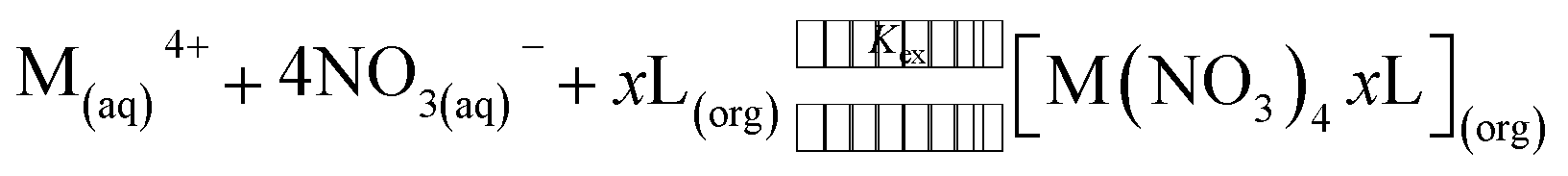DOI:
10.1039/C9RA05977F
(Paper)
RSC Adv., 2019,
9, 31928-31935
Evaluation of two aza-crown ether-based multiple diglycolamide-containing ligands for complexation with the tetravalent actinide ions Np4+ and Pu4+: extraction and DFT studies†
Received
1st August 2019
, Accepted 23rd September 2019
First published on 8th October 2019
Abstract
Two multiple diglycolamide (DGA)-containing extractants where the DGA arms are tethered to the nitrogen atoms of two aza-crown ether scaffolds, a 9-membered aza-crown ether containing three ‘N’ atoms (LI) and a 12-membered aza-crown ether containing four ‘N’ atoms (LII), were evaluated for the extraction of the tetravalent actinide ions Np4+ and Pu4+. The tripodal ligand with three DGA arms (LI) was relatively inferior in its metal ion extraction properties as compared to the tetrapodal ligand with four DGA arms (LII) and Pu4+ ion was better extracted than Np4+ ion with both the ligands. A solvation extraction mechanism, where species of the type ML(NO3)4 are extracted, was found to be operative for both the ligands involving both the tetravalent actinide ions. While the extraction of the metal ions increased with the feed nitric acid concentration up to 4 M, a sharp decline in the extraction was seen after that. Quantitative extraction (>99%) of the actinide ions was observed with LII from 4 M HNO3, suggesting the possible application of the ligands for actinide partitioning of high-level waste. The structure and the composition of the complexes were optimized by DFT computations.
Introduction
Spent fuel reprocessing using TBP (tri-n-butyl phosphate) selectively separates U and Pu from the dissolver solution which contains actinides viz. U, Np, Pu, Am, Cm, etc, a host of fission product nuclides and also structural materials viz. Zr, Fe, Cr, Ni, etc., in 3–4 M nitric acid.1 In view of the difficulties faced in the separation of trivalent minor actinides which contributes to the major part of the alpha dose emanating from the raffinate stream after the spent fuel reprocessing, several specific extractants such as CMPO (carbamoylmethylphosphine oxide), TRPO (trialkylphosphine oxide), DIDPA (diisodecyl phosphoric acid) were developed by American, Chinese and Japanese researchers, respectively, for this purpose.2–4 However, all these extractants are phosphorus based which result in large volumes of secondary waste and hence, needed to be replaced with suitable ‘green’ alternatives.5 Further research on eco-friendly extractants led to the discovery of the tetraalkyl malonamides by research groups at the European Union by the end of last century.6 Subsequently, diglycolamide (DGA) extractants were reported to be highly efficient in view of the significantly lower extractant inventory and high extraction efficiency.7 Typically, a 0.1 M solution of TODGA (N,N,N′,N′-tetra-n-octyl diglycolamide) in n-dodecane gives a DAm (distribution ratio) value of 30 with 1 M HNO3 as the aqueous feed and increases sharply with the feed acid concentration.8 Under identical conditions, the DEu value was reported to be ca. nine times larger suggesting the possibility of the separation of the lanthanides, which act as neutron poison and also create problems during vitrification of the radioactive wastes, from the trivalent actinides9,10 under suitable conditions. This has been quite promising as compared to other extractants reported before and hence, has been employed for counter-current extraction runs.11–13 Though most reported studies involving TODGA were found to deal with trivalent actinides viz. Am3+ and Cm3+, there is scant data on the extraction of Np, a very important minor actinide.14 The separation of Np, major constituent being 237Np (t1/2: 2.1 × 106 years), from the acidic radioactive wastes including the high level waste (a concentrate of the raffinate emanating from the spent fuel reprocessing) can ultimately lead to the production of 238Pu for use as a power source.
The selectivity observed in the case of TODGA is quite different from that seen with other extractants, i.e., trivalent actinides are better extracted than the tetravalent actinide ions, while the hexavalent actinides are extracted to a much lower extent. Different research groups have made attempts to understand the unusual extraction behaviour. A size selective extraction of metal ions of 100 pm ionic radii has been proposed by Zhu et al.,15 which was supported subsequently by a theory of aggregate formation as confirmed from SANS (small angle neutron scattering) as well as dynamic light scattering studies.16 These studies indicated that 3–4 TODGA molecules form a reverse micellar aggregate, which facilitates the extraction of the metal ions.17 Though this explains the size selective extraction of the metal ions, the formation of aggregates is limited to non-polar diluents such as n-dodecane. It was thought appropriate to synthesize multiple DGA ligands where several DGA groups are appended to a scaffold to enhance the efficiency of extraction. These ligands were expected to give the same or better extraction efficiency without any diluent property limitation. In this connection, multiple DGA extractants such as DGA-functionalized calix[4]arenes18 and pillar[5]arenes19 containing four and five DGA arms, respectively, were synthesized and evaluated for their extraction efficiency. Other multiple DGA ligands, containing three DGA arms, have also been studied over the years, which include those with a central carbon or nitrogen atom or a set of benzene-centered tripodal DGA ligands.20–22
Recently, we reported the first time study on the extraction of actinides using a tripodal DGA ligand where the DGA arms are tethered to a triazamacrocycle scaffold (LI; Fig. 1).23 The promising results prompted us to explore the possibility of synthesizing a tetrapodal DGA ligand with a ‘cyclen’ (1,4,7,10-tetrazacyclododecane) scaffold (LII; Fig. 1). The ligand was synthesized and studied for the separation of the trivalent f-cations viz. Am3+ and Eu3+.24 The major advantage of such multiple DGA ligands with a macrocyclic scaffold was to add some degree of flexibility to the otherwise pre-organized structures, which could result in a better binding. Results of our recent studies24 using the tetrapodal DGA ligand LII with trivalent actinide ions were indeed quite spectacular, where a nearly 20 times lower ligand concentration was used to achieve a reasonably high extraction efficiency. In view of the lack of detailed research on the extraction of tetravalent actinides with these multiple DGA extractants, the present attempt was made for a detailed investigation.
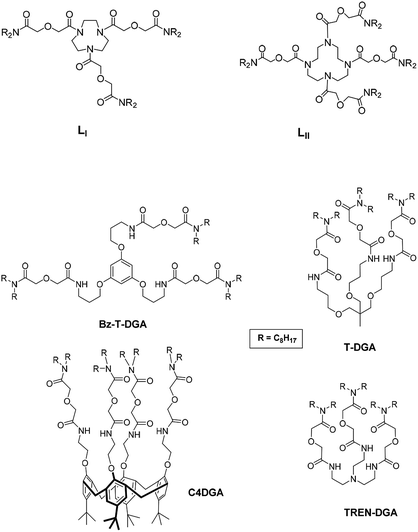 |
| | Fig. 1 Structural formulae of the ligands used in the present study (LI and LII) and other relevant ligands. | |
The present work deals with the extraction of Np4+ and Pu4+ from nitric acid feed solutions using millimolar concentrations of LI and LII in a diluent mixture containing 95% n-dodecane + 5% isodecanol. The diluent mixture was chosen in view of the poor solubility of such multiple DGA ligands as reported earlier25 and to make a proper comparison with other analogous extractants. Apart from the solvent extraction studies, DFT computational studies were carried out to get structural information.
Experimental
Materials
The multiple DGA-containing ligand LI was prepared as reported.23LII was prepared following an analogous method, details of which are given in the ESI.† The purity of the ligands was checked by elemental analysis, NMR and HR-MS techniques. The diluents, n-dodecane and isodecanol (>99% purity), were procured from Lancaster, UK and SRL, Mumbai, respectively, and were used as obtained. HTTA (2-thenoyltrifluoroacetone) was obtained from Sigma-Aldrich (USA) and was used after recrystallization. Suprapur nitric acid (Merck, Germany) was used for the preparation of dilute nitric acid solutions applied in this study, which were standardized using volumetric methods using AR grade NaOH (BDH) with phenolphthalein (Fluka, Switzerland) as the indicator.
Radiotracers.
Pu (mainly 239Pu) was used from the laboratory stock after fresh purification from 241Am using a HTTA extraction method reported in the literature.26239Np was prepared by the neutron activation of natural uranium in the Dhruva reactor at BARC, Mumbai, at a neutron flux of 5.0 × 1013 n per cm2 per s for 5 days. The radiotracer (239Np) was subsequently separated from the bulk of uranium and the fission products as per a reported method which used HTTA extraction.27
Methods
Oxidation state adjustment of actinides.
The oxidation state of Pu was adjusted to the +4 state by first adding a few drops of 5.0 × 10−3 M NaNO2 solution to the Pu tracer solution in 1 M HNO3 followed by extraction using 0.5 M HTTA in xylene. The organic phase contained the converted Pu4+, which was subsequently stripped using 8 M HNO3. Np4+ conversion was done as mentioned above and the aqueous stock solution was found to be stable for over one month. The stability of the oxidation state for both Pu and Np was tested by a HTTA concentration variation experiment (distribution ratio (D) was measured at varying HTTA concentrations) where the linear log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) D vs. log[HTTA] plot yielded a positive slope of ca. 4.
D vs. log[HTTA] plot yielded a positive slope of ca. 4.
Solvent extraction.
The solvent extraction experiments were carried out by equilibrating equal volumes (usually 1 mL) of the organic phase containing 1.0 × 10−3 M of the multiple DGA-containing ligands LI and LII (or as specified) and the aqueous phase containing the required radiotracer in the acidity of interest. The equilibrations were done in leak tight Pyrex glass tubes (10 mL capacity), in a thermostated water bath at 25 ± 0.1 °C. The time of equilibration was maintained at about 1 h after confirming the extraction kinetics was over much before that (vide infra). Subsequently, the tubes were rested, centrifuged and 100 μL aliquots were taken out from both phases for subsequent radiometric assay. While the radiometric assay of 239Np was done using a well type NaI(Tl) scintillation counter (Para Electronics) coupled with a multi-channel analyzer (ECIL, India), Pu was assayed using a liquid scintillation counting system (Hidex, Finland) where an Ultima Gold (PerkinElmer, USA) scintillation cocktail was used. The distribution ratio of the metal ions (DM) was calculated as the ratio of counts per unit time per unit volume in the organic phase to that in the aqueous phase. Each distribution experiment was carried out in triplicate and the accepted data were within the relative standard deviation of 5%.
DFT methodology.
We considered the corresponding methyl derivatives of the DGA-containing ligands LI and LII, respectively, to avoid difficulties in convergence due to the C–C single bond rotation of the long hydrocarbon chains. All the DFT based studies were performed using the TURBOMOLE 7.2 program package.28–30 The geometries of LI and its Am3+ and Eu3+ complexes were optimized by using Becke's exchange functional31 in conjunction with Perdew's correlation functional32 (BP86) with generalized gradient approximation (GGA). The geometries obtained by energy minimization were used for the calculation of the vibrational frequencies. 60 Electron core pseudo-potentials (ECPs) along with the corresponding def-SV(P) basis set as implemented in the TURBOMOLE suit of program were selected for the Am3+ ion, whereas 28 electron core potentials (ECPs) along with the corresponding def-SV(P) basis set were chosen for Eu3+. All the other lighter atoms were treated at the all electron (AE) level. Natural population analysis (NPA)33 has improved numerical stability as compared to the conventional “Mulliken Population Analysis (MPA)”. The electron distribution in compounds of high ionic character, such as metal complexes, is also described in a much superior way by the NPA. NPA analysis was, therefore, performed in the present work to calculate the natural charges on different atoms in the Am3+ and Eu3+ complexes. Natural population analysis (NPA)33 was performed on the complexes of Am3+ and Eu3+ using the hybrid B3LYP density functional31,34,35 employing the triple zeta valence plus polarization (TZVP) basis set36,37 using equilibrated structures obtained at the BP86/SVP level of theory as implemented in the TURBOMOLE suit of program. In case of Am3+ and Eu3+, the high spin septet was found to be the ground state configuration. In the present chemical system, the close matching of the 〈S2〉 values with the S(S + 1) ideal values indicated negligible spin contamination. In order to have a quantitative idea about the comparative ‘M–O’ bond strength for the Np4+ and Pu4+ complexes the molecular orbitals were analyzed to calculate the two-center Wiberg's bond order using the AOMix program.38,39
Results & discussion
Solvent extraction studies
Extractions of Np4+ and Pu4+ were carried out using the valency adjusted stock solutions as mentioned above. The extraction studies were carried out with a holding oxidant (ammonium metavanadate27) for Pu4+ and a holding reductant (hydroxylamine hydrochloride and ferrous sulphamate40) for Np4+. In the absence of any extractant in the diluent mixture, the extraction of the metal ions was negligible (D < 0.001). On the other hand, the presence of 1 × 10−3 M ligands resulted in very high extraction of the metal ions (Table 1). Table 1 also includes the reported D values with other analogous extractants with Pu4+ and Am3+ at 3 M HNO3 as Np4+ ion extraction data is not available. The relative extraction of M4+ and M3+ ions does not show any clear trend in the previously reported multiple DGA-containing extractants: while Am3+ ion extraction is more than that of Pu4+ ion for TREN-DGA and Bz-T-DGA, an opposite trend was reported for T-DGA and C4DGA. On the other hand, the extraction of the metal ions was nearly comparable (within experimental error limits) when LI and LII were used (Table 1). The extraction of Np4+ was found to be significantly inferior as compared to that of Pu4+, which may be attributed to the smaller ionic size and hence, higher complexation of the latter. This suggests that the extraction of Np4+ ensures that of Pu4+ as well. Knowing very well about the poor extraction of UO22+ ion with multiple DGA-containing ligands,18,21–23 decontamination of the actinides can be easily achieved from U. The following sections include results of the other extraction studies.
Table 1 Actinide ion extraction data with multiple DGA ligands from aqueous phase containing 3 M HNO3 using 1 × 10−3 M ligand solution in 95% n-dodecane + 5% isodecanol
| Ligand |
D
Np(IV)
|
D
Pu(IV)
|
D
Am(III)
|
Reference |
|
Diluent: 90% n-dodecane + 10% isodecanol.
3 × 10−3 M ligand solution in n-dodecane.
Data taken from ref. 23.
Between brackets: 5 × 10−5 M ligand solution in 95% n-dodecane + 5% isodecanol.
|
|
T-DGA
|
— |
19.0 ± 1.1a |
11.1 ± 0.06a |
21
|
|
TREN-DGA
|
— |
0.25 ± 0.01a |
0.36 ± 0.02a |
21
|
|
Bz-T-DGA
|
— |
103 ± 10.6 |
235 ± 19.4 |
22
|
|
C4DGA
|
— |
67.9 ± 1.40b |
26.5 ± 1.40b |
18
|
|
LI
|
42.7 ± 4.1 |
51.4 ± 3.7 |
71.1 ± 6.1c |
This work |
|
LII
|
163 ± 11 |
183 ± 8.5 (12.4 ± 0.52)d |
4.83 ± 0.26d |
This work |
Extraction kinetics.
Extraction kinetics is one of the important parameters needed to be assured in solvent extraction to obtain equilibrium D values, particularly with bulky ligands where the mass transfer of the bulky metal/ligand complex is slow. This could be due to slow binding of the multiple ligating groups with the metal ion. On the other hand, if the metal ion is coordinated to only a few donor atoms present in the ligand, the extraction can be fast. Scheme 1 gives a pictorial representation of the complexation of the metal ion with the multiple DGA-containing ligand (LI) for (a) one (b) two and (c) three DGA arm binding. A tetravalent metal ion, as in the present case, is always associated with four nitrate counter anions to result in a charge neutralized complex. However, association with a ligand is mandatory for a better partitioning into the organic phase. As the ligand is assumed to be pre-organized, we started with interaction of the metal ion with one of the DGA groups. Subsequently, another DGA may interact due to its proximity to the metal ion. In view of the coordination of four nitrate ions to the metal ion in a ‘solvation’ type extraction mechanism (vide infra), the number of coordination sites available for binding to the DGA groups is limited (considered in the range of 9 to 10) and hence, not more than two DGA arms should be binding to the metal ion at one time if one considers inner-sphere nitrate ion binding. If one considers three DGA arm binding, all four nitrate ions should be present in the outer sphere (Scheme 1(c)). For the sake of simplicity, we discount outer-sphere coordination in the present study and hence consider binding by either one or two DGA arms. It may be considered that binding to two DGA arms can result in sluggishness due to more steric crowding, while binding to a single DGA arm may result in relatively faster extraction. Similar model can be presented for LII which has four DGA arms. However, binding to all four DGA arms does not appear logical if one needs to restrict the coordination number in the range of 9.
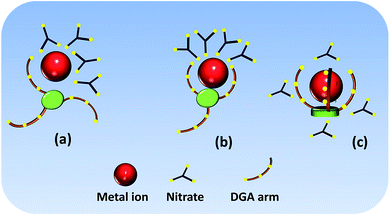 |
| | Scheme 1 Representative binding of the M4+ ion with multiple DGA-containing ligands and nitrate ions in case of LI: (a) one DGA arm binding, (b) two DGA arm binding; (c) all three DGA arm binding. | |
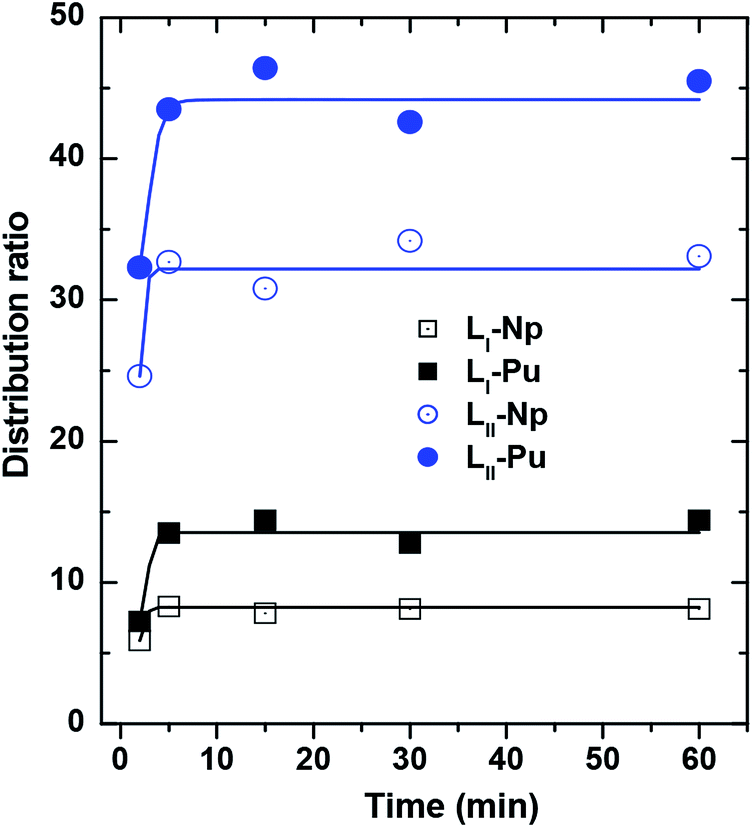
In the present case, the extraction equilibrium could be reached within 5 minutes of equilibration for both the ligands (Fig. 2). This suggests that the binding of the metal ions to the ligands may be quite simple in the sense that only one DGA arm might be binding out of the three available arms of LI and four available arms of LII. However, for all the subsequent experiments, 30 minutes of equilibration time were kept for the sake of convenience.
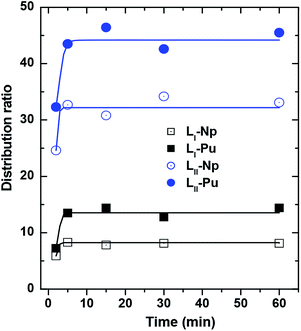 |
| | Fig. 2 Distribution ratio of actinides by DGA ligands LI and LII showing the extraction kinetics for reaching the equilibrium condition. Organic phase: 1.0 mmol L−1 ligands in 95% n-dodecane + 5% isodecanol; aqueous phase: 1 M HNO3. | |
Effect of HNO3 concentration.
The equilibrium reaction for the extraction of tetravalent actinides from nitric acid medium with the above two neutral ligands LI and LII can be written as:| |  | (1) |
where M4+ represents either Np4+ or Pu4+, and x represents the number of ligand (L is either LI or LII) molecules associated in the extracted complex. The subscripts (aq) and (org) represent the species present in the aqueous and the organic phases, respectively. The two-phase extraction equilibrium constant (Kex) for the above reaction can be written as:| | 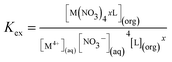 | (2) |
Since the distribution ratio (D) is experimentally obtained as the ratio of the total metal ion concentration in the organic phase to that in the aqueous phase, the above equation can be simplified to:
| | | D = Kex[NO3−](aq)4[L](org)x | (3) |
As per the above equation, the distribution ratio must increase with the nitric acid concentration due to increased nitrate ion concentration. This is also reflected in Fig. 3, for both the ligands, up to 4 M HNO3. However, beyond 4 M HNO3, a decrease in the distribution ratio indicates that the following equilibrium reaction, which facilitates the adduct formation of the ligand with one or more molecules of nitric acid, is also operational paralell to eqn (1):
| | 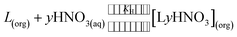 | (4) |
where
KH is the equilibrium constant referred to as acid uptake constant for the ligand or basicity of the ligand. From
Fig. 3, it is very obvious that at higher nitric acid concentration,
eqn (4) is dominating and that most of the ligand molecules are complexed with nitric acid, and therefore, result in a decrease of the distribution ratio of the metal ions. Such features are common for neutral ligands where a ‘solvation’ type of extraction mechanism is operating. While studying the extraction profiles of trivalent actinide/lanthanide ions with
TREN-DGA or
Bz-T-DGA a monotonous increase in the
D values was seen up to 6 M HNO
3.
21,22 On the other hand, extraction with
T-DGA20 showed a profile similar to the one shown here.
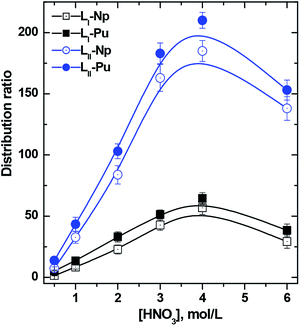 |
| | Fig. 3 Effect of nitric acid concentration on the distribution ratio of actinides with DGA ligands LI and LII. Organic phase: 1 mmol L−1 ligands in 95% n-dodecane + 5% isodecanol. | |
Nature of the extracted complex.
The stoichiometry of the extracted complex in solvent extraction is generally obtained by ‘slope analysis’ as detailed below. Eqn (3) can be rearranged after taking logarithms as follows:| | | logD = logKex + 4log[NO3](aq)− + xlog[L](org) | (5) |
Dependence of the extraction of Np4+ and Pu4+ was studied as a function of the concentration of the extractant (LI or LII) at a fixed nitrate ion concentration and vice versa. The slopes of the straight-line logD vs. log ligand concentration plots (Fig. 4) are used to determine the number of ligand molecules present in the extracted species. As shown in Fig. 4, linear plots of slope values of 1 (approx.) were obtained for the tetravalent actinides with both the ligands. Considering the fact that LI and LII have 9 and 12 coordinating atoms, respectively, and four nitrate ions are required to coordinate with the metal ions (for charge neutralization purpose), the possibility of participation of more than one extractant molecule in the extracted species appears to be improbable. As per eqn (4), four nitrate ions are required to be present in the complex to maintain the charge neutrality on the metal ion. The logD vs. log[NO3−] plots (the experiments were carried out using varying concentrations of NaNO3 along with a fixed amount of HNO3, required to prevent hydrolysis of the metal ions) yielded slope values of ca. 3, which after corrections due to nitrate ion complexation in a manner similar to that reported by Horwitz et al.41 resulted in slope values close to 4 (Fig. 5). The slope values of the logD vs. log[NO3−] straight-line plots from the nitrate ion concentration variation experiments are listed in Table S1.† The slope analysis suggested that the extracted species conform to ML(NO3)4, where M = Np and Pu, while L = LI and LII. The extraction constants were obtained from the intercept values and are listed in Table 2. The extraction constant value for the extraction of Pu4+ with TODGA (using n-dodecane as the diluent) is also included in the table for comparison purpose. The values obtained with the present ligands are higher than that reported for TODGA. TODGA operates as a reverse micelle-based extraction system16,17 where the metal ion sits inside the aggregate based on its size. Pu4+, its ionic size being very close to 100 pm,42 gets extracted favourably by TODGA.15 However, the ligands used in the present study apparently form a stronger complex with the metal ion and in view of the pre-organized structure favour the extraction in a more effective way. The higher extraction constant with LII can be attributed to a higher lypophilicity of the complex due to the presence of eight n-octyl groups vis-à-vis six n-octyl groups present in LI.
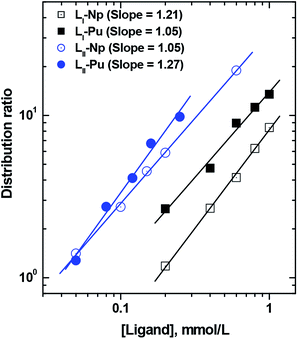 |
| | Fig. 4 Effect of ligand concentration on the distribution ratio of actinides with DGA ligands LI and LII. Aqueous phase: 1 M HNO3; diluent: 95% n-dodecane + 5% isodecanol. | |
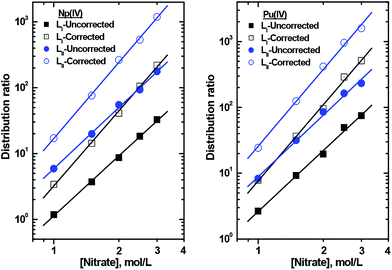 |
| | Fig. 5 Effect of nitrate concentration on the distribution ratio of actinides with DGA ligands LI and LII. Organic phase: 0.2 mmol L−1 ligands in 95% n-dodecane + 5% isodecanol; aqueous phase: 1 M H+ + varying nitrate ions. | |
Table 2 Two-phase extraction constant values for the extraction of Np4+ and Pu4+ ions using ligands LI and LII and TODGA
| Metal ion |
Log Kex values |
|
LI
|
LII
|
TODGA |
|
Data taken from ref. 6.
|
| Np4+ |
3.98 ± 0.02 |
4.58 ± 0.01 |
— |
| Pu4+ |
4.21 ± 0.03 |
4.86 ± 0.02 |
4.0 ± 0.4a |
However, the position of the nitrates in the complex, outer sphere or inner sphere, will be decided by how many coordination sites of the ligands are directly bonded to the metal ions and the vacant coordination positions on the metal ions. Considering nine ligating sites through ‘O’ atoms in LI (three arms) and 12 in LII (four arms), and also the possibility of coordination by the four nitrate ions (either by mono- or bidentate mode), it is logical that only very few DGA arms are coordinating to the metal ion. For an in-depth understanding of the bonding in the metal/ligand complex, DFT calculations were done as described below.
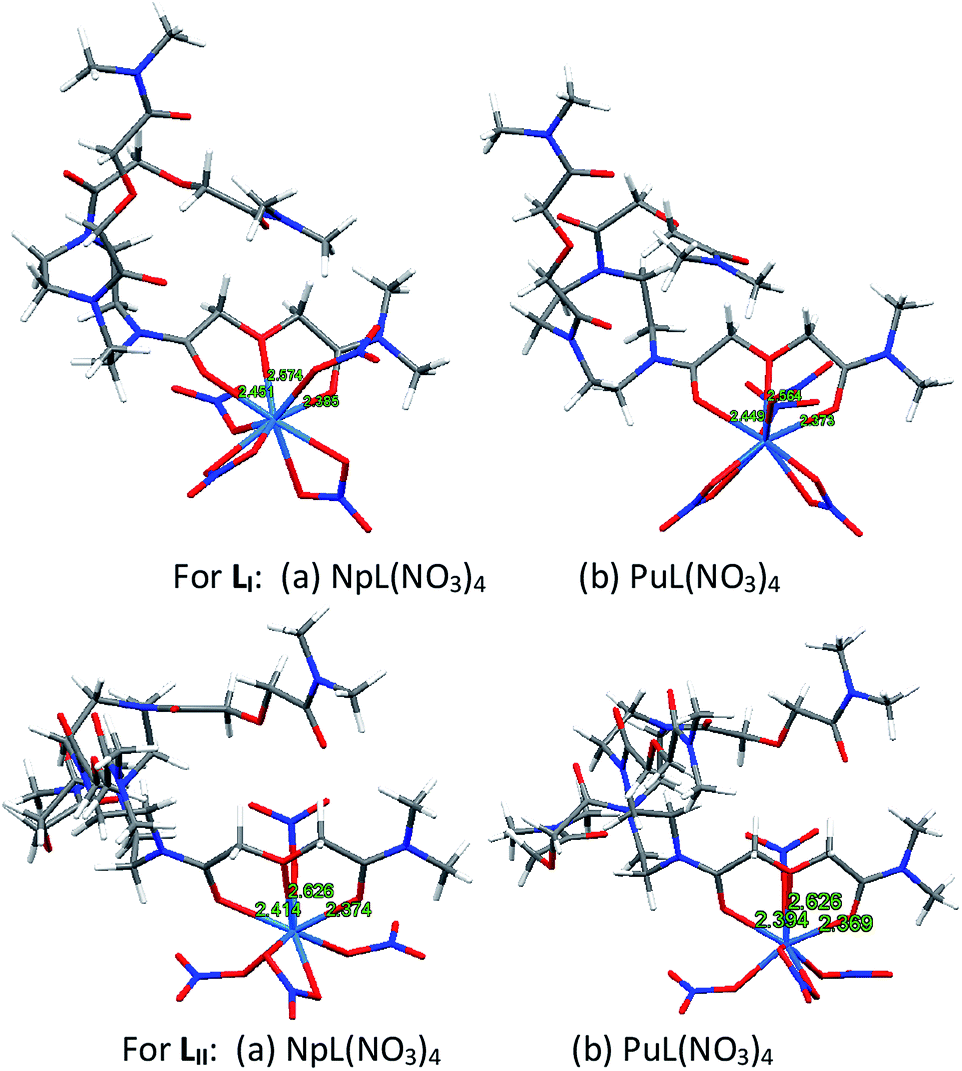
DFT calculations
For both the metal ions, viz. Pu and Np, a ML type of complex was found to be the most stable for both the ligands (LI and LII). The energy minimized structures of the complexes of Np4+ and Pu4+ with both ligands are presented in Fig. 6. In case of the complexes with LI, the metal ions were found to be nine-coordinated, where one DGA arm of the ligand coordinated in tridentate fashion. It should be noted that, out of three DGA arms, only one arm is coordinating via two ![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) C
C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups and one ethereal group. Out of four nitrate ions present in the complex, two were found to be bidentate and the other two were monodentate.
O groups and one ethereal group. Out of four nitrate ions present in the complex, two were found to be bidentate and the other two were monodentate.
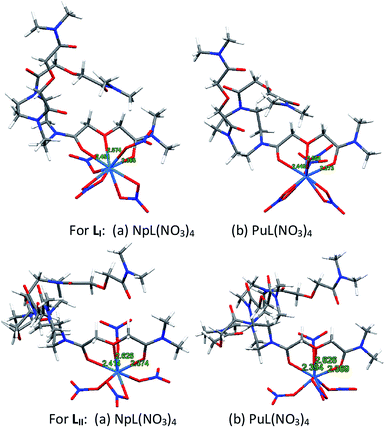 |
| | Fig. 6 DFT optimized structures of the Np4+ and Pu4+ complexes with LI and LII (with methyl instead of octyl chains). | |
In case of LII, on the other hand, the metal ions were eight-coordinated, where one DGA arm of the ligand coordinated in tridentate fashion, and out of four nitrate ions, three coordinated as monodentate and one as bidentate mode. Similar to LI, out of four DGA arms, only one arm is coordinating via two CO groups and one ethereal group. In case of both the ligands, in spite of having multiple DGA arms, only one arm is coordinating to the metal ions which could be due to the fact that bringing multiple DGA arms to coordinate a metal ion is energy intensive. Significant increase in the metal ion extraction employing these ligands as compared to ligands having only one DGA arm is probably due to the increase in lipophilicity of the metal ion complexes of these multiple DGA ligands due to the presence of six and eight n-octyl groups per ligand molecule.
For both the ligands, the carbonyl bond of a DGA site with Pu is shorter than that with Np, implying that the former complex is stronger than the latter (Table 3). Bond order calculations also indicated that the Pu complex is more stable than the Np complex by 0.85 eV and 0.76 eV for LI and LII, respectively. This feature has also been observed experimentally from the solvent extraction studies. The natural charges on the central metal ions (QM) in the respective complexes of plutonium and neptunium are also shown in Table 3, which shows that neptunium contains lesser positive charges as compared to plutonium in the complexes of both the ligands (LI and LII) indicating lesser ligand to metal charge transfer in case of plutonium complexes as compared to that in case of neptunium complexes. The higher stability of plutonium complexes in spite of the lesser ligand to metal charge transfer, suggests that the metal–ligand interactions here are mainly dominated by ionic interactions.
Table 3 Different ‘M–O’ bond distances along with the Wiberg's bond order (numbers in parenthesis) and the natural charges on the metal ion (QM) obtained for Pu(IV) and Np(IV) complexes
| Bond/QM |
LI
|
LII
|
| PuL(NO3)4 |
NpL(NO3)4 |
PuL(NO3)4 |
NpL(NO3)4 |
|
d(M–Ocarb) |
2.451 (0.435) |
2.449 (0.422) |
2.394 (0.372) |
2.414 (0.352) |
| 2.385 (0.572) |
2.373 (0.546) |
2.369 (0.421) |
2.374 (0.409) |
|
d(M–Oether) |
2.574 (0.132) |
2.564 (0.118) |
2.626 (0.105) |
2.626 (0.100) |
|
d(M–ONO3(m)) |
2.290 (0.580) |
2.313 (0.626) |
2.169 (0.740) |
2.170 (0.782) |
| 2.213 (0.662) |
2.224 (0.674) |
2.189 (0.725) |
2.189 (0.757) |
| |
|
2.294 (0.551) |
2.286 (0.587) |
|
d(M–ONO3(b)) |
2.486 (0.335) |
2.504 (0.334) |
|
|
| 2.407 (0.532) |
2.373 (0.530) |
2.411 (0.489) |
2.419 (0.481) |
| 2.441 (0.463) |
2.437 (0.474) |
2.452 (0.459) |
2.468 (0.456) |
| 2.425 (0.512) |
2.406 (0.512) |
|
|
|
Q
M
|
1.678 |
1.630 |
1.829 |
1.761 |
Conclusions
The extraction of tetravalent actinide ions, viz. Np4+ and Pu4+, was investigated using the two aza-macrocycle-based multiple DGA-containing ligands LI and LII. The extraction of Pu4+ was significantly larger than that of Np4+, while that using LII was superior to that with LI. The extracted species contained four nitrate ions for charge neutralization purpose, while one ligand molecule was associated with the complex, possibly by bonding to a single DGA arm. The structures of the complexes were optimized by DFT computations and conformed to two bidentate nitrates, two monodentate nitrates and a multiple DGA-containing ligand coordinating through a single DGA arm. The D values of both Pu4+ as well as Np4+ were found to be reasonably good for possible application of these ligands for their recovery from acidic wastes such as the HLW. Published data on trivalent actinide ion such as Am3+ suggested its co-extraction along with Np4+ and Pu4+ and a good decontamination from UO22+ ion.
Conflicts of interest
The authors have no conflicts of interest to declare.
Acknowledgements
The authors (SAA, AB and PKM) thank Dr P. K. Pujari, Assoc. Director, RC&I Group and Head, Radiochemistry Division, BARC for his keen interest in this work.
References
-
P. A. Baisden and G. R. Choppin, Nuclear waste management and the nuclear fuel cycle, radiochemistry and nuclear chemistry. in Encyclopedia of Life Support Systems (EOLSS), ed. S. Nagyl, Eolss Publishers, Oxford, U.K, 2007, https://www.eolss.net/Sample-Chapters/C06/E6-104-11.pdf Search PubMed
 .
.
- E. P. Horwitz, D. G. Kalina, H. Diamond, G. H. Vandegrift and W. W. Schulz, Solvent Extr. Ion Exch., 1985, 3, 75–109 CrossRef CAS .
-
Y. Zhu, R. Jiao, S. Wang, S. Fan, B. Liu, H. Zheng, S. Zhou and S. Chen, An extractant (TRPO) for the removal and recovery of actinides from high level radioactive liquid waste, ISEC'83, Denver, Colorado USA, 1983 Search PubMed .
- Y. Morita and M. Kubota, Solvent Extr. Ion Exch., 1988, 6, 233–246 CrossRef CAS .
- A. Leoncini, J. Huskens and W. Verboom, Chem. Soc. Rev., 2017, 46, 7229–7273 RSC .
- O. Courson, M. Lebrun, R. Malmbeck, G. Pagliosa, K. Romer, B. Satmark and J. P. Glatz, Radiochim. Acta, 2000, 88, 857–863 CAS .
- S. A. Ansari, P. N. Pathak, P. K. Mohapatra and V. K. Manchanda, Chem. Rev., 2012, 112, 1751–1772 CrossRef CAS .
- Y. Sasaki, Y. Sugo, S. Suzuki and S. Tachimori, Solvent Extr. Ion Exch., 2001, 19, 91–103 CrossRef CAS .
- G. J. Lumetta, A. V. Gelis, J. C. Carter, C. M. Niver and M. R. Smoot, Solvent Extr. Ion Exch., 2014, 32, 333–347 CrossRef CAS .
- Z. Kolarik, Chem. Rev., 2008, 108, 4208–4252 CrossRef CAS .
- S. A. Ansari, D. R. Prabhu, R. B. Gujar, A. S. Kanekar, M. S. Murali, S. Rajeswari, T. G. Srinivasan and V. K. Manchanda, Sep. Purif. Technol., 2009, 66, 118–124 CrossRef CAS .
- R. B. Gujar, S. A. Ansari, D. R. Prabhu, P. N. Pathak, A. Sengupta, S. K. Thulasidas, P. K. Mohapatra and V. K. Manchanda, Solvent Extr. Ion Exch., 2012, 30, 156–170 CrossRef CAS .
- D. Whittaker, A. Geist, G. Modolo, R. Taylor, M. Sarsfield and A. Wilden, Solvent Extr. Ion Exch., 2018, 36, 223–256 CrossRef CAS .
- S. A. Ansari, R. B. Gujar, D. R. Prabhu, P. N. Pathak and P. K. Mohapatra, Solvent Extr. Ion Exch., 2012, 30, 457–468 CrossRef CAS .
- Z. X. Zhu, Y. Sasaki, H. Suzuki, S. Suzuki and T. Kimura, Anal. Chim. Acta, 2004, 527, 163–168 CrossRef CAS .
- P. N. Pathak, S. A. Ansari, S. Kumar, B. S. Tomar and V. K. Manchanda, J. Colloid Interface Sci., 2010, 342, 114–118 CrossRef CAS .
- M. P. Jensen, T. Yaita and R. Chiarizia, Langmuir, 2007, 23, 4765–4774 CrossRef CAS .
- P. K. Mohapatra, M. Iqbal, D. R. Raut, J. Huskens, W. Verboom and S. V. Godbole, Dalton Trans., 2012, 41, 360–363 RSC .
- L. Wu, Y. Y. Fang, Y. M. Jia, Y. Y. Yang, J. L. Liao, N. Liu, X. S. Yang, W. Feng, J. L. Ming and L. H. Yuan, Dalton Trans., 2014, 43, 3835–3838 RSC .
- K. Matloka, A. Gelis, M. Regalbuto, G. Vandegrift and M. J. Scott, Dalton Trans., 2005, 3719–3721 RSC .
- A. Leoncini, P. K. Mohapatra, A. Bhattacharyya, D. R. Raut, A. Sengupta, P. K. Verma, N. Tiwari, D. Bhattacharyya, S. N. Jha, A. M. Wouda, J. Huskens and W. Verboom, Dalton Trans., 2016, 45, 2476–2484 RSC .
- A. Leoncini, S. A. Ansari, P. K. Mohapatra, A. Boda, S. M. Ali, A. Sengupta, J. Huskens and W. Verboom, Dalton Trans., 2017, 46, 1431–1438 RSC .
- A. Bhattacharyya, A. Leoncini, R. J. M. Egberink, P. K. Mohapatra, P. K. Verma, A. S. Kanekar, A. K. Yadav, S. N. Jha, D. Bhattacharyya, J. Huskens and W. Verboom, Inorg. Chem., 2018, 57, 12987–12998 CrossRef CAS .
-
A. Bhattacharyya, R. J. M. Egberink, P. K. Mohapatra, P. K. Verma, A. S. Kanekar, A. K. Yadav, S. N. Jha, D. Bhatacharyya, J. Huskens and W. Verboom, DOI:10.1021/acs.inorgchem.9b02605.
- M. Iqbal, P. K. Mohapatra, S. A. Ansari, J. Huskens and W. Verboom, Tetrahedron, 2012, 68, 7840–7847 CrossRef CAS .
- S. Bagawde, V. V. Ramakrishna and S. K. Patil, J. Inorg. Nucl. Chem., 1976, 38, 1669–1672 CrossRef CAS .
- P. K. Mohapatra, P. B. Ruikar and V. K. Manchanda, Radiochim. Acta, 2002, 90, 323–327 CAS .
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS .
- O. Treutler and R. Ahlrichs, J. Chem. Phys., 1995, 102, 346–354 CrossRef CAS .
- TURBOMOLE V6.6, a development of the University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989−2007, TURBOMOLE GmbH, since 2007, available from http://www.turbomole.com.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS .
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef .
- A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735–746 CrossRef CAS .
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS .
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS .
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC .
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef .
-
S. I. Gorelsky, AOMix: Program for Molecular Orbital Analysis; Version 6.X, University of Ottawa, 2013, http://www.sgchem.net Search PubMed .
- S. I. Gorelsky and A. B. P. Lever, J. Organomet. Chem., 2001, 635, 187–196 CrossRef CAS .
- B. Mahanty, A. Bhattacharyya and P. K. Mohapatra, J. Chromatogr. A, 2018, 1564, 94–101 CrossRef CAS .
- E. P. Horwitz, A. C. Muscatello, D. G. Kalina and L. Kaplan, Sep. Sci. Technol., 1981, 16, 417–437 CrossRef CAS .
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef .
Footnote |
| † Electronic supplementary information (ESI) available: Purification radiotracers, coordinates complexes DFT calculations, and synthesis of LII. See DOI: 10.1039/c9ra05977f |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Arunasis
Bhattacharyya
a,
Prasanta K.
Mohapatra
a,
Arunasis
Bhattacharyya
a,
Prasanta K.
Mohapatra