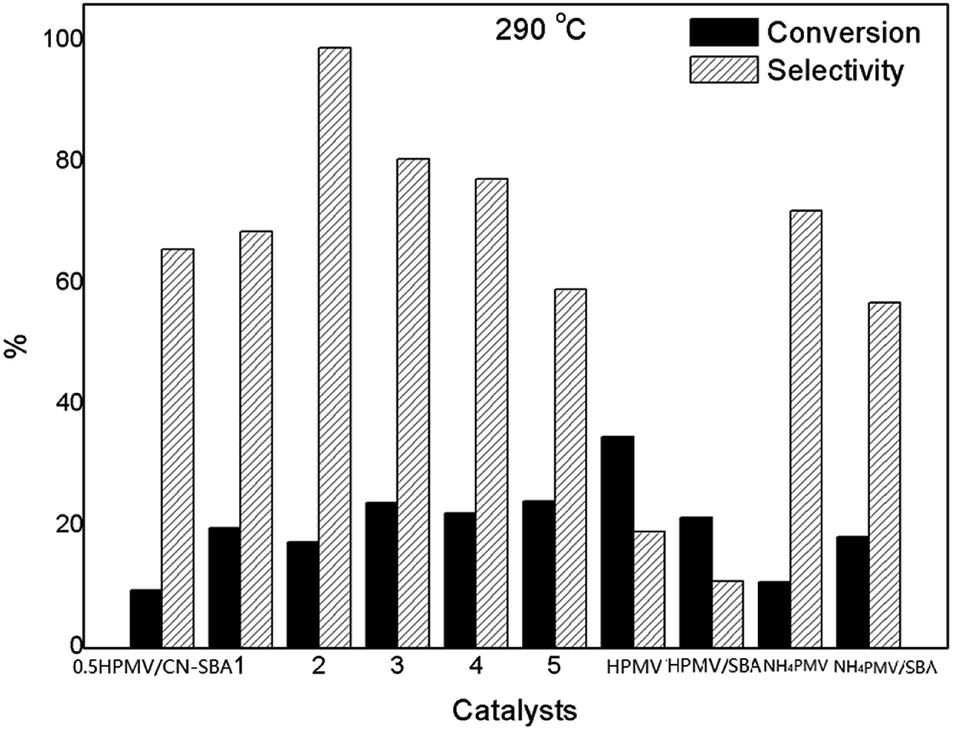 | ||
| Fig. 9 MAL conversion and MAA selectivity over 0.5HPMV/CN-SBA, 1HPMV/CN-SBA (1), 2HPMV/CN-SBA (2), 3HPMV/CN-SBA (3), 4HPMV/CN-SBA (4), 5HPMV/CN-SBA (5), and HPMV, HPMV/SBA-15, NH4PMV, and NHPMV/SBA at 290 °C. (The volume ratios of MAL, O2, and H2O in the reactant stream were 4.4 vol%, 11.1 vol%, and 17.8 vol%, respectively, with a balance of N2). | ||
When NH4PMV was supported on SBA-15, MAA selectivity decreased to 56.9%. When the reaction was performed at 310 °C, MAL conversion on the supported catalysts increased considerably, while MAA selectivity decreased (Fig. S28†). Additionally, at 310 °C, both MAL conversion and MAA selectivity increased for HPMV and NH4PMV. Similarly, when the reaction temperature was further increased to 320 °C (Fig. S30†), MAL conversion on all the catalysts increased, while MAA selectivity on all the catalysts decreased. HPMV was also successfully supported by KIT-6, HY zeolite, TiO2, Al2O3, SiO2, CNTs, and NH3-modified CNTs. Although HMPV supported on these supports showed increased catalytic activity, MAA selectivity over HPMV on these supports was much lower than that on CN-SBA (Fig. S29†). The results showed that CN-SBA is the most favourable porous support for HPMV.
Catalyst preparation temperature and mixing time during synthesis were also examined, using 2HPMV/CN-SBA with the conversion and selectivity results at 290 °C shown in Fig. 10, S29 and S30†. Increasing the preparation temperature (Fig. 10a) provided a small increase in MAL conversion, while MAA selectivity was the most optimal (98.9%) at a preparation temperature of 80 °C. The most suitable mixing time synthesis was found to be 2 h (Fig. 10b), with longer times resulting in poorer catalyst selectivity, in particular.
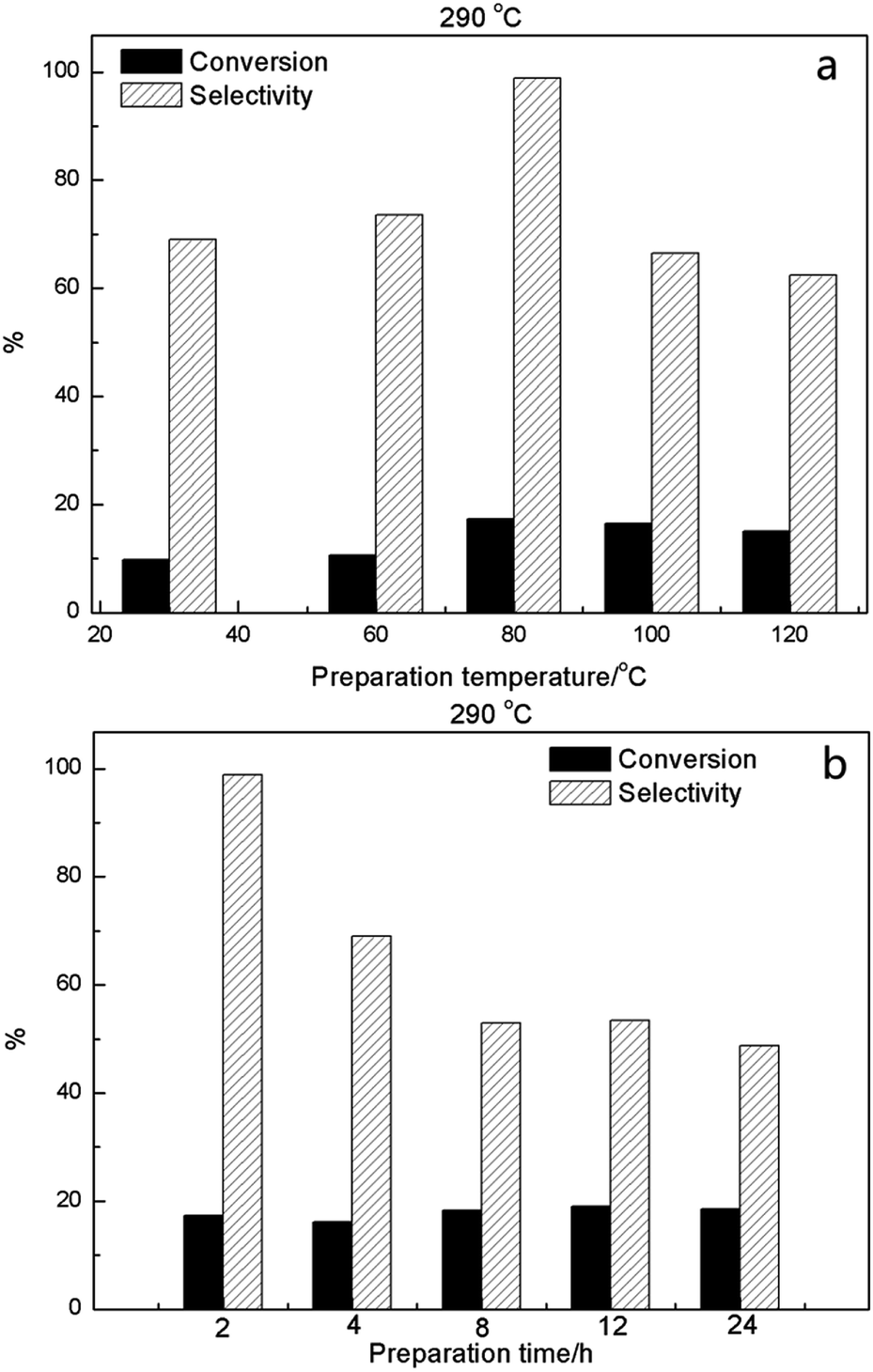 | ||
| Fig. 10 MAL conversion and MAA selectivity at 290 °C for 2HPMV/CN-SBA prepared at (a) different temperatures and (b) using different synthesis (mixing) times. | ||
Discussion
Relative to all the control cases, the HPMV/CN-SBA delivered a generally better catalytic performance (Fig. 8), either in terms of conversion or MAA selectivity. Loading HPMV on a silica support resulted in a decrease in both activity and selectivity relative to the neat HPMV. The poorer catalytic performance of HPMV/SBA arose from the decomposition of HPMV into MoO3 and V2O5, due to interaction between the HPMV and the silica support 17. The poor selectivity is thought to have originated from further MAA oxidation on the MoO3 and V2O5 to other byproducts.37 Including NH4+ in the HPMV structure is particularly beneficial for MAA selectivity, although loading it into the SBA-15 mildly counteracted the benefit. FT-IR and XRD results (Fig. S33 and S34†) showed that part of the NH4PMV on SBA-15 degraded to MoO3 and V2O5 when supported on the SBA-15. Consequently, it is apparent that it is necessary to separate HPMV from silica to minimize their interaction and avoid partial decomposition of the HPA. For almost all the HPMV catalysts supported on CN-SBA, a comparable or higher MAA selectivity relative to the NH4PMV/SBA was observed. Importantly, no MoO3 or V2O5 was detected for the HPMV/CN-SBA by any of the characterization techniques, demonstrating that the CN is crucial for keeping the HPMV from the silica and minimizing HPA decomposition. The CN barrier strategy adopted here is an effective approach for improving and maintaining catalyst performance.From a MAL conversion perspective, the most optimal performance was exhibited by the neat HPMV because it was able to process the strongest acidity (Fig. S35 and Table S2†). The oxidation of MAL to MAA on HPAs follows the Van-Marvel mechanism, whereby strong acidity is positive for MAL adsorption on the catalyst surface, which then promotes higher MAL conversion.43 Consequently, MAL conversion on both NH4PMV and NH4PMV/SBA was lower than that for HPMV due to their lower acidity levels. When HPMV was loaded onto the CN-SBA, overall, the acidity decreased compared to the neat HPMV, although the effect was moderated with increasing HPMV loading. Interaction of the HPMV with basic amino groups on the CN-SBA surface effectively lessened HPA acidity, and in turn, its capacity for MAL conversion. Additional HPMV acted to partially offset the neutralizing influence of the amino groups, which appeared to reach (and remain at) a maximum HPMV loading of 3. The findings demonstrate that although the surface area of the supported catalysts was considerably larger than that for bulk HPAV (Table 1), any beneficial impact on MAL conversion was overshadowed by the influence of the basic amino groups on the CN-SBA. These findings demonstrate that acidity is the key driver that influences MAL conversion, and while the CN is important for preserving the HPA structure, its amino groups have a mildly negative impact on catalytic activity.
The presence of ammonium in the catalyst system resulted in a significant increase in selectivity towards MAA. Previous studies showed that ammonia addition decreased the chemical valence of vanadyl and molybdenum species, such that the oxidation susceptibility decreased. This hampered MAL conversion on the CN-SBA-supported HPMV, NH4PMV, and NH4PMV/SBA.43 However, restricting oxidation susceptibility could also prevent deep oxidation of the MAL and increase MAA selectivity.43,44 Here, with the increase in the HPMV loading, MAA initially increased to reach a maximum for 2HPMV/CN-SBA, after which it began to decrease. As the HPMV loading increased, the surface area decreased (Table 1), although this was eclipsed by a parallel increase in catalyst acidity (Table S2†), while the ammonia content remained constant.
The low acidity (e.g., 0.5HPMV/CN-SBA) was not positive for MAL adsorption and MAA desorption, resulting in the observed low MAL conversion and MAA selectivity. However, when acidity was too strong (5HPMV/CN-SBA), deep oxidation of the MAA occurred, which decreased the MAA selectivity while retaining reasonable MAA conversion. Additionally, if the HPMV loading was too low or too high, the decomposition of the HPA to MoO3 or V2O5 was evident (Fig. S10†). Decomposition of HPMV on CN-SBA when at a low is envisaged to relate to the small size of supported HPA, rendering it unstable at an elevated temperature. In contrast, it is thought that higher HPMV loadings lead to direct interaction between the SBA-15 and the HPA, promoting its partial decomposition. It is suspected that the SBA-15 surface in not entirely covered with C3N4. When the HPA is loaded onto CN-SBA, it would preferentially react with and be immobilized by amino groups on the C3N4 surface. However, when the C3N4 surface area became saturated with HPMA at high loading, excess HPMA was found directly on the silica surface. The ideal HPMV loading appears to be approximately 66.7 wt% (i.e., 2HPMV/CN-SBA).
Consideration of the catalyst preparation temperature and synthesis (mixing) time showed that they both influenced the performance in terms of MAL conversion and MAA selectivity, with the effect being considerably more pronounced for selectivity. An optimum synthesis temperature of 80 °C was observed. Catalysts prepared at a temperature below 80 °C exhibited smaller crystal size, which was detrimental to performance. At synthesis temperatures above 80 °C, FTIR and XRD illustrated the presence of MoO3 in the catalysts (Fig. S8 and S12†), indicating that partial thermal decomposition of the HPMV had occurred, which was responsible for the diminished performance. In relation to synthesis time, a shorter stirring time (2 h, Fig. 10b and S32†) delivered more optimal catalyst performance. The XRD spectra again showed an MoO3 presence for synthesis times of 4 h or more, highlighting a decomposition of the HPMV and explaining the poorer performance.
Experimental
Catalyst preparation
HPMV was synthesized according to a previously published procedure described by Kanno et al.11 A mixture comprising 5 g of molybdenum trioxide (Sinopharm Chemical Reagent Co., Ltd.) and 0.287 g of vanadic anhydride (Tianjin Guangfu Fine Chemical Research Institute) in 100 mL of deionized water was initially heated to reflux. An 85% solution of H3PO4 (Sinopharm Chemical Reagent Co., Ltd.), equal to 0.364 g of acid, was added dropwise into the turbid liquid. The mixture was vigorously stirred and refluxed for 5 h, and a deep-orange solution was formed. The solid was recovered via filtration, and then, the solution was evaporated at 80 °C to obtain a bright orange powder. The structure of HPMV was confirmed by FT-IR (Fig. S1†).SBA-15 was prepared by a process described elsewhere.32 In a typical experiment, P123 (4 g, poly(ethylene glycol)-block-poly(propylene glycol)-block-poly(ethylene glycol), Aldrich Chemical Co.) was dissolved in 125 mL of 1.8 wt% hydrochloric acid (Sinopharm Chemical Reagent Co. Ltd.) solution at 40 °C. Tetraethoxysilane (8.16 g, TEOS, Sinopharm Chemical Reagent Co. Ltd.) was then added dropwise into the solution, which was hydrolyzed for 24 h at 40 °C. Following hydrolysis, the mixture was hydrothermally treated at 100 °C for 24 h. The resulting solid was recovered using filtration, washed with water and alcohol, and calcined at 550 °C for 6 h. It was characterized by small angle XRD (Fig. S2†).
Carbon nitride (CN)-modified SBA-15 (CN-SBA) was prepared by dissolving 0.8 g of melamine into 30 mL of deionized water at 80 °C. After the melamine was completely dissolved, 2 g of SBA-15 was added to the solution with the mixture vigorously stirring, and the stirring continued for 2 h. Water was then evaporated from the mixture at 110 °C, and the solid dried in an oven at 80 °C for 12 h. The dried solid was calcined in an airtight magnetic boat at a ramp rate of 3 °C min−1 to 550 °C, where the temperature was held for 3 h. The amino group content of CN-SBA was 2.2 mmol g−1, which was evaluated using CO2-TPD and energy dispersive X-ray spectroscopy (EDS) (Fig. S3 and S4†).
HPMV was loaded onto the CN-SBA using the following procedure. First, 2 g of HPMV was dissolved in 10 mL of deionized water and added dropwise to 1 g of CN-SBA powder. The mixture was then stirred at 80 °C for 2 h. The remaining water was evaporated, and the solid dried in an oven at 80 °C for 12 h. Catalyst nomenclature is in the form of nHPMV/CN-SBA, where n denotes the HPMV loading (n = (weight of HPMV, g)/(weight of CN-SBA, g)). HPMV was also loaded onto neat SBA-15 (nHPMV/SBA) following the same procedure described above.
As a control to evaluate the HPMV/CN-SBA, NH4H3PMo11VO40 (NHPMV) was synthesized by mixing 2 g of HPMV in 10 mL of deionized water with 0.06 g of NH4Cl (Sinopharm Chemical Reagent Co., Ltd.) in 10 mL of deionized water and stirring at 80 °C for 2 h. The remaining water was evaporated, and the resulting yellow solid was dried in an oven at 80 °C for 12 h.
NHPMV supported on SBA-15 (NHPMV/SBA) was prepared by dissolving 0.06 g NH4Cl in 10 mL deionized water, and the solution was added dropwise onto 3 g of dried HPMV/SBA. The mixture was then stirred at 80 °C for 2 h. Any remaining water was evaporated, and the recovered yellow solid was dried in an oven at 80 °C for 12 h.
Following synthesis, all catalysts were calcined at 360 °C in air at a flow rate of 80 mL min−1 in a pipe furnace (SK-G03123K, Zhonghuan Co., Ltd., Tianjin).
Catalyst characterization
Fourier transform infrared (FT-IR) spectrograms of the catalysts were recorded from 4000 to 400 cm−1 on a Nicolet 380 FT-IR spectrometer (Thermal Electron Corporation) with anhydrous KBr as the standard. The crystal phase of the catalysts was evaluated by X-ray diffraction (XRD, Bruker D8 Advance X-ray powder diffractometer) with Cu Ka radiation (λ = 0.154 nm) as the X-ray source. Thermal stability of the samples was analyzed using a thermogravimetric differential thermal analyzer (TG/DTA, DTG-60H, Shimadzu, Japan). Approximately 10 mg of the sample was heated from 25 to 700 °C at 5 °C min−1 in an air atmosphere (flow rate 10 mL min−1). Catalyst acidity was determined using NH3 temperature-programmed desorption analysis on a Micromeritics AutoChem II Chemisorption Analyzer. NH3 desorption was recorded over the temperature range of 50 to 650 °C at a ramp rate of 10 °C min−1. Catalyst morphology was observed using scanning electron microscopy (SEM, SU8020, Hitachi, Japan). Oxidation states of the catalyst components were evaluated by X-ray photoelectron spectroscopy (XPS, Kratos Axis Ultra DLD spectrometer) with monochromatic Al Kα radiation as the source. Surface area and pore structure of the catalysts were analyzed using N2 physisorption on a Micromeritics ASAP 2460 (US) via the BET and Barrett–Joyner–Halenda (BJH) methods, respectively. Prior to analysis, the samples were pretreated by heating at 200 °C for 4 h under vacuum. Silicon and phosphorus species in the catalysts were detected by 29Si NMR and 31P NMR [Bruker Avance III (400M)] analyses at 12 kHz, respectively.Catalyst performance
Catalyst performance was evaluated at atmospheric pressure in a 400 mm-long fixed bed reactor with an internal diameter of 6 mm. Prior to performance assessment, the bulk catalysts were crushed and screened to a mesh size fraction of 20 to 40. Next, 0.8 g of the catalyst was loaded into the reactor. Air was fed through the catalyst bed at a rate of 10 mL min−1, and the bed temperature was increased to 310 °C. The reactant gas stream, comprising N2 that had passed through a bubbler containing MAL at 19 °C with water added via an advection pump, was passed through the catalyst bed at 19 mL min−1. The volume percent (vol%) of MAL, O2, and H2O in the reactant stream was 4.4, 11.1, and 17.8, respectively, with the balance composed of N2. Once the reaction had proceeded for 1 h, the products were analyzed by a gas chromatograph (7890B, Agilent Technologies Co. Ltd.) with a flame ionization detector and a DB-FFAP capillary column. The column tank temperature increased from 40 to 240 °C at a rate of 25 °C min−1 and was maintained at 240 °C for 5 min; the injection temperature was 180 °C. The MAA selectivity was calculated by the molar amount of MAA being divided by the converted MAL molar amount (Scheme 1).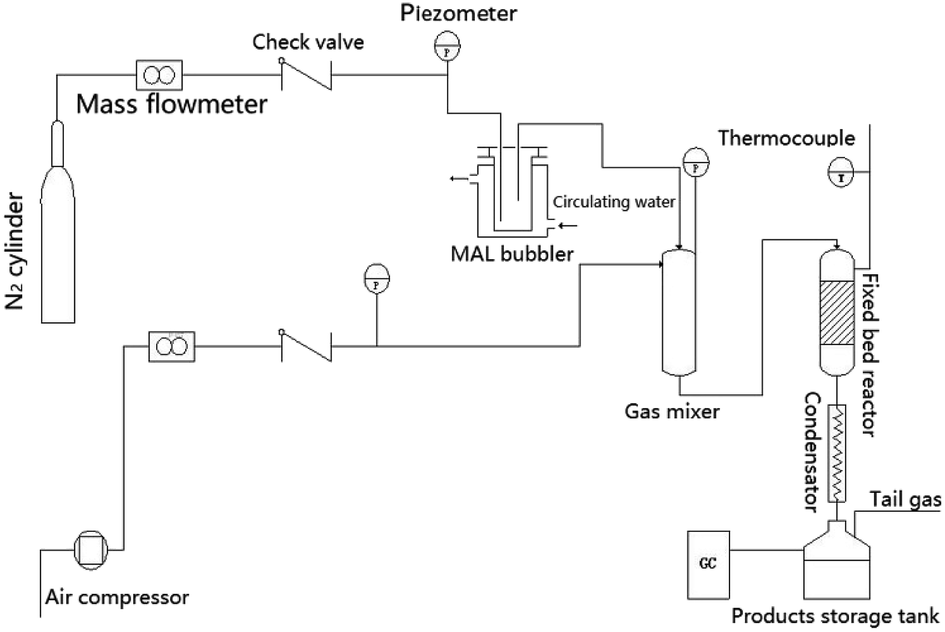 | ||
| Scheme 1 Catalytic process for oxidation of MAL to MAA. | ||
MAL conversion was calculated by eqn (1), and selectivity toward MAA was calculated by eqn (2).
| MAL conversion = (1 − F(MAL)tail)/F(MAL) | (1) |
| MAA selectivity = F(MAA)/[F(MAL) × Conversion] | (2) |
Conclusions
In this work, modified SBA-15 was used as a support for HPMV for the oxidation of MAL to MAA. The CN modification offered twin benefits in terms of improving catalyst performance: (i) it acted as a barrier to restrict direct interaction between HPMV and SBA-15, improving HPMV stability; (ii) it furnished the catalyst support with amino groups, which were advantageous for MAA selectivity. In the absence of C3N4, HPMV was partially decomposed to MoO3 and V2O5 by SBA-15, which was detrimental to performance. At an optimum HPMV catalyst formulation of 2HPMV/CN-SBA, the MAA selectivity was improved by more than five times over neat HPMV. Lower and higher HMPV loadings resulted in partial decomposition of the HPMV, which was attributed to smaller less thermally stable HPMV deposits and excess HPMV spillover onto the SBA-15, respectively. Synthesis temperature and mixing time were found to be important for catalyst performance, with the optimized conditions identified to be 80 °C and 2 h stirring, respectively. These findings reveal a new approach that overcomes selectivity and stability challenges associated with using supported heteropoly acids as catalysts for oxidation reactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge financial support from the Youth Foundation of the Education Department of Hebei Province (No. QN2019230) and the Natural Science Foundation of Hebei Province Youth Project (No. B2019208333).Notes and references
- J. W. Bauer, Ultmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2002
.
- K. Nagai, Appl. Catal., A, 2001, 221, 367–377 CrossRef CAS
- N. Mizuno and M. Misono, Chem. Rev., 1998, 98, 199–217 CrossRef CAS
- M. Misono, ChemComm, 2001, 1141–1152 RSC
- L. M. G. Sainero, S. Damyanova and J. L. G. Fierro, Appl. Catal., A, 2001, 208, 63–75 CrossRef
- N. K. Kala Raj, S. S. Deshpande, R. H. Ingle, T. Raja and P. Manikandan, Catal. Lett., 2004, 98, 217–224 CrossRef
- A. Popa, V. Sasca and J. Halasz, Appl. Surf. Sci., 2008, 255, 1830–1835 CrossRef CAS
- A. A. Spojakina, N. G. Kostova, B. Sow, M. W. Stamenova and K. Jiratova, Catal. Today, 2001, 65315–65321 Search PubMed
- N. Legagneux, J. M. Basset, A. Thomas, F. Lefebvre, A. Goguet, J. Sab and C. Hardacre, Dalton Trans., 2009, 2235–2240 RSC
- N. Ballarini, F. Candiracci, F. Cavani, H. Degrand, J. L. Dubois, G. Lucarelli, M. Margotti, A. Patinet, A. Pigamo and F. Trifirò, Appl. Catal., A, 2007, 325, 263–269 CrossRef CAS
- M. Kanno, T. Yasukawa, W. Ninomiya, K. Ooyachi and Y. Kamiya, J. Catal., 2010, 273, 1–8 CrossRef CAS
- M. Kanno, Y. Miura, T. Yasukawa, T. Hasegawa, W. Ninomiya, K. Ooyachi, H. Imai, T. Tatsumi and Y. Kamiya, Catal. Commun., 2011, 13, 59–62 CrossRef CAS
- M. Hino and K. Arata, Green Chem., 2001, 3, 170–172 RSC
- J. M. Tatibout, C. Montalescot and K. Brückman, Appl. Catal., A, 1996, 138, 11–16 Search PubMed
- Y. Guo, K. Li, X. Yu and J. H. Clark, Appl. Catal., B, 2008, 81, 182–191 CrossRef CAS
- A. Gaurav, F. T. T. Ng and G. L. Rempel, Green Energy & Environment, 2016, 1, 62–74 Search PubMed
- L. Zhou, L. Wang, H. Wang, Y. Cao, R. Yan and S. Zhang, J. Thermodyn. Catal., 2016, 7(4), 176–181 CrossRef
- A. Y. Liu and M. L. Cohen, Science, 1989, 245, 841–842 CrossRef CAS
- M. Kawaguchi, S. Yagi and H. Enomoto, Carbon, 2004, 423, 45–350 Search PubMed
- Y. Qiu and L. Gao, Chem. Commun., 2003, 9, 2378–2379 RSC
- Q. Guo, Q. Yang, L. Zhu, C. Yi, S. Zhang and Y. Xie, Solid State Commun., 2004, 132, 369–374 CrossRef CAS
- E. Kroke and M. Schwarz, Coord. Chem. Rev., 2004, 248, 493–532 CrossRef CAS
- Y. J. Bai, B. Lu, Z. G. Liu, L. Li, D. L. Cui, X. G. Xu and Q. L. Wang, J. Cryst. Growth, 2003, 247, 505–508 CrossRef CAS
- J. L. Zimmerman, R. Williams, V. N. Khabashesku and J. L. Margrave, Nano Lett., 2001, 1731–1734 Search PubMed
- V. N. Khabashesku, J. L. Zimmerman and J. L. Margrave, Chem. Mater., 2000, 12, 3264–3270 CrossRef CAS
- J. Wang, D. R. Miller and E. G. Gillan, Carbon, 2003, 41, 2031–2037 CrossRef CAS
- D. R. Miller, J. Wang and E. G. Gillan, J. Mater. Chem., 2002, 12, 2463–2469 RSC
- M. Kim, S. Hwang and J. S. Yu, J. Mater. Chem., 2007, 17, 1656–1659 RSC
- J. Kouvetakis, A. Bondari, M. Todd, B. Wilkens and N. Cave, Chem. Mater., 1994, 6, 811–814 CrossRef CAS
- X. Chen, Y. S. Jun, K. Takanabe, K. Maeda, K. Domen, X. Fu, M. Antonietti and X. Wang, Chem. Mater., 2009, 21, 4093–4095 CrossRef CAS
- D. Zhao, J. Feng, Q. Huo, N. Melosh, N. Fredrickson, B. F. Chmelka and G. D. Stucky, Science, 1998, 279, 548–552 CrossRef CAS
- L. Marosi and C. O. Areán, J. Catal., 2003, 213, 235–240 CrossRef CAS
- D. Zhao, Q. Huo, J. Feng, B. F. Chmelka and G. D. Stucky, J. Am. Chem. Soc., 1998, 120, 6024–6036 CrossRef CAS
- H. Zhang, R. Yan, L. Yang, Y. Diao, L. Wang and S. Zhang, Ind. Eng. Chem. Res., 2013, 52, 4484–4490 CrossRef CAS
- B. Lei, B. Li, H. Zhang, L. Zhang and W. Li, J. Phys. Chem. C, 2007, 111(30), 11291–11301 CrossRef CAS
- X. Fan, L. Zhang, R. Cheng, M. Wang, M. Li, Y. Zhou and J. Shi, ACS Catal., 2015, 5(9), 5008–5015 CrossRef CAS
- L. Zhou, L. Wang, S. Zhang, R. Yan and Y. Diao, J. Catal., 2015, 329, 431–440 CrossRef CAS
- K. Mukhopadhyay, A. Ghoshb and R. Kumar, Chem. Commun., 2002, 2404–2405 RSC
- Y. L. Cao, L. Wang, L. L. Zhou, G. J. Zhang, B. H. Xu and S. J. Zhang, Ind. Eng. Chem. Res., 2017, 56(3), 653–664 CrossRef CAS
- F. Jing, B. Katryniok, F. Dumeignil, E. Bordes-Richard and S. Paul, J. Catal., 2014, 309, 121–135 CrossRef CAS
- I. V. Kozhevnikov, J. Mol. Catal. A: Chem., 2009, 305, 104–111 CrossRef CAS
- S. Ganapathy, M. Fournier, J. F. Paul, L. Delevoye, M. Guelton and J. P. Amoureux, J. Am. Chem. Soc., 2002, 124, 7821–7828 CrossRef CAS
- Y. L. Cao, L. Wang, L. L. Zhou, B. H. Xu, Y. Y. Diao and S. J. Zhang, J. Ind. Eng. Chem., 2018, 65, 254–263 CrossRef CAS
- L. Zhou, L. Wang, Y. Diao, R. Yan and S. Zhang, Mol. Catal., 2017, 433, 153–161 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra06021a |
| This journal is © The Royal Society of Chemistry 2019 |