
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular interaction between MeOH and genistein during soy extraction
Hailiang Zhao ab,
Xue Songb,
Yingming Zhangb and
Xia Sheng*b
ab,
Xue Songb,
Yingming Zhangb and
Xia Sheng*b
aProvince Key Laboratory of Cereal Resource Transformation and Utilization, Henan University of Technology, Lianhua Street 100, 450001 Zhengzhou, China
bCollege of Chemistry, Chemical and Environmental Engineering, Henan University of Technology, Lianhua Street 100, 450001 Zhengzhou, China. E-mail: xia.sheng@haut.edu.cn
First published on 29th November 2019
Abstract
Genistein has received great attention due to its possible anti-oxidant properties. The interaction between genistein and the extraction solvent helps in understanding the extraction efficiency. Hydrogen bonding plays a crucial role in liquid systems. Density functional theory quantum chemical computations in both gas phase and solution were performed to investigate the molecular interaction between genistein and methanol. All the resulting complexes (MeOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :genistein = 1:1, 2:1, 3:1, 6:1) were studied using the B3LYP-D3 computational level and the cc-pVTZ basis set. Binding energies demonstrate that more MeOH molecules surrounding genistein could stabilize the system more. Geometry optimizations show that there are strong O–H⋯O interactions between MeOH and genistein. The electron density and the corresponding Laplacian of charge density at bond critical points were also calculated using AIM theory, and the results are in line with the structural and energetic analysis of the studied system. Moreover, energy decomposition analysis shows that the exchange energy term has the largest contribution to the attraction interaction energy as compared with other energy terms. Meanwhile, this study shows that the MeOH–genistein system is more stable under basic conditions. This study could help increase the efficiency of extraction.
:genistein = 1:1, 2:1, 3:1, 6:1) were studied using the B3LYP-D3 computational level and the cc-pVTZ basis set. Binding energies demonstrate that more MeOH molecules surrounding genistein could stabilize the system more. Geometry optimizations show that there are strong O–H⋯O interactions between MeOH and genistein. The electron density and the corresponding Laplacian of charge density at bond critical points were also calculated using AIM theory, and the results are in line with the structural and energetic analysis of the studied system. Moreover, energy decomposition analysis shows that the exchange energy term has the largest contribution to the attraction interaction energy as compared with other energy terms. Meanwhile, this study shows that the MeOH–genistein system is more stable under basic conditions. This study could help increase the efficiency of extraction.
1 Introduction
Natural plant components have received great attention due to their possible anti-oxidant properties in the field of medicine, such as against cancer, diabetes, and cardiovascular disease.1,2 Many studies have been carried out to investigate the chemical structures and the activity of anti-oxidants. In particular, isoflavonoids have been widely found in plants, and almost exclusively in legumes (Leguminosae family), for instance, soybeans, chickpeas, etc.3Genistein [C15H10O5, 4′,5,7-trihydroxyisoflavone or 5,7-dihydroxy-3-(4-hydroxyphenyl)chromen-4-one] is a member of a multifunctional natural isoflavonoid class of flavonoids with a 15-carbon skeleton which consists of two phenyl rings and one heterocyclic ring.4 It has a 3-phenylchromen-4-one skeleton, but there is no hydroxyl group substitution on position 2.5 The concentration of genistein in soy foods are about 1.9–229 mg g−1, and it is the major anti-cancer component in soybean.6,7 The analyses of isoflavone aglycones of soybean including daidzein, glycitein, and genistein were evaluated by various extraction methods. To extract isoflavonoid from plants is a crucial task, and solvents, such as methanol, ethanol, acetonitrile, etc. are normally used.8–10 In addition, there are hydroxyl and carbonyl groups in isoflavonoids. Thus, strong hydrogen bonding interactions between solvent and isoflavonoids could be expected. Meanwhile, the hydrogen bonding interaction has strong components of electrostatic and charge-transfer, then the polar solvent will affect the hydrogen bond.11,12 Furthermore, the alcohols such as methanol and ethanol could improve the extraction efficiency.13 More study demonstrated that the mixture of 80% MeOH with HCl and water was the best solvent for extraction.14 However, the best solvents for isoflavone extraction from these studies have not been conclusive. On the other hand, to understand the extraction mechanism from molecular level is a proper way to determine an appropriate solvent. In addition, density functional theory (DFT) calculations can deliver a better understanding of the conformational parameters, electronic structures, hydrogen bonding interactions of isoflavonoids, such as daidzein, genistein, etc.15,16 The inter-molecular interaction in solvent, namely, hydrogen bonding interaction, has a great impact on the extraction process.17 Methanol (MeOH) is a polar organic compound, and this makes it a good industrial solvent.18 In this study, MeOH was used to understand the extraction mechanism. The main purpose is to calculate the hydrogen bonding interaction between genistein and methanol in the extraction reaction from a theoretical point of view to determine an appropriate solvent for isoflavone extraction.
2 Methodology
The geometry optimization and vibrational frequencies were carried out on the basis of the Gaussian09 (RevE.01) suit program.19 The calculations of harmonic frequencies were carried out to ensure that the optimized structures were indeed energy minima with no imaginary frequencies. Moreover, the correlation-consistent polarized valence triple-zeta basis set (cc-pVTZ) basis sets were used for the calculations. All MeOH, genistein, and MeOH–genistein geometries in the gas phase and MeOH solvent (static dielectric constant, ε = 32.613) were optimized using the density functional theory (DFT) method with the hybrid exchange–correlation B3LYP functional. The D3 dispersion corrections, designed by Grimme in 2010,20 was included as well. The reason to choose the B3LYP-D3 functional is because that this method was shown to give reliable interaction energies, infrared vibrational frequencies, structural geometries, etc. on small hydrogen bonded clusters.21–26 All optimizations performed in solution were carried out by utilizing the default self-consistent reaction field (SCRF) method with the integral equation formalism variant model (the IEFPCM procedure) in Gaussian09.27The structure is stabilized, and it is due to immersion in solution (ΔES) and the total inter-molecular interaction energy (ΔEINT) were calculated by making use eqn (1) and (2), respectively.
| ΔES = E(solvent) − E(gas) | (1) |
| ΔEINT = Ecomplex − Emonomer(A) − Emonomer(B) | (2) |
In addition, the energy decomposition analysis (EDA) is a useful technique to decompose of chemical bonds in terms of six expressions: electrostatic energy (ES), repulsion energy (REP), polarization energy (POL), exchange energy (EX), DFT correlation (CORR), and Grimme dispersion energy (DC) by using GKS-EDA method as implemented in GAMESS-US program.29,30 Moreover, it is a challenging task to analyze weak molecular bonds, the atoms in molecules (AIM) approach was used to calculate the topological properties of electron density. Wave functions were generated using the corresponding optimized structures in the gas phase from single point calculations using B3LYP-D3/cc-pVTZ. The critical points including bond critical points (BCPs) and ring critical points (RCPs), molecular graph generation as well as calculation of charge transfer within the AIM approach were carried out using AIM2000 package.31
3 Results and discussion
3.1 Conformational analysis
The framework and atom numbering of genistein is presented in Fig. 1. Genistein has a non-planar arrangement of the three rings (A, B, C). There is a torsion angle between the rings B and C. The torsion angle of the B and C rings for the most stable genistein conformer was calculated to be 41.1° (B3LYP/6-31+G(d)) is in good agreement with our study (42.0–43.3°).32 In addition, the calculated values are smaller than the experimental value (53.2°) determined by X-ray crystallography.33 The difference is likely due to the inter-molecular forces in the crystal packing, such as, hydrogen bonding interactions.33,34 Changes in the orientations of the hydroxyl groups (namely, OaHa, ObHb, and OeHe) results in eight conformers with significant energy difference of less than 61 kJ mol−1 as shown in Fig. 2. In particularly, the ObHb reorientation increases the energy by nearly 60 kJ mol−1. This is due to the formation of a strong intra-molecular hydrogen bond between the oxygen Oc and the ObHb hydrogen of genistein. In the most stable conformer genistein (h), the intra-molecular hydrogen bond length was calculated to be 1.6753 Å, which is shorter than a typical hydrogen bond length (1.97 Å).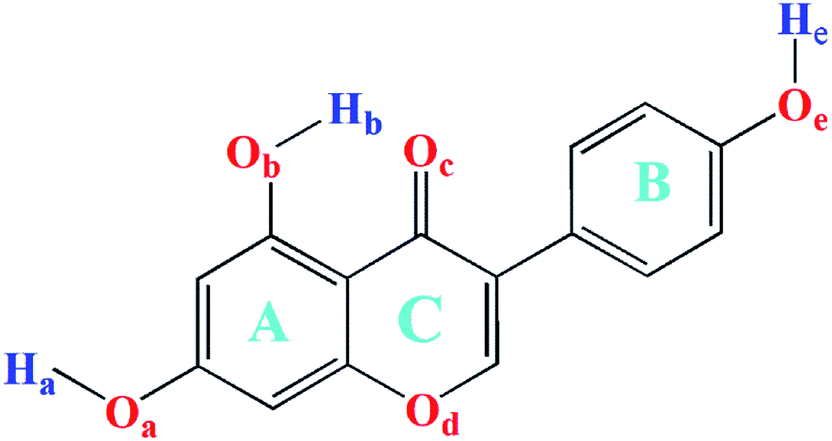 | ||
| Fig. 1 Chemical structure with atom numbering of genistein. | ||
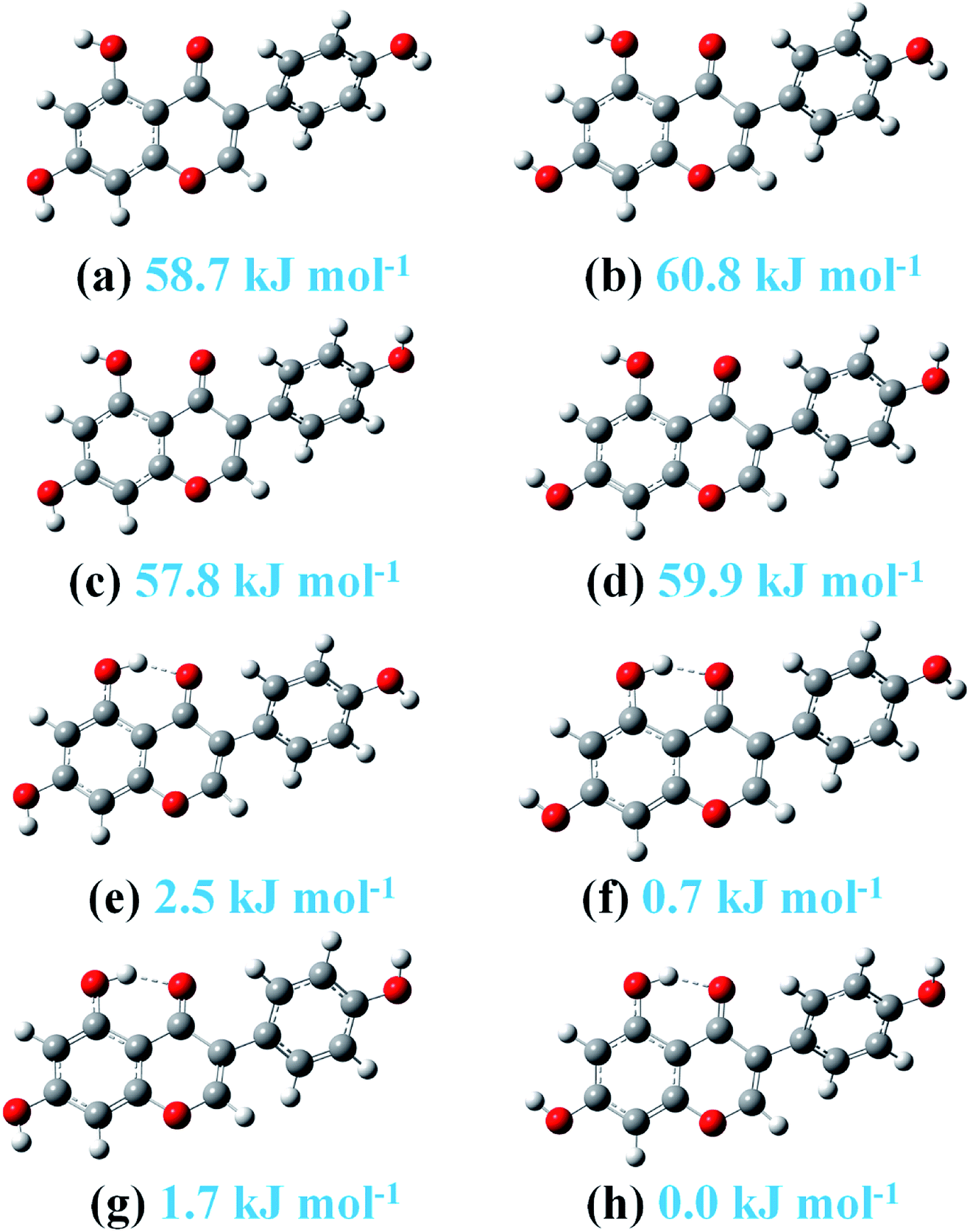 | ||
| Fig. 2 The eight stable genistein conformers optimized at the B3LYP-D3/cc-pVTZ level. Relative energies are listed. | ||
Isoflavonoid is a group of hydroxylated phenolic substance. MeOH is one of the most widely used and efficient solvent to extract phenolic compounds.35 This is because that MeOH can enhance the capacity to establish hydrogen bonds with isoflavones, since MeOH is not only a proton acceptor but also a proton donor. In this study, genistein was treated in the MeOH solution for extraction. Moreover, electrostatic potential (ESP) maps of MeOH and genistein (gas phase) are displayed in Fig. 3. The ESP maps can present the distribution of the electron density.36 The hydrogen atom of the OH group of genistein (Mulliken spin densities on Ha, Hb, He = 0.21–0.24 e−) and MeOH (Mulliken spin density on H = 0.20 e−) show positive ESPs (blue color in Fig. 3), and these enables genistein, and MeOH to be a hydrogen bond donor. Meanwhile, the negative ESP region of genistein (red color in Fig. 3) reveals the nucleophilicity, and this demonstrates that the carbonyl oxygen (Mulliken spin density on Oc = −0.39 e−) is the best hydrogen bond acceptor than the ether oxygen (Mulliken spin density on Od = −0.16 e−) and the oxygen atom of the OH groups (Mulliken spin densities on Oa, Ob, Oe = −0.29 to −0.27 e−).
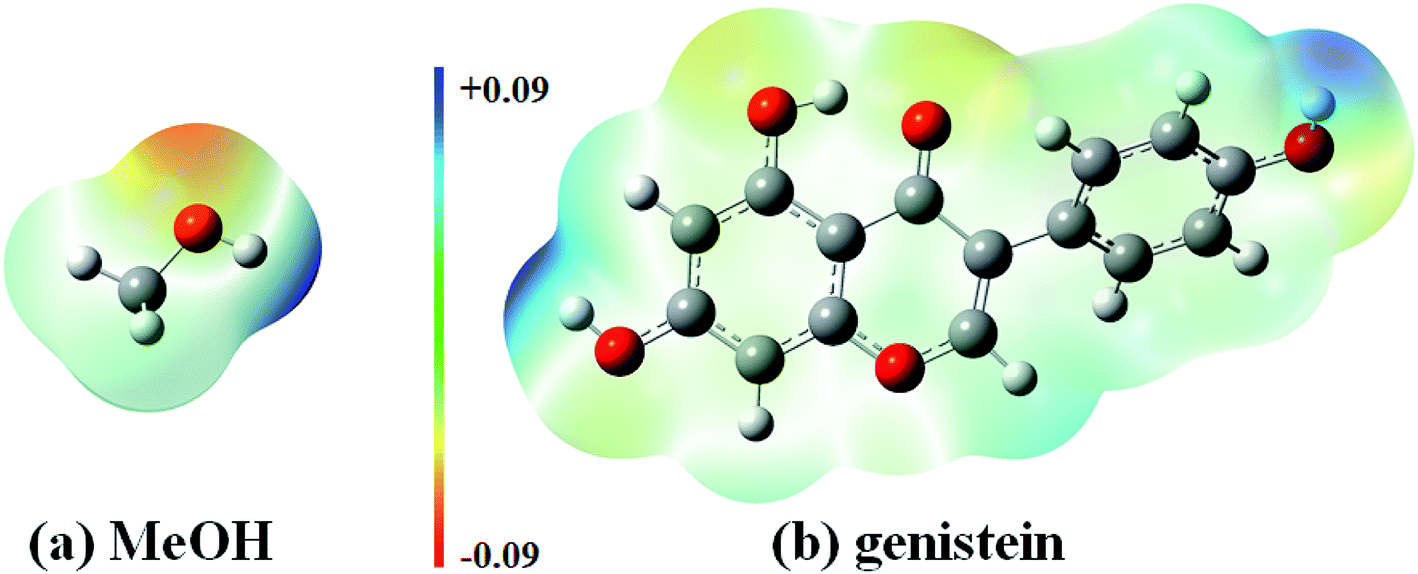 | ||
| Fig. 3 Electrostatic potential surfaces of MeOH and genistein calculated at the B3LYP-D3/cc-pVTZ level of theory. | ||
Hydrogen bond is normally an electrostatic force of attraction between a hydrogen atom and a more electronegative atom or group, such as oxygen.37 Thus, MeOH and genistein can approach to each other forming hydrogen bonded structures via O–H⋯O hydrogen bonding interaction. The twelve most stable conformers of MeOH–genistein are presented in Fig. 4. The corresponding binding energy (BE), zero-point vibrational energy (ZPVE), basis set superposition error (BSSE), and Gibbs energy of formation (ΔGθ298 K) are summarized in Table 1. ZPVE varies from 4.8 to 6.6 kJ mol−1 for the bimolecular MeOH–genistein complexes. Meanwhile, BSSE is necessary to better describe weak molecular interactions.38–41 BSSE ranges from 6.7 to 7.9 kJ mol−1. Thus, the binding energies were corrected with ZPVE and BSSE. When MeOH acts a hydrogen bond donor, it donates its proton to Oa, Ob, Oc, Od, and Oe. When genistein is a hydrogen bond donor, it donates the Ha or He atom to MeOH. Since genistein is non-planar, the rotation of MeOH can bring some difference in energy. The difference between MeOH–genistein (a–f) and MeOH–genistein (a′–f′) is the orientation of MeOH. The calculations reveal that the energy differences between MeOH–genistein (a–f) and MeOH–genistein (a′–f′) are very small, and former ones are slightly more stable with less than 2 kJ mol−1 in binding energy. Therefore, the MeOH–genistein (a–f) conformers were used for further analysis. Meanwhile, basing on the binding energies, it is clearly seen that the strength of the hydrogen bond docking sites is Oc > Ob > Oe > Oa. In addition, the strength of hydrogen bond donating sites is OaHa > OeHe. When MeOH is the hydrogen bond donor, the MeOH–genistein (a–d) conformers are much less stable (BEs = −27.8 to −16.2 kJ mol−1) than the ones with genistein as the hydrogen donor (MeOH–genistein (e and f), BEs = −32.8 to −30.3 kJ mol−1). This means that genistein prefers to be a hydrogen bond donor rather than a hydrogen bond acceptor.
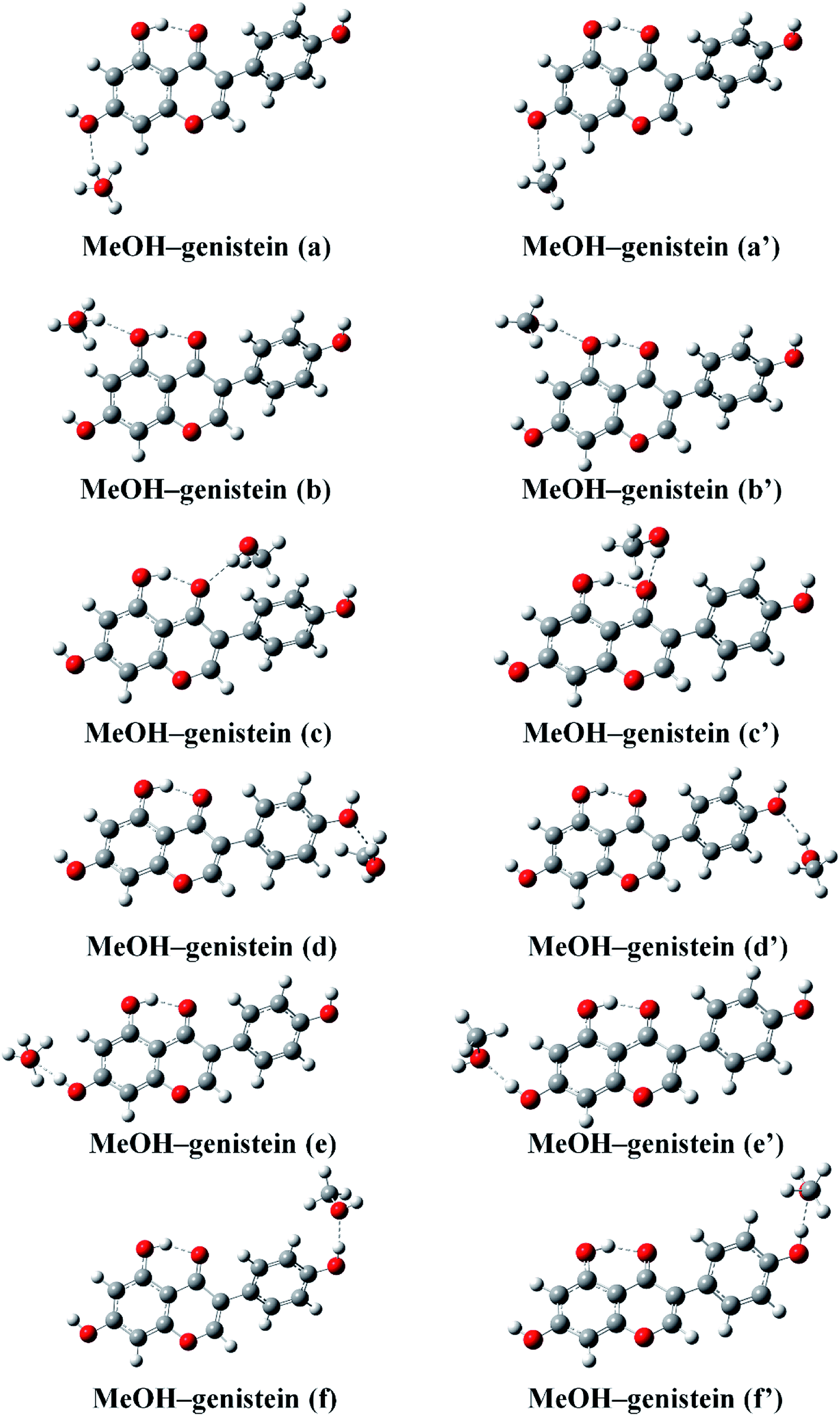 | ||
| Fig. 4 The twelve stable MeOH–genistein molecular clusters optimized at the B3LYP-D3/cc-pVTZ level. Hydrogen bonds between MeOH and genistein are represented by dashed lines. | ||
| Conformer | BE | ZPVE | BSSE | ΔGθ298 K |
|---|---|---|---|---|
| a Energies are given in kJ mol−1. Binding energies are corrected with BSSE and ZPVE. | ||||
| MeOH–genistein (a) | −16.2 | 4.8 | 7.1 | 13.6 |
| MeOH–genistein (a′) | −16.2 | 4.9 | 7.1 | 13.8 |
| MeOH–genistein (b) | −22.8 | 5.3 | 7.4 | 7.3 |
| MeOH–genistein (b′) | −22.7 | 5.3 | 7.4 | 7.5 |
| MeOH–genistein (c) | −27.8 | 5.5 | 6.7 | 6.6 |
| MeOH–genistein (c′) | −27.2 | 5.4 | 6.7 | 5.2 |
| MeOH–genistein (d) | −17.5 | 5.1 | 7.2 | 12.2 |
| MeOH–genistein (d′) | −17.3 | 5.0 | 7.2 | 12.2 |
| MeOH–genistein (e) | −32.8 | 6.2 | 7.9 | −2.8 |
| MeOH–genistein (e′) | −32.7 | 6.4 | 7.9 | −2.2 |
| MeOH–genistein (f) | −30.3 | 6.6 | 7.4 | 1.2 |
| MeOH–genistein (f′) | −28.5 | 5.9 | 7.9 | 0.4 |
Meanwhile, the hydrogen bonding interaction between genistein and caffeine was obtained in the their 1:1 cocrystalline phase. The HaOa hydroxyl group of genistein interacts with the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group of caffeine. The distance between the two O atoms was measured to be 2.7136 Å and the O–H⋯O hydrogen bond angle was obtained to be 179.4° in the X-ray single-crystal and powder diffraction.42 Moreover, genistein also reacts with amine by transferring the proton of HaOa hydroxyl group to amine in the crystal packing.43 All these are in line with our study that HaOa forms the strongest hydrogen bond with the hydrogen bond acceptor.
O group of caffeine. The distance between the two O atoms was measured to be 2.7136 Å and the O–H⋯O hydrogen bond angle was obtained to be 179.4° in the X-ray single-crystal and powder diffraction.42 Moreover, genistein also reacts with amine by transferring the proton of HaOa hydroxyl group to amine in the crystal packing.43 All these are in line with our study that HaOa forms the strongest hydrogen bond with the hydrogen bond acceptor.
Since genistein has several docking sites for the proton of MeOH and it also could act as a hydrogen bond donor, the systems containing multiple MeOH molecules (MeOH:genistein = 2:1, 3:1, and 6:1) were further studied. The selected stable structures of the MeOH:genistein = 2:1, 3:1, and 6:1 complexes at the B3LYP-D3/cc-pVTZ level are displayed in Fig. 5. Hydrogen bond donor is presented in blue shaded ring, and hydrogen bond acceptor is presented in red shaded square. In (MeOH)2–genistein (a) and (MeOH)2–genistein (c), one MeOH is the hydrogen bond donor and another MeOH is the hydrogen bond acceptor. The hydrogen bonding interaction in (MeOH)2–genistein (a) can be treated as the combination of MeOH–genistein (c) and MeOH–genistein (e). In (MeOH)2–genistein (b), the two MeOH molecules are the hydrogen bond acceptors. In (MeOH)2–genistein (d), the genistein HaOa group acts as both hydrogen bond donor and acceptor. It can be noticed that the binding energy of (MeOH)2–genistein (a) was calculated to −76.6 kJ mol−1, which is much lower than the summation (−60.6 kJ mol−1) of MeOH–genistein (c) and MeOH–genistein (e). It is the same for (MeOH)2–genistein (b–d), (MeOH)3–genistein (e) and (MeOH)6–genistein (f). This means that the more MeOH molecules surrounding the genistein molecule could give extra stability for the whole system.
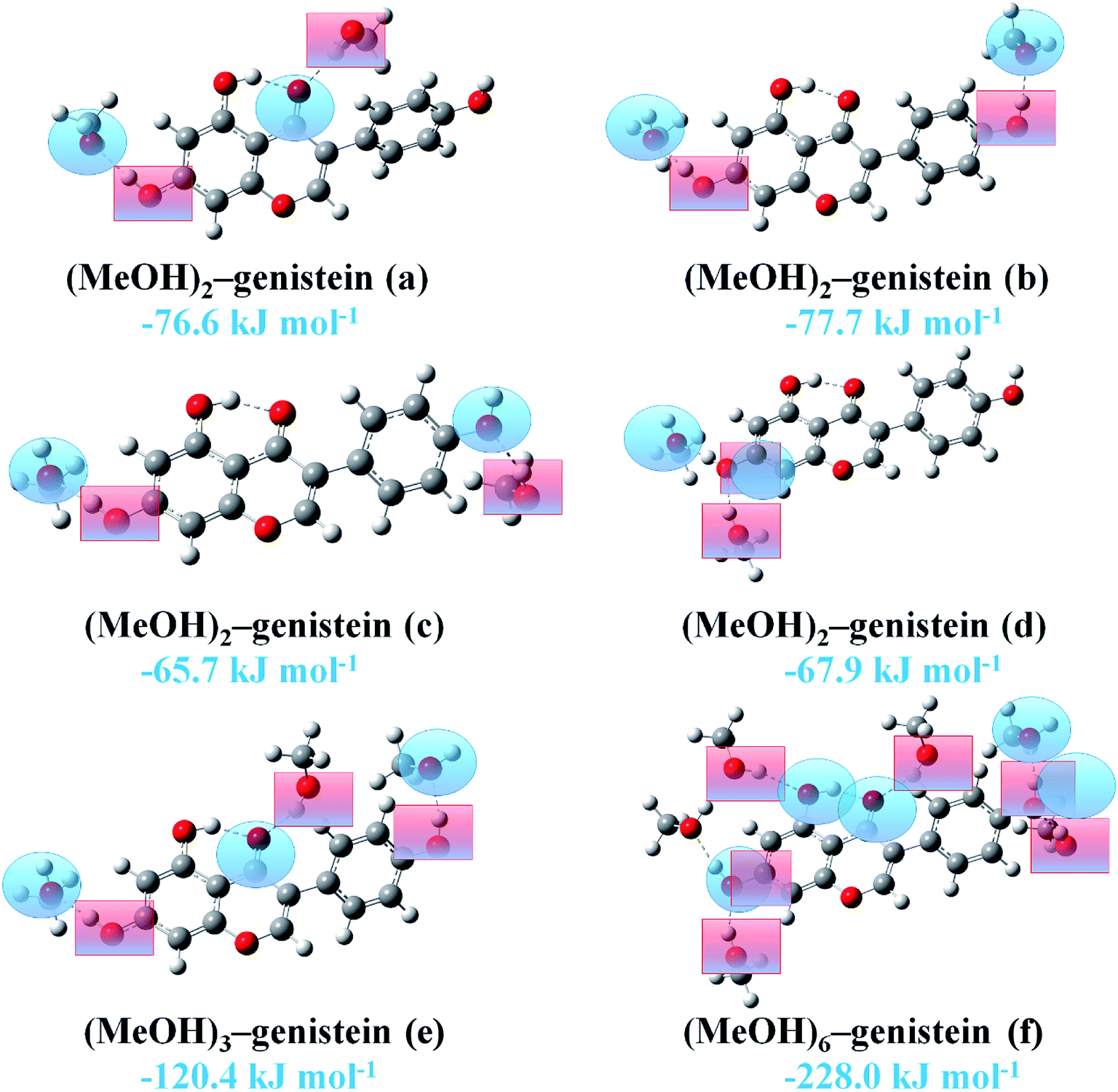 | ||
| Fig. 5 The selected stable (MeOH)2–genistein, (MeOH)3–genistein and (MeOH)6–genistein molecular clusters optimized at the B3LYP-D3/cc-pVTZ level. Hydrogen bonds between MeOH and genistein are represented by dashed lines. Hydrogen bond donor is presented in blue shaded ring, and hydrogen bond acceptor is presented in red shaded square. Binding energies are listed. | ||
On the other hand, the HaOa hydroxyl group could readily lose the Ha proton under basic condition to form an anion. Previous study shown that there was hydrogen bonding interaction between MeOH and Oa of genistein anion in a crystal structure, and the distance between the two O atoms was measured to be 2.753(3) Å.44 In this study, the interactions between genistein anion and MeOH were also investigated. The six most stable conformers of MeOH–genistein anion are presented in Fig. 6. The BEs are displayed in the figure, and they are in the range of −68.5 to −30.9 kJ mol−1. This means that genistein anion forms much more stable complexes with MeOH than genistein does. In addition, the BEs shows that the interaction between Oa and MeOH is the strongest one. The distance between the O atoms was calculated to be 2.6594 Å (MeOH–genistein anion (a)), which is slightly shorter than the value in crystal (2.753(3) Å). This is because there was lack of crystal packing interaction in our calculation.
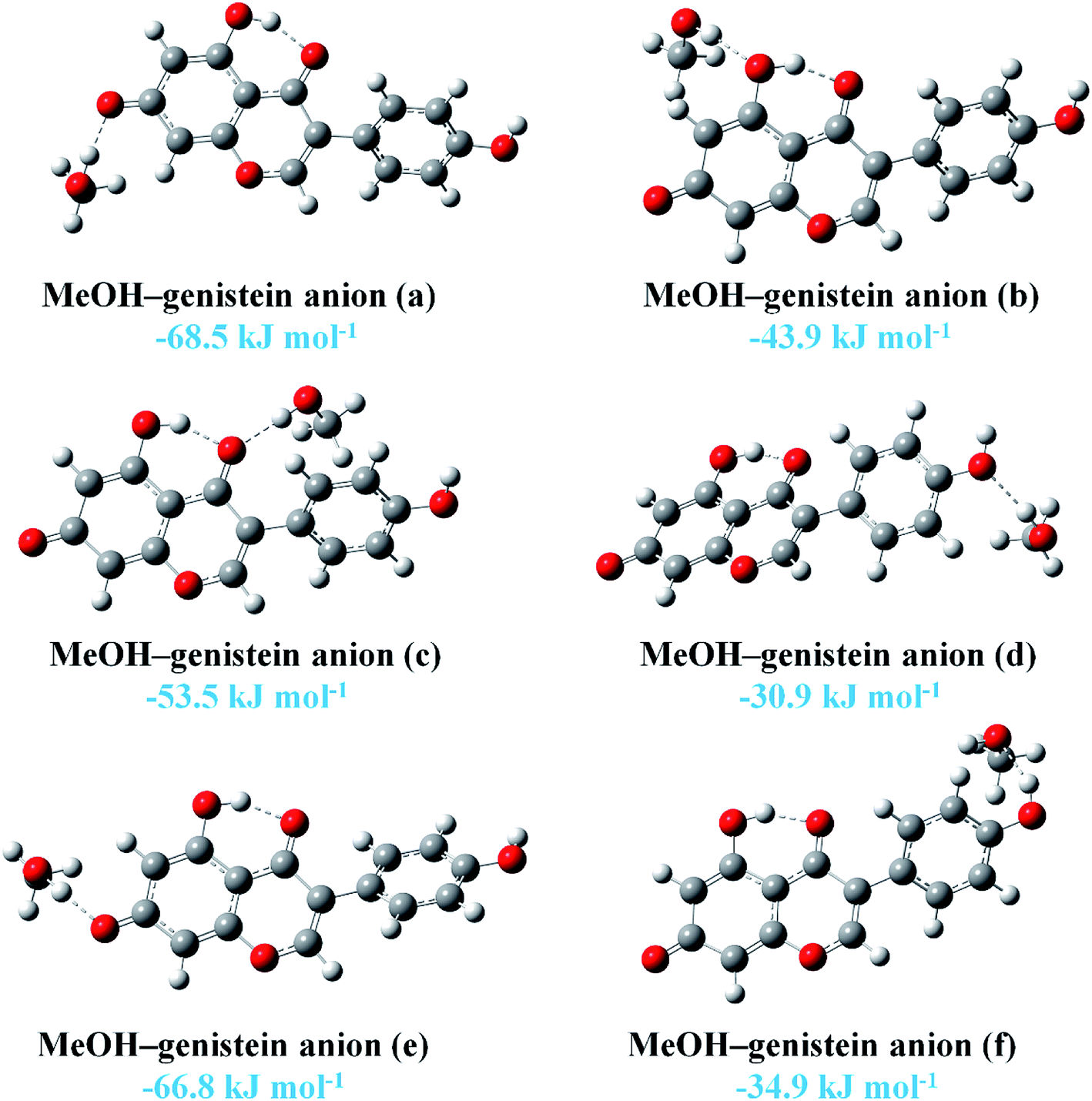 | ||
| Fig. 6 The six stable MeOH–genistein anion molecular clusters optimized at the B3LYP-D3/cc-pVTZ level. Hydrogen bonds between MeOH and genistein are represented by dashed lines. Binding energies are listed. | ||
3.2 Solvent effects and its influence on hydrogen bond
The interaction energies, geometrical structures of MeOH–genistein in MeOH solution were fully optimized within the Polarizable Continuum Model (PCM) model at B3LYP-D3/cc-pVTZ. The corresponding geometrical parameters, and interaction energies are presented in Table 2. The stabilized energies (ΔES) were calculated by using eqn (1). Since hydrogen bonding interactions contain a large amount of electrostatic and charge-transfer interactions, the influence of the polar solution on hydrogen bond is expected.11 The relative permittivity (ε) value of methanol is 32.613.19 In the MeOH solution, the monomers and complexes were stabilized. The polarity of solvent plays an important role in extraction, and this is due to the hydroxyl groups of genistein. The dipole moments of the genistein, MeOH–genistein (gas), and MeOH–genistein (in MeOH) were calculated to be 1.41 D, 0.59–3.56 D, and 1.63–4.22 D at the B3LYP-D3/cc-pVTZ level of theory, respectively. The MeOH–genistein conformers were stabilized by −45.0 to −40.6 kJ mol−1 in MeOH solution. Moreover, the red shifts of the OH-stretching transition were also much larger (100–501 cm−1) in MeOH solution than the ones (53–316 cm−1) in the gas phase. The changes of the OH bond demonstrate that the hydrogen bond is stronger in the MeOH solution and they become even shorter under the solvent effect.| Conformer | ΔEsa | Gas phase | MeOH solution | ||||
|---|---|---|---|---|---|---|---|
Δ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) b b |
Δr(OH)c | Dipole | Δb |
Δr(OH)c | Dipole | ||
| a ΔES = E(solvent) − E(gas).b Δ = monomer − dimer.c Δr(OH) = rdimer − rmonomer, is the change in the OH bond length upon complexation. |
|||||||
| MeOH–genistein (a) | −45.0 | 53 | 0.003 | 0.59 | 100 | 0.005 | 1.63 |
| MeOH–genistein (b) | −40.6 | 95 | 0.006 | 2.34 | 139 | 0.007 | 2.80 |
| MeOH–genistein (c) | −45.4 | 126 | 0.007 | 3.56 | 212 | 0.011 | 3.05 |
| MeOH–genistein (d) | −44.8 | 68 | 0.004 | 2.16 | 130 | 0.007 | 2.74 |
| MeOH–genistein (e) | −44.9 | 316 | 0.016 | 2.35 | 501 | 0.024 | 4.22 |
| MeOH–genistein (f) | −44.6 | 126 | 0.013 | 2.28 | 235 | 0.021 | 3.04 |
3.3 Nature of hydrogen bond: QTAIM analysis
The property of the Laplacian of the electron density ∇2ρ(BCP) can be used to identify the regions either concentration or depletion of the electron charge density, leading to catalogue the atomic interactions.45 Then, the atomic interactions can be classified into two classes: shared interactions and closed-shell interactions. The former ones are covalent and polar bonds, and they are formed by a charge density contraction towards the line of interaction linking the nuclei. Then, the electronic charge is concentrated and ∇2ρ(BCP) < 0.45 The closed-shell interactions are hydrogen bonds, ionic bonds, van der Waals' interactions, etc. These interactions are the charge density contractions towards each to the interacting nuclei. Therefore, the electronic charge is depleted and ∇2ρ(BCP) > 0.45The calculated topological parameters, such as electron density ρ(BCP), its Laplacian of charge density ∇2ρ(BCP) at bond critical points, changes of the atomic charge (Δq(H)) and energy (ΔE(H)) differences on the H atoms for MeOH–genistein (a–f) are given in Table 3. The AIM molecular graphs with BCPs and RCPs of the MeOH–genistein conformers are shown in Fig. 7. The hydrogen bond can be defined if values of the electron density ρ(BCP) and the Laplacian of charge density ∇2ρ(BCP) at bond critical points fall within 0.002–0.040 a.u. and 0.014–0.139 a.u., respectively.45,46 The calculated electron density properties show that the studied hydrogen bonds have low ρ(BCP) (0.020 to 0.038 a.u.), and positive Laplacian of charge density ∇2ρ(BCP) (0.073 to 0.095 a.u.). These properties indicate that these interactions are typical for closed-shell interactions as hydrogen bond. They demonstrate the electrostatic character of the interactions.
| Conformer | Δq(H) | ΔE(H) | ρ(BCP) | ∇2ρ(BCP) |
|---|---|---|---|---|
| a All values are in atomic unit (a.u.). | ||||
| MeOH–genistein (a) | 0.049 | 0.013 | 0.020 | 0.073 |
| MeOH–genistein (b) | 0.064 | 0.024 | 0.022 | 0.080 |
| MeOH–genistein (c) | 0.070 | 0.028 | 0.026 | 0.085 |
| MeOH–genistein (d) | 0.055 | 0.017 | 0.022 | 0.077 |
| MeOH–genistein (e) | 0.051 | 0.034 | 0.038 | 0.097 |
| MeOH–genistein (f) | 0.047 | 0.031 | 0.035 | 0.095 |
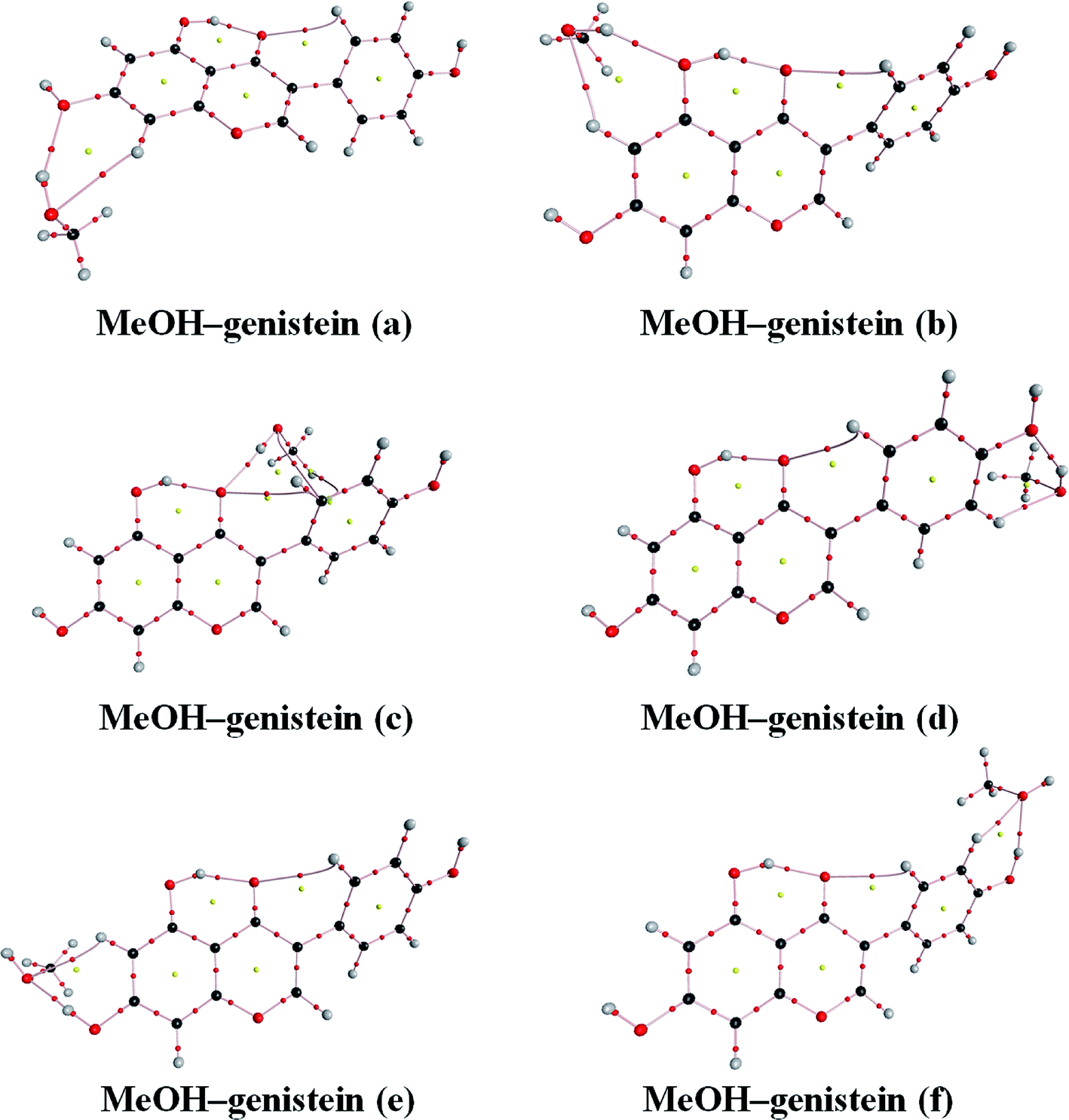 | ||
| Fig. 7 AIM graphic plots of the MeOH–genistein complexes obtained at the B3LYP-D3/cc-pVTZ level. The bond critical points and ring critical points are presented by the red and yellow balls, respectively. | ||
When two molecules form a hydrogen bonded complex, there will be a charge transfer (CT) from the hydrogen bond acceptor to the donor. This leads a charge decrease in the hydrogen atom involved in the hydrogen bond.47 The AIM atomic charge and energy differences on the H atoms (Δq(H) and ΔE(H)) upon complexation are listed in Table 3. The atomic charges at the H atoms (Δq(H)) are increased by 0.047–0.070 a.u. upon complexation. This causes the atomic energies rise up by 0.013–0.034 a.u. In our previous study on the carboxylic acid–sulfuric acid hydrogen bonded systems, a larger Δq(H) brings a greater red shift of the OH-stretching transitions.48
The nature of the hydrogen bond was further interpreted by the generalized Kohn–Sham energy decomposition analysis (GKS-EDA).30 The interaction energies were divided into six components, including electrostatic energy (ES), repulsion energy (REP), polarization energy (POL), exchange energy (EX), DFT correlation (CORR), and Grimme dispersion energy (DC). Table 4 summarizes the GKS-EDA results. The five components, namely, EX, ES, POL, DC and CORR, are attractive terms with negative values. These results show that the EX component has large negative values (−93.9 to −55.5 kJ mol−1). This implies that there is a great overlapping of molecular orbitals and consequently indicates strong orbital interactions in the studied system. The values of the ES components are larger than ones of POL, DC, and CORR. In general, the decomposition of the total interaction energy shows that the contributions of ES and EX to the attraction energies are very large. Meanwhile, the values of the REP components are positive, and this means that REP destabilizes the hydrogen bond. REP in the studied system has a negative contribution to the total interaction energy.
| Conformer | ES | EX | REP | POL | DC | CORR | INT |
|---|---|---|---|---|---|---|---|
| a All values are in kJ mol−1. | |||||||
| MeOH–genistein (a) | −30.6 | −55.5 | 93.2 | −9.4 | −12.3 | −6.7 | −21.3 |
| MeOH–genistein (b) | −41.1 | −65.9 | 111.5 | −13.1 | −12.6 | −7.3 | −28.6 |
| MeOH–genistein (c) | −43.1 | −81.3 | 136.6 | −15.6 | −21.3 | −9.4 | −34.2 |
| MeOH–genistein (d) | −35.5 | −57.0 | 96.4 | −11.4 | −10.9 | −6.3 | −24.7 |
| MeOH–genistein (e) | −58.6 | −93.9 | 158.8 | −18.7 | −18.0 | −10.4 | −40.7 |
| MeOH–genistein (f) | −57.1 | −85.9 | 150.3 | −24.0 | −12.1 | −9.2 | −38.0 |
4 Concluding remark
Genistein is poorly water soluble, and this affects its broad application in food and medicine. Genistein dissolves in polar organic solvent, but the solute interacts with solvent via hydrogen bonds. This influences the chemical and physical properties of the genistein. We have investigated the hydrogen bonding interaction between genistein and MeOH, and the solvation effect for the clusters formed. Genistein can act as either a hydrogen bond donor or acceptor to interact with the MeOH solvent. The binding energies of the formed clusters range from −32.8 to −16.2 kJ mol−1. The geometric parameters and topological analysis also confirm that genistein prefers to be a hydrogen bond donor rather than a hydrogen bond acceptor. The title systems were stabilized in the MeOH solution. The decomposition of the total interaction energy show that the contributions of ES and EX to the attraction interaction energies are very large. In addition, the more MeOH molecules surrounding the genistein molecule could give extra stability for the whole system. On the other hand, that genistein anion forms much more stable complexes with MeOH than the neutral genistein does.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by a grant from the Province Key Laboratory of Cereal Resource Transformation and Utilization, Henan University of Technology under grant number: PL2018010. We also thank the High Performance Computing Centre of Shandong University for providing GAUSSIAN 09 Revision E.01 software package and high-performance computation.References
- M. Messina and V. Messina, J. Am. Diet. Assoc., 1991, 91, 836–840 CAS.
- M. J. Messina, V. Persky, K. D. Setchell and S. Barnes, Nutr. Cancer, 1994, 21, 113–131 CrossRef CAS.
- J. L. Ingham, Progress in the Chemistry of Organic Natural Products, Springer Verlag GMBH, Vienna, Austria, 2012 Search PubMed.
- H. Wang and P. A. Murphy, J. Agric. Food Chem., 1994, 42, 1666–1673 CrossRef CAS.
- L. Coward, N. C. Barnes, K. D. R. Setchell and S. Barnes, J. Agric. Food Chem., 1993, 41, 1961–1967 CrossRef CAS.
- M. Fukutake, M. Takahashi, K. Ishida, H. Kawamura, T. Sugimura and K. Wakabayashi, Food Chem. Toxicol., 1996, 34, 457–461 CrossRef CAS.
- C. Spagnuolo, G. L. Russo, I. E. Orhan, S. Habtemariam, M. Daglia, A. Sureda, S. F. Nabavi, K. P. Devi, M. R. Loizzo, R. Tundis and S. M. Nabavi, Adv. Nutr., 2015, 6, 408–419 CrossRef CAS.
- Q. Wu, M. Wang and J. E. Simon, J. Chromatogr. A, 2003, 1016, 195–209 CrossRef CAS.
- M. Bustamante-Rangel, M. M. Delgado-Zamarreño, R. Carabias-Martínez and J. Domínguez-Álvarez, Anal. Chim. Acta, 2012, 709, 113–119 CrossRef CAS.
- A. Zafra-Gómez, A. Garballo, L. E. García-Ayuso and J. C. Morales, Food Chem., 2010, 123, 872–877 CrossRef.
- A. J. A. Aquino, D. Tunega, G. Haberhauer, M. H. Gerzabek and H. Lischka, J. Phys. Chem. A, 2002, 106, 1862–1871 CrossRef CAS.
- L. S. Hutabarat, H. Greenfield and M. Mulholland, J. Food Compos. Anal., 2001, 14, 43–58 CrossRef CAS.
- M. A. Rostagno, M. Palma and C. G. Barroso, Anal. Chim. Acta, 2004, 522, 169–177 CrossRef CAS.
- L. Y. Yoshiara, T. B. Madeira, F. Delaroza, J. B. da Silva and E. I. Ida, Int. J. Food Sci. Nutr., 2012, 63, 978–986 CrossRef CAS.
- H. Singh, S. Singh, A. Srivastava, P. Tandon, P. Bharti, S. Kumar and R. Maurya, Spectrochim. Acta, Part A, 2014, 120, 405–415 CrossRef CAS.
- H. Singh, S. Singh, A. Srivastava, P. Tandon, P. Bharti, S. Kumar, K. Dev and R. Maurya, J. Mol. Struct., 2017, 1130, 929–939 CrossRef CAS.
- S. Scheiner, Hydrogen Bonding: A Theoretical Perspective, Oxford University Press, New York, 1997 Search PubMed.
- Y.-Z. Zheng, G. Deng, R. Guo, D.-F. Chen and L.-M. Wu, J. Mol. Model., 2019, 25, 67 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision E.01, Gaussian, Inc., Wallingford, CT, USA, 2013 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef.
- S. Cheng, S. Tang, N. T. Tsona and L. Du, Sci. Rep., 2017, 7, 11310 CrossRef PubMed.
- Q. Zhang and L. Du, Comput. Theor. Chem., 2016, 1078, 123–128 CrossRef CAS.
- S. Li, H. G. Kjaergaard and L. Du, J. Environ. Sci., 2016, 40, 51–59 CrossRef.
- S. Tang and L. Du, Spectrochim. Acta, Part A, 2019, 217, 237–246 CrossRef CAS.
- S. Tang, N. T. Tsona and L. Du, Sci. Rep., 2018, 8, 1553 CrossRef.
- X. Jiang, N. T. Tsona, S. Tang and L. Du, Spectrochim. Acta, Part A, 2018, 191, 155–164 CrossRef CAS.
- V. Barone, M. Cossi and J. Tomasi, J. Comput. Chem., 1998, 19, 404–417 CrossRef CAS.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. Su, T. L. Windus, M. Dupuis and J. A. Montgomery, J. Comput. Chem., 1993, 14, 1347–1363 CrossRef CAS.
- P. Su and H. Li, J. Chem. Phys., 2009, 131, 014102 CrossRef.
- J. R. Lane, J. Contreras-Garcia, J.-P. Piquemal, B. J. Miller and H. G. Kjaergaard, J. Chem. Theory Comput., 2013, 9, 3263–3266 CrossRef CAS.
- R. Sekine, E. G. Robertson and D. McNaughton, Vib. Spectrosc., 2011, 57, 306–314 CrossRef CAS.
- M. Breton, G. Precigoux, C. Courseille and M. Hospital, Acta Crystallogr., Sect. B: Struct. Sci., 1975, 31, 921–923 CrossRef.
- E. J. Yearley, E. A. Zhurova, V. V. Zhurov and A. A. Pinkerton, J. Am. Chem. Soc., 2007, 129, 15013–15021 CrossRef CAS.
- J. S. Boeing, E. O. Barizão, B. C. E Silva, P. F. Montanher, V. de Cinque Almeida and J. V. Visentainer, Chem. Cent. J., 2014, 8, 48 CrossRef.
- P. Wang, N. Zhao and Y. Tang, J. Phys. Chem. A, 2017, 121, 5045–5055 CrossRef CAS.
- E. Arunan, G. R. Desiraju, R. A. Klein, J. Sadlej, S. Scheiner, I. Alkorta, D. C. Clary, R. H. Crabtree, J. J. Dannenberg, P. Hobza, H. G. Kjaergaard, A. C. Legon, B. Mennucci and D. J. Nesbitt, Pure Appl. Chem., 2011, 83, 1637–1641 CAS.
- M. Hippler, J. Chem. Phys., 2005, 123, 204311 CrossRef.
- S. Chung and M. Hippler, J. Chem. Phys., 2006, 124, 214316 CrossRef.
- S. Simon, M. Duran and J. J. Dannenberg, J. Phys. Chem. A, 1999, 103, 1640–1643 CrossRef CAS.
- M. Hippler, S. Hesse and M. A. Suhm, Phys. Chem. Chem. Phys., 2010, 12, 13555–13565 RSC.
- M. Sowa, K. Ślepokura and E. Matczak-Jon, J. Mol. Struct., 2014, 1076, 80–88 CrossRef CAS.
- A. P. Mazurek, L. Kozerski, J. Sadlej, R. Kawę, E. Bednarek, J. Sitkowski, J. C. Dobrowolski, J. K. Maurin, K. Biniecki, J. Witowska, P. Fiedor and J. Pachecka, J. Chem. Soc., Perkin Trans. 2, 1998, 1223–1230 RSC.
- Y. Zhang, B. Lou, Y. Huang, G. Zheng and Q. Lin, Z. Kristal. New Cryst. Struct., 2017, 232, 681 CAS.
- U. Koch and P. Popelier, J. Phys. Chem., 1995, 99, 9747–9754 CrossRef CAS.
- S. J. Grabowski, J. Phys. Org. Chem., 2004, 17, 18–31 CrossRef CAS.
- I. S. Bushmarinov, K. A. Lyssenko and M. Y. Antipin, Russ. Chem. Rev., 2009, 78, 283–302 CrossRef CAS.
- H. Zhao, Q. Zhang and L. Du, RSC Adv., 2016, 6, 71733–71743 RSC.
| This journal is © The Royal Society of Chemistry 2019 |