
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNovel camphor-based pyrimidine derivatives induced cancer cell death through a ROS-mediated mitochondrial apoptosis pathway†
Yan Zhanga,
Yunyun Wanga,
Yuxun Zhaoa,
Wen Gu ac,
Yongqiang Zhu*b and
Shifa Wang*ac
ac,
Yongqiang Zhu*b and
Shifa Wang*ac
aCollege of Chemical Engineering, Nanjing Forestry University, Nanjing, Jiangsu 210037, People's Republic of China. E-mail: wangshifa65@163.com; Fax: +86 25 85427812; Tel: +86 25 85427812
bJiangsu Chia Tai Fenghai Pharmaceutical Co. Ltd, Nanjing, 210046, P. R. China
cCo-Innovation Center of Efficient Processing and Utilization of Forest Resources, Nanjing Forestry University, Nanjing, 210037, P. R. China
First published on 19th September 2019
Abstract
A series of novel camphor-based pyrimidine derivatives (3a–3x) have been synthesized; their structures were determined by using conventional methods and compound 3f was further confirmed through single crystal XRD analysis. The cytotoxic activity of the target compounds against a panel of human normal (GES-1) and cancer cell lines (MDA-MB-231, RPMI-8226, A549) was evaluated by MTS assay. Here we found that compound 3f exhibited the strongest anti-tumor activity, comparable to that of etoposide, and had much lower cytotoxicity to normal GES-1 cells (IC50 > 50 μM) than the reference drug (IC50 = 8.89 μM). Subsequent mechanism studies in MDA-MB-231 cells revealed that compound 3f caused G0/G1 phase arrest and apoptosis in a dose dependent manner. Moreover, the loss of mitochondrial membrane potential and enhancement of cellular ROS levels were also observed upon 3f treatment, which indicated that 3f exerted cytotoxic activity by a ROS-mediated mitochondrial apoptosis pathway. This result was confirmed by a significant increase in the expression of pro-apoptotic proteins Bax, cytochrome C and caspase-3, and downregulation of anti-apoptosis protein Bcl-2. Overall, 3f can be adopted for further investigation in the development of antitumor agents based on natural products.
1. Introduction
Cancer is one of leading causes of death globally. According to the statistics released by the World Health Organization (WHO), millions of new cancer patients are diagnosed each year worldwide.1 Although there has been steady progress made in reducing the mortality of cancer, the successful treatment of cancer remains a challenge. Mostly, therapeutic effectiveness of anti-cancer drugs is encumbered by some adverse side effects including high toxicity and drug resistance. Therefore, it is urgent to search for effective and safe agents for cancer treatment.Recently, it has become increasingly evident that cancer therapy is mainly realized through the induction of apoptosis.2–4 Hence, developing reliable anticancer drugs, which can successfully induce apoptosis, has become the new focus of anti-tumor research. It's well-known that there are two major semi-interdependent pathways contributing to apoptosis: the death receptor pathway and the mitochondria apoptotic pathway.5 In the latter case, an early trigger of the apoptotic pathway is an increase in the permeability of the mitochondria membrane which is defined as sudden loss of the membrane potential (ΔΨm), causing release of apoptogenic factors including cytochrome C.6 Under normal circumstances, cytochrome C is bound to the cardiolipin from inner mitochondria membrane, which is susceptible to reactive oxygen species (ROS).7 Thus, the level of ROS could affect the dissociation of cytochrome C from the mitochondrial membrane, and further mediate the mitochondria apoptosis. Additionally, the release of cytochrome C is controlled by the expression of Bcl-2 family proteins that contains pro-apoptotic proteins (Bcl-2, Bcl-xL, etc.) and anti-apoptotic proteins (Bax, Bak, etc.).8 Upon entering into the cytosol from mitochondria, cytochrome C would activate a cascade of caspases – a family of killer proteases, and eventually, result in cell death.9
The majority of the commercially available drugs are built on heterocyclic scaffolds and these chemical fragments are responsible for desired pharmacological activity.10 Pyrimidine, a six membered heterocyclic ring with nitrogen atom, exhibits various pharmacological activities such as anticancer,11–13 antimicrobial,14,15 antiviral,16–18 anti-tubercular,19,20 anti-inflammatory,21,22 antimalarial,23,24 anti-diabetic25 and antipyretic.26 Among these diverse pyrimidine derivatives, amino-pyrimidine plays a vital role in the therapeutic activities against cancers, meanwhile, a large number of studies have been reported in recent years.27–30 Natural compounds are the most promising source for the synthesis of biologically active substances in medicinal chemistry. Camphor, as one of the important monoterpenoids, is widespread in plants and has been used as chiral, enantiopure starting materials.31–33 Moreover, camphor-based derivatives exhibit a number of biological activities. For instance, N-arylpiperazine camphor sulfonamides were discovered as novel CXCR3 antagonists,34 camphor-based cyanopyrrolidines were used as inhibitors for the treatment of type 2 diabetes mellitus,35 the camphane-based amides presented the promising anti-mycobacterial activity.36 Aliphatic and alicyclic camphor imines showed significant activity against influenza virus H1N1,37 the platinum(II) complex possessing camphoryl had antiproliferative activity.38 From the view of the chemical structure, camphor is a small molecule with chirality and rigidity property which may bind to some active site, together with its low cost and convenience to transform into synthetically useful derivatives, it can serve as a good building block of bioactive leading compounds.
In view of the above-mentioned facts, it was envisaged that combining these two pharmacological scaffolds would construct novel molecular templates which are likely to show interesting biological properties. Based on the previous studies of anticancer drug in our research team, in this study, we coupled amino-pyrimidine fragments to camphor scaffold by a cyclization reaction to synthesize 24 novel camphor-based pyrimidine derivatives. Besides, we evaluated the cytotoxic activities of the target compounds by MTS in three different human-derived cancer cell lines including breast cancer cells (MDA-MB-231), multiple myeloma cells (RPMI-8226) and non-small cell lung cancer cells (A549), and explored the possible preliminary mechanism of the most potent compound 3f in MDA-MB-231 cells.
2. Results and discussion
2.1. Synthesis and characterization
With commercially available camphor 1 as the starting material, a series of pyrimidine derivatives were synthesized via two simple steps, following the route outlined in Scheme 1. Initially, aldol condensation of the compound 1 with various substituted aromatic aldehydes was carried out to prepare intermediates 2a–2x. Subsequently, without purification, intermediates 2a–2x reacted respectively with guanidine hydrochloride under base-catalyzed cyclization conditions to obtain target compounds 3a–3x in excellent yields. All desired pyrimidine analogs were purified by recrystallization and characterized by 1H NMR, 13C NMR and HRMS analysis. As exemplified with the 1H NMR analysis of 4-(4-chlorophenyl)-8,9,9-trimethyl-5,6,7,8-tetrahydro-5,8-methanoquinazolin-2-amine (3f), 1H NMR spectra revealed appearance of NH2 signal at δ 5.27 ppm, meanwhile, two symmetric doublets at δ 7.41–7.73 ppm were corresponding to aromatic protons. The 13C NMR spectrum of 3f displayed all characteristic peaks corresponding to molecular structure. For the pyrimidine ring, three aromatic carbons adjacent to N atom at δ 182.11, 161.75 and 154.87 ppm were found, respectively. The presence of two high peaks at δ 128.67 and 129.63 ppm confirmed the para-substituted structure of p-chlorophenyl moiety. Besides, some representative spectra are given in ESI.†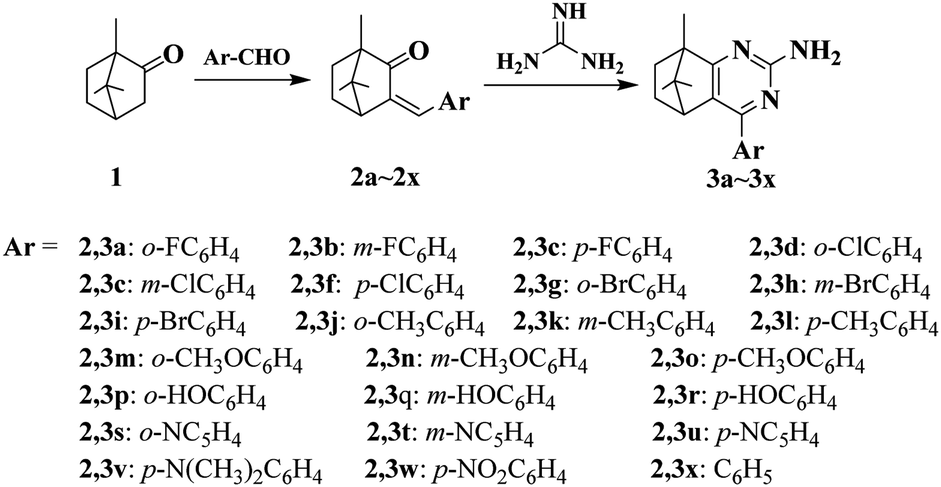 | ||
| Scheme 1 Synthesis of compounds 3a–3x. | ||
2.2. Crystal structure of the compound 3f
The single crystal of 3f was grown from chloroform solvent by the slow evaporation at room temperature. The compound 3f belongs to triclinic system with space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 6.9895(2) Å, b = 8.6676(3) Å, c = 13.6291(5) Å, α = 76.606(2)°, β = 81.336(2)°, γ = 79.948(2)°, Z = 2, V = 785.64(5) Å3. Molecular structure of compounds 3f together with the atom labeling scheme is given in Fig. 1, and the crystal data and structure refinement details are summarized in Table S1.† Crystallographic data of the compound 3f was deposited with the Cambridge Crystallographic Data Centre (CCDC) No 1838718.
, a = 6.9895(2) Å, b = 8.6676(3) Å, c = 13.6291(5) Å, α = 76.606(2)°, β = 81.336(2)°, γ = 79.948(2)°, Z = 2, V = 785.64(5) Å3. Molecular structure of compounds 3f together with the atom labeling scheme is given in Fig. 1, and the crystal data and structure refinement details are summarized in Table S1.† Crystallographic data of the compound 3f was deposited with the Cambridge Crystallographic Data Centre (CCDC) No 1838718.
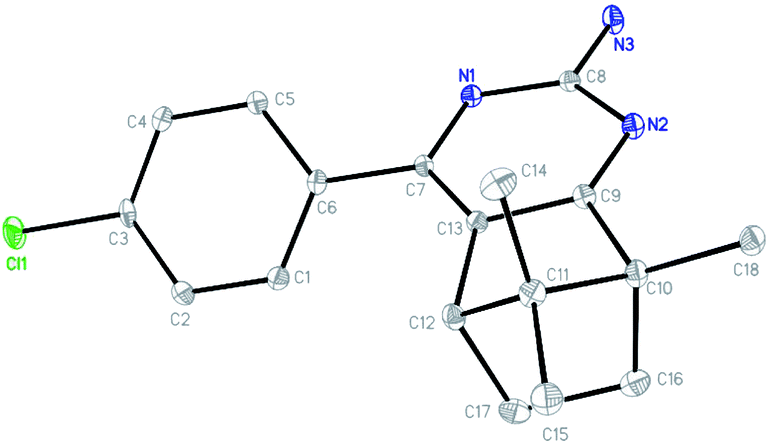 | ||
| Fig. 1 X-ray crystal structure of compound 3f. | ||
2.3. Biological evaluation of compounds assay
All target compounds were appraised for the anticancer activities against three human malignant cell lines (lung adenocarcinoma A549, breast cancer MDA-MB-231, multiple myeloma RPMI-8226) by using MTS assay. The preliminary screening results were presented graphically in Fig. 2.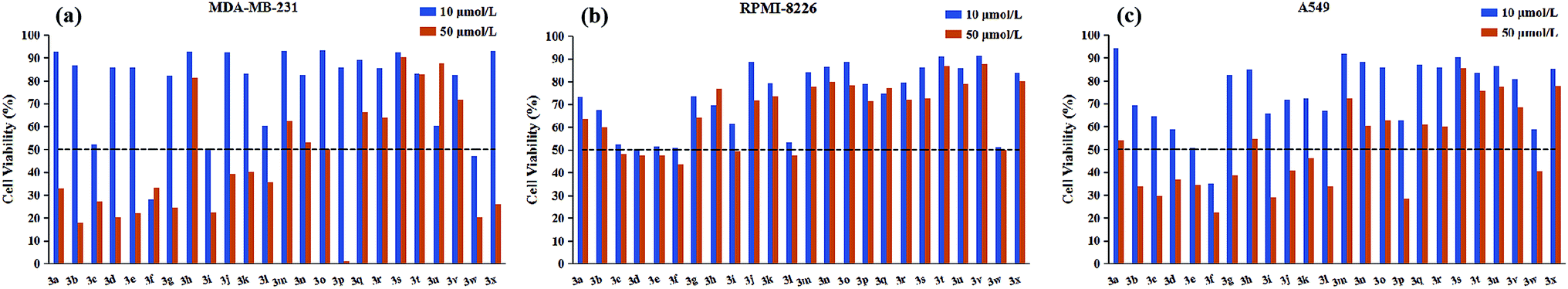 | ||
| Fig. 2 The cell viability of synthesized compounds against (a) MDA-MB-231 (b) RPMI-8226 (c) A549 cancer cell line at testing concentrations of 10 μM and 50 μM. The dashed line across the graph represents the threshold percentage for further IC50 determination. Cell viability was measured by MTS assay. | ||
As depicted in Fig. 2, it is noticeable that most of compounds displayed little cytotoxic effect against all tested cancer cell lines at testing concentrations (50 μM and 10 μM), in contrast, only several compounds showed strong growth inhibition (above 50% inhibition). Interestingly, for RPMI-8226 cell line, there is no obvious difference in the growth inhibition with the decrease of testing concentration from 50 μM to 10 μM, but this trend was not observed in the other two cell lines. On the other hand, considering the inhibitory selectivity of designed compounds on selected cancer cell lines, these pyrimidine derivatives present a preferable MDA-MB-231 targeting structure because the majority of tested analogs showed higher MDA-MB-231 inhibition compared with their respective activity on the other two cancer cell lines. In order to gain better insight into the role of different structural functionalities on cytotoxic effect, IC50 determination study was performed in triplicate and the results of the mean values were summarized in Table 1, in which etoposide was used as a positive control.
| Compound | Ar | IC50 (μM) | |||
|---|---|---|---|---|---|
| MBA-MD-231 | RPMI-8226 | A549 | GES-1 | ||
| 3a | o-FC6H4 | 32.85 ± 0.31 | >50 | >50 | >50 |
| 3b | m-FC6H4 | 28.56 ± 0.26 | >50 | 24.35 ± 0.01 | >50 |
| 3c | p-FC6H4 | 12.59 ± 0.04 | 9.30 ± 0.34 | 12.63 ± 0.02 | >50 |
| 3d | o-ClC6H4 | 25.48 ± 0.39 | 11.42 ± 0.18 | 16.93 ± 0.50 | >50 |
| 3e | m-ClC6H4 | 23.26 ± 0.44 | 10.36 ± 0.35 | 10.63 ± 0.09 | >50 |
| 3f | p-ClC6H4 | 5.00 ± 0.14 | 8.64 ± 0.24 | 7.65 ± 0.21 | >50 |
| 3g | o-BrC6H4 | 18.28 ± 0.26 | >50 | 23.32 ± 0.03 | >50 |
| 3h | m-BrC6H4 | >50 | >50 | >50 | >50 |
| 3i | p-BrC6H4 | 8.66 ± 0.17 | 18.94 ± 0.08 | 16.45 ± 0.04 | >50 |
| 3j | o-CH3C6H4 | 26.20 ± 0.40 | >50 | 22.54 ± 0.15 | >50 |
| 3k | m-CH3C6H4 | 20.00 ± 0.09 | >50 | 17.97 ± 0.18 | >50 |
| 3l | p-CH3C6H4 | 15.68 ± 0.14 | 13.86 ± 0.09 | 15.32 ± 0.09 | >50 |
| 3m | o-CH3OC6H4 | >50 | >50 | >50 | >50 |
| 3n | m-CH3OC6H4 | >50 | >50 | >50 | >50 |
| 3o | p-CH3OC6H4 | >50 | >50 | >50 | >50 |
| 3p | o-HOC6H4 | 16.28 ± 0.26 | >50 | 13.98 ± 0.16 | >50 |
| 3q | m-HOC6H4 | >50 | >50 | >50 | >50 |
| 3r | p-HOC6H4 | >50 | >50 | >50 | >50 |
| 3s | o-NC5H4 | >50 | >50 | >50 | >50 |
| 3t | m-NC5H4 | >50 | >50 | >50 | >50 |
| 3u | p-NC5H4 | >50 | >50 | >50 | >50 |
| 3v | p-N(CH3)2C6H4 | >50 | >50 | >50 | >50 |
| 3w | p-NO2C6H4 | 6.21 ± 0.14 | 9.58 ± 0.11 | 10.84 ± 0.09 | >50 |
| 3x | p-C6H5 | 29.88 ± 0.16 | >50 | >50 | >50 |
| Etoposide | — | 3.97 ± 0.45 | 1.92 ± 0.17 | 10.89 ± 0.79 | 8.89 ± 0.70 |
As illustrated with Table 1, the majority of compounds (3a–3i) with halogen group showed improved inhibitory activity, especially, of which compound 3f bearing chlorine atom was identified as the most promising candidates due to their lowest overall IC50 values (ranging from 5.00 to 8.64 μM) comparable to etoposide. Meanwhile, introduction of nitro group on the phenyl ring (3w) also resulted in good cytotoxicity with IC50 values similar to compound 3f. However, the substituents (3j–3r and 3v) with the electron-donating group showed decreased or complete loss of activity. Besides, the replacement of phenyl ring with pyridine ring (3s–3u) also largely weakened the activity against three cancer cell lines. These results indicated that the electron deficiency of the phenyl ring is a favorable feature to increase anticancer potential. In addition to substituting functional group, substitution position was also found to play a role in altering the cytotoxicity of synthesized compounds. Close observation of results from Table 1, para-substitution of phenyl ring (3c, 3f, 3i, 3l) exerted the most potent activity compared to their respective ortho- and meta-substituted analogs regardless to the substituting functional groups, except 3p which dramatically increased the potency in two solid tumor cell lines (A549 and MDA-MB-231). On top of this, meta-substituted derivatives (3b, 3e, 3k) performed better in reducing cancer cells viability than that of ortho-substitutions (3a, 3d, 3j), but the trend was reversed when only meta-brominated compound 3h was being considered. According to above findings, we can conclude that the tested pyrimidine derivatives with phenyl ring bearing electron withdrawing group at p-position have a positive influence on antitumor activity. Furthermore, all compounds were examined for possible toxicity against GES-1 (normal human gastric epithelial cell line). We found that none of compounds showed significant toxicity toward GES-1, whereas the reference compound (etoposide) exhibited severe cytotoxicity (IC50 = 8.89 μM).
2.4. Cell cycle assay
The cytotoxicity results, which were discussed earlier, suggest 3f is the most potent compound, especially in MDA-MB-231 cells. The perturbation of cell cycle is closely associated to the cytotoxicity, to determine whether 3f might trigger the arrest of cell cycle, a flow-cytometric analysis of MDA-MB-231 cells stained with propidium iodide (PI) was conducted. After the cells were treated with various concentrations of 3f (2.5, 5 and 10 μM) for 72 h, significant alternations in the cell phases were observed. In Fig. 3, compared with 56.86% in the control, the percentage of cells in the G0/G1 phase after treatment with 2.5, 5, 10 μM of compound 3f steadily increased to 64.89%, 78.02%, and 81.72%, accompanied by decreasing the proportion of cells at G2/M and S phase of cell cycle. Therefore, these results illustrated that 3f induced an obvious G0/G1 arrest in a concentration-dependent manner.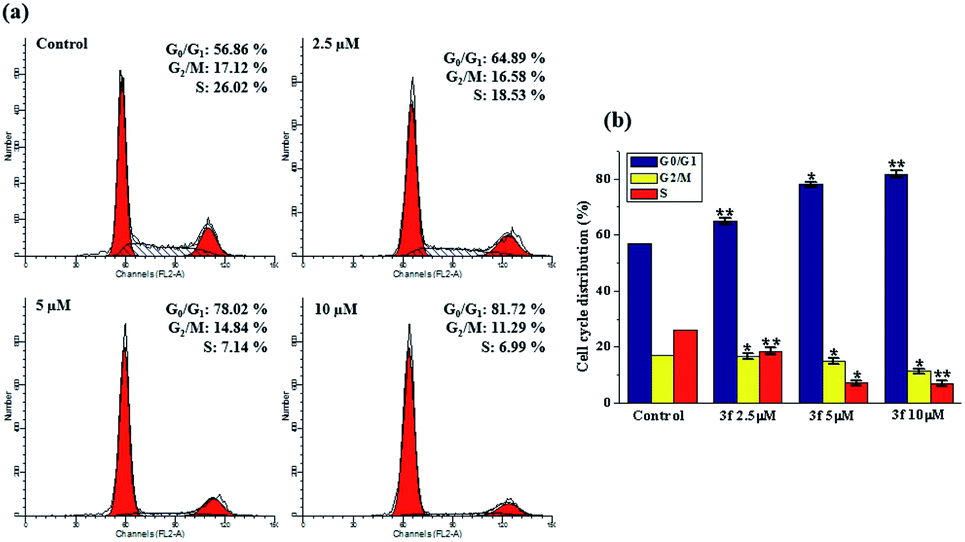 | ||
| Fig. 3 Influence of compound 3f on cell cycle distribution in MDA-MB-231 cells. (a) Cells were treated with different concentrations (2.5, 5 and 10 μM) of compound 3f for 72 h, which then were stained with propidium iodide (PI), and the DNA content were analyzed by flow cytometry. (b) Percentages of cells in various phases of the cell cycle. These data are from three independent experiments. *p < 0.05; **p < 0.01, verse the negative control. | ||
2.5. Apoptosis analysis
To get further insight into the mechanism of cell death induced by compound 3f, apoptosis in treated MDA-MB-231 cells was quantified by the Annexin V-APC/7-AAD double staining flow cytometric assay, in which Annexin V may specifically bind to the early apoptosis marker phosphatidylserine (PS) exposed on the outside of the cell (green fluorescence) and 7-AAD (7-amino-actinomycin D) can stain DNA and enter only dead and late apoptotic cells (red fluorescence). As shown in dot plot (Fig. 4), a high percentage of the control (untreated) MDA-MB-231 cells were negative for both Annexin V and 7-AAD (97.4%). After the completion of 72 h incubation with 2.5 μM compound 3f, a little amount of cell lines (3.78%) were in early stage of the apoptosis, and 5.69% of cancer cells were appeared in late apoptosis. A similar pattern was observed in MDA-MB-231 cells treated with 5 μM of compound 3f as well. Exposure of MDA-MB-231 cells to 10 μM of 3f resulted in significant increase in percentage of late apoptotic cells by 38.4%, in contrast, caused no changes in the levels of early apoptotic cells. In a word, the total (early and late) ratios of apoptosis enhanced from 9.47% to 57.9%, indicating that anti-proliferative effect of 3f depended on apoptosis.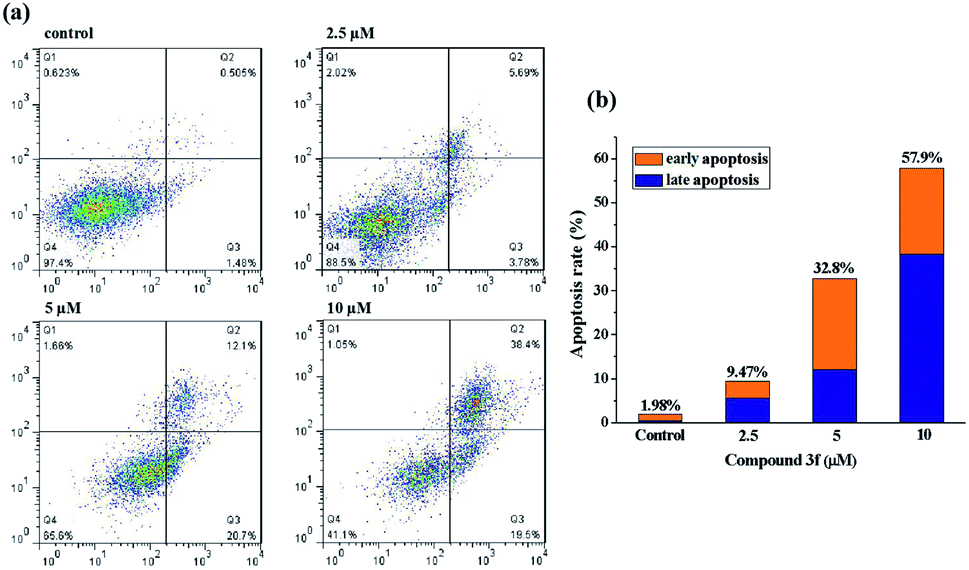 | ||
| Fig. 4 Compound 3f induced apoptosis in MDA-MB-231 cells. (a) Flow cytometry analysis of MDA-MB-231 cells were treated with compound 3f (2.5, 5 and 10 μM) for 72 h, respectively, and stained with Annexin V-APC/7-AAD. (b) Quantitative analysis of apoptotic cells in histogram. These data are from three independent experiments. | ||
2.6. Intracellular ROS level
Reactive oxygen species (ROS) are recognized as a pivotal factor in intracellular signaling and regulation. Cell growth and proliferation are encouraged with a condition of a modest rise in intracellular ROS, on the other hand, the elevation of ROS is consequent to disruption of intracellular redox homeostasis and inhibition of biomacromolecules function, resulting in promoting cell death. Since cancer cells are more sensitive to enhanced intracellular ROS than the nontransformed cells, the regulation of intracellular ROS levels could be exploited for selective therapeutic approach of cancer. To determine whether the anticancer effect of active compound 3f is involved in ROS-mediated therapy, the level of ROS in MDA-MB-231 cells treated with compound 3f at 10 μM concentration increased by almost 12 times (63.4%) in comparison to the control group (5.13%) (Fig. 5). However, the promoted intracellular ROS accumulation induced by 3f was distinctly suppressed by N-acetyl cysteine (NAC), which is one of the ROS scavengers. On the basis of these results, it clearly indicated that compound 3f can modulate the generation of intracellular ROS, and at least partly, explain the anticancer activity of compound 3f.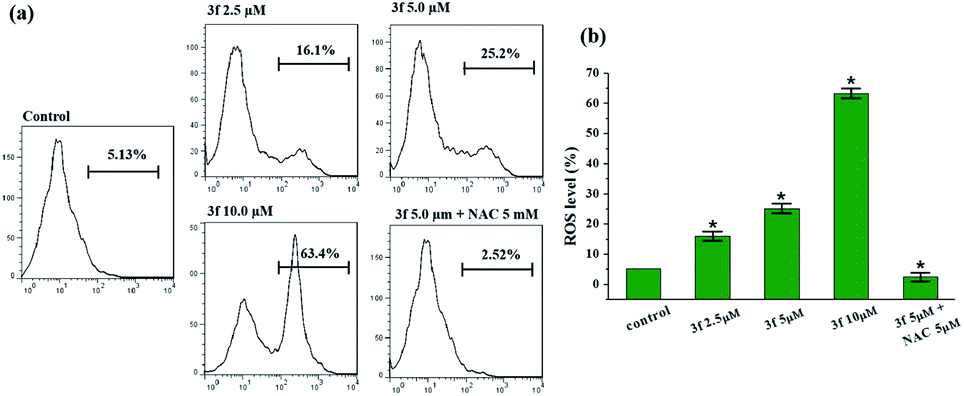 | ||
| Fig. 5 Effect of compound 3f on ROS production. (a) Analysis of the ROS levels by flow cytometry after the MDA-MB-231 cells were treated with compound 3f at indicated concentrations or in presence of NAC for 72 h and stained with DCFH-DA. (b) Quantification of the flow cytometric results. These data are from three independent experiments. *p < 0.05 verse the negative control. | ||
2.7. Mitochondrial membrane potential (MMP) analysis
It's well-known that there are two major semi-interdependent routes contributing to apoptosis. One is initiated by ligand binding to the death receptors at the cell surface (extrinsic apoptotic pathway), the other involves mitochondria (intrinsic apoptotic pathway). In the latter case, an early trigger of the apoptotic pathway is an increase in the permeability of the outer membrane, which leads to a preliminary reduction of the mitochondrial membrane potential (ΔΨm). Hence, in order to understand the mechanism of apoptosis, we used JC-1 as a fluorescent probe, a liquid crystal-forming cationic dye which can reversibly change its color from red to green as membrane potential decreases, to observe the effect of compound 3f on ΔΨm. The results are presented in Fig. 6, in the control, the percentage of green fluorescent cells is 3.9%, whereas MDA-MB-231 cells were exposed to 3f at different concentration (2.5 μM, 5 μM and 10 μM) for 72 h, there is a gradual increase of the percentage of cells with a collapsed ΔΨm (from 3.9% to 10.4%, 33.6%, 53.2%). Based on such study, we concluded that the compound 3f could mediate perturbation of the mitochondrial membrane integrity with a consequent concentration-dependent collapse of ΔΨm, and its antiproliferative activities might be associated with mitochondrial apoptosis pathway.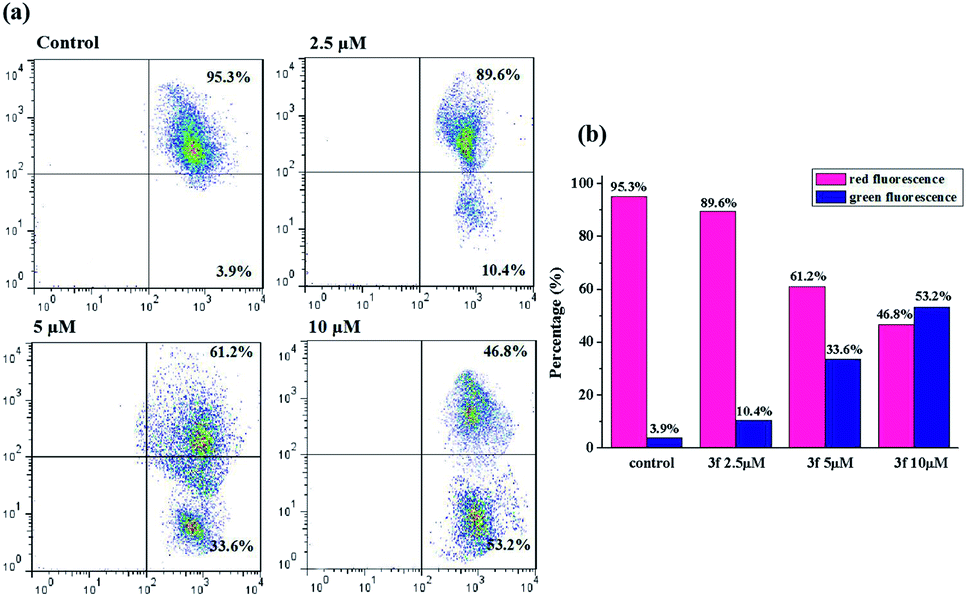 | ||
| Fig. 6 Determination of mitochondrial membrane potential in MDA-MB-231 cells. (a) Cells were treated with compound 3f (2.5, 5 and 10 μM) for 72 h and observed the changes in mitochondrial membrane potential by flow cytometry. (b) Bar graph represents the percentage distribution of cells with red fluorescence or green fluorescence. These data are from three independent experiments. | ||
2.8. Western blot analysis
Aforementioned findings indicated that compound 3f gave rise to a preliminary disruption of the mitochondrial transmembrane potential (ΔΨm), which would be the consequence of mitochondrial membrane permeabilization. This permeabilization is chiefly mediated and controlled by Bcl-2 family members which mainly includes pro-survival group (Bcl-2, Bcl-xL, etc.), pro-apoptotic group (Bax, Bak, etc.) and apoptosis initiator group (Bid, Bim, etc.). The ratio of anti- to pro-apoptotic molecules constitutes a rheostat that sets the threshold of susceptibility to apoptosis for the intrinsic pathway. In the presence of apoptotic stimuli, the expression of pro-apoptotic proteins increased and the release of pro-survival proteins is inhibited, which leads to cytochrome C release from the intermembrane space of mitochondria to the cytoplasm, likely by forming a channel in the outer mitochondrial membrane. The released cytochrome C binds to apoptosis protease-activating factor 1 (Apaf-1) and procaspase-9, generating apoptosome. Consecutively, the executioner caspase-3 is recruited to the apoptosome, where it is activated by the resident caspase-9. Then, caspase-3 cleaves key substrates in the cell to produce many of the cellular and biochemical events of apoptosis. In the present study, several apoptosis associated molecules, such as Bcl-2, Bax, cytochrome C, and caspase-3, were evaluated. After treatment with compound 3f (2.5 μM, 5 μM and 10 μM) for 72 h, the levels of apoptosis inducing proteins in MDA-MB-231 cells were tested via western blot assays. As depicted in Fig. 7, the expression of anti-apoptosis protein Bcl-2 was downregulated dose-dependently, whereas the expression of pro-apoptotic proteins Bax was up-regulated. Besides, the upregulation of cytochrome C and caspase-3 were also observed compared with the control. Thus, these results confirmed that compound 3f can induce mitochondria-related apoptosis.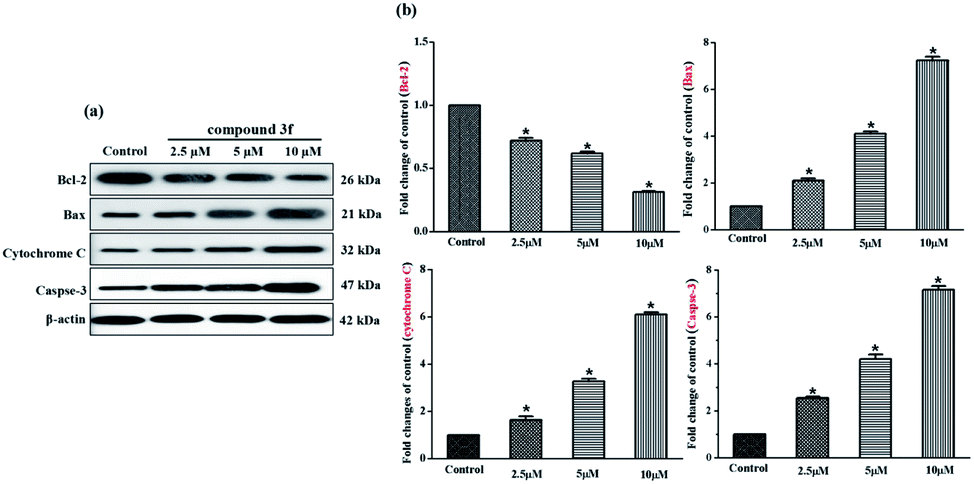 | ||
| Fig. 7 Expression changes of apoptosis related proteins. (a) Western blot analysis of Bcl-2, Bax, cytochrome C, caspase-3 in MDA-MB-231 cells treated with compound 3f (2.5, 5 and 10 μM) for 72 h, β-actin was used as the internal reference. (b) Statistical analysis of protein expression levels. These data are from three independent experiments. *p < 0.05 verse the negative control. | ||
3. Experimental
3.1. Chemistry
Starting materials and chemical reagents were purchased from Aladdin or Energy, and used without further purification. Solvents were purchased from common commercial suppliers. Thin-layer chromatography (TLC) was utilized to monitor the reaction progress, and components were visualized under 254 nm illumination. The purity of target compounds evaluated biologically was greater than 98% by America Agilent 1260 high performance liquid chromatography (HPLC) analysis. 1H (400 MHz) and 13C NMR (100 MHz) spectra were recorded on Bruker Advance spectrometer in CDCl3, DMSO-d6 solutions. The proton and carbon chemical shifts (ppm) are given with respect to tetramethylsilane (TMS). High resolution mass spectra (HRMS) were obtained by using Agilent 6540 high-resolution mass spectrometer. Melting points were measured on Optimelt-MPA100 melting point apparatus (uncorrected). X-ray diffraction analysis was determined on a Bruker D8 Venture area diffractometer.3.1.2.1. 4-(2-Fluorophenyl)-8,9,9-trimethyl-5,6,7,8-tetrahydro-5,8-methanoquinazolin-2-amine (3a). Yield 92%; mp 166.2–166.4 °C. 1H NMR (400 MHz, CDCl3) δ: 7.57 (s, 1H), 7.40 (s, 1H), 7.13 (s, 2H), 5.39 (s, 2H), 2.79 (s, 1H), 2.28 (s, 1H), 2.18 (s, 1H), 1.90 (s, 1H), 1.32 (s, 1H), 1.25 (s, 3H), 0.97 (s, 3H), 0.66 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 181.40, 161.89, 158.46, 151.97, 131.02, 130.70, 127.79, 125.87, 124.34, 116.03, 56.00, 54.64, 49.55, 31.78, 25.77, 19.83, 18.97, 10.04; HR-MS (ESI+): m/z calculated for C18H20FN3 [M + H]+ 298.1720, found 298.1717.
3.1.2.2. 4-(3-Fluorophenyl)-8,9,9-trimethyl-5,6,7,8-tetrahydro-5,8-methanoquinazolin-2-amine (3b). Yield 88%; mp 155.4–155.9 °C. 1H NMR (400 MHz, CDCl3) δ: 7.49–7.55 (m, 2H), 7.39–7.44 (m, 1H), 7.09–7.14 (m, 1H), 5.31 (s, 2H), 3.06 (d, J = 4 Hz, 1H), 2.18–2.23 (m, 1H), 1.86–1.96 (m, 1H), 1.32–1.37 (m, 2H), 1.25 (s, 3H), 0.99 (s, 3H), 0.63 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 182.24, 164.11, 161.75, 154.70, 140.38, 129.98, 125.48, 123.96, 116.18, 115.33, 55.96, 54.25, 49.65, 31.76, 25.95, 19.96, 18.98, 10.03; HR-MS (ESI+): m/z calculated for C18H20FN3 [M + H]+ 298.1720, found 298.1717.
3.1.2.3. 4-(4-Fluorophenyl)-8,9,9-trimethyl-5,6,7,8-tetrahydro-5,8-methanoquinazolin-2-amine (3c). Yield 91%; mp 197.0–197.9 °C. 1H NMR (400 MHz, CDCl3) δ: 7.75–7.79 (m, 2H), 7.14 (t, J = 8 Hz, 2H), 5.27 (s, 2H), 3.03 (d, J = 3.9 Hz, 1H), 2.16–2.22 (m, 1H), 1.86–1.95 (m, 1H), 1.32–1.36 (m, 2H), 1.25 (s, 3H), 0.99 (s, 3H), 0.63 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 181.98, 164.66, 161.74, 155.07, 134.17, 130.24, 125.17, 115.32, 55.97, 54.21, 49.70, 31.76, 25.97, 19.95, 19.01, 10.03; HR-MS (ESI+): m/z calculated for C18H20FN3 [M + H]+ 298.1720, found 298.1717.
3.1.2.4. 4-(2-Chlorophenyl)-8,9,9-trimethyl-5,6,7,8-tetrahydro-5,8-methanoquinazolin-2-amine (3d). Yield 92%; mp 185.5–185.8 °C. 1H NMR (400 MHz, CDCl3) δ: 7.45–7.48 (m, 3H), 7.31–7.36 (m, 3H), 5.20 (s, 2H), 2.69 (d, J = 4 Hz, 1H), 1.97–2.04 (m, 1H), 1.84–1.87 (m, 1H), 1.30–1.33 (m, 1H), 1.27 (s, 3H), 1.14–1.20 (m, 1H), 0.97 (s, 3H), 0.74 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 181.27, 161.59, 155.60, 137.18, 132.15, 130.30, 129.82, 129.73, 127.53, 126.81, 56.14, 54.66, 48.82, 31.86, 25.87, 19.91, 18.93, 9.94; HR-MS (ESI+): m/z calculated for C18H20ClN3 [M + H]+ 314.1424, found 314.1420.
3.1.2.5. 4-(3-Chlorophenyl)-8,9,9-trimethyl-5,6,7,8-tetrahydro-5,8-methanoquinazolin-2-amine (3e). Yield 87%; mp 162.8–163.2 °C. 1H NMR (400 MHz, CDCl3) δ: 7.78 (s, 1H), 7.61–7.64 (m, 1H), 7.38–7.39 (m, 1H), 5.25 (s, 2H), 3.05 (d, J = 4 Hz, 1H), 2.17–2.23 (m, 1H), 1.86–1.96 (m, 1H), 1.34 (t, J = 8 Hz, 2H), 1.25 (s, 3H), 0.99 (s, 3H), 0.63 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 182.24, 161.73, 154.59, 139.87, 134.55, 129.66, 129.22, 128.39, 126.42, 125.56, 55.99, 54.25, 49.61, 31.73, 25.96, 19.96, 18.98, 10.02; HR-MS (ESI+): m/z calculated for C18H20ClN3 [M + H]+ 314.1424, found 314.1423.
3.1.2.6. 4-(4-Chlorophenyl)-8,9,9-trimethyl-5,6,7,8-tetrahydro-5,8-methanoquinazolin-2-amine (3f). Yield 94%; mp 192.3–192.6 °C. 1H NMR (400 MHz, CDCl3) δ: 7.71 (d, J = 12 Hz, 2H), 7.42 (d, J = 12 Hz, 2H), 5.27 (s, 2H), 3.0 (d, J = 4 Hz, 1H), 2.16–2.22 (m, 1H), 1.86–1.96 (m, 1H), 1.29–1.35 (m, 2H), 1.25 (s, 3H), 0.99 (s, 3H), 0.62 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 182.11, 161.75, 154.87, 136.48, 135.29, 129.63, 128.67, 125.34, 77.36, 77.04, 76.72, 55.98, 54.22, 49.70, 31.74, 25.96, 19.95, 19.00, 10.03; HR-MS (ESI+): m/z calculated for C18H20ClN3 [M + H]+ 314.1424, found 314.1420.
3.1.2.7. 4-(2-Bromophenyl)-8,9,9-trimethyl-5,6,7,8-tetrahydro-5,8-methanoquinazolin-2-amine (3g). Yield 88%; mp 172.5–172.9 °C. 1H NMR (400 MHz, CDCl3) δ: 7.75–7.77 (m, 2H), 7.41–7.48 (m, 3H), 5.28 (s, 2H), 3.07 (d, J = 4 Hz, 1H), 2.14–2.22 (m, 1H), 1.87–1.94 (m, 1H), 1.32–1.39 (m, 2H), 1.25 (s, 3H), 0.98 (s, 3H), 0.63 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 181.86, 161.79, 156.18, 138.05, 129.18, 128.44, 128.30, 125.48, 55.97, 54.23, 49.67, 31.80, 26.05, 19.97, 19.04, 10.06; HR-MS (ESI+): m/z calculated for C18H20BrN3 [M + H]+ 358.0919, found 358.0911.
3.1.2.8. 4-(3-Bromophenyl)-8,9,9-trimethyl-5,6,7,8-tetrahydro-5,8-methanoquinazolin-2-amine (3h). Yield 83%; mp 187.2–187.8 °C. 1H NMR (400 MHz, CDCl3) δ: 7.94 (t, J = 4 Hz, 1H), 7.68, 7.65–7.68 (m, 1H), 7.53–7.56 (m, 1H), 7.32 (t, J = 8 Hz, 1H), 5.24 (s, 2H), 3.04 (d, J = 4 Hz, 1H), 2.17–2.23 (m, 1H), 1.93, 1.91, 1.86–1.96 (m, 1H), 1.29–1.36 (m, 2H), 1.25 (s, 3H), 0.99 (s, 3H), 0.63 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 182.24, 161.72, 154.48, 140.11, 132.14, 131.31, 129.91, 126.87, 125.57, 122.72, 56.00, 54.25, 49.60, 31.73, 25.96, 19.97, 18.99, 10.02; HR-MS (ESI+): m/z calculated for C18H20BrN3 [M + H]+ 358.0919, found 358.0911.
3.1.2.9. 4-(4-Bromophenyl)-8,9,9-trimethyl-5,6,7,8-tetrahydro-5,8-methanoquinazolin-2-amine (3i). Yield 89%; mp 191.4–191.8 °C. 1H NMR (400 MHz, CDCl3) δ: 7.65 (d, J = 8 Hz, 1H), 7.58 (d, J = 8 Hz, 1H), 5.25 (s, 2H), 3.02 (d, J = 2 Hz, 1H), 2.16–2.22 (m, 1H), 1.91 (t, J = 12 Hz, 1H), 1.28–1.35 (m, 2H), 1.25 (s, 3H), 0.99 (s, 3H), 0.62 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 182.15, 161.73, 154.90, 136.91, 131.64, 129.90, 125.37, 123.64, 77.36, 77.04, 76.73, 55.99, 54.23, 49.68, 31.73, 25.95, 19.95, 19.01, 10.04; HR-MS (ESI+): m/z calculated for C18H20BrN3 [M + H]+ 358.0919, found 358.0913.
3.1.2.10. 8,9,9-Trimethyl-4-(o-tolyl)-5,6,7,8-tetrahydro-5,8-methanoquinazolin-2-amine (3j). Yield 88%; mp 171.9–172.5 °C. 1H NMR (400 MHz, CDCl3) δ: 7.20–7.30 (m, 3H), 7.15 (d, J = 8 Hz, 1H), 5.39 (s, 2H), 2.61 (d, J = 4 Hz, 1H), 2.27 (s, 3H), 1.98–2.05 (m, 1H), 1.82–1.88 (m, 1H), 1.27–1.33 (m, 1H), 1.24 (s, 3H), 1.12–1.18 (m, 1H), 0.93 (s, 3H), 0.70 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 181.13, 161.53, 158.38, 137.51, 135.64, 130.53, 128.41, 126.91, 125.55, 55.99, 54.61, 48.71, 31.76, 26.26, 19.97, 19.87, 18.94, 9.97; HR-MS (ESI+): m/z calculated for C19H23N3 [M + H]+ 294.1970, found 294.1967.
3.1.2.11. 8,9,9-Trimethyl-4-(m-tolyl)-5,6,7,8-tetrahydro-5,8-methanoquinazolin-2-amine (3k). Yield 86%; mp 156.8–157.3 °C. 1H NMR (400 MHz, CDCl3) δ: 7.60 (s, 1H), 7.50–7.52 (d, J = 7.6 Hz, 1H), 7.34 (t, J = 8 Hz, 1H), 7.23 (d, J = 8 Hz, 1H), 5.24 (s, 2H), 3.05 (d, J = 4 Hz, 1H), 2.42 (s, 3H), 2.15–2.21 (m, 1H), 1.87–1.94 (m, 1H), 1.32–1.36 (m, 2H), 1.25 (s, 3H), 0.98 (s, 3H), 0.63 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 181.76, 161.75, 156.39, 138.17, 137.98, 129.97, 128.87, 128.25, 125.52, 125.46, 55.96, 54.23, 49.69, 31.80, 26.04, 21.57, 19.97, 19.04, 10.06; HR-MS (ESI+): m/z calculated for C19H23N3 [M + H]+ 294.1970, found 294.1967.
3.1.2.12. 8,9,9-Trimethyl-4-(p-tolyl)-5,6,7,8-tetrahydro-5,8-methanoquinazolin-2-amine (3l). Yield 90%; mp 197.5–197.9 °C. 1H NMR (400 MHz, CDCl3) δ: 7.67 (d, J = 8 Hz, 2H), 7.26 (d, J = 8 Hz, 2H), 5.24 (s, 2H), 3.06 (d, J = 4 Hz, 1H), 2.40 (s, 3H), 2.14–2.21 (m, 1H), 1.86–1.92 (m, 1H), 1.32–1.35 (m, 2H), 1.24 (s, 3H), 0.98 (s, 3H), 0.62 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 181.70, 161.73, 156.20, 139.27, 135.22, 129.15, 128.24, 125.26, 77.36, 77.04, 76.72, 55.94, 54.18, 49.74, 31.81, 26.01, 21.38, 19.96, 19.05, 10.07; HR-MS (ESI+): m/z calculated for C19H23N3 [M + H]+ 294.1970, found 294.1967.
3.1.2.13. 4-(2-Methoxyphenyl)-8,9,9-trimethyl-5,6,7,8-tetrahydro-5,8-methanoquinazolin-2-amine (3m). Yield 91%; mp 175.2–175.8 °C. 1H NMR (400 MHz, CDCl3) δ: 7.34–7.41 (m, 2H), 7.03 (t, J = 8 Hz, 1H), 6.95 (d, J = 8 Hz, 1H), 5.32 (s, 2H), 3.78 (s, 3H), 2.63 (d, J = 4 Hz, 1H), 1.97–2.03 (m, 1H), 1.81–1.87 (m, 1H), 1.27–1.33 (m, 1H), 1.23 (s, 3H), 0.94 (s, 3H), 0.65 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 180.39, 161.87, 156.61, 154.90, 130.60, 130.08, 128.08, 127.44, 120.80, 111.00, 55.91, 55.17, 54.47, 49.71, 32.01, 25.65, 19.85, 19.07, 10.07; HR-MS (ESI+): m/z calculated for C19H23N3O [M + H]+ 310.1919, found 310.1915.
3.1.2.14. 4-(3-Methoxyphenyl)-8,9,9-trimethyl-5,6,7,8-tetrahydro-5,8-methanoquinazolin-2-amine (3n). Yield 90%; mp 183.2–183.4 °C. 1H NMR (400 MHz, CDCl3) δ: 7.31–7.38 (m, 3H), 6.95–6.98 (m, 1H), 5.27 (s, 2H), 3.87 (s, 3H), 3.07 (d, J = 4 Hz, 1H), 2.15–2.21 (m, 1H), 1.87–1.92 (m, 1H), 1.32–1.36 (m, 2H), 1.25 (s, 3H), 0.98 (s, 3H), 0.63 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 181.91, 161.74, 159.72, 155.99, 139.43, 129.40, 125.52, 120.78, 115.07, 113.58, 55.94, 55.32, 54.24, 49.74, 31.80, 25.99, 19.97, 19.02, 10.05; HR-MS (ESI+): m/z calculated for C19H23N3O [M + H]+ 310.1919, found 310.1919.
3.1.2.15. 4-(4-Methoxyphenyl)-8,9,9-trimethyl-5,6,7,8-tetrahydro-5,8-methanoquinazolin-2-amine (3o). Yield 93%; mp 131.7–131.9 °C. 1H NMR (400 MHz, CDCl3) δ: 7.75 (d, J = 8 Hz, 2H), 6.98 (d, J = 8 Hz, 2H), 5.27 (s, 2H), 3.85 (s, 3H), 3.06 (d, J = 4 Hz, 1H), 2.14–2.21 (m, 1H), 1.86–1.94 (m, 1H), 1.31–1.35 (m, 2H), 1.24 (s, 3H), 0.98 (s, 3H), 0.63 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 181.59, 161.72, 160.54, 155.82, 130.61, 129.76, 124.81, 113.85, 55.91, 55.33, 54.12, 49.83, 31.83, 25.97, 19.96, 19.06, 10.08; HR-MS (ESI+): m/z calculated for C19H23N3O [M + H]+ 310.1919, found 310.1916.
3.1.2.16. 2-(2-Amino-8,9,9-trimethyl-5,6,7,8-tetrahydro-5,8-methanoquinazolin-4-yl)phenol (3p). Yield 89%; mp 167.9–168.3 °C. 1H NMR (400 MHz, CDCl3) δ: 7.65 (d, J = 8 Hz, 1H), 7.31 (t, J = 8 Hz, 1H), 7.0 (d, J = 8 Hz, 1H), 6.9 (d, J = 16 Hz, 1H), 5.21 (s, 2H), 3.27 (d, J = 4 Hz, 1H), 2.21–2.29 (m, 1H), 1.90–1.96 (m, 1H), 1.31–1.43 (m, 2H), 1.24 (s, 3H), 1.00 (s, 3H), 0.63 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 183.33, 159.85, 159.01, 155.66, 131.66, 129.11, 123.93, 119.21, 118.64, 118.22, 55.72, 54.22, 50.86, 31.88, 25.35, 19.94, 18.99, 10.03; HR-MS (ESI+): m/z calculated for C18H21N3O [M + H]+ 296.1763, found 296.1758.
3.1.2.17. 3-(2-Amino-8,9,9-trimethyl-5,6,7,8-tetrahydro-5,8-methanoquinazolin-4-yl)phenol (3q). Yield 87%; mp 262.0–262.8 °C. 1H NMR (400 MHz, DMSO-d6) δ: 9.32 (s, 1H), 6.92–7.04 (m, 3H), 6.59 (d, J = 8 Hz, 1H), 6.10 (s, 2H), 2.78 (d, J = 4 Hz, 1H), 1.89–1.94 (m, 1H), 1.60–1.65 (m, 1H), 0.94–1.03 (m, 2H), 0.90 (s, 3H), 0.70 (s, 3H), 0.31 (s, 3H); 13C NMR (100 MHz, DMSO-d6): δ 180.89, 161.97, 157.31, 154.84, 139.11, 129.28, 123.15, 118.75, 116.10, 114.81, 55.00, 53.62, 49.26, 31.62, 25.84, 19.65, 18.68, 10.19; HR-MS (ESI+): m/z calculated for C18H21N3O [M + H]+ 296.1763, found 296.1761.
3.1.2.18. 4-(2-Amino-8,9,9-trimethyl-5,6,7,8-tetrahydro-5,8-methanoquinazolin-4-yl)phenol (3r). Yield 89%; mp 264.1–264.8 °C. 1H NMR (400 MHz, DMSO-d6) δ: 9.79 (s, 1H), 7.65 (d, J = 8 Hz, 2H), 6.87 (d, J = 8 Hz, 2H), 6.26 (s, 2H), 3.04 (d, J = 4 Hz, 1H), 2.13–2.18 (m, 1H), 1.83–1.89 (m, 1H), 1.17–1.25 (m, 2H), 1.14 (s, 3H), 0.94 (s, 3H), 0.56 (s, 3H); 13C NMR (100 MHz, DMSO-d6): δ 180.56, 161.87, 158.52, 154.75, 129.49, 128.62, 122.24, 115.12, 54.95, 53.50, 49.33, 31.59, 25.77, 19.66, 18.72, 10.22; HR-MS (ESI+): m/z calculated for C18H21N3O [M + H]+ 296.1763, found 296.1759.
3.1.2.19. 8,9,9-Trimethyl-4-(pyridin-2-yl)-5,6,7,8-tetrahydro-5,8-methanoquinazolin-2-amine (3s). Yield 89%; mp 174.7–175.2 °C. 1H NMR (400 MHz, CDCl3) δ: 8.72 (d, J = 8 Hz, 1H), 8.10 (d, J = 8 Hz, 1H), 7.76–7.80 (m, 1H), 7.29–7.31 (m, 1H), 5.18 (s, 2H), 3.62 (d, J = 4 Hz, 1H), 2.15–2.21 (m, 1H), 1.85–1.93 (m, 1H), 1.28–1.36 (m, 2H), 1.26 (s, 3H), 1.00 (s, 3H), 0.65 (s, 3H); 13C NMR (100 MHz, DMSO-d6): δ 177.83, 156.26, 151.19, 148.60, 144.14, 131.07, 121.59, 118.33, 117.43, 72.10, 71.78, 71.46, 50.06, 48.96, 44.84, 26.70, 20.51, 14.84, 13.70, 4.80; HR-MS (ESI+): m/z calculated for C17H20N4 [M + H]+ 281.1766, found 281.1760.
3.1.2.20. 8,9,9-Trimethyl-4-(pyridin-3-yl)-5,6,7,8-tetrahydro-5,8-methanoquinazolin-2-amine (3t). Yield 85%; mp 197.2–197.7 °C. 1H NMR (400 MHz, CDCl3) δ: 8.99 (s, 1H), 8.66, 8.65 (d, J = 4 Hz, 1H), 8.13 (d, J = 8 Hz, 1H), 7.39–7.42 (m, 1H), 5.25 (s, 2H), 3.07 (d, J = 4 Hz, 1H), 2.19–2.29 (m, 1H), 1.90–1.97 (m, 1H), 1.34 (d, J = 8 Hz, 1H), 1.26 (s, 3H), 1.00 (s, 3H), 0.64 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 182.34, 161.82, 153.16, 150.10, 149.46, 135.60, 133.74, 125.83, 123.45, 56.07, 54.28, 49.59, 31.65, 26.04, 19.96, 18.97, 9.99; HR-MS (ESI+): m/z calculated for C17H20N4 [M + H]+ 281.1766, found 281.1762.
3.1.2.21. 8,9,9-Trimethyl-4-(pyridin-4-yl)-5,6,7,8-tetrahydro-5,8-methanoquinazolin-2-amine (3u). Yield 90%; mp 207.9–208.1 °C. 1H NMR (400 MHz, CDCl3) δ: 8.72 (d, J = 4 Hz, 2H), 7.66 (d, J = 4 Hz, 2H), 5.35 (s, 2H), 3.08 (d, J = 4 Hz, 1H), 2.19–2.25 (m, 1H), 1.89–1.98 (m, 1H), 1.30–1.37 (m, 2H), 1.26 (s, 3H), 1.01 (s, 3H), 0.63 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 182.70, 161.85, 153.26, 150.16, 145.40, 126.10, 122.52, 56.00, 54.27, 49.55, 31.67, 25.93, 19.96, 18.94, 9.96; HR-MS (ESI+): m/z calculated for C17H20N4 [M + H]+ 281.1766, found 281.1763.
3.1.2.22. 4-(4-(Dimethylamino)phenyl)-8,9,9-trimethyl-5,6,7,8-tetrahydro-5,8-methanoquinazolin-2-amine (3v). Yield 93%; mp 198.1–198.6 °C. 1H NMR (400 MHz, CDCl3) δ: 7.76 (d, J = 8 Hz, 2H), 6.79 (d, J = 8 Hz, 2H), 5.24 (s, 2H), 3.12 (d, J = 4 Hz, 1H), 3.03 (s, 2H), 2.17–2.22 (m, 1H), 1.87–1.92 (m, 1H), 1.30–1.41 (m, 2H), 1.25 (s, 3H), 0.99 (s, 3H), 0.64 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 181.14, 161.65, 156.35, 151.15, 129.47, 125.73, 124.28, 111.77, 55.84, 54.00, 50.01, 40.29, 31.92, 25.94, 19.97, 19.11, 10.12; HR-MS (ESI+): m/z calculated for C20H26N4 [M + H]+ 323.2236, found 323.2231.
3.1.2.23. 8,9,9-Trimethyl-4-(4-nitrophenyl)-5,6,7,8-tetrahydro-5,8-methanoquinazolin-2-amine (3w). Yield 82%; mp 138.7–139.1 °C. 1H NMR (400 MHz, CDCl3) δ: 8.33 (d, J = 8 Hz, 2H), 7.97 (d, J = 8 Hz, 2H), 5.07 (s, 2H), 3.07 (d, J = 4 Hz, 1H), 2.22–2.25 (m, 1H), 1.94 (m, 1H), 1.33–1.40 (m, 1H), 1.28 (s, 3H), 1.03 (s, 3H), 0.65 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 182.76, 161.66, 153.40, 145.72, 142.31, 130.14, 126.27, 123.90, 57.19, 49.33, 46.65, 30.46, 25.92, 20.64, 18.19, 9.21; HR-MS (ESI+): m/z calculated for C18H20N4O2 [M + H]+ 325.1665, found 325.1659.
3.1.2.24. 8,9,9-Trimethyl-4-phenyl-5,6,7,8-tetrahydro-5,8-methanoquinazolin-2-amine (3x). Yield 94%; mp 172.9–173.2 °C. 1H NMR (400 MHz, CDCl3) δ: 7.75–7.77 (m, 2H), 7.41–7.48 (m, 3H), 5.33 (s, 2H), 3.06 (d, J = 4 Hz, 1H), 2.16–2.21 (m, 1H), 1.87–1.92 (m, 1H), 1.32–1.35 (m, 2H), 1.25 (s, 3H), 0.98 (s, 3H), 0.63 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 181.85, 161.81, 156.19, 138.05, 129.18, 128.43, 128.30, 125.45, 55.96, 54.24, 49.67, 31.80, 26.05, 19.97, 19.04, 10.07. HR-MS (ESI+): m/z calculated for C18H21N3 [M + H]+ 280.1814, found 280.1810.
3.2. Cell imaging
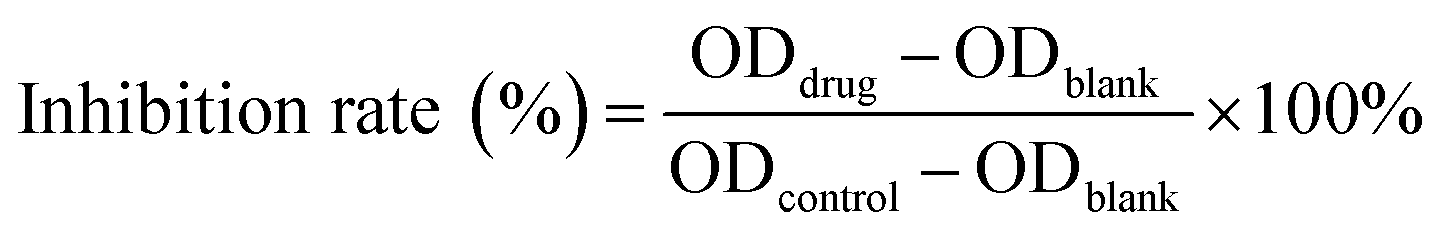
4. Conclusion
In summary, we synthesized a series of novel pyrimidine derivatives based on camphor and evaluated their anticancer activities against three human cancer cell lines (MDA-MB-231, RPMI-8226, and A549). The majority of synthesized compounds possessed moderate to good growth inhibition, particularly, compound 3f appeared to be the most active comparable to etoposide, and had much lower cytotoxicity to normal GES-1 cells (IC50 > 50 μM) than the reference drug (IC50 = 8.89 μM). Meanwhile, the overall result suggested that electron-withdrawing group at para-position on aromatic rings is crucial for cytotoxicity. Subsequently, compound 3f was selected for further anticancer mechanisms in a battery of assays. Flow cytometry experiments demonstrated that compound 3f induced G0/G1 phase arrest and apoptosis in MDA-MB-231 cells, and the DCFH-DA assays revealed that apoptosis was mediated through intracellular ROS stimulation. Moreover, compound 3f could also cause the depolarization of mitochondrial membrane potentials, which indicated that compound 3f could activate the mitochondrial apoptosis pathway. This mechanistic pathway was confirmed by upregulation of pro-apoptotic proteins Bax, cytochrome C and caspase-3, whereas the expression of anti-apoptosis protein Bcl-2 was downregulated. On the basis of above studies, it can be concluded that compound 3f exhibited potent anticancer efficiency via ROS-mediated mitochondrial apoptosis pathway. So far, plenty of designed compounds against breast cancer cell, including ones based on pyrimidine rings, have been reported, and their anti-tumor mechanism is mostly related to mitochondrial apoptosis pathway.39–41 However, there is no effective drug for triple-negative breast cancer (MDA-MB-231) because of its high metastasis and drug resistance. We hope these findings in this paper will provide some valuable hints for exploring more effective drugs for breast cancer therapy.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This study was supported by the National Natural Science Foundation of China (No. 31470592), Major Project of Jiangsu Province University Natural Science (No.14KJ220001) and Open Funding of Jiangsu Key Laboratory of Biomass Energy and Materials (No. JSBEM2014010). Besides, sincere gratitude to Jiangsu Chia Tai Fenghai Pharmaceutical Co. Ltd for support in cytotoxicity evaluation assay.References
- B. Vainshelboim, J. Müller, R. M. Lima, K. T. Nead, C. Chester, K. Chan, P. Kokkinos and J. Myers, Prev. Med., 2017, 100, 89–94 CrossRef PubMed.
- K. Yang, Y. Li, Q. Tang, L. Zheng and D. He, Eur. J. Med. Chem., 2019, 170, 45–54 CrossRef CAS PubMed.
- D. Wan, B. Tang, Y. J. Wang, B. H. Guo, H. Yin, Q. Y. Yi and Y. J. Liu, Eur. J. Med. Chem., 2017, 139, 180–190 CrossRef CAS PubMed.
- S. Gupta, G. E. Kass, E. Szegezdi and B. Joseph, J. Cell. Mol. Med., 2009, 13, 1004–1033 CrossRef CAS PubMed.
- Z. Jin and W. S. El-Deiry, Cancer Biol. Ther., 2005, 4, 139–163 CAS.
- J. C. Martinou, S. Desagher and B. Antonsson, Nat. Cell Biol., 2000, 2, 41–43 CrossRef PubMed.
- P. Giuseppe, F. M. Ruggiero and P. Giuseppe, FASEB J., 2003, 17, 2202 CrossRef PubMed.
- S. Xiong, T. Mu, G. Wang and X. Jiang, Protein Cell, 2014, 5, 737–749 CrossRef CAS PubMed.
- E. Finkel, Science, 2001, 292, 624–626 CrossRef CAS PubMed.
- S. Cherukupalli, G. A. Hampannavar, S. Chinnam, B. Chandrasekaran, N. Sayyad, F. Kayamba, A. R. Reddy and R. Karpoormath, Bioorg. Med. Chem., 2018, 26, 309–339 CrossRef CAS PubMed.
- A. M. Fargualy, N. S. Habib, K. A. Ismail, A. M. M. Hassan and M. T. M. Sarg, ChemInform, 2013, 66, 276–295 CAS.
- Y. Ouyang, W. Zou, L. Peng, Z. Yang, Q. Tang, M. Chen, S. Jia, H. Zhang, Z. Lan and P. Zheng, Eur. J. Med. Chem., 2018, 154, 29–43 CrossRef CAS PubMed.
- M. Allam, A. K. D. Bhavani, A. Mudiraj, N. Ranjan, M. Thippana and P. P. Babu, Eur. J. Med. Chem., 2018, 156, 43–52 CrossRef CAS PubMed.
- Y. Yi, G. Yang, Z. Chao, J. Chen, J. Liang and R. Shang, Eur. J. Med. Chem., 2015, 101, 179–184 CrossRef CAS PubMed.
- E. Scocchera, S. M. Reeve, S. Keshipeddy, M. N. Lombardo, B. Hajian, A. E. Sochia, J. B. Alverson, N. D. Priestley, A. C. Anderson and D. L. Wright, ACS Med. Chem. Lett., 2016, 7, 692–696 CrossRef CAS PubMed.
- P. Gao, L. Zhang, L. Sun, T. Huang, J. Tan, J. Zhang, Z. Zhou, T. Zhao, L. Menéndez-Arias and C. Pannecouque, Bioorg. Med. Chem., 2017, 25, 5779–5789 CrossRef CAS PubMed.
- M. Krečmerová, M. Dračínský, R. Snoeck, J. Balzarini, K. Pomeisl and G. Andrei, Bioorg. Med. Chem., 2017, 25, 4637–4648 CrossRef PubMed.
- J. Wang, L. Zhang, J. Zhao, Y. Zhang, Q. Liu, C. Tian, Z. Zhang, J. Liu and X. Wang, Bioorg. Med. Chem., 2018, 100, 89–94 Search PubMed.
- E. S. Zuniga, A. Korkegian, S. Mullen, E. J. Hembre, P. L. Ornstein, G. Cortez, K. Biswas, N. Kumar, J. Cramer and T. Masquelin, Bioorg. Med. Chem., 2017, 25, 3922–3946 CrossRef CAS PubMed.
- P. S. Shirude, B. Paul, C. N. Roy, C. Kedari, B. Bandodkar and B. G. Ugarkar, ACS Med. Chem. Lett., 2012, 3, 736–740 CrossRef CAS PubMed.
- K. Shah, X. Lin, S. F. Queener, V. Cody, J. Pace and A. Gangjee, Bioorg. Med. Chem., 2018, 26, 2640–2650 CrossRef CAS PubMed.
- D. Giles, K. Roopa, F. R. Sheeba, P. M. Gurubasavarajaswamy, G. Divakar and T. Vidhya, Eur. J. Med. Chem., 2012, 58, 478–484 CrossRef CAS PubMed.
- H. Kaur, J. Balzarini, C. D. Kock, P. J. Smith, K. Chibale and K. Singh, Eur. J. Med. Chem., 2015, 101, 52–62 CrossRef CAS PubMed.
- S. Manohar, U. C. Rajesh, S. I. Khan, B. L. Tekwani and D. S. Rawat, ACS Med. Chem. Lett., 2012, 3, 555–559 CrossRef CAS PubMed.
- Z. Yang, Y. Fang and H. Park, Bioorg. Med. Chem. Lett., 2017, 27, 2515–2519 CrossRef CAS PubMed.
- R. S. Keri, K. M. Hosamani, R. V. Shingalapur and M. H. Hugar, ChemInform, 2010, 45, 2597–2605 CAS.
- Y. Addepalli, X. Yang, M. Zhou, D. P. Reddy, S. L. Zhang, Z. Wang and Y. He, Eur. J. Med. Chem., 2018, 151, 214–225 CrossRef CAS PubMed.
- G. Joshi, H. Nayyar, S. Kalra, P. Sharma, A. Munshi, S. Singh and R. Kumar, Chem. Biol. Drug Des., 2017, 90, 995–1006 CrossRef CAS PubMed.
- M. Murahari, K. V. Prakash, G. J. Peters and Y. C. Mayur, Eur. J. Med. Chem., 2017, 139, 961–981 CrossRef CAS PubMed.
- G. Luo, Z. Tang, K. Lao, X. Li, Q. You and H. Xiang, Eur. J. Med. Chem., 2018, 150, 783–795 CrossRef CAS PubMed.
- M. Coffinet, F. Massicot, J. Joseph, J. B. Behr, F. Jaroschik and J. L. Vasse, Chem. Commun., 2016, 53, 111–114 RSC.
- G. Blay, V. Hernández-Olmos and J. R. Pedro, ChemInform, 2008, 40, 4840–4842 Search PubMed.
- F. W. Lewis, T. C. Mccabe and D. H. Grayson, Tetrahedron, 2011, 67, 7517–7528 CrossRef CAS.
- Y. Wang, J. Busch-Petersen, F. Wang, T. J. Kiesow, T. L. Graybill, J. Jin, Z. Yang, J. J. Foley, G. E. Hunsberger and D. B. Schmidt, Bioorg. Med. Chem. Lett., 2009, 19, 114–118 CrossRef CAS PubMed.
- S. O. Kuranov, I. P. Tsypysheva, M. V. Khvostov, L. F. Zainullina, S. S. Borisevich, Y. V. Vakhitova, O. A. Luzina and N. F. Salakhutdinov, Bioorg. Med. Chem., 2018, 26, 4402–4409 CrossRef CAS PubMed.
- G. Stavrakov, V. Valcheva, I. Philipova and I. Doytchinova, Eur. J. Med. Chem., 2013, 70, 372–379 CrossRef CAS PubMed.
- A. M. Montana, F. J. Bernal, L. Julia, F. Carlos, B. Consuelo, M. J. Prieto, M. Virtudes, F. X. Avilés, J. M. Mesas and A. María-Teresa, Bioorg. Med. Chem., 2008, 16, 1721–1737 CrossRef CAS PubMed.
- M. L. Circu and T. Y. Aw, Free Radicals Biol. Med., 2010, 48, 749–762 CrossRef CAS PubMed.
- M. Zhao, H. Ren, J. Chang, D. Zhang, Y. Yang, Y. He, C. Qi and H. Zhang, Eur. J. Med. Chem., 2016, 119, 183–196 CrossRef CAS PubMed.
- E. F. Abdelhaleem, M. K. Abdelhameid, A. E. Kassab and M. M. Kandeel, Eur. J. Med. Chem., 2017, 143, 1807–1825 CrossRef PubMed.
- D. Yao, Y. Zhou, L. Zhu, L. Ouyang, J. Zhang, Y. Jiang, Y. Zhao, D. Sun, S. Yang and Y. Yu, Eur. J. Med. Chem., 2017, 140, 155–171 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1838718. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra05900h |
| This journal is © The Royal Society of Chemistry 2019 |