
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIodine-mediated C–N and N–N bond formation: a facile one-pot synthetic approach to 1,2,3-triazoles under metal-free and azide-free conditions†
Geeta Sai Mania,
Kavitha Donthiboinaa,
Siddiq Pasha Shaikb,
Nagula Shankaraiah *a and
Ahmed Kamal*abc
*a and
Ahmed Kamal*abc
aDepartment of Medicinal Chemistry, National Institute of Pharmaceutical Education and Research (NIPER), Hyderabad 500037, India. E-mail: shankar@niperhyd.ac.in
bMedicinal Chemistry and Pharmacology, CSIR-Indian Institute of Chemical Technology, Hyderabad 500007, India. E-mail: ahmedkamal@iict.res.in
cSchool of Pharmaceutical Education and Research (SPER) Jamia Hamdard, New Delhi, 110062, India
First published on 28th August 2019
Abstract
A novel strategy towards the synthesis of 1,4-disubstituted 1,2,3-triazoles via C–N and N–N bond formation has been demonstrated under transition metal-free and azide-free conditions. These 1,2,3-triazoles were obtained in a regioselective manner from commercially available anilines, aryl alkenes/aryl alkynes and N-tosylhydrazines using I2 under O2 atmosphere. Broad substrate scope, milder reaction conditions, good to moderate yields and clean protocol are the notable features of the method. Moreover, this protocol is amenable for the generation of a library of medicinally important key building blocks.
Introduction
Nitrogen-containing heterocyclics constitute a major class of bioactive organic compounds which are essential for life and widely distributed in natural products and synthetic pharmaceuticals.1 They form a structural part of vitamins, hormones, etc and are fundamental for metabolism in living cells.2 Their unique ability to serve as biomimetics and reactive pharmacophores has facilitated their use as low molecular weight lead molecules in drug design.3 Particularly, 1,2,3-triazole contributes a noteworthy role among the pharmaceutically important nitrogen-containing heterocyclic scaffolds due to its broad spectrum of biological activities. Active pharmaceuticals based on 1,2,3-triazoles including tert-butyldimethylsilylspiroaminooxathioledioxide (TSAO, non-nucleoside reverse transcriptase inhibitor),4a carboxyamidotriazole (CAI, anticancer),4b cefatrizine4c and tazobactum (β-lactam antibiotic)4d are shown in Fig. 1. Over the past few years, our research group has significantly developed many novel 1,2,3-triazole bearing molecules with anticancer activities.4e–k Moreover, they are also reported to have anti-inflammatory,5a anticonvulsant,5b antiviral,5c antihistaminic,5d antileishmanial,5e peptidomimetic tyrosinase inhibitory,5f hepatitis C virus inhibitory,5g neuroleptic5h and adenosine receptor inhibitory5i properties, as well as being conditioning agents for plant growth.5j Additionally, the 1,2,3-triazole moiety is an ideal linker that has been widely utilized in the recent past to bridge the two components in order to get the synergistic effect of the combined molecules and it is relatively resistant to hydrolysis reactions.6 Beyond pharmaceutical applications, the structural features of 1,2,3-triazole facilitate to mimic various functional groups justifying its use as a bioisostere in the synthesis of biologically active molecules.7 Moreover, due to the hydrogen-bonding ability of 1,2,3-triazole connecting to a poorly soluble drug could enhance their solubility aspects and accordingly a better pharmacokinetic profile is achieved.8 Owing to their utility, a wide range of applications and many novel synthetic protocols have been developed by many researchers for the regioselective formation of this five-membered heterocycle and it is highly exploring motif in organic synthesis. | ||
| Fig. 1 Representative biologically active molecules containing 1,2,3-triazole. | ||
Numerous methodologies have been reported previously and still, many researchers are working for the development of effective new routes for the synthesis of 1,2,3-triazoles due to their valuable drug-like nature. The classical methods include Huisgen's thermally induced 1,3-dipolar cycloaddition of alkynes and azides.9 However, it is well-known that this method suffers from the limited substrate scope and poor regioselectivity. Subsequently, Sharpless and Meldel groups independently proposed a copper-catalyzed azide–alkyne cycloaddition (CuAAC) for the synthesis of 1,2,3-triazoles which is a highly regioselective and straight forward method.10 This approach made 1,2,3-triazole more accessible in organic synthesis. Later, numerous methods have been developed for the synthesis of 1,2,3-triazoles comprising the use of metal triflates,11 expensive metal catalyst complexes,12 organocatalysts,13 ionic liquids,14 microwaves,15 polymers,16 bases,17 etc. However, most of these reported methods utilized heavy metals (Cu, Pd, Ru, Ir) which limit their use in the biological, environmental point of views due to their hazardous nature and toxicity profile. Moreover, organic azides are also exploited which are likely to explode and difficult to handle on large scale. Consequently, a great deal of interest was diverted towards the development of simple, metal-free and azide-free approaches for the synthesis of these attractive scaffolds,18 because metal free approaches are very lucrative on account of being inexpensive and environmentally benign nature. However, regioselective construction of C–N, N–N bonds is quite difficult in the absence of transition metals. In this context, molecular iodine has grabbed much attention of researchers owing to its advantages viz. relatively inexpensive, efficient and a green alternative to transition metals.19 On the other hand, iodine is also reported to provide a mild and efficient approach involved in numerous oxidative transformations for the formation of C–C, C–N and N–N bonds.20 As a part of our research program in the development of medicinally active molecules in an eco-friendly manner herein, we developed a novel method for the synthesis of 1,2,3-triazoles regioselectively in a one-pot manner by using I2/O2 and DMSO as a solvent from readily accessible anilines, aryl alkynes or aryl alkenes and N-tosylhydrazine as substrates.
Results and discussion
At the onset to test the feasibility of the reaction, a selected model reaction was performed using 4-methyl aniline (1p), phenylacetylene (2a) and N-tosyl hydrazine (3a) in DMSO with iodine (1.0 equiv.) at room temperature for 24 h, resulting in no desired product 4p was observed (entry 1, Table 1). To our delight, 4p was obtained in low yield (10%) when we performed the reaction at 100 °C for 12 h (entry 2, Table 1). In order to improve the yield of 4p, we further screened different oxidants, solvents, catalysts, amount of catalyst as well as reaction temperature. Interestingly, reaction yield was increased to a greater extent providing the formation of 4p with 76% yield, when the reaction mixture was heated to 100 °C for 6 h in the presence of oxygen (entry 3, Table 1). Encouraged by the above result, common oxidants namely TBHP, oxone, PhI(OAc)2, K2S2O8 (entries 4–7, Table 1) were screened and O2 was found to be a suitable oxidant. DMSO played a crucial role in this reaction and found to be the best solvent when compared to other tested solvents such as DMF, 1,4-dioxane and CH3CN found to deliver the required product with inferior yields (entries 9–11, Table 1). In non-polar solvent such as toluene, under optimized reaction conditions, product 4p was not formed (entry 8, Table 1). Next, we screened various iodine-containing catalysts such as potassium iodide (KI), N-iodosuccinimide (NIS) and tetrabutylammonium iodide (TBAI) in DMSO to check the feasibility of the reaction (entry 12–14, Table 1).
|
||||||
|---|---|---|---|---|---|---|
| Entry | Additive (equiv.) | Oxidant (equiv.) | Solvent | Temperature (°C) | Time (h) | Yield (%)b |
| a All reactions were performed with 1p (1 mmol), 2a (1.1 mmol), and 3a (1.5 mmol) in presence of I2 (1.2 equiv.) and under O2 at 120 °C for 6 h.b Isolated yields.c Iodine was added to a solution of 1p (1 mmol), 2a (1.1 mmol) and allowed to stir for 1 h followed by addition of 3a (1.5 mmol) under O2 at 120 °C for 4 h. NF – Not Formed. TBHP = tert-butyl hydroperoxide (70% in water), DMSO = dimethyl sulfoxide, DMF – dimethyl formamide. | ||||||
| 1 | I2 (1.0) | — | DMSO | rt | 24 | NF |
| 2 | I2 (1.0) | — | DMSO | 100 | 12 | 10 |
| 3 | I2 (1.0) | O2 | DMSO | 100 | 6 | 76 |
| 4 | I2 (1.0) | TBHP (1) | DMSO | 100 | 6 | 68 |
| 5 | I2 (1.0) | Oxone (1) | DMSO | 100 | 6 | 56 |
| 6 | I2 (1.0) | PhI(OAc)2(1) | DMSO | 100 | 6 | 26 |
| 7 | I2 (1.0) | K2S2O8 (1) | DMSO | 100 | 6 | 38 |
| 8 | I2 (1.0) | O2 | toluene | 100 | 6 | NF |
| 9 | I2 (1.0) | O2 | DMF | 100 | 6 | 56 |
| 10 | I2 (1.0) | O2 | 1,4-Dioxane | 100 | 6 | 32 |
| 11 | I2 (1.0) | O2 | CH3CN | 80 | 6 | 12 |
| 12 | KI (1.0) | O2 | DMSO | 100 | 6 | 10 |
| 13 | NIS (1.0) | O2 | DMSO | 100 | 6 | 34 |
| 14 | TBAI (1.0) | O2 | DMSO | 100 | 6 | 28 |
| 15 | — | O2 | DMSO | 100 | 24 | NF |
| 16 | I2 (1.0) | O2 | — | 100 | 6 | trace |
| 17 | I2 (1.2) | O2 | DMSO | 100 | 6 | 82 |
| 18 | I2 (1.5) | O2 | DMSO | 100 | 6 | 78 |
| 19 | I2 (0.8) | O2 | DMSO | 100 | 6 | 71 |
| 20 | I2 (1.2)c | O2 | DMSO | 100 | 6 | 86 |
| 21 | I2 (1.2)c | O2 | DMSO | 120 | 4 | 89 |
| 22 | I2 (1.2)c | O2 | DMSO | 80 | 12 | 68 |
| 23 | I2 (1.2)c | O2 | DMSO | 150 | 4 | 87 |
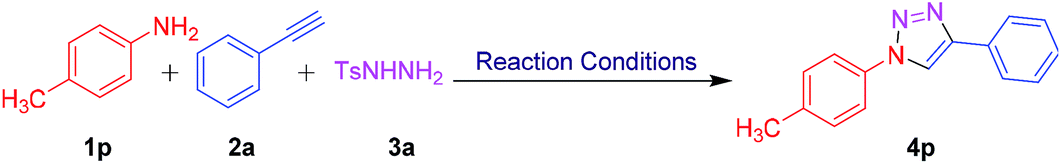
However, all the tested iodine sources were found to be ineffective as they deliver the product 4p with reduced yields. While in the absence of iodine, product 4p was not produced (entry 15, Table 1). The desired product 4p was obtained in trace amounts in the absence of solvent (entry 16, Table 1). The reduced yield of 4p is due to the low solubility of reactants. After establishing a suitable oxidant, solvent, and catalyst for the synthesis of 1,2,3-triazole, we focussed our interest in the quantity of iodine. Interestingly, improvement in the yield of 4p was observed when the stoichiometric amount of iodine was increased to 1.2 equiv. (entry 17, Table 1). The molecular iodine was revived in situ from iodide anions. Hence, the reaction proceeded well only with 1.2 equiv. of iodine. However, a further increase in the amount of iodine (entry 18, Table 1) did not improve the yield. Further, reaction efficiency was improved to a smaller extent providing an increased yield of 4p with 86%, when iodine (1.2 equiv.) was added to a solution of 1p (1 mmol), 2a (1.1 mmol) and stirred at 100 °C for 1 h under oxygen atmosphere followed by addition of 3a (1.5 mmol) (entry 20, Table 1). To our delight, the yield of 4p was increased to 89%, when the reaction was performed with 1.2 equiv. of iodine at 120 °C for 4 h (entry 21, Table 1). Further, increase or decrease in the temperature leads to a negative impact on the yield of 4p (entries 22–23, Table 1). Therefore, systematic screening of the reaction conditions revealed that iodine (1.2 equiv.)/O2 atmosphere in DMSO at 120 °C is the optimal choice for the formation of 1,2,3-triazoles.
After achieving the optimized conditions, we have explored the generality and substrate scope of the present protocol. Hence, a diverse range of anilines 1a–t were treated with phenylacetylene (2a) and N-tosyl hydrazine (3a) under optimized reaction conditions to afford the corresponding products 4a–t in a moderate to good yields (25–89%, Table 2). As shown in Table 2, it was observed that electron with-drawing group bearing anilines including halo groups like fluoro, chloro, bromo, and iodo (4b–h) were well tolerated to furnish the desired products with good yields. It was noted that the corresponding products obtained in these reactions act as potential substrates for further functionalizations. Substrates having electron deficient trifluoromethyl (4i), nitro (4j), and ester (4k) groups displayed good tolerance towards the reaction yields provided 62%, 65%, and 56% respectively. However, an electron donating groups like phenoxy, methyl, and methoxy containing anilines 4l–q showed high reactivity with excellent yields as shown in Table 2. Moreover, the position of substituents on the aromatic ring of anilines had a little influence on the yield of the products. 2-Naphthylamine (1r) was also readily participated in the reaction to afford product 4r in 68% yield. Interestingly, the reaction proceeded with benzylamine (4s) under optimized condition albeit at lower yield (25%). Unluckily, the complex mixture was observed when we attempted this protocol with aliphatic and propargyl amine. Furthermore, heteroaryl aniline such as 8-aminoquinoline (1t) was also served well to generate the corresponding product in good yield (4t, 65%). In addition, 8-aminoquinoline is a core structure in many antimalarial drugs (primaquine, pamaquine, etc).21 The antimalarial properties of a molecule can be readily modulated with respect to structure–activity parameters by attaching new fragments on a conventional 8-aminoquinoline pharmacophore, for example, compound 4t. By applying this efficient method, a tail fragment can be easily incorporated on established pharmacophore in a one-pot manner.
| a All reactions were performed with 1 (1 mmol), 2a (1.1 mmol), 3 (1.5 mmol) and I2 (1.2 mmol) in DMSO (3 mL) at 120 °C for 4–6 h. |
|---|
 |
With the consistent results of this protocol, we have also applied this new approach towards a range of aryl alkynes 2a–j (Table 2). To our delight, aromatic ring of aryl alkyne bearing electron rich and electron deficient groups reacted smoothly under these optimized conditions to furnish the corresponding 1,2,3-triazoles 4u–ad with moderate to good yields as shown in Table 2. Aryl alkynes substituted with electron donating groups like hydroxyl (2f), methoxy (2g), methyl (2h) and ethyl (2i) participated efficiently to offer the corresponding products 4z (70%), 4aa (82%), 4ab (81%), 4ac (83%) respectively in good yields (Table 2). However, the substrates with electron withdrawing fluoro, chloro, bromo, trifluoromethyl (2a–e) groups delivered the corresponding 1,2,3-triazoles with moderate yields and longer reaction times (4u–y, Table 2). Investigation with 2-ethynyl pyridine (2j) was also found to be fruitful with the current strategy but the reaction proceeded sluggishly to offer the corresponding product 4ad in low yield i.e. 32% (Table 2). However, the extension of the reaction towards aliphatic alkynes and propargyl derivatives were failed to yield the desired products. Subsequently, we performed the reaction with substituted anilines, substituted aryl alkynes, and N-tosyl hydrazine. The reaction proceeded efficiently as expected to deliver the corresponding products 4ae–ag in decent yields (Table 2).
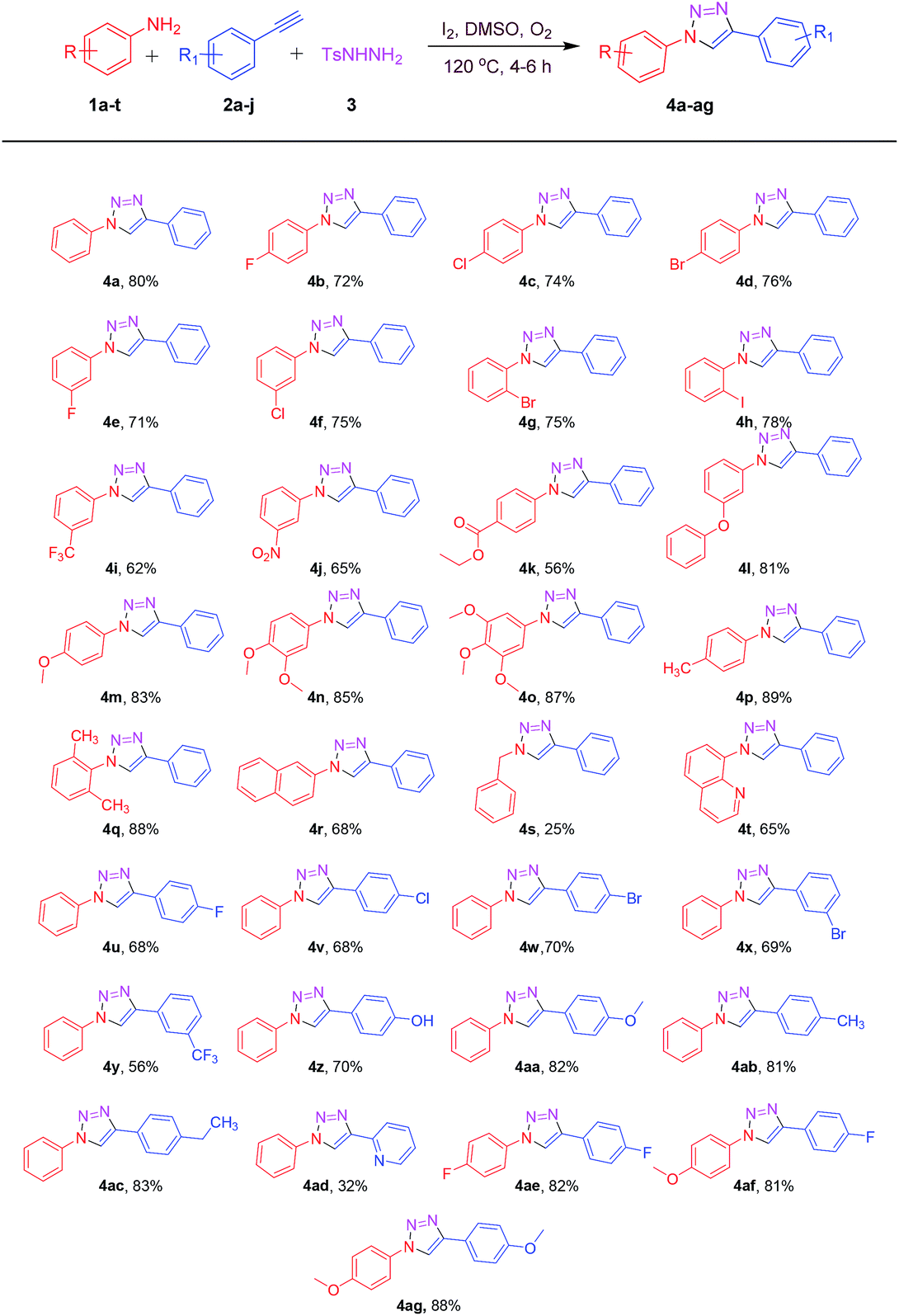
Encouraged by the above results, in order to expand the synthetic applicability of the present protocol, we replaced the terminal aryl alkyne functionality with terminal aryl alkene. To our delight, the reactions proceeded successfully under standard conditions. Aryl alkenes and anilines bearing electron donating and electron withdrawing groups reacted efficiently to deliver a valuable 1,2,3-triazoles 4a, 4c, 4m, 4u, 4ab and 4af as shown in Table 3. Electronic nature of aryl alkenes had a little influence on the reaction efficiency, by affording all the desired products in a moderate to good yields (62–76%).
| a All reactions were performed with 1 (1 mmol), 5 (1.1 mmol), 3 (1.5 mmol) and I2 (1.2 mmol) in DMSO (3 mL) at 120 °C for 4–6 h. |
|---|
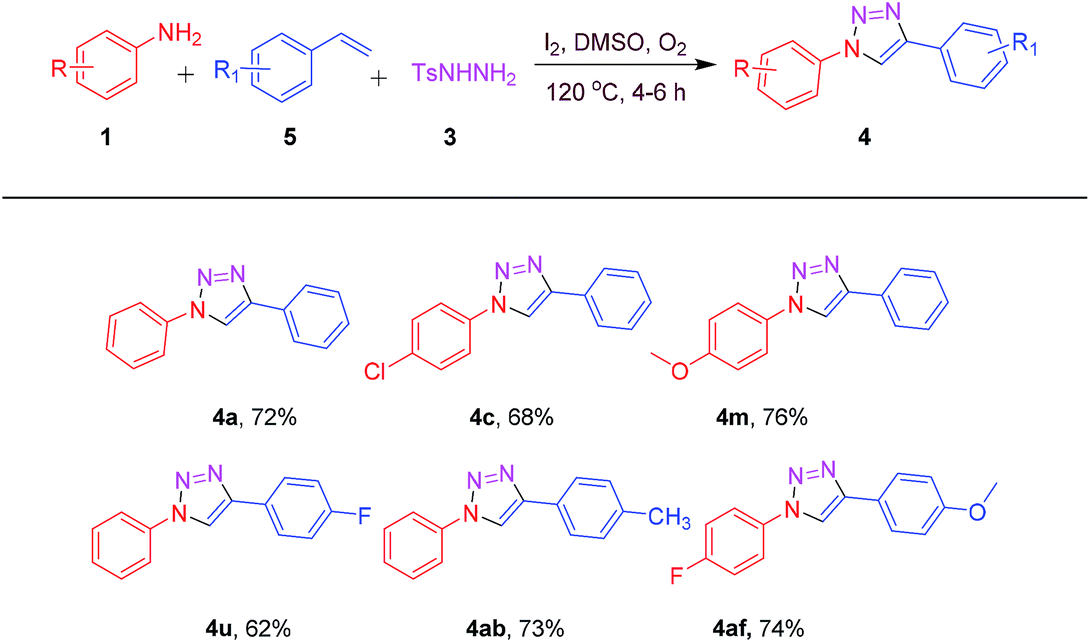 |
To further elucidate the synthetic competence of our protocol, a gram scale reaction was performed using 1a (2 g, 21.27 mmol), 3 (5.93 g, 31.91 mmol) with 2a (2.38 g, 23.40 mmol) and 5a (2.43 g, 23.40 mmol) under standard optimal conditions. To our delight, reaction forwarded smoothly to afford the corresponding product 4a in 75% (3.52 g) and 67% (3.16 g) isolated yields with 2a and 5a respectively (Scheme 1). Scalability of this protocol has the potential for utility in industrial scale-up operations.
 | ||
| Scheme 1 Gram scale synthesis of 4a. | ||
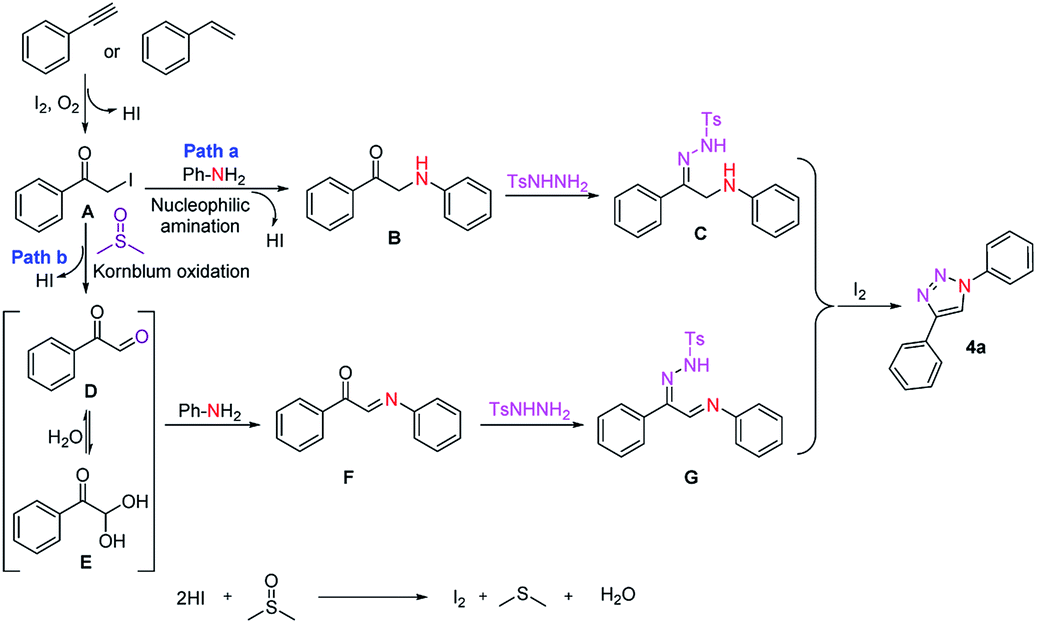
In order to understand the possible reaction mechanism, a series of control experiments were carried out subsequently (Scheme 2). In this context, we performed the reaction with phenylacetylene (2a) or styrene (7a) under optimized reaction conditions in the absence of aniline (1a) and tosyl hydrazine (3) to afford phenylglyoxal (D) in a quantitative yield (Scheme 2a). Assuming that the reaction proceeds through α-iodo ketone (A) and phenylglyoxal (D) intermediates.22 Next, they were treated with aniline (1a), N-tosyl hydrazine (3) under standard reaction conditions, which deliver the corresponding product 4a with good yields (Scheme 2b and c). Moreover, aniline (1a) reacted with an α-iodo ketone (A) under standard optimised conditions to afford a phenylaminoethanone (B) intermediate which was cyclized to afford 4a in the presence of tosyl hydrazine (3) under optimized reaction conditions (Scheme 2d). It was found that phenyl acetylene (2a) or styrene (7a) with molecular iodine and DMSO in the presence of oxygen affords phenylglyoxal (D), subsequent addition of aniline affords a condensed intermediate F. The reaction of intermediate F with N-tosyl hydrazine affords 1,2,3-triazole with 80% yield (Scheme 2e). Furthermore, intermediate C which was synthesized by a literature report,23 could be smoothly cyclized and aromatized through N–N bond formation to afford the triazole product 4a with a 90% yield in the presence of iodine and 25% in the absence of iodine (Scheme 2f). However, these reactions were proceeded well under optimised reaction temperature and not fruitful when these reactions were performed at lower temperatures i.e. at rt and 60 °C.
 | ||
| Scheme 2 Control experiments (a)–(f). | ||
Based on the control experiments and literature reports, a plausible mechanistic pathway for the formation of 1,4-substituted 1,2,3-triazole is depicted in Scheme 3. Initially, phenylacetylene (2a) or styrene (7a) is converted into α-iodoacetophenone (A) through consequent iodination followed by oxidation with I2/O2 system. Next, in the pathway a, intermediate A undergoes nucleophilic amination directly with aniline to give an intermediate B. Subsequently, intermediate B undergoes condensation with N-tosylhydrazine (3) and afforded the intermediate C which upon oxidative cyclization and aromatization in the presence of iodine and molecular oxygen gave the desired triazole 4a. Alternatively, in pathway b, α-iodoacetophenone (A) converted into phenylglyoxal (D) via Kornblum oxidation24 in DMSO which act as oxygen donor in the reaction process then upon condensation with aniline (1a) provided a C-acylimine intermediate followed by a subsequent condensation with N-tosylhydrazine to afford an intermediate G, which further undergo cyclization and aromatization in the presence of iodine and oxygen to afford the desired triazole 4a. Molecular iodine can be readily regenerated in situ by the oxidation of iodide anions with DMSO.
 | ||
| Scheme 3 Plausible mechanism for the iodine-catalyzed synthesis of 1,2,3-triazoles. | ||
Conclusion
In summary, we have developed an efficient I2/O2 mediated protocol for the synthesis of 1,4-disubstituted 1,2,3-triazoles in a regioselective manner from commercially available anilines, aryl alkynes or aryl alkenes, and N-tosyl hydrazine through C–N, N–N bond formation. Advantages of the presented methodology are broad substrate scope, milder reaction conditions, operational simplicity and cost-effective. Importantly, gram scale synthesis was also accomplished which makes this protocol for the utilization in the synthesis of biologically important molecules.Experimental section
General information
All the reagents and chemicals were obtained from commercial sources and used without further purification. Analytical thin layer chromatography (TLC) was performed with silica gel MERCK silica gel 60-F254 (0.5 mm) precoated glass plates. TLC plates were visualized by exposure to ultraviolet light. Column chromatography was performed using 100–200 mesh silica gel, and the eluent was as a mixture of ethyl acetate and n-hexane. 1H and 13C NMR spectra were recorded with a Bruker 500 MHz NMR instrument in CDCl3 or DMSO-d6 with tetramethylsilane (TMS) as an internal standard. Chemical shifts (δ) and coupling constants (J) are reported in parts per million (ppm), Hertz (Hz) respectively. The peak patterns are indicated as follows: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; dd, doublet of doublet; dt, doublet of triplet; td, triplet of doublet; ddd, doublet of doublet of doublet. Melting points were determined using an electrothermal apparatus (Model IA9200) and are uncorrected. High-resolution mass spectra (HRMS) were recorded on LC-QTOF mass spectrometer.General procedure for the synthesis of 1,4-disubstituted 1,2,3-triazoles (4a–ag)
In a 25 mL round-bottomed flask, aryl alkyne (1.1 mmol)/aryl alkene (1.1 mmol), arylamine (1 mmol) and iodine (1.2 mmol) in DMSO were refluxed at 120 °C under molecular dioxygen atmosphere for 1 h followed by an addition of N-tosyl hydrazine (1.5 mmol), continued to reflux at 120 °C for 3–5 h. After completion of the reaction (monitored with TLC), the reaction mixture was quenched with a saturated solution of sodium thiosulfate and extracted with ethyl acetate. The combined organic layers were washed with water and brine, dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue obtained was purified by silica gel column chromatography using ethyl acetate–hexane as an eluent in increasing polarity to afford the desired 1,4-disubstituted 1,2,3-triazoles 4a–ag.1,4-Diphenyl-1H-1,2,3-triazole (4a)
176 mg (80%) of 4a was obtained as a white solid; Rf = 0.420 (ethyl acetate/n-hexane, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1); mp. 173–175 °C (lit. 181–183 °C);23a 1H NMR (500 MHz, CDCl3): δ 8.21 (s, 1H), 7.96–7.90 (m, 2H), 7.80 (dd, J = 8.5, 1.0 Hz, 2H), 7.56 (dd, J = 10.7, 5.0 Hz, 2H), 7.49–7.44 (m, 3H), 7.38 (t, J = 7.4 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ 137.09, 130.13, 129.80, 128.94, 128.84, 128.50, 125.92, 120.58, 117.66; HRMS (ESI): m/z calcd for C14H12N3 [M + H]+ 222.1031, found 222.1031.
:1); mp. 173–175 °C (lit. 181–183 °C);23a 1H NMR (500 MHz, CDCl3): δ 8.21 (s, 1H), 7.96–7.90 (m, 2H), 7.80 (dd, J = 8.5, 1.0 Hz, 2H), 7.56 (dd, J = 10.7, 5.0 Hz, 2H), 7.49–7.44 (m, 3H), 7.38 (t, J = 7.4 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ 137.09, 130.13, 129.80, 128.94, 128.84, 128.50, 125.92, 120.58, 117.66; HRMS (ESI): m/z calcd for C14H12N3 [M + H]+ 222.1031, found 222.1031.
1-(4-Fluorophenyl)-4-phenyl-1H-1,2,3-triazole (4b)
172 mg (72%) of 4b was obtained as a white solid; Rf = 0.587 (ethyl acetate/n-hexane, 1:1); mp. 203–205 °C (lit. 209–210 °C);23a 1H NMR (500 MHz, CDCl3): δ 8.16 (s, 1H), 7.92 (d, J = 7.5 Hz, 2H), 7.78 (dd, J = 8.6, 4.5 Hz, 2H), 7.47 (t, J = 7.4 Hz, 2H), 7.37 (dd, J = 24.3, 17.0 Hz, 1H), 7.27 (d, J = 2.8 Hz, 1H), 7.24 (d, J = 8.2 Hz, 1H); 13C NMR (125 MHz, DMSO): δ 162.18 (d, J = 234.4 Hz), 147.79, 133.71 (d, J = 2.7 Hz), 130.69, 129.49, 128.75, 125.82, 122.86 (d, J = 8.8 Hz), 120.39, 117.28 (d, J = 23.2 Hz), HRMS (ESI): m/z calcd for C14H11FN3 [M + H]+ 240.0937, found 240.0943.
1-(4-Chlorophenyl)-4-phenyl-1H-1,2,3-triazole (4c)
178 mg (70%) of 4c was obtained as a white solid; Rf = 0.516 (ethyl acetate/n-hexane, 1:1); mp. 221–224 °C (lit. 222–224 °C);23a 1H NMR (500 MHz, DMSO): δ 9.35 (s, 1H), 8.05–7.99 (m, 2H), 7.95 (dd, J = 8.2, 1.2 Hz, 2H), 7.77–7.70 (m, 2H), 7.52 (dd, J = 10.6, 4.7 Hz, 2H), 7.44–7.37 (m, 1H); 13C NMR (125 MHz, DMSO): δ 147.94, 135.93, 133.45, 130.62, 130.42, 129.52, 128.82, 125.84, 122.16, 120.21; HRMS (ESI): m/z calcd for C14H11ClN3 [M + H]+ 256.0642, found 256.0641.
1-(4-Bromophenyl)-4-phenyl-1H-1,2,3-triazole (4d)
227 mg (76%) of 4d was obtained as a white solid; Rf = 0.586 (ethyl acetate/n-hexane, 1:1); mp. 222–224 °C (lit. 216–217 °C);23a 1H NMR (500 MHz, CDCl3): δ 8.18 (s, 1H), 7.97–7.85 (m, 2H), 7.75–7.66 (m, 4H), 7.47 (dt, J = 12.5, 3.2 Hz, 2H), 7.42–7.36 (m, 1H); 13C NMR (125 MHz, CDCl3): δ 141.64, 136.07, 132.97, 130.02, 128.98, 128.61, 125.91, 122.43, 121.91, 117.34; HRMS (ESI): m/z calcd for C14H1181BrN3 [M + H]+ 302.0116, found 302.0118.
1-(3-Fluorophenyl)-4-phenyl-1H-1,2,3-triazole (4e)
169 mg (71%) of 4e was obtained as a solid; Rf = 0.586 (ethyl acetate/n-hexane, 1:1); mp. 179–182 °C (lit. 185–187 °C);25a 1H NMR (500 MHz, CDCl3): δ 8.19 (s, 1H), 7.92 (dd, J = 10.2, 8.9 Hz, 2H), 7.62–7.57 (m, 2H), 7.56–7.50 (m, 1H), 7.47 (t, J = 7.6 Hz, 2H), 7.38 (td, J = 7.1, 1.1 Hz, 1H), 7.19–7.14 (m, 1H); 13C NMR (125 MHz, CDCl3): δ 163.15 (d, J = 252.5 Hz), 148.66, 138.25 (d, J = 10.3 Hz), 131.23 (d, J = 9.0 Hz), 129.95, 128.97, 128.63, 125.93, 117.44, 115.81 (d, J = 3.3 Hz), 115.67 (d, J = 21.2 Hz), 108.29 (d, J = 26.2 Hz); HRMS (ESI): m/z calcd for C14H11FN3 [M + H]+ 240. 0937, found 240.0941.
1-(3-Chlorophenyl)-4-phenyl-1H-1,2,3-triazole (4f)
191 mg (75%) of 4f was obtained as an yellow solid; Rf = 0.500 (ethyl acetate/n-hexane, 1:1); mp. 172–174 °C (lit. 167–170 °C);25a 1H NMR (500 MHz, CDCl3): δ 8.19 (s, 1H), 7.93–7.88 (m, 2H), 7.84 (t, J = 2.0 Hz, 1H), 7.71 (ddd, J = 8.0, 2.0, 1.1 Hz, 1H), 7.49 (dd, J = 7.3, 4.8 Hz, 1H), 7.47 (d, J = 1.7 Hz, 1H), 7.46–7.42 (m, 2H), 7.40–7.36 (m, 1H); 13C NMR (125 MHz, CDCl3): δ 148.70, 137.85, 135.66, 130.87, 129.96, 128.97, 128.82, 128.63, 125.92, 120.75, 118.48, 117.44; HRMS (ESI): m/z calcd for C14H11ClN3 [M + H]+ 256. 0642, found 256.0645.
1-(2-Bromophenyl)-4-phenyl-1H-1,2,3-triazole (4g)
224 mg (75%) of 4g was obtained as a solid; Rf = 0.482 (ethyl acetate/n-hexane, 1:1); mp. 99–100 °C (100–101 °C);25b 1H NMR (500 MHz, CDCl3): δ 8.17 (s, 1H), 7.95–7.91 (m, 2H), 7.78 (dd, J = 8.1, 1.3 Hz, 1H), 7.62 (dd, J = 7.9, 1.6 Hz, 1H), 7.51 (td, J = 7.7, 1.4 Hz, 1H), 7.49–7.45 (m, 2H), 7.43–7.40 (m, 1H), 7.39–7.35 (m, 1H); 13C NMR (125 MHz, CDCl3): δ 147.02, 136.93, 132.28, 131.32, 130.51, 129.87, 129.00, 128.83, 124.40, 123.05, 120.59, 118.02; HRMS (ESI): m/z calcd for C14H11BrN3 [M + H]+ 300.0136, found 300.0099.
1-(2-Iodophenyl)-4-phenyl-1H-1,2,3-triazole (4h)
269 mg (78%) of 4h was obtained as a pale yellow solid; Rf = 0.550 (ethyl acetate/n-hexane, 1:1); mp. 159–162 °C (lit. 156–158 °C);25b 1H NMR (500 MHz, CDCl3): δ 8.08 (d, J = 5.0 Hz, 1H), 8.02 (dd, J = 8.0, 1.0 Hz, 1H), 7.95–7.91 (m, 2H), 7.55–7.51 (m, 1H), 7.50 (d, J = 2.0 Hz, 1H), 7.49–7.44 (m, 2H), 7.39–7.35 (m, 1H), 7.28–7.23 (m, 1H); 13C NMR (125 MHz, CDCl3): δ 147.65, 140.32, 140.10, 131.55, 130.26, 129.33, 128.96, 128.37, 127.88, 125.94, 121.61, 93.91; HRMS (ESI): m/z calcd for C14H11IN3 [M + H]+ 347.9998, found 348.0010.
:1); mp. 152–154 °C (lit. 144–146 °C);25c 1H NMR (500 MHz, CDCl3): δ 8.26 (s, 1H), 8.08 (s, 1H), 8.04 (d, J = 7.5 Hz, 1H), 7.92 (d, J = 7.3 Hz, 2H), 7.71 (m, J = 7.8 Hz, 2H), 7.47 (t, J = 7.6 Hz, 2H), 7.39 (t, J = 7.4 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ 137.43, 132.50 (q, J = 33.3 Hz), 130.61, 129.87, 129.00, 128.71, 125.94, 125.35 (q, J = 3.3 Hz), 123.56, 123.38 (q, J = 272.7 Hz), 117.34 (q, J = 3.8 Hz), HRMS (ESI): m/z calcd for C15H11F3N3 [M + H]+ 290.0905, found 290.0916.1-(3-Nitrophenyl)-4-phenyl-1H-1,2,3-triazole (4j)
172 mg (65%) of 4j was obtained as an yellow solid; Rf = 0.379 (ethyl acetate/n-hexane, 1:1); mp. 198–200 °C (198–200 °C);25b 1H NMR (500 MHz, DMSO): δ 9.57 (s, 1H), 8.80 (t, J = 2.1 Hz, 1H), 8.48 (ddd, J = 8.1, 2.1, 0.7 Hz, 1H), 8.40–8.34 (m, 1H), 7.97 (dt, J = 14.8, 4.7 Hz, 3H), 7.54 (dd, J = 10.6, 4.8 Hz, 2H), 7.45–7.39 (m, 1H); 13C NMR (125 MHz, DMSO): δ 149.08, 148.09, 137.71, 132.12, 130.40, 129.56, 128.97, 126.44, 125.87, 123.62, 120.53, 115.08; HRMS (ESI): m/z calcd for C14H11N4O2 [M + H]+ 267.0882, found 267.0885.
Ethyl 4-(4-phenyl-1H-1,2,3-triazol-1-yl)benzoate (4k)
164 mg (56%) of 4k was obtained as a white solid; Rf = 0.448 (ethyl acetate/n-hexane, 1:1); mp. 175–178 °C (lit. 181–183 °C)18c; 1H NMR (500 MHz, CDCl3): δ 8.34–8.17 (m, 3H), 7.92 (dd, J = 7.6, 6.3 Hz, 4H), 7.48 (t, J = 7.5 Hz, 2H), 7.39 (t, J = 7.4 Hz, 1H), 4.43 (q, J = 7.1 Hz, 2H), 1.44 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3): 13C NMR (125 MHz, CDCl3): δ 165.44, 148.77, 140.04, 131.30, 130.63, 129.92, 129.00, 128.68, 125.96, 119.84, 117.35, 61.35, 14.59; HRMS (ESI): m/z calcd for C17H16N3O2 [M + H]+ 294.1240, found 294.1243.
1-(3-Phenoxyphenyl)-4-phenyl-1H-1,2,3-triazole (4l)
253 mg (81%) of 4l was obtained as a white solid; Rf = 0.519 (ethyl acetate/n-hexane, 1:1); mp. 137–139 °C; 1H NMR (500 MHz, CDCl3): δ 8.15 (s, 1H), 7.90 (d, J = 7.4 Hz, 2H), 7.53–7.43 (m, 5H), 7.42–7.35 (m, 3H), 7.19 (t, J = 7.4 Hz, 1H), 7.08 (dd, J = 10.9, 7.9 Hz, 3H); 13C NMR (125 MHz, CDCl3): δ 158.85, 156.11, 148.40, 138.38, 130.92, 130.09, 128.86, 128.53, 125.93, 124.34, 119.61, 118.33, 117.68, 114.72, 110.62; HRMS (ESI): m/z calcd for C20H16N3O [M + H]+ 314.1293, found 314.1300.
1-(4-Methoxyphenyl)-4-phenyl-1H-1,2,3-triazole (4m)
208 mg (83%) of 4m was obtained as a off white solid; Rf = 0.206 (ethyl acetate/n-hexane, 1:1); mp. 163–165 °C (lit. 162–164 °C)23a; 1H NMR (500 MHz, CDCl3): δ 8.24 (s, 1H), 7.93 (s, 2H), 7.69 (d, J = 8.8 Hz, 2H), 7.47 (dd, J = 16.9, 10.5 Hz, 2H), 7.37 (t, J = 7.4 Hz, 1H), 7.03 (d, J = 8.7 Hz, 2H), 3.87 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 159.96, 130.58, 130.22, 128.94, 128.74, 128.45, 126.70, 125.88, 122.25, 114.87, 55.46; HRMS (ESI): m/z calcd for C15H14N3O [M + H]+ 252.1137, found 252.1137.
1-(3,4-Dimethoxyphenyl)-4-phenyl-1H-1,2,3-triazole (4n)
238 mg (85%) of 4n was obtained as a pale brown solid; Rf = 0.160 (ethyl acetate/n-hexane, 1:1); mp. 147–149 °C (lit. 151–153 °C);25d 1H NMR (500 MHz, CDCl3): δ 8.16 (s, 1H), 7.91 (d, J = 7.5 Hz, 2H), 7.45 (t, J = 7.4 Hz, 2H), 7.40 (d, J = 2.4 Hz, 1H), 7.36 (t, J = 7.4 Hz, 1H), 7.21 (dd, J = 8.6, 2.4 Hz, 1H), 6.96 (d, J = 8.6 Hz, 1H), 3.97 (s, 3H), 3.94 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 165.66, 163.65, 161.12, 150.17, 143.51, 137.47, 125.86, 125.03, 123.36, 105.05, 55.82; HRMS (ESI): m/z calcd for C16H16N3O2 [M + H]+ 282.1243, found 282.1231.
4-Phenyl-1-(3,4,5-trimethoxyphenyl)-1H-1,2,3-triazole (4o)
270 mg (87%) of 4o was obtained as pale yellow solid; Rf = 0.152 (ethyl acetate/n-hexane, 1:1); mp. 119–122 °C (110–112 °C);25d 1H NMR (500 MHz, CDCl3): δ 8.16 (s, 1H), 7.91 (d, J = 7.5 Hz, 2H), 7.46 (t, J = 7.5 Hz, 2H), 7.36 (dd, J = 18.3, 11.1 Hz, 1H), 7.02 (d, J = 20.3 Hz, 2H), 3.95 (s, 6H), 3.90 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 153.96, 138.35, 132.92, 130.16, 128.94, 128.49, 125.86, 118.14, 98.55, 61.06, 56.47; HRMS (ESI): m/z calcd for C17H18N3O3 [M + H]+ 312.1348, found 312.1357.
4-Phenyl-1-(p-tolyl)-1H-1,2,3-triazole (4p)
209 mg (89%) of 4p was obtained as a white solid; Rf = 0.580 (ethyl acetate/n-hexane, 1:1); mp. 173–175 °C (lit. 169–171 °C)23a; 1H NMR (500 MHz, CDCl3): δ 8.16 (s, 1H), 7.91 (d, J = 7.5 Hz, 2H), 7.66 (d, J = 8.3 Hz, 2H), 7.45 (t, J = 7.6 Hz, 2H), 7.37 (d, J = 7.4 Hz, 1H), 7.33 (d, J = 8.2 Hz, 2H), 2.43 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 148.12, 138.99, 134.81, 130.29, 130.21, 128.93, 128.43, 125.91, 120.49, 117.91, 21.10; HRMS (ESI): m/z calcd for C15H14N3 [M + H]+ 236.1188, found 236.1188.
1-(2,6-Dimethylphenyl)-4-phenyl-1H-1,2,3-triazole (4q)
219 mg (88%) of 4q was obtained as a pale white solid; Rf = 0.519 (ethyl acetate/n-hexane, 1:1); mp. 122–124 °C; 1H NMR (500 MHz, CDCl3): δ 7.94 (s, 1H), 7.93–7.90 (m, 2H), 7.45 (t, J = 7.6 Hz, 2H), 7.36 (dd, J = 10.9, 3.9 Hz, 1H), 7.28–7.20 (m, 3H), 2.39 (s, 3H), 2.22 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 147.55, 136.88, 136.33, 131.32, 130.61, 130.47, 130.42, 128.92, 128.29, 126.48, 125.83, 121.12, 20.82, 17.57; HRMS (ESI): m/z calcd for C16H16N3 [M + H]+ 250.1344, found 250.1346.
1-(Naphthalen-2-yl)-4-phenyl-1H-1,2,3-triazole (4r)
184 mg (68%) of 4r was obtained as a white solid; Rf = 0.500 (ethyl acetate/n-hexane, 1:1); mp. 186–189 °C (lit. 182–184 °C);25d 1H NMR (500 MHz, CDCl3): δ 8.35 (s, 1H), 8.24 (s, 1H), 8.03 (d, J = 8.6 Hz, 1H), 8.01–7.91 (m, 5H), 7.64–7.55 (m, 2H), 7.49 (t, J = 7.3 Hz, 2H), 7.39 (t, J = 7.3 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ 134.38, 133.26, 133.02, 130.16, 129.87, 129.04, 128.68, 128.37, 128.00, 127.56, 127.12, 126.09, 118.98, 118.58, 118.23; HRMS (ESI): m/z calcd for C18H14N3 [M + H]+ 272.1188, found 272.1194.
1-Benzyl-4-phenyl-1H-1,2,3-triazole (4s)
58 mg (25%) of 4s was obtained as a white solid; Rf = 0.360 (ethyl acetate/n-hexane, 1:1); mp. 132–133 °C (lit. 128–130 °C);25b 1H NMR (500 MHz, CDCl3): δ 7.81 (t, J = 7.2 Hz, 2H), 7.68 (s, 1H), 7.43–7.34 (m, 5H), 7.35–7.27 (m, 3H), 5.56 (t, J = 6.1 Hz, 2H); 13C NMR (125 MHz, CDCl3): δ 148.15, 134.69, 130.47, 129.16, 128.82, 128.79, 128.22, 128.08, 125.76, 119.69, 54.47; HRMS (ESI): m/z calcd for C15H14N3 [M + H]+ 236.1188, found 236.1197.
8-(4-Phenyl-1H-1,2,3-triazol-1-yl)quinoline (4t)
176 mg (65%) of 4t was obtained as a yellow solid; Rf = 0.230 (ethyl acetate/n-hexane, 1:1); mp. 120–123 °C (lit. 115–117 °C)23a; 1H NMR (500 MHz, CDCl3): δ 9.06 (d, J = 43.6 Hz, 2H), 8.35 (d, J = 46.8 Hz, 2H), 7.98 (d, J = 22.4 Hz, 3H), 7.73 (s, 1H), 7.47 (t, J = 46.1 Hz, 4H); 13C NMR (125 MHz, CDCl3): δ 150.86, 147.31, 140.30, 136.54, 133.93, 130.89, 129.18, 128.81, 128.05, 126.49, 125.98, 124.51, 123.65, 122.03; HRMS (ESI): m/z calcd for C17H13N4 [M + H]+ 273.1140, found 273.1139.
4-(4-Fluorophenyl)-1-phenyl-1H-1,2,3-triazole (4u)
162 mg (68%) of 4u was obtained as white solid; Rf = 0.466 (ethyl acetate/n-hexane, 1:1); mp. 205–207 °C (lit. 198–200 °C)23a; 1H NMR (500 MHz, CDCl3): δ 8.15 (s, 1H), 7.91–7.87 (m, 2H), 7.81–7.77 (m, 2H), 7.58–7.53 (m, 2H), 7.50–7.43 (m, 1H), 7.19–7.13 (m, 2H); 13C NMR (125 MHz, CDCl3): δ 162.91 (d, J = 255.5 Hz), 147.53, 137.01, 129.86, 129.01, 127.72 (d, J = 8.2 Hz); 126.35 (d, J = 3.0 Hz), 120.61, 117.57, 115.99 (d, J = 21.8 Hz) HRMS (ESI): m/z calcd for C14H11FN3 [M + H]+ 240.0937, found 240.0943.
4-(4-Chlorophenyl)-1-phenyl-1H-1,2,3-triazole (4v)
173 mg (68%) of 4v was obtained as a white solid; Rf = 0.820 (ethyl acetate/n-hexane, 1:1); mp. 209–211 °C (lit. 207–209 °C)23a; 1H NMR (500 MHz, DMSO): δ 9.37 (d, J = 11.2 Hz, 1H), 8.03–7.91 (m, 4H), 7.65 (t, J = 7.8 Hz, 2H), 7.59 (d, J = 8.4 Hz, 2H), 7.54 (t, J = 7.4 Hz, 1H); 13C NMR (125 MHz, DMSO): δ 146.71, 137.02, 133.21, 130.44, 129.66, 129.57, 129.31, 127.51, 120.54, 120.50; HRMS (ESI): m/z calcd for C14H11ClN3 [M + H]+ 256.0642, found 256.0645.
4-(4-Bromophenyl)-1-phenyl-1H-1,2,3-triazole (4w)
210 mg of 4w was obtained as a white solid; Rf = 0.630 (ethyl acetate/n-hexane, 1:1); mp. 205–208 °C (lit. 212–214 °C)22a; 1H NMR (500 MHz, DMSO): δ 9.37 (s, 1H), 7.96 (d, J = 7.7 Hz, 2H), 7.92 (d, J = 8.5 Hz, 2H), 7.73 (d, J = 8.4 Hz, 2H), 7.65 (t, J = 7.9 Hz, 2H), 7.54 (t, J = 7.4 Hz, 1H); 13C NMR (125 MHz, DMSO): δ 146.75, 139.16, 137.05, 132.48, 130.45, 130.01, 129.32, 127.78, 122.36, 121.74, 120.54; HRMS (ESI): m/z calcd for C14H11BrN3 [M + H]+ 300.0136, found 300.0140.
4-(3-Bromophenyl)-1-phenyl-1H-1,2,3-triazole (4x)
206 mg (69%) of 4x was obtained as a white solid; Rf = 0.655 (ethyl acetate/n-hexane, 1:1); mp. 148–151 °C (lit. 148–150 °C)23a; 1H NMR (500 MHz, CDCl3): δ 8.20 (s, 1H), 8.06 (dd, J = 5.7, 4.0 Hz, 1H), 7.88–7.84 (m, 1H), 7.81–7.76 (m, 2H), 7.58–7.53 (m, 2H), 7.52–7.45 (m, 2H), 7.33 (t, J = 7.9 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ 146.99, 136.92, 132.27, 131.36, 130.51, 129.86, 128.99, 128.83, 124.42, 123.04, 120.58, 118.12; HRMS (ESI): m/z calcd for C14H11BrN3 [M + H]+ 300.0136, found 300.0141.
1-Phenyl-4-(3-(trifluoromethyl)phenyl)-1H-1,2,3-triazole (4y)
161 mg (56%) of 4y was obtained as white solid; Rf = 0.519 (ethyl acetate/n-hexane, 1:1); mp. 223–225 °C; 1H NMR (500 MHz, DMSO): δ 9.39 (s, 1H), 8.25–8.10 (m, 2H), 8.02–7.91 (m, 2H), 7.80 (d, J = 8.1 Hz, 2H), 7.71–7.57 (m, 2H), 7.50 (ddt, J = 11.5, 10.4, 5.2 Hz, 1H); 13C NMR (125 MHz, DMSO): δ 146.41, 135.88 (q, J = 286.7 Hz), 130.47, 129.43, 128.97, 126.51 (q, J = 3.3 Hz), 126.42, 126.36; 126.28, 125.80, 121.48, 120.62; HRMS (ESI): m/z calcd for C15H11F3N3 [M + H]+ 290.0905, found 290.0909.
4-(1-Phenyl-1H-1,2,3-triazol-4-yl)phenol (4z)
165 mg (70%) of 4z was obtained as a pale white solid; Rf = 0.137 (ethyl acetate/n-hexane, 1:1); mp. 194–196 °C; 1H NMR (500 MHz, DMSO): δ 9.62 (s, 1H), 9.24 (s, 1H), 7.97 (d, J = 7.8 Hz, 2H), 7.64 (t, J = 7.6 Hz, 2H), 7.52 (t, J = 7.2 Hz, 1H), 7.41 (s, 1H), 7.37 (d, J = 7.4 Hz, 1H), 7.29 (t, J = 7.7 Hz, 1H), 6.80 (d, J = 7.3 Hz, 1H); 13C NMR (125 MHz, DMSO): δ 158.32, 147.92, 137.16, 131.94, 130.49, 130.38, 129.14, 120.48, 119.99, 116.67, 115.77, 112.63; HRMS (ESI): m/z calcd for C14H12N3O [M + H]+ 238.0980, found 238.0987.
4-(4-Methoxyphenyl)-1-phenyl-1H-1,2,3-triazole (4aa)
205 mg (82%) of 4aa was obtained as a white solid; Rf = 0.206 (ethyl acetate/n-hexane, 1:1); mp. 137–139 °C (lit. 142–143);23a 1H NMR (500 MHz, CDCl3): δ 8.10 (s, 1H), 7.83 (d, J = 8.5 Hz, 2H), 7.77 (d, J = 7.9 Hz, 2H), 7.52 (t, J = 7.7 Hz, 2H), 7.43 (t, J = 7.4 Hz, 1H), 6.98 (d, J = 8.4 Hz, 2H), 3.84 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 159.86, 148.28, 137.14, 129.78, 128.70, 127.24, 122.91, 120.44, 116.95, 114.38, 55.47; HRMS: m/z calcd for C15H14N3O [M + H]+ 252.1137, found 252.1106.
1-Phenyl-4-(p-tolyl)-1H-1,2,3-triazole (4ab)
190 mg (81%) of 4ab was obtained as a white solid; Rf = 0.551 (ethyl acetate/n-hexane, 1:1); mp. 148–150 °C (lit. 152–154 °C)23a; 1H NMR (500 MHz, CDCl3): δ 8.08 (s, 1H), 7.85–7.66 (m, 4H), 7.54–7.42 (m, 2H), 7.38 (t, J = 7.2 Hz, 1H), 7.20 (d, J = 5.6 Hz, 2H), 2.33 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 148.48, 138.41, 137.12, 129.84, 129.68, 128.78, 127.33, 125.92, 120.65, 117.83, 21.46; HRMS (ESI): m/z calcd for C15H14N3 [M + H]+ 236.1188, found 236.1184.
4-(4-Ethylphenyl)-1-phenyl-1H-1,2,3-triazole (4ac)
206 mg (83%) of 4ac was obtained as a white solid; Rf = 0.866 (ethyl acetate/n-hexane, 1:1); mp. 123–125 °C (128–130 °C)23a; 1H NMR (500 MHz, CDCl3): δ 8.16 (s, 1H), 7.90–7.74 (m, 4H), 7.61–7.50 (m, 2H), 7.48–7.43 (m, 1H), 7.30 (d, J = 8.3 Hz, 2H), 2.70 (q, J = 7.6 Hz, 2H), 1.28 (t, J = 7.6 Hz, 3H); 13C NMR (125 MHz, CDCl3): δ 148.51, 144.72, 137.12, 129.78, 128.72, 128.45, 127.65, 125.88, 120.51, 117.32, 28.64, 15.52; HRMS (ESI): m/z calcd for C16H16N3 [M + H]+ 250.1344, found 250.1353.
2-(1-Phenyl-1H-1,2,3-triazol-4-yl)pyridine (4ad)
71 mg (32%) of 4ad was obtained as a light yellow solid; Rf = 0.510 (ethyl acetate/n-hexane, 1:1); mp. 85–88 °C (lit. 89–92 °C);25e 1H NMR (500 MHz, CDCl3): δ 8.71 (s, 1H), 8.64 (t, J = 10.2 Hz, 1H), 8.29 (d, J = 7.9 Hz, 1H), 7.91–7.79 (m, 3H), 7.57 (dd, J = 17.5, 9.9 Hz, 2H), 7.47 (t, J = 7.4 Hz, 1H), 7.33–7.28 (m, 1H); 13C NMR (125 MHz, CDCl3): δ 149.63, 148.94, 148.30, 137.58, 136.97, 129.86, 128.95, 123.21, 120.70, 120.49, 120.36; HRMS (ESI): m/z calcd for C13H11N4 [M + H]+ 223.0984, found 223.0980.
1,4-Bis(4-fluorophenyl)-1H-1,2,3-triazole (4ae)
210 mg (82%) of 4ae was obtained as a off white solid; Rf = 0.866 (ethyl acetate/n-hexane, 1:1); mp. 228–229 °C; 1H NMR (500 MHz, DMSO): δ 9.28 (s, 1H), 8.07–7.92 (m, 4H), 7.58–7.46 (m, 2H), 7.42–7.29 (m, 2H); 13C NMR (125 MHz, CDCl3 + DMSO): δ 162.57 (d, J = 249.6 Hz), 162.14 (d, J = 260.0 Hz), 147.03, 133.60 (d, J = 2.9 Hz), 127.72 (d, J = 8.2 Hz), 127.15 (d, J = 3.1 Hz), 122.47 (d, J = 8.6 Hz), 119.60, 116.94 (d, J = 23.2 Hz), 116.08 (d, J = 21.7 Hz); HRMS (ESI): m/z calcd for C14H10F2N3 [M + H]+ 258.0843, found 258.0850.
4-(4-Fluorophenyl)-1-(4-methoxyphenyl)-1H-1,2,3-triazole (4af)
215 mg (81%) of 4af was obtained as a off white solid; Rf = 0.666 (ethyl acetate/n-hexane, 1:1); mp. 178–180 °C; 1H NMR (500 MHz, DMSO): δ 8.06 (s, 1H), 7.94–7.81 (m, 2H), 7.74–7.62 (m, 2H), 7.20–7.10 (m, 2H), 7.09–6.97 (m, 2H), 3.88 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 162.63 (d, J = 239.0 Hz), 159.92, 147.38, 130.51, 127.58 (d, J = 8.2 Hz), 126.66 (d, J = 3.3 Hz), 115.90 (d, J = 21.8 Hz), 114.86, 55.66; HRMS (ESI): m/z calcd for C15H13FN3O [M + H]+ 270.0143, found 270.1054.
1,4-Bis(4-methoxyphenyl)-1H-1,2,3-triazole (4ag)
247 mg (88%) of 4ag was obtained as a white solid; Rf = 0.563 (ethyl acetate/n-hexane, 1:1); mp. 198–200 °C; 1H NMR (500 MHz, CDCl3): δ 8.02 (s, 1H), 7.86–7.80 (m, 2H), 7.70–7.65 (m, 2H), 7.05–7.01 (m, 2H), 7.01–6.96 (m, 2H), 3.87 (s, 3H), 3.85 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 159.83, 148.13, 130.70, 127.16, 123.16, 122.23, 117.11, 114.81, 114.35, 55.65, 55.35; HRMS (ESI): m/z calcd for C16H16N3O2 [M + H]+ 282.1243, found 282.1255.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
G. S. M and K. D. is thankful to the Department of Pharmaceuticals, the Ministry of Chemicals and Fertilizers Govt. of India, New Delhi for the award of a Research Fellowships.References
- (a) P. Thansadote and M. Lautens, Chem.–Eur. J., 2009, 24, 5874 CrossRef PubMed; (b) A. Majunder, R. Gupta and A. Jain, Green Chem. Lett. Rev., 2013, 6, 151 CrossRef; (c) M. A. Asif, Int. J. Bioorg. Chem. Mol. Biol., 2017, 2, 146 Search PubMed; (d) E. C. Taylor and J. E. Saxton, The Chemistry of Heterocyclic Compounds, Wiley-Interscience, New York, 1983/1994 Search PubMed; (e) J. A. Joule and K. Mills, Heterocyclic Chemistry; Blackwell Science, Oxford, 2000 Search PubMed; (f) T. Eicher, S. Hauptmann and A. Speicher, The Chemistry of Heterocycles, Wiley-VCH Verlag GmbH&Co, Weinheim, 2nd edn, 2003 CrossRef; (g) N. Saracoglu, Top. Heterocycl. Chem., 2007, 11, 145 CrossRef; (h) B. Zhang and A. Studer, Chem. Soc. Rev., 2015, 44, 3505 RSC; (i) A. Kamal, S. Prabhakar, M. J. Ramaiah, P. V. Reddy, Ch. R. Reddy, A. Mallareddy, N. Shankaraiah, T. L. N. Reddy, S. N. C. V. L. Pushpavalli and M.-P. Bhadra, Eur. J. Med. Chem., 2011, 46, 3820 CrossRef CAS PubMed.
- (a) R. D. Carpenter and M. J. Kurth, Nat. Protoc., 2010, 5, 1731 CrossRef CAS PubMed; (b) M. A. Siracusa, L. Salerno, M. N. Modica, V. Pittala, G. Romeo, M. E. Amato, M. Nowak, A. J. Bojarski, I. Mereghetti, A. Cagnotto and T. Mennini, J. Med. Chem., 2008, 51, 4529 CrossRef CAS PubMed; (c) C. G. Mortimer, G. Wells, J.-P. Crochard, E. L. Stone, T. D. Bradshaw, M. F. G. Stevens and A. D. Westwell, J. Med. Chem., 2006, 49, 179 CrossRef CAS PubMed; (d) M. Balasubramanian, J. G. Keay, in Comprehensive Heterocyclic Chemistry II, ed. A. R. Katritzky, C. W. Rees and E. V. F. Scriven, Pergamon Press, London, 1996, vol. 5, ch. 6, p. 245 Search PubMed; (e) A. Z. Halimehjani, I. N. N. Namboothiri and S. E. Hosshmand, RSC Adv., 2014, 4, 51794 RSC.
- (a) R. Dua, S. Shrivastava, S. K. Sonwane and S. K. Shrivastava, Adv. Biomed. Res., 2011, 5, 120 CAS; (b) D. Dalvie, P. Kang, C.-M. Loi, L. Goulet and S. Nair, Influence of Heteroaromatic Rings on ADME Properties of Drugs, ed. D. E. Smith, RSC Publishing, Cambridge, U.K., 1st edn, 2010, p. 328 Search PubMed; (c) A. Gomtsyan, Chem. Heterocycl. Comp., 2012, 48, 7 CrossRef CAS.
- (a) M.-J. Camarasa, A. San-Felix, S. velázquez, M.-J. Perez-Perez, F. Gago and J. Balzarini, Curr. Top. Med. Chem., 2004, 4, 671 CrossRef; (b) M. J. Soltis, H. J. Yeh, K. A. Cole, N. Whittaker, R. P. Wersto and E. C. Kohn, Drug Metab. Dispos., 1996, 24, 799 CAS; (c) P. Actor, D. H. Pitkin, G. Lucyszyn, J. A. Weisbach and J. L. Bran, Antimicrob. Agents Chemother., 1976, 9, 800 CrossRef CAS PubMed; (d) H. M. Bryson and R. N. Brogden, Drugs, 1994, 47, 506 CrossRef CAS PubMed; (e) A. Kamal, N. Shankaraiah, V. Devaiah, K. L. Reddy, A. Juvekar, S. Sen, N. Kurian and S. Zingde, Bioorg. Med. Chem. Lett., 2008, 18, 1468 CrossRef CAS PubMed; (f) K. R. Senwar, P. Sharma, T. S. Reddy, M. K. Jeengar, V. L. Nayak, V. G. M. Naidu, A. Kamal and N. Shankaraiah, Eur. J. Med. Chem., 2015, 102, 413 CrossRef CAS PubMed; (g) N. Shankaraiah, C. Jadala, S. Nekkanti, K. R. Senwar, N. Nagesh, S. Shrivastava, V. G. M. Naidu, M. Sathish and A. Kamal, Bioorg. Chem., 2016, 64, 42 CrossRef CAS PubMed; (h) A. Kamal, N. Shankaraiah, C. R. Reddy, S. Prabhakar, N. Markandeya, H. K. Srivastava and G. N. Sastry, Tetrahedron, 2010, 66, 5498 CrossRef CAS; (i) N. P. Kumar, S. Nekkanti, S. S. Kumari, P. Sharma and N. Shankariah, Bioorg. Med. Chem. Lett., 2017, 27, 2369 CrossRef CAS PubMed; (j) T. S. Reddy, H. Kulhari, V. G. Reddy, A. V. S. Rao, V. Bansal, A. Kamal and R. Shukla, Org. Biomol. Chem., 2015, 13, 10136 RSC; (k) K. R. Senwar, P. Sharma, S. Nekkanti, M. Sathish, A. Kamal, B. Sridhar and N. Shankaraiah, New J. Chem., 2015, 39, 3973 RSC.
- (a) T. W. Kim, Y. Yong, S. Y. Shin, H. Jung, K. H. Park, Y. H. Lee, Y. Lim and K.-Y. Jung, Bioorg. Chem., 2015, 59, 1 CrossRef CAS PubMed; (b) P. K. Kadaba, J. Med. Chem., 1988, 31, 196 CrossRef CAS PubMed; (c) M. De Lourdes, G. Ferreria, L. C. S. Pinheiro, O. A. Santos-Filho, M. D. S. Pecanha, C. Q. Sacramento, V. Machado, V. F. Ferreria, T. M. L. Souza and N. Boechat, Med. Chem. Res., 2014, 23, 1501 CrossRef; (d) D. R. Buckle, C. J. M. Rockell, H. Smith and B. A. Spicer, J. Med. Chem., 1986, 29, 2262 CrossRef CAS PubMed; (e) P. Dwivedi, K. B. Mishra, B. B. Mishra, N. Singh, R. K. Singh and V. K. Tiwari, Glycoconj. J., 2015, 32, 127 CrossRef CAS PubMed; (f) V. D. Bock, R. Perciaccante, T. P. Jansen, H. Hiemstra and J. H Van Maarseveen, Org. Lett., 2006, 8, 919–922 CrossRef CAS PubMed; (g) P. Wang, J. Du, S. Rachakonda, C. Byoung-Kwon, Ph. M. Tharnish and L. J. Stuyver, et al., J. Med. Chem., 2005, 48, 6454 CrossRef CAS PubMed; (h) J. K. Chakrabarti, T. M. Hotten, I. A. Pullar and D. J. Steggles, J. Med. Chem., 1989, 32, 2375 CrossRef CAS PubMed; (i) P. G. Baraldi, S. Manfredini, D. Simoni, L. Zappaterra, C. Zocchi, S. Dionisotti and E. Ongini, Bioorg. Med. Chem. Lett., 1994, 4, 2539 CrossRef CAS; (j) J. Huo, H. Hu, M. Zhang, X. Hu, M. Chen, D. Chen, J. Liu, G. Xiao, Y. Wang and Z. Wen, RSC Adv., 2017, 7, 2281 RSC.
- (a) G. de Miguel, M. Wielopolski, D. I. Schuster, M. A. Fazio, O. P. Lee, C. K. Haley, A. L. Ortiz, L. Echegoyen, T. Clark and D. M. Guldi, J. Am. Chem. Soc., 2011, 133, 13036 CrossRef CAS PubMed; (b) S. B. Ferreira, A. C. Sodero, M. F. Cardoso, E. S. Lima, C. R. Kaiser, F. P. Silva and V. F. Ferreira, J. Med. Chem., 2010, 53, 2364 CrossRef CAS PubMed; (c) A. Kamal, S. Prabhakar, N. Shankaraiah, Ch. R. Reddy and P. V. Reddy, Tetrahedron Lett., 2008, 49, 3620 CrossRef CAS.
- E. Bonandi, M. S. Christodoulou, G. Famagalli, D. Perdicchia, G. Rastelli and D. Passarella, Drug Discov. Today, 2017, 22, 1572 CrossRef CAS PubMed.
- (a) M. Juricek, P. H. J. Kouwer and A. E. Rowan, Chem. Commun., 2011, 47, 8740 RSC; (b) S. G. Agalave, S. R. Maujan and V. S. Pore, Chem.–Eur. J., 2011, 6, 2696 CAS.
- (a) R. Huisgen, Angew. Chem., Int. Ed., 1963, 75, 565 CrossRef; (b) R. Huisgen, Angew. Chem., Int. Ed., 1963, 2, 633 CrossRef.
- (a) C. W. Tornoe, C. Christensen and M. Meldel, J. Org. Chem., 2002, 67, 3057 CrossRef CAS PubMed; (b) V. V. Rostovev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef.
- (a) Y.-C. Wang, Y.-Y. Xie, H.-E. Qu, H.-S. Wang, Y.-M. Pan and F.-P. Huang, J. Org. Chem., 2014, 79, 4463 CrossRef CAS PubMed; (b) Y. Chen, G. Nie, Q. Zhang, S. Ma, H. Li and Q. Hu, Org. Lett., 2015, 17, 1118 CrossRef CAS PubMed.
- (a) S. Kamijo, T. Jin, Z. Huo and Y. Yamamoto, J. Am. Chem. Soc., 2003, 125, 7786 CrossRef CAS PubMed; (b) B. C. Boren, S. Narayan, L. K. Rasmussen, L. Zhang, H. Zhao, Z. Lin, G. Jia and V. V. Fokin, J. Am. Chem. Soc., 2008, 130, 8923 CrossRef CAS PubMed; (c) F. Wei, H. Li, C. Song, Y. Ma, L. Zhou, C.-H. Tung and Z. Xu, Org. Lett., 2015, 17, 2860 CrossRef CAS PubMed; (d) W. G. Kim, M. E. Kang, J. B. Lee, M. H. Jeon, S. Lee, J. Lee, B. Choi, P. M. S. D. Cal, S. Kang, J.-M. Kee, G. J. L. Bernardes, J.-U. Rohde, W. Choe and S. Y. Hong, J. Am. Chem. Soc., 2017, 139, 12121 CrossRef CAS PubMed; (e) S. Lal and S. Díez-González, J. Org. Chem., 2011, 76, 2367 CrossRef CAS PubMed.
- J. John, J. Thomas and W. Dehaen, Chem. Commun., 2015, 51, 10797 RSC.
- (a) S. Koguchi and K. Izawa, ACS Comb. Sci., 2014, 16, 381 CrossRef CAS PubMed; (b) A. De Nino, P. Merino, V. Algieri, M. Nardi, M. L. Di Giona, B. Russo, M. A. Tallarida and L. Maiuolo, Catalysts, 2018, 8, 364 Search PubMed.
- (a) P. Appukuttan, W. Dehaen, V. V. Fokin and E. V. Eycken, Org. Lett., 2004, 6, 4223 CrossRef PubMed; (b) F. Friscourt and G.-J. Boons, Org. Lett., 2010, 12, 4936 CrossRef CAS PubMed; (c) L. Jing, L. Hongwei, M. Fanyu, Y. Liuqing, S. Yanpeng, Z. Yumin and G. Qiang, Chem. Res. Chin. Univ., 2018, 34, 197 CrossRef.
- (a) R. U. Islam, A. Taher, M. Choudhary, S. Siwal and K. Mallick, Sci. Rep., 2015, 5, 9632 CrossRef PubMed; (b) L. Wang and C. Cai, Green Chem. Lett. Rev., 2010, 3, 121 CrossRef CAS.
- (a) X.-J. Wang, K. Sidhu, L. Zhang, S. Campbell, N. Haddad, D. C. Reeves, D. Krishnamurthy and C. H. Senanayake, Org. Lett., 2009, 11, 5460 Search PubMed; (b) L. Wu, X. Chen, M. Tang, X. Song, G. Chen, X. Song and Q. Lin, Synlett, 2012, 23, 1529 CrossRef CAS.
- (a) K. Sakai, N. Hida and K. Kondo, Bull. Chem. Soc. Jpn., 1986, 59, 179 CrossRef CAS; (b) S. S. Van Berkel, S. Brauch, L. Gabriel, M. Henze, S. Stark, D. Vasilev, L. A. Wessjohann, M. Abbas and B. Westermann, Angew. Chem., Int. Ed., 2012, 51, 5343 CrossRef CAS PubMed; (c) Z.-J. Cai, X.-M. Lu, Y. Zi, C. Yang, L.-J. Shen, J. Li, S.-Y. Wang and S.-J. Ji, Org. Lett., 2014, 16, 5108 CrossRef CAS PubMed; (d) Z. Chen, Q. Yan, Z. Liu, A. Lei and Y. Zhang, Chem.–Eur. J., 2014, 20, 13692 CrossRef CAS PubMed.
- (a) Y.-M. Ren, C. Cai and R.-C. Yang, RSC Adv., 2013, 3, 7182 RSC; (b) M. S. Yusubov and V. V. Zhdankin, Resour.-Effic. Technol., 2015, 1, 49 Search PubMed.
- (a) K. Donthiboina, H. K. Namballa, S. P. Shaik, J. B. Nanubolu, N. Shankaraiah and A. Kamal, Org. Biomol. Chem., 2018, 16, 1720 RSC; (b) G. S. Mani, S. P. Shaik, Y. Tangella, S. Bale, C. Godugu and A. Kamal, Org. Biomol. Chem., 2017, 15, 6780 RSC; (c) G. S. Mani, A. V. S. Rao, Y. Tangella, S. Sunkari, F. Sultana, H. K. Namballa, N. Shankaraiah and A. Kamal, New J. Chem., 2018, 42, 15820 RSC; (d) L. Ren, M. Wang, B. Fang, W. Yu and J. Chang, Org. Biomol. Chem., 2019, 17, 3446 RSC.
- (a) S. Bawa, S. Kumar, S. Drabu and R. Kumar, J. Pharm. BioAllied Sci., 2010, 2, 64 CrossRef CAS PubMed; (b) X. Nqoro, N. Tobeka and B. A. Aderibigbe, Molecules, 2017, 22, 2268 CrossRef PubMed; (c) J. L. Vennerstrom, E. O. Nuzum, R. E. Miller, A. Dorn, L. Gerena, P. A. Dande, W. Y. Ellis, R. G. Ridley and W. K. Milhous, Antimicrob. Agents Chemother., 1999, 43, 598 CrossRef CAS PubMed.
- (a) K. K. D. R. Viswanadham, M. P. Reddy, P. Satyanarayana, O. Ravi, R. Kant and S. R. Bathula, Chem. Commun., 2014, 50, 13517 RSC; (b) S. P. Shaik, F. Sultana, A. R. Kumar, S. Sunkari, A. Alarifi and A. Kamal, Org. Biomol. Chem., 2017, 15, 7696 RSC.
- (a) Z. Chen, Q. Yan, Z. Liu, Y. Xu and Y. Zhang, Angew. Chem., Int. Ed., 2013, 52, 13324 (Angew. Chem., Int. Ed., 2013, 125, 13566) CrossRef CAS PubMed; (b) Z. Chen, Q. Yan, H. Yi, Z. Liu, A. Lei and Y. Zheng, Chem.–Eur. J., 2014, 20, 13692 CrossRef CAS PubMed.
- (a) N. Kornblum, W. J. Jones and G. J. Anderson, J. Am. Chem. Soc., 1959, 81, 4113 CrossRef CAS; (b) N. Kornblum, J. W. Powers, G. J. Anderson, W. J. Jones, H. O. Larson, O. Levand and W. M. Weaver, J. Am. Chem. Soc., 1957, 79, 6562 CrossRef CAS.
- (a) J.-Q. Shang, H. Fu, Y. Li, T. Yang, C. Gao and Y.-M. Li, Tetrahedron, 2019, 75, 253 CrossRef CAS; (b) D. Wang, N. Li, M. Zhao, W. Shi, C. Ma and B. Chen, Green Chem., 2010, 12, 2120 RSC; (c) M. A. Topchiy, A. A. Ageshina, P. S. Gribanov, S. M. Masoud, T. R. Akmalov, S. E. Nefedov, S. N. Osipov, M. S. Nechaev and A. F. Asachenko, Eur. J. Org. Chem., 2019, 2019, 1016 CrossRef CAS; (d) A. S. Kumar, M. A. Reddy, M. Knorn, O. Reiser and B. Sreedhar, Eur. J. Org. Chem., 2013, 21, 4674 CrossRef; (e) A. Bolje, D. Urankar and J. Kosmrlj, Eur. J. Org. Chem., 2014, 36, 8167 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra06005g |
| This journal is © The Royal Society of Chemistry 2019 |