
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Functionalized polycarbonates via triphenylborane catalyzed polymerization-hydrosilylation†
Kori A. Andrea and
Francesca M. Kerton*
and
Francesca M. Kerton*
Department of Chemistry, Memorial University of Newfoundland, St. John's, NL A1B 3X7, Canada. E-mail: fkerton@mun.ca
First published on 23rd August 2019
Abstract
Triphenylborane catalyzes the copolymerization and terpolymerization of epoxides and CO2 to yield polycarbonates with excellent dispersity. Via assisted tandem catalysis, these materials could be hydrosilylated in a one-pot fashion yielding modified polymeric materials. Using only a few reagents, materials with glass transition temperatures ranging from 37–110 °C were obtained.
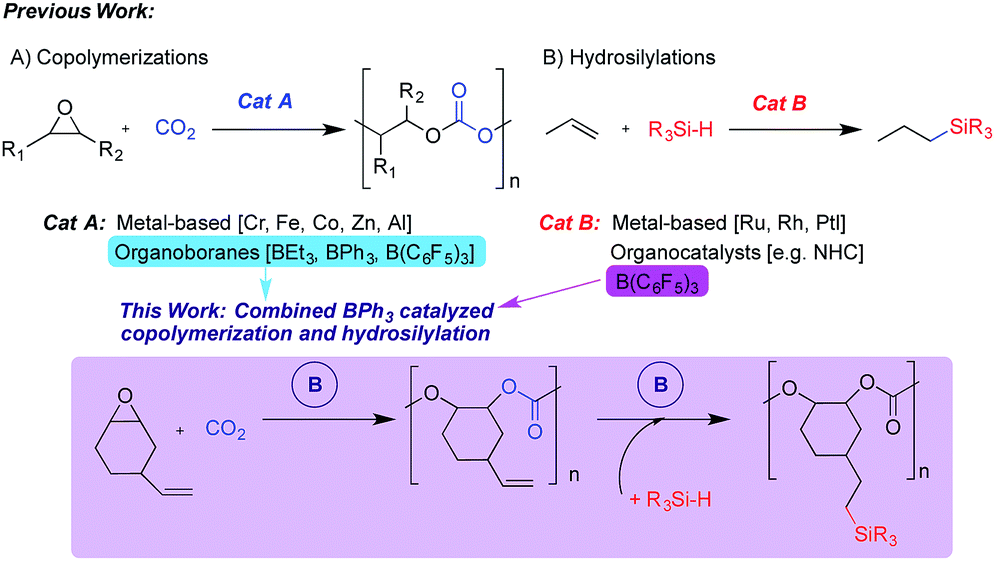
Transformation of carbon dioxide (CO2) into useful organic materials is important from an economic and environmental viewpoint.1,2 Specifically, the reaction of CO2 and epoxides can yield either cyclic carbonates or polycarbonates, with product selectivity relying on several factors such as temperature, pressure, substrate and catalyst design. The polycarbonate product is attractive as it paves a new road towards the development of new sustainable polymeric materials that may serve as alternatives to the traditional petroleum-based products that dominate society today.3–5 The use of catalysts that can incorporate a mixture of epoxide monomers into the final product has evolved in recent years, which can allow renewable functional epoxides to be incorporated into a biorenewable end product.6,7 Furthermore, such functional epoxides including unsaturated building blocks allow for subsequent modification and tailoring of the polymer and its properties. This has been achieved previously,8 for example, via olefin metathesis and thiol–ene crosslinking reactions.8–14
Copolymerization of epoxides and CO2 is usually facilitated by metal-based catalytic systems,6,15 but recently the use of organo- and non-metal catalysts has emerged,16 including two examples making use of organoboranes. The first used triethylborane to yield polycarbonates with high carbonate content when either propylene oxide (PO) or cyclohexene oxide (CHO) were used as the substrate.17 We recently reported the use of arylboranes, both triphenylborane (BPh3) and the more Lewis acidic tris(pentafluorophenyl)borane (BCF), as catalysts for the production of either cyclic carbonate or polycarbonate products with substrate dependent selectivity.18
Triarylboranes, particularly BCF, either alone or as a Frustrated Lewis Pair (FLP) or within a complex ion-pair are known to catalyze a broad range of reactions,19–24 including hydroelementations that possess enormous potential for production of chemicals in a sustainable manner.25,26 Specifically, hydrosilylation involves the addition of Si–H groups across C–C, C–O and C–N multiple bonds.27 As hydrosilylation of alkenes by BCF had been reported,21 along with our recent report of BPh3 catalyzed copolymerization of CO2 and vinylcyclohexene oxide (VCHO), we were motivated to combine these two reactions in one-pot to yield silylated-polycarbonates. Herein, we report the first example of an alkene hydrosilylation catalyzed by the less Lewis acidic BPh3. Building on our previous findings regarding the ability of BPh3 to produce perfectly alternating polycarbonates, we report sequential copolymerization-hydrosilylation in a one-pot manner via assisted tandem catalysis (Scheme 1). We anticipate that such processes can lead to CO2-derived polymers with tailorable physical properties including glass transition temperatures. Also, these polymers may show enhanced solubility in organic solvents, which will facilitate film-casting, and if some Si–H bonds remain, it may allow polymers to be attached to surfaces via covalent bonding or grafted to other macromolecular species to form more complex architectures.
 | ||
| Scheme 1 Catalytic copolymerization and hydrosilylation. | ||
The vinyl groups of the polyvinylcyclohexene carbonate (PVCHC) provide several potential routes to polymer modification, which could result in tuning of its physical and chemical properties. This has been done previously using methods such as thiol–ene click chemistry28 and metathesis.10,18 BCF is known to activate Si–H bonds and facilitate their addition across unsaturated substrates,21,29,30 whereas BPh3 has been studied to a lesser extent. We envisioned that BPh3 would be able to catalyze the addition of Si–H groups onto a vinyl-substituted polycarbonate. Therefore, we performed the following ‘one-pot’ sequence (Fig. 1): BPh3 was used to catalyze the copolymerization of VCHO and CO2, the CO2 was vented and phenyldimethylsilane added to the reaction mixture so the BPh3 present could then catalyze the hydrosilylation of the alkene within the polycarbonate. We did not attempt to perform the hydrosilylation reaction in the presence of CO2, or prior to or during the copolymerization, as BPh3 is able to hydrosilylate CO2 but does not react with propylene carbonate,31 and we presume other carbonates. We monitored the one-pot process via in situ IR spectroscopy. The formation of PVCHC was monitored via growth of the carbonate stretch at 1747 cm−1. We observed no induction period and signal saturation occurred within approximately 1 h. After 24 h, we cooled and depressurized the vessel before injecting a mixture of phenyldimethylsilane in dichloromethane and heating to 40 °C. A trial hydrosilylation reaction (NMR scale) on isolated PVCHC was successful at this temperature. However, for the one pot process after 4 days, no new bands were observed in the IR spectrum. Upon increasing the temperature to 60 °C, within hours we saw a decrease in intensity of bands at 2122 and 882 cm−1 (PhMe2SiH), and an increase in intensity of bands at 834 and 791 cm−1 demonstrating the successful addition of the silane across the alkene of the polycarbonate. The higher temperature for the one-pot process is likely needed to displace the Cl− anion from the boron centre and allow activation of the Si–H bond by the borane. Cl− is used as a co-catalyst in the CO2 epoxide copolymerization process and the NMR scale trial reaction was performed in the absence of PPNCl. The successful one-pot reaction was confirmed with 1H, 13C, HSQC and refocused INEPT 29Si NMR spectroscopy (Fig. S1–S6†), and integration of 1H NMR signals for the residual vinyl protons and the silyl protons (Si–(CH3)2, Si–ArH) showed 10% of the vinyl groups had been modified. In the refocused INEPT 29Si NMR spectrum of the product, a new signal appeared at δ = −1.26 ppm characteristic of a Si–C saturated bond cf. PhMe2SiH δ = −17.27 ppm. Gel permeation chromatography (GPC) traces show an increase in Mn for the product, while calculated Mark–Houwink–Sakurada (MHS) confirmation plots show an increased degree of branching in the product (a = 0.697 in PVCHC vs. silyl-PVCHC a = 0.479), further confirming successful functionalization (Fig. S7 and S8†).32 As anticipated the silyl-modified polymer exhibited a lower glass transition temperature (Tg) 71.5 °C compared with PVCHC, 99.0 °C. This Tg may possibly be further decreased if a larger proportion of vinyl groups are converted or a different silane employed. Polycarbonates with relatively low Tg includes commercially available polypropylene carbonate that finds applications in films and coatings. This one-pot copolymerization-silylation process is an example of assisted tandem catalysis,33 as the silane reagent triggers the mode of catalysis to change, and represents a new approach to functionalized polycarbonate.
 | ||
| Fig. 1 One-pot assisted tandem catalysis to yield silane modified polycarbonates. (A) One-pot catalytic formation of silylated-PVCHC. (B) Refocused INEPT 29Si NMR spectra of PhMe2SiH and product. (C) In situ IR spectra of reaction mixture over time. (D) Reaction profile obtained from in situ IR data. | ||
To obtain more examples of functional polycarbonates, while building upon past examples of mixed epoxide/CO2 terpolymerizations,34–38 we sought to investigate our BPh3/PPNCl catalytic system for similar activity. In our previous research, when propylene oxide (PO) was used as the substrate neither polypropylene oxide nor polypropylene carbonate (PPC) was formed.18 However, when an initial monomer mixture of 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 CHO:PO was used, we saw an incorporation ratio of 4:1 CHO:PO in the resulting polycarbonate i.e. 20% PPC linkages (Table 1, entry 1). Moving to a 10:90 CHO:PO monomer mixture, a terpolymer with 50% PPC linkages and a lower Tg, 37.3 °C, was obtained (Table 1, entry 2). From in situ IR monitoring, in addition to terpolymer, a notable amount of cyclic propylene carbonate formed. However, traces showed from a kinetic standpoint while the cyclic product formed quickly, once polymerization began there was no further cyclic formation (Fig. S13†). Instead the starting PO monomer continued to insert into the growing polymeric chain. When these ratios were reversed 90:10 CHO:PO (Table 1, entry 3), the polymer contained mostly PCHC linkages with a Tg similar to polycyclohexene carbonate. When CHO was replaced with VCHO in combination with PO (Table 1, entries 1 and 4), a larger proportion of PO was incorporated into the terpolymer. The BPh3/PPNCl system did not give cyclic or polymer product when glycidol was used (Table 1, entry 6). Allyl glycidyl ether (AGE) in the presence of CHO or VCHO (Table 1, entries 7 and 9) could be incorporated into terpolymers but only modest amounts of AGE were found in the resulting polymer. All obtained terpolymers were characterized by 1H and 13C NMR spectroscopy, GPC and DSC (Fig. S9–S26†). DOSY NMR spectroscopy confirmed the incorporation of both epoxides within the same polymeric chain. The terpolymers with alkene functionality (i.e. those containing VCHO and AGE) are attractive as they introduce the potential to further modify the polymers.
:50 CHO:PO was used, we saw an incorporation ratio of 4:1 CHO:PO in the resulting polycarbonate i.e. 20% PPC linkages (Table 1, entry 1). Moving to a 10:90 CHO:PO monomer mixture, a terpolymer with 50% PPC linkages and a lower Tg, 37.3 °C, was obtained (Table 1, entry 2). From in situ IR monitoring, in addition to terpolymer, a notable amount of cyclic propylene carbonate formed. However, traces showed from a kinetic standpoint while the cyclic product formed quickly, once polymerization began there was no further cyclic formation (Fig. S13†). Instead the starting PO monomer continued to insert into the growing polymeric chain. When these ratios were reversed 90:10 CHO:PO (Table 1, entry 3), the polymer contained mostly PCHC linkages with a Tg similar to polycyclohexene carbonate. When CHO was replaced with VCHO in combination with PO (Table 1, entries 1 and 4), a larger proportion of PO was incorporated into the terpolymer. The BPh3/PPNCl system did not give cyclic or polymer product when glycidol was used (Table 1, entry 6). Allyl glycidyl ether (AGE) in the presence of CHO or VCHO (Table 1, entries 7 and 9) could be incorporated into terpolymers but only modest amounts of AGE were found in the resulting polymer. All obtained terpolymers were characterized by 1H and 13C NMR spectroscopy, GPC and DSC (Fig. S9–S26†). DOSY NMR spectroscopy confirmed the incorporation of both epoxides within the same polymeric chain. The terpolymers with alkene functionality (i.e. those containing VCHO and AGE) are attractive as they introduce the potential to further modify the polymers.
| Entry | Epoxide A equiv. (%) | Epoxide B equiv. (%) | Monomer incorporationb (A:B) |
Mnc (g mol−1) | Đc | Tgd (°C) |
|---|---|---|---|---|---|---|
| a General reaction conditions unless otherwise indicated: total epoxide (A + B) (0.025 mol), PPNCl (0.124 mmol), BPh3 (0.124 mmol), 60 °C, 40 bar CO2. All obtained terpolymers contained >99% CO3 linkages, no evidence of polyether formation.b Determined by 1H NMR spectroscopy.c Đ, dispersity = Mw/Mn. Determined in THF by GPC equipped with a multiangle light-scattering detector.d Determined from DSC. | ||||||
| 1 | CHO (50) | PO (50) | 4:1 |
7990 | 1.03 | 79.9 |
| 2 | CHO (10) | PO (90) | 1:2 |
7310 | 1.06 | 37.3 |
| 3 | CHO (90) | PO (10) | 25:1 |
3760 | 1.03 | 110.2 |
| 4 | VCHO (50) | PO (50) | 1.5:1 |
9660 | 1.08 | 78.4 |
| 5 | CHO (50) | Glycidol (50) | No reaction | — | — | — |
| 6 | CHO (50) | AGE (50) | 1:0.1 |
6080 | 1.07 | 72.5 |
| 7 | CHO (50) | VCHO (50) | 1:0.5 |
5120 | 1.10 | 109.1 |
| 8 | VCHO (50) | AGE (50) | 2.2:1 |
7540 | 1.09 | 76.6 |
Building upon our initial polycarbonate hydrosilylation results, we then performed ‘one-pot’ hydrosilylation using the CHO/VCHO terpolymer (Table 1, entry 8) following similar procedures to those discussed above (Fig. 2). For the copolymerization step, a catalyst loading of 2.5 mol% BPh3 was used, which corresponds to 5 mol% BPh3 for the hydrosilylation step (as only 50% VCHO was present). After 24 h the vessel was cooled, depressurized and a mixture of diphenylsilane in dichloromethane was injected into the vessel. The mixture was then heated to 60 °C for 24 h. Via in situ IR spectroscopy, we observed a decreased in intensity of the silane bands (2144 and 845 cm−1), which plateaued after approx. 12 h and growth of a band at 830 cm−1 corresponding to the hydrosilylated product. The hydrosilylated polymer was further characterized by 1H, 13C, HSQC and refocused INEPT 29Si NMR spectroscopy (Fig. S27–S32†). From 1H NMR integration of signals for the residual vinyl protons and the aromatic protons (–SiPh2), 36% of vinyl groups had been modified. The refocused INEPT 29Si NMR spectrum of the product had a resonance at δ = −19.32 ppm, cf. δ = −33.18 ppm (Ph2SiH2). From both 1H and refocused INEPT 29Si NMR it is evident that only one Si–H bond added across the alkene of the terpolymer and hence no cross-linking occurred. From DSC data, a decline in Tg from 111.6 °C to 47.8 °C was observed for the silylated product (Fig. S33†). From GPC, an increase in molecular weight from 4.31 × 103 g mol−1 (Đ = 1.07) to 6.20 × 103 g mol−1 (Đ = 1.12) was observed as well as increased branching in MHS confirmation plots (Fig. S34†).
 | ||
| Fig. 2 One-pot assisted tandem catalysis to yield silylated-terpolymers. General reaction scheme (top) and three-dimensional plots obtained via in situ IR spectroscopy showing a decreased for silane bands and growth of product bands (bottom). | ||
Finally, we set out to evaluate the substrate scope of these transformations by evaluating the reactivity of a hydride terminated polydimethylsiloxane. For the hydride terminated polydimethylsiloxane (DMS-HO3) and PVCHC, the resulting polymer was characterized by 1H, 13C, refocused INEPT 29Si, and H–Si HMQC NMR spectroscopy (Fig. S35–S37†). Via integration of the 1H NMR spectrum, hydrosilylation has occurred to a similar extent to other hydrosilylations reported herein. We note that only one Si–H group per DMS-HO3 has undergone reaction and no cross-linking between polycarbonate chains was indicated by NMR and DSC data. IR spectra of the hydrosilylated-polycarbonate showed new bands at ν = 1013, 907 and 788 cm−1 corresponding to O–Si–O, Si–H and Si–CH3 groups respectively (Fig. S38†). From GPC, there was a moderate increase in molecular weight and no significant change in Đ. There was a decline in the slope of the MHS plot indicating a higher degree of branching in the final product (Fig. S39†). DSC analysis demonstrated a slight increase in Tg from 99.0 °C to 104.6 °C. The residual unreacted Si–H bonds in the functionalized polymer introduces further functionality potential. For example, BCF has been reported to catalyze the addition of Si–H bonds onto silica derived materials.39
In summary, we report the first example of BPh3 catalyzed hydrosilylation of perfectly alternating PVCHC in a tandem catalytic manner. These reactions were monitored by in situ IR spectroscopy, which demonstrated the addition of the Si–H bond across the pendent alkenes in the polymer. In an attempt to build new classes of polymeric materials, we showed the ability of BPh3 to catalyze the terpolymerization of CO2 and several epoxide combinations, yielding products with Tg values from 37.3 °C to 110.2 °C, which we could then functionalize in a similar one-pot manner as above. Finally, we evaluated the reactivity of a polymeric hydride terminated siloxane which can serve as a precursor for silica surface modification. Using the results in-hand, we will work towards developing sustainable surface functionalized materials in the future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Dr Celine Schneider (Memorial University) for assistance and discussions regarding NMR studies. NSERC of Canada, Memorial University, CFI and RDC-NL are thanked for operating and infrastructure grants. K. A. A is supported by a Vanier Canada Graduate Scholarship.Notes and references
- J. Artz, T. E. Müller, K. Thenert, J. Kleinekorte, R. Meys, A. Sternberg, A. Bardow and W. Leitner, Sustainable Conversion of Carbon Dioxide: An Integrated Review of Catalysis and Life Cycle Assessment, Chem. Rev., 2018, 118, 434–504 CrossRef CAS PubMed
.
- M. Aresta, A. Dibenedetto and A. Angelini, Catalysis for the Valorization of Exhaust Carbon: from CO2 to Chemicals, Materials, and Fuels. Technological Use of CO2, Chem. Rev., 2014, 114, 1709–1742 CrossRef CAS PubMed
- M. Cokoja, C. Bruckmeier, B. Rieger, W. A. Herrmann and F. E. Kühn, Transformation of Carbon Dioxide with Homogeneous Transition-Metal Catalysts: A Molecular Solution to a Global Challenge?, Angew. Chem., Int. Ed., 2011, 50, 8510–8537 CrossRef CAS PubMed
- Q. Liu, L. Wu, R. Jackstell and M. Beller, Using Carbon Dioxide as a Building Block in Organic Synthesis, Nat. Commun., 2015, 6, 5933 CrossRef PubMed
- M. North and P. Styring, Perspectives and Visions on CO2 Capture and Utilisation, Faraday Discuss., 2015, 183, 489–502 RSC
- G. Trott, P. K. Saini and C. K. Williams, Catalysts For CO2/epoxide Ring-opening Copolymerization, Philos. Trans. R. Soc., A, 2016, 374, 20150085 CrossRef PubMed
- O. Hauenstein, S. Agarwal and A. Greiner, Bio-based Polycarbonate as Synthetic Toolbox, Nat. Commun., 2016, 7, 11862 CrossRef CAS PubMed
- Y. Wang and D. J. Darensbourg, Carbon Dioxide-based Functional Polycarbonates: Metal Catalyzed Copolymerization of CO2 and Epoxides, Coord. Chem. Rev., 2018, 372, 85–100 CrossRef CAS
- S. Liu, X. Zhao, H. Guo, Y. Qin, X. Wang and F. Wang, Construction of Well-Defined Redox-Responsive CO2-Based Polycarbonates: Combination of Immortal Copolymer-ization and Prereaction Approach, Macromol. Rapid Commun., 2017, 38, 1600754 CrossRef PubMed
- A. E. Cherian, F. C. Sun, S. S. Sheiko and G. W. Coates, Formation of Nanoparticles by Intramolecular Cross-Linking: Following the Reaction Progress of Single Polymer Chains by Atomic Force Microscopy, J. Am. Chem. Soc., 2007, 129, 11350–11351 CrossRef CAS PubMed
- D. J. Darensbourg, W.-C. Chung, C. J. Arp, F.-T. Tsai and S. J. Kyran, Copolymerization and Cycloaddition Products Derived from Coupling Reactions of 1,2-Epoxy-4-cyclohexene and Carbon Dioxide. Postpolymerization Functionalization via Thiol–Ene Click Reactions, Macromolecules, 2014, 47, 7347–7353 CrossRef CAS
- G.-W. Yang, Y.-Y. Zhang, Y. Wang, G.-P. Wu, Z.-K. Xu and D. J. Darensbourg, Construction of Autonomic Self-Healing CO2-Based Polycarbonates via One-Pot Tandem Synthetic Strategy, Macromolecules, 2018, 51, 1308–1313 CrossRef CAS
- C. Martín and A. W. Kleij, Terpolymers Derived from Limonene Oxide and Carbon Dioxide: Access to Cross-Linked Polycarbonates with Improved Thermal Properties, Macromolecules, 2016, 49, 6285–6295 CrossRef
- T. Stößer, C. Li, J. Unruangsri, P. K. Saini, R. J. Sablong, M. A. R. Meier, C. K. Williams and C. Koning, Bio-derived Polymers for Coating Applications: Comparing Poly(Limonene Carbonate) and Poly(Cyclohexadiene Carbonate), Polym. Chem., 2017, 8, 6099–6105 RSC
- C. M. Kozak, K. Ambrose and T. S. Anderson, Copolymerization of Carbon Dioxide and Epoxides by Metal Coordination Complexes, Coord. Chem. Rev., 2018, 376, 565–587 CrossRef CAS
- R. R. Shaikh, S. Pornpraprom and V. D'Elia, Catalytic Strategies for the Cycloaddition of Pure, Diluted, and Waste CO2 to Epoxides under Ambient Conditions, ACS Catal., 2018, 8, 419–450 CrossRef CAS
- D. Zhang, S. K. Boopathi, N. Hadjichristidis, Y. Gnanou and X. Feng, Metal-Free Alternating Copolymerization of CO2 with Epoxides: Fulfilling “Green” Synthesis and Activity, J. Am. Chem. Soc., 2016, 138, 11117–11120 CrossRef CAS PubMed
- K. A. Andrea and F. M. Kerton, Triarylborane-Catalyzed Formation
of Cyclic Organic Carbonates and Polycarbonates, ACS Catal., 2019, 9, 1799–1809 CrossRef CAS
- D. W. Stephan, The Broadening Reach of Frustrated Lewis Pair Chemistry, Science, 2016, 354, aaf7229 CrossRef PubMed
- J. R. Lawson and R. L. Melen, Tris(pentafluorophenyl)-borane and Beyond: Modern Advances in Borylation Chemistry, Inorg. Chem., 2017, 56, 8627–8643 CrossRef CAS PubMed
- M. Rubin, T. Schwier and V. Gevorgyan, Highly efficient B(C6F5)3-catalyzed hydrosilylation of olefins, J. Org. Chem., 2002, 67, 1936–1940 CrossRef CAS PubMed
- M. Pérez, L. J. Hounjet, C. B. Caputo, R. Dobrovetsky and D. W. Stephan, Olefin Isomerization and Hydrosilylation Catalysis by Lewis Acidic Organofluorophosphonium Salts, J. Am. Chem. Soc., 2013, 135, 18308–18310 CrossRef PubMed
- M. H. Holthausen, M. Mehta and D. W. Stephan, The Highly Lewis Acidic Dicationic Phosphonium Salt: [(SIMes)PFPh2][B(C6F5)4]2, Angew. Chem., Int. Ed., 2014, 53, 6538–6541 CrossRef CAS PubMed
- A. Simonneau and M. Oestreich, 3-Silylated Cyclohexa-1,4-dienes as Precursors for Gaseous Hydrosilanes: The B(C6F5)3-Catalyzed Transfer Hydrosilylation of Alkenes, Angew. Chem., Int. Ed., 2013, 52, 11905–11907 CrossRef CAS PubMed
- M. Oestreich, Transfer Hydrosilylation, Angew. Chem., Int. Ed., 2016, 55, 494–499 CrossRef CAS PubMed
- X. Zeng, Recent Advances in Catalytic Sequential Reactions Involving Hydroelement Addition to Carbon-Carbon Multiple Bonds, Chem. Rev., 2013, 113, 6864–6900 CrossRef CAS PubMed
- Y. Nakajima and S. Shimada, Hydrosilylation Reaction of Olefins: Recent Advances And Perspectives, RSC Adv., 2015, 5, 20603–20616 RSC
- D. J. Darensbourg and Y. Wang, Terpolymerization of Propylene Oxide and Vinyl Oxides With CO2: Copolymer Cross-Linking and Surface Modification via Thiol–Ene Click Chemistry, Polym. Chem., 2015, 6, 1768–1776 RSC
- A. Berkefeld, W. E. Piers and M. Parvez, Tandem Frustrated Lewis Pair/Tris(Pentafluorophenyl)Borane-Catalyzed Deoxygenative Hydrosilylation of Carbon Dioxide, J. Am. Chem. Soc., 2010, 132, 10660–10661 CrossRef CAS PubMed
- L. C. Wilkins, J. L. Howard, S. Burger, L. Frentzel-Beyme, D. L. Browne and R. L. Melen, Exploring Multistep Continuous-Flow Hydrosilylation Reactions Catalyzed by Tris(Pentafluorophenyl)Borane, Adv. Synth. Catal., 2017, 359, 2580–2584 CrossRef CAS
- D. Mukherjee, D. F. Sauer, A. Zanardi and J. Okuda, Selective Metal-Free Hydrosilylation of CO2 Catalyzed by Triphenylborane in Highly Polar, Aprotic Solvents, Chem.–Eur. J., 2016, 22, 7730–7733 CrossRef CAS PubMed
- S. Mori and H. G. Barth, Size Exclusion Chromatography, Springer-Verlag, Berlin-Heidelberg-New York, 1999 Search PubMed
- D. E. Fogg and E. N. dos Santos, Tandem Catalysis: A Taxonomy and Illustrative Review, Coord. Chem. Rev., 2004, 248, 2365–2379 CrossRef CAS
- H. Li and Y. Niu, Alternating Copolymerization of CO2 with Propylene Oxide and Terpolymerization with Aliphatic Epoxides by Bifunctional Cobalt Salen Complex, Polym. J., 2010, 43, 121–125 CrossRef
- J. E. Seong, S. J. Na, A. Cyriac, B. W. Kim and B. Y. Lee, Terpolymerizations of CO2, Propylene Oxide, and Various Epoxides Using a Cobalt(III) Complex of Salen-Type Ligand Tethered by Four Quaternary Ammonium Salts, Macromolecules, 2010, 43, 903–908 CrossRef CAS
- G. P. Wu, P. X. Xu, Y. P. Zu, W. M. Ren and X. B. Lu, Cobalt(III)-Complex-Mediated Terpolymerization of CO2, Styrene Oxide, and Epoxides with an Electron-Donating Group, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 874–879 CrossRef CAS
- D. J. Darensbourg, R. R. Poland and A. L. Strickland, (Salan)CrCl, An Effective Catalyst for the Copolymerization and Terpolymerization of Epoxides and Carbon Dioxide, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 127–133 CrossRef CAS
- K. Deng, S. Wang, S. Ren, D. Han, M. Xiao and Y. Meng, A Novel Single-Ion-Conducting Polymer Electrolyte Derived from CO2-Based Multifunctional Polycarbonate, ACS Appl. Mater. Interfaces, 2016, 8, 33642–33648 CrossRef CAS PubMed
- N. Moitra, S. Ichii, T. Kamei, K. Kanamori, Y. Zhu, K. Takeda, K. Nakanishi and T. Shimada, Surface Functionalization of Silica by Si–H Activation of Hydrosilanes, J. Am. Chem. Soc., 2014, 136, 11570–11573 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Detailed information on experimental procedures and characterization data. See DOI: 10.1039/c9ra05947d |
| This journal is © The Royal Society of Chemistry 2019 |