
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and anion recognition studies of new oligomethylene bis(nitrophenylureylbenzamide) receptors†
José García-Elíasab,
Adrián Ochoa-Terán *a,
Anatoli K. Yatsimirskyc,
Hisila Santacruz Ortegad,
Karen Ochoa-Lara*d,
Luis Miguel López-Martíneza,
Christian L. Castro-Riquelmea,
Ángel L. Garcíaa,
Domingo Madrigal-Peraltaa,
Victoria Labastida-Galváne and
Mario Ordoñeze
*a,
Anatoli K. Yatsimirskyc,
Hisila Santacruz Ortegad,
Karen Ochoa-Lara*d,
Luis Miguel López-Martíneza,
Christian L. Castro-Riquelmea,
Ángel L. Garcíaa,
Domingo Madrigal-Peraltaa,
Victoria Labastida-Galváne and
Mario Ordoñeze
aCentro de Graduados e Investigación en Química, Tecnológico Nacional de México/IT de Tijuana, Tijuana, México. E-mail: ochoa@tectijuana.mx
bUnidad Académica de Ciencias de la Tierra, Universidad Autónoma de Zacatecas, Zacatecas, México
cFacultad de Química, Universidad Nacional Autónoma de México, Ciudad de México, México
dDepartamento de Investigación en Polímeros y Materiales, Universidad de Sonora, Hermosillo, Sonora, México. E-mail: karenol@polimeros.unison.mx
eCentro de Investigaciones Químicas-(IICBA), Universidad Autónoma del Estado de Morelos, Cuernavaca, Mexico
First published on 28th November 2019
Abstract
A new series of oligomethylene bis(nitrophenylureylbenzamide) receptors were synthesized varying the relative position of the urea and amide groups (ortho 4 and meta 8) and the length of the oligomethylene chain (C2 to C8). An anion recognition study was performed with TBAX salts (X = AcO−, BzO−, F−, H2PO4−, and HP2O73−) by UV-vis and 1H NMR. The flexibility of these receptors allows a cooperative effect of both ureylbenzamide units in the receptors. Noteworthy, the ortho position favored the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry in the complexes with the carboxylates. The formation of 2:1 receptor–anion complexes with both types of receptors 4 and 8 and with hydrogen pyrophosphate and high logK values obtained were very significant in this work. The NMR studies evidenced the formation of supramolecular complexes, even in a competitive solvent, such as DMSO.
:1 stoichiometry in the complexes with the carboxylates. The formation of 2:1 receptor–anion complexes with both types of receptors 4 and 8 and with hydrogen pyrophosphate and high logK values obtained were very significant in this work. The NMR studies evidenced the formation of supramolecular complexes, even in a competitive solvent, such as DMSO.
Introduction
Artificial receptors capable of interacting with anions is a very important subject in supramolecular chemistry because of their participation in biological processes, like function, detection, recognition, catalysis, and transport through the cellular membrane, among others.1–3 There are many reports related to anion recognition by urea-based organic receptors. The urea group is a good hydrogen bond donor due to the presence of N–H bonds, and due to their functionality and directionality.4,5 These characteristics play a very important role in the selective recognition of anions with different geometries. The selectivity is related to the energy of the receptor–anion interaction. The strongest hydrogen bond interactions are established with the most basic of anions, such as fluoride, carboxylates, and inorganic oxoanions. From a recent point of view, all the hydrogen bonds behave as proton–transfer reactions.6 Therefore, for a receptor that contains an acid N–H, the affinity is mainly related to the anion basicity or the pKα of its conjugated acid.7In this context, nitrophenylureas have been prepared due to the major acidity caused by the resonance effect of the nitro group.8 Also, this group allows the study of anion recognition study by spectrophotometric changes; whereby the absorption bands shift to higher wavelengths in complexes with anions, or a new band appears when an acid–base reaction occurs.5 The effect in the analytical response and of the interaction with anions of nitrophenylurea receptors when varying the position of the nitro group has been studied, and it was found that the position of the nitro group influences the acidity of the urea hydrogens and the relative affinity toward different anions.6
Gunnlaugsson and coworkers9 prepared receptors with urea and acetamide groups by changing the relative position of both groups (ortho, meta, and para). The acetamide position had a clear effect over the selectivity and stoichiometry in the complexes with anions. The interaction occurred only in the urea group with one equivalent of the anion in the meta and para receptors, while the interaction with the amide hydrogen occurred forming 1:2 receptor:anion complexes. They proposed this behavior as a “positive allosteric effect” promoted by conformational changes. Interestingly, the ortho receptor formed only 1:1 complexes and the 1H NMR spectra showed the participation of the three hydrogens, which indicated a cooperative interaction of the amide hydrogen with the anions.
Bis(urea) receptors are excellent hosts for anions with diverse geometries due to the high number of N–H bonds in their structure. In addition, these receptors display high preorganization and cooperativity, which enhance the complexation process,10 and exhibit a limited number of stable configurations for the interaction with the anion.11,12 Each group forms two hydrogen bonds with the anion, the strength of which may differ depending on the ligand nature, the distance between each urea group, and the size and geometry of the anion.8,13,14
Several bis(urea) receptors for the molecular recognition of anions are reported in the literature. Chauhan and coworkers13 prepared four azo bis(urea/tiourea) receptors bearing phenyl or nitrophenyl substituents in the urea group for the colorimetric detection of anions. The affinity of these receptors was in the order F− > AcO− > H2PO4− with a change of color from yellow to red. The 1H NMR studies demonstrated that the hydrogen bond in the urea group induced different effects in the aromatic signals, whereby the enhancement of the electronic density in the phenyl ring generated a shift of the signals to upfield due to the electronic enrichment, while the polarization of the C–H bonds through the space caused a deprotecting effect and shifted the signals to downfield.
The most studied bis(urea) receptors are the N,N′-1,3-phenylenbis(N′-urea/tiourea) derivatives, initially studied by Nishizawa.14 Complexation studies by 1H NMR in DMSO revealed association constant values of 110 M−1 and 43 M−1 with dihydrogen phosphate and acetate, respectively.15 Then, these receptors showed high affinity toward anions by electrochemical methods.16 The complex stoichiometry with acetate was found to be 1:1 by UV-vis measurements and the reported association constant was 210 M−1.17 A colorimetric anion sensor was developed with a bis(urea) receptor synthetized from 4,5-dimethyl-1,2-phenylenediamine and nitrophenyl isocyanate.18 Spectroscopic changes were observed only with fluoride and acetate anions, indicating selectivity over the other studied anions.
Caltagirone and coworkers19 synthesized a series of six bis(urea) receptors using the diamines 1,3-bis(aminomethyl)benzene and 2,6-bis(aminomethyl)pyridine, and the isocianates phenyl:2-nitrophenyl and 1-naphthyl isocyanates. The binding properties of these receptors were studied in DMSO with acetate, benzoate, glutarate, malonate, dihydrogen phosphate hydrogenpyrophosphate, triphosphate, AMP, and ADP by UV-vis spectroscopy and 1H NMR. These receptors showed high affinity toward acetate, benzoate, and dihydrogen phosphate. The values of the association constants were in the range of 88 to 299 M−1 for acetate, 65 to 109 M−1 for benzoate, and 167 to 698 M−1 for dihydrogenphosphate. Noteworthy, the hydrogen pyrophosphate caused a partial or complete deprotonation to most of the receptors, allowing the colorimetric detection of this anion in DMSO. The association constants evaluated for two of these receptors were 5840 and >104 M−1, respectively.
All the receptors previously described have a rigid connection between the two urea units, mainly alkenyl or aromatic backbones, which confers some structural preorganization in the anion recognition. Continuing with our interest in the synthesis of new receptors and the study of anion recognition,20 a series of new oligomethylene bis(4-nitrophenylureylbenzamide) receptors bearing the urea and amide groups in the ortho or meta position, were synthesized and evaluated as anion receptors. We envisioned that these bifunctional receptors with flexible oligomethylene chains may be modulated in their acidity, with the cooperativity of the binding groups and the size of the receptors cavities allowing interaction with anions of different sizes and geometries (Fig. 1).
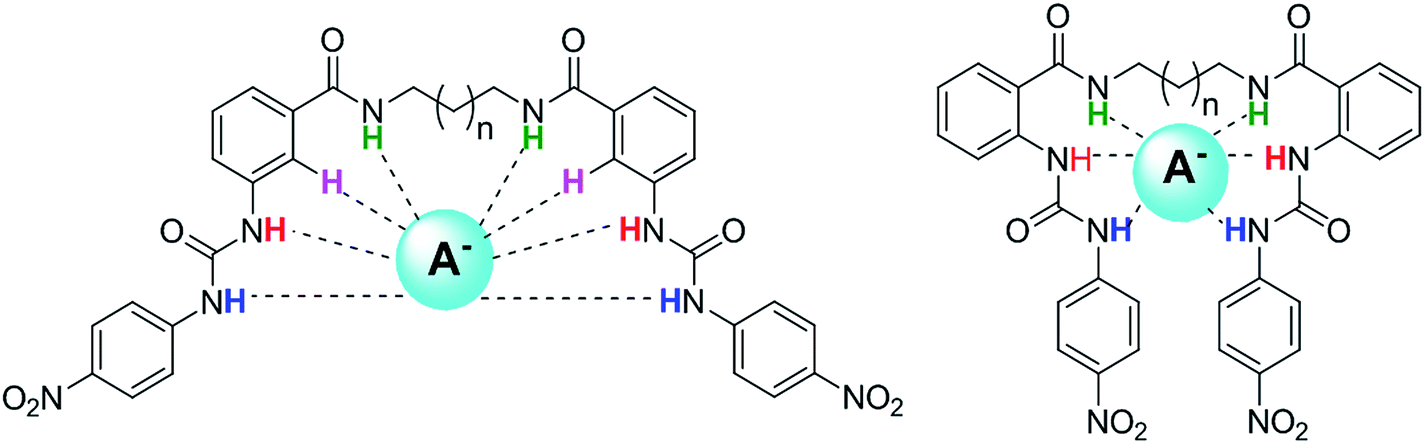 | ||
| Fig. 1 Plausible interaction of oligomethylene bis(nitrophenylureylbenzamide) receptors with anions. | ||
Experimental
Materials and equipment
All chemicals used in the study were obtained from commercial suppliers and used without further purification. 1H NMR spectra were recorded using a Bruker 400 MHz NMR spectrometer at a probe temperature of 30 °C with TMS as the internal standard. Electronic impact mass spectra were obtained by direct insertion in an Agilent 5975C mass spectrometer; and the intensities are reported as a relative percentage to the base peak corresponding to an m/z value. FAB mass spectra and HRMS were recorded in an MStation JMS-700 JEOL. UV-vis absorption spectra were obtained using a Cary 300 spectrophotometer.Procedure for the spectrophotometric titrations
The effect of anions upon the absorbance of receptors 4 and 8 was examined by adding 3 μL of a 5 × 10−3 M TBAX solution to a known volume (3 mL) of a 1 × 10−5 M receptor solution. Some titrations were repeated adding 3 μL of a 1 × 10−3 M TBAX (X = H2PO4− and HP2O3−) solution.Procedure for the NMR titrations
The effect of anions upon the chemical shift in the signals of the receptors was examined by adding 5 μL of 0.1 M TBAX stock solution to a known volume (0.5 mL) of a 5 × 10−3 M receptor solution contained in an NMR tube. The addition was limited to 0.050 mL, so that the dilution remained insignificant.Synthesis
N,N'-(Ethane-1,2-diyl)bis(2-aminobenzamide) (2a). Beige solid, 80.6% yield. Mp 235–237 °C. FTIR: 3473, 3367, 3277, 3057, 2944, 1626, 1580, 1540 cm−1. 1H-NMR (400 MHz, DMSO-d6): δ 8.29 (s, 2H), 7.48 (d, J = 8.0 Hz, 2H), 7.13 (t, J = 8.0 Hz, 2H), 6.69 (d, J = 8.0 Hz, 2H), 6.50 (t, J = 8.0 Hz, 2H), 6.38 (s, 4H), 3.47–3.22 (m, 4H). 13C-NMR (101 MHz, DMSO-d6): δ 169.1, 149.6, 131.6, 128.1, 116.3, 114.8, 114.5, 36.0. HRMS (FAB+) calculated for C16H19N4O2: 299.1508; found 299.1509.
N,N'-(Propane-1,3-diyl)bis(3-aminobenzamide) (2b). White solid, 40.8% yield. Mp 158–160 °C. FTIR: 3470, 3355, 3294, 3055, 2967, 2931, 1625, 1576, 1531 cm−1. 1H-NMR (400 MHz, DMSO-d6): δ 8.21 (s, 2H), 7.47 (d, J = 7.7 Hz, 2H), 7.13 (t, J = 7.2 Hz, 2H), 6.69 (d, J = 7.7 Hz, 2H), 6.51 (t, J = 7.2 Hz, 2H), 6.37 (s, 4H), 3.27 (brs, 4H), 1.81–1.61 (m, 2H). 13C-NMR (101 MHz, DMSO-d6): δ 168.8, 149.4, 131.4, 127.8, 116.2, 114.8, 114.5, 36.5, 29.2. HRMS (FAB+) calculated for C17H21N4O2: 313.1665; found 313.1699.
N,N'-(Butane-1,4-diyl)bis(3-aminobenzamide) (2c). White solid, 72.2% yield. Mp 190–192 °C. FTIR: 3469, 3557, 3284, 3056, 2930, 2860, 1621, 1578, 1534 cm−1. 1H-NMR (400 MHz, DMSO-d6): δ 8.19 (t, J = 4.9 Hz, 2H), 7.46 (d, J = 7.85 Hz, 2H), 7.12 (t, J = 7.4 Hz, 2H), 6.68 (d, J = 7.85 Hz, 2H), 6.49 (t, J = 7.4 Hz, 2H), 6.36 (brs, 4H), 3.40–3.18 (m, 4H), 1.59–1.49 (m, 4H). 13C-NMR (101 MHz, DMSO-d6): δ 168.7, 149.4, 131.3, 127.9, 116.2, 114.9, 114.4, 38.4, 26.7. HRMS (FAB+) calculated for C18H23N4O2: 327.1821; found 327.1820.
N,N'-(Hexane-1,6-diyl)bis(3-aminobenzamide) (2d). White solid, 54.8% yield. Mp 158–160 °C. FTIR: 3473, 3668, 3292, 3057, 2926, 2858, 1622, 1581, 1538 cm−1. 1H-NMR (400 MHz, DMSO-d6): δ 8.16 (t, J = 4.4 Hz, 2H), 7.45 (d, J = 7.9 Hz, 2H), 7.11 (t, J = 7.4 Hz, 2H), 6.67 (d, J = 7.9 Hz, 2H), 6.50 (t, J = 7.4 Hz, 2H), 6.34 (brs, 4H), 3.28–3.10 (m, 4H), 1.59–1.41 (m, 4H), 1.41–1.22 (m, 4H). 13C-NMR (101 MHz, DMSO-d6): δ 169.2, 150.0, 131.9, 128.5, 116.7, 115.6, 115.0, 39.2, 29.7, 26.8.
N,N'-(Octane-1,8-diyl)bis(3-aminobenzamide) (2e). Pearl solid, 36.1% yield. Mp 164–166 °C. FTIR: 3475, 3670, 3292, 3057, 2926, 2858, 1622, 1581, 1538 cm−1. 1H-NMR (400 MHz, DMSO-d6): δ 8.15 (t, J = 4.9 Hz, 2H), 7.45 (d, J = 7.9 Hz, 2H), 7.11 (t, J = 7.4 Hz, 2H), 6.67 (d, J = 7.9 Hz, 2H), 6.49 (t, J = 7.4 Hz, 2H), 6.35 (brs, 4H), 3.19 (q, J = 6.6 Hz, 4H), 1.56–1.41 (m, 4H), 1.36–1.19 (m, 8H). 13C-NMR (101 MHz, DMSO-d6): δ 169.2, 150.0, 131.9, 128.5, 116.7, 115.6, 115.0, 39.3, 29.6, 29.3, 27.0. EM (FAB+): 383 [M + H]+. HRMS (FAB+) calculated for C22H31N4O2: 383.2447; found: 383.2469.
N,N'-(Ethane-1,2-diyl)bis(2-(3-(4-nitrophenyl)ureyl)benzamide) (4a). Yellow solid, 92.9% yield. Mp 214–216 °C. FTIR: 3290, 3218, 3157, 3096, 2934, 2872, 1699, 1629, 1562, 1497 cm−1. 1H-NMR (400 MHz, DMSO-d6): δ 10.55 (s, 2H), 10.43 (s, 2H), 8.85 (brs, 2H), 8.20 (d, J = 9.3 Hz, 4H), 8.20 (dd, J = 8.1, 1.0 Hz, 2H), 7.75 (d, J = 9.3 Hz, 4H), 7.71 (dd, J = 8.1, 1.5 Hz, 2H), 7.43 (t, J = 8.3 Hz, 2H), 7.06 (t, J = 8.3 Hz, 2H), 3.57–3.49 (m, 4H). 13C-NMR (101 MHz, DMSO-d6): δ 168.4, 151.8, 146.6, 140.9, 139.0, 131.4, 128.0, 124.9, 121.6, 121.2, 120.7, 117.6, 25.0. HRMS (FAB+) calculated for C30H27N8O8: 627.5940; found 627.5919.
N,N'-(Propane-1,3-diyl)bis(3-(3-(4-nitrophenyl)ureyl)benzamide) (4c). Yellow solid, 84.8% yield. Mp 218–220 °C. FTIR: 3278, 3218, 3155, 3092, 2924, 2855, 1688, 1616, 1560, 1493 cm−1. 1H-NMR (400 MHz, DMSO-d6): δ 10.58 (s, 2H), 10.41 (s, 2H), 8.77 (t, J = 5.3 Hz, 2H), 8.24 (dd, J = 7.8, 1.2 Hz, 2H), 8.18 (d, J = 9.3 Hz, 4H), 7.75 (d, J = 9.3 Hz, 4H), 7.70 (dd, J = 7.8, 1.2 Hz, 2H), 7.48 (t, J = 7.3 Hz, 2H), 7.10 (t, J = 7.5 Hz, 2H), 3.40 (q, J = 6.4 Hz, 4H), 1.88 (qnt, J = 6.9 Hz, 2H). 13C-NMR (101 MHz, DMSO-d6): δ 168.2, 151.8, 146.5, 140.9, 139.0, 131.4, 127.8, 124.9, 121.6, 121.1, 120.7, 117.6, 28.7, 26.0. HRMS (FAB+) calculated for C31H29N8O8: 641.2108; found: 641.2122.
N,N'-(Butane-1,4-diyl)bis(3-(3-(4-nitrophenyl)ureyl)benzamide) (4c). Yellow solid, 81.6% yield. Mp 230–232 °C. FTIR: 3268, 3213, 3154, 3087, 2949, 2871, 1693, 1617, 1561, 1496 cm−1. 1H-NMR (400 MHz, DMSO-d6): δ 10.62 (s, 2H), 10.42 (s, 2H), 8.76 (t, J = 5.5 Hz, 2H), 8.24 (dd, J = 8.5, 0.9 Hz, 2H), 8.19 (d, J = 9.3 Hz, 4H), 7.76 (d, J = 9.3 Hz, 4H), 7.68 (dd, J = 7.8, 1.5 Hz, 2H), 7.46 (t, J = 8.5 Hz, 1H), 7.08 (t, J = 7.8 Hz, 1H), 3.43–3.23 (m, 4H), 1.69–1.55 (m, 4H). 13C-NMR (101 MHz, DMSO-d6): δ 168.2, 151.9, 146.7, 141.0, 139.1, 131.5, 127.8, 125.0, 121.6, 121.2, 120.7, 117.7, 26.5, 25.1. HRMS (FAB+) calculated for C32H31N8O8: 655.2265; found: 655.2276.
N,N'-(Hexane-1,6-diyl)bis(3-(3-(4-nitrophenyl)ureyl)benzamide) (4d). Yellow solid, 91.9% yield. Mp 236–238 °C. FT-IR: 3276, 3216, 3155, 3090, 2964, 2868, 1697, 1621, 1564, 1497 cm−1. 1H-NMR (400 MHz, DMSO-d6): δ 10.60 (s, 2H), 10.41 (s, 2H), 8.72 (t, J = 5.5 Hz, 2H), 8.23 (d, J = 7.8 Hz, 2H), 8.18 (d, J = 9.3 Hz, 4H), 7.76 (d, J = 9.3 Hz, 4H), 7.67 (dd, J = 7.8, 1.2 Hz, 2H), 7.46 (t, J = 8.1 Hz, 2H), 7.09 (t, J = 8.1 Hz, 2H), 3.29 (q, J = 6.6 Hz, 4H), 1.65–1.49 (m, 4H), 1.44–1.30 (m, 4H). 13C-NMR (101 MHz, DMSO-d6): δ 168.2, 151.9, 146.7, 141.1, 139.1, 131.4, 127.8, 125.0, 121.7, 121.4, 120.8, 117.7, 28.9, 26.3, 25.1.
N,N'-(Octane-1,8-diyl)bis(3-(3-(4-nitrophenyl)ureyl)benzamide) (4e). Brown solid, 96.0% yield. Mp 236–238 °C. FTIR: 3278 3218, 3155, 3092, 2924, 2855, 1688, 1616, 1560 cm−1. 1H-NMR (400 MHz, DMSO-d6): δ 10.59 (s, 2H), 10.41 (s, 2H), 8.70 (t, J = 5.6 Hz, 2H), 8.23 (dd, J = 8.4, 1.0 Hz, 2H), 8.19 (d, J = 9.3 Hz, 4H), 7.76 (d, J = 9.3 Hz, 4H), 7.66 (dd, J = 8.0, 1.3 Hz, 2H), 7.47 (t, J = 8.4 Hz, 2H), 7.09 (t, J = 8.0 Hz, 2H), 3.27 (q, J = 6.8 Hz, 4H), 1.62–1.47 (m, 4H), 1.40–1.22 (m, 8H). 13C-NMR (101 MHz, DMSO-d6): δ 168.1, 151.9, 146.7, 141.0, 139.0, 131.4, 127.8, 125.0, 121.7, 121.4, 120.7, 117.7, 39.2, 28.8, 26.5. HRMS (FAB+) calculated for C36H39N8O8: 711.2891; found: 711.2901.
N,N'-(Ethane-1,2-diyl)bis(3-(3-(4-nitrophenyl)benzamide)) (6a). White solid, 88.7% yield. Mp 216–218 °C. FTIR: 3300, 3079, 2947, 2869, 1637, 1551, 1453 cm−1. 1H-NMR (400 MHz, DMSO-d6): δ 9.08 (s, 2H), 8.69 (t, J = 2.2 Hz, 2H), 8.38 (ddd, J = 8.2, 2.2, 1.2 Hz, 2H), 8.32 (td, J = 7.8, 1.2 Hz, 2H), 7.79 (t, J = 8.2 Hz, 2H), 3.57–3.48 (m, 4H). 13C-NMR (101 MHz, DMSO-d6): δ 164.3, 147.6, 135.9, 133.6, 130.0, 125.7, 121.9, 36.5. MS (FAB+) m/z: 359 [M + H]+. HRMS (FAB+) calculated for C16H15N4O6: 359.0992; found: 359.0981.
N,N'-(Propane-1,3-diyl)bis(3-(3-(4-nitrophenyl)benzamide)) (6b). White solid, 90.5% yield. Mp 210–212 °C. FTIR: 3363, 3098, 2945, 2874, 1637, 1548, 1524, 1471 cm−1. 1H-NMR (400 MHz, DMSO-d6): δ 8.93 (t, J = 5.5 Hz, 2H), 8.68 (t, J = 2.0 Hz, 2H), 8.38 (ddd, J = 8.0, 2.0, 1.0 Hz, 2H), 8.31 (td, J = 8.0, 2.0 Hz, 2H), 7.78 (t, J = 8.0 Hz, 2H), 3.46–3.36 (m, 4H), 1.87 (qnt, J = 6.9 Hz, 2H). 13C-NMR (101 MHz, DMSO-d6): δ 164.0, 147.7, 135.9, 133.5, 130.0, 125.7, 121.8, 37.4, 28.7. MS (FAB+) m/z: 373 [M + H]+. HRMS (FAB+) calculated for C17H17N4O6: 373.1148; found: 373.1143.
N,N'-(Butane-1,4-diyl)bis(3-(3-(4-nitrophenyl)benzamide)) (6c). White solid, 80.3% yield. Mp 242–244 °C. FTIR: 3361, 3091, 2937, 2861, 1637, 1551, 1523, 1478 cm−1. 1H-NMR (400 MHz, DMSO-d6): δ 8.87 (t, J = 5.4 Hz, 2H), 8.67 (t, J = 2.0 Hz, 2H), 8.37 (ddd, J = 0.7, 2.0, 8.1 Hz, 1H), 8.29 (td, J = 1.2, 8.1 Hz, 2H), 7.77 (t, J = 8.1 Hz, 2H), 3.39–3.26 (m, 4H), 1.66–1.56 (m, 4H). 13C-NMR (101 MHz, DMSO-d6): δ 163.8, 147.6, 135.9, 133.5, 130.0, 125.6, 121.7, 33.6, 26.4. MS (FAB+) m/z: 387 [M + H]+. HRMS (FAB+) calculated for C18H19N4O6: 387.1305; found: 387.1310.
N,N'-(Hexane-1,6-diyl)bis(3-(3-(4-nitrophenyl)benzamide)) (6d). White solid, 59% yield. Mp 192–194 °C. FTIR: 3309, 3100, 2935, 2873, 1629, 1577, 1530, 1478 cm−1. 1H-NMR (400 MHz, DMSO-d6): δ 8.83 (t, J = 5.4 Hz, 2H), 8.67 (t, J = 2.0 Hz, 2H), 8.37 (ddd, J = 8.1, 2.3, 1.0 Hz, 2H), 8.28 (td, J = 8.1, 1.3 Hz, 2H), 7.77 (t, J = 8.1 Hz, 2H), 3.38–3.24 (m, 4H), 1.63–1.47 (m, 4H), 1.46–1.30 (m, 4H). 13C-NMR (101 MHz, DMSO-d6): δ 163.8, 147.7, 135.9, 133.5, 130.0, 125.6, 121.8, 39.3, 28.8, 26.1. MS (FAB+) m/z: 415 [M + H]+. HRMS (FAB+) calculated for C20H23N4O6: 415.4260; found: 415.4268.
N,N'-(Octane-1,8-diyl)bis(3-(3-(4-nitrophenyl)benzamide)) (6e). White solid, 59.0% yield. Mp 178–180 °C. FTIR: 3312, 3100, 2932, 2872, 1629, 1577, 1530, 1477 cm−1. 1H-NMR (400 MHz, DMSO-d6): δ 8.81 (s, 2H), 8.66 (s, 2H), 8.28 (d, J = 7.3 Hz, 2H), 8.37 (d, J = 8.1 Hz, 2H), 7.77 (t, J = 7.3 Hz, 2H), 1.55 (m, 4H), 1.43–1.17 (m, 8H).
N,N'-(Ethane-1,2-diyl)bis(3-aminobenzamide) (7a). Yellow solid, 42.0% yield. Mp 280–282 °C. FTIR: 3456, 3330, 3216, 3056, 2949, 2912, 1632, 1579, 1537, 1486 cm−1. 1H-NMR (200 MHz, DMSO-d6): δ 8.33 (brs, 2H), 7.07 (t, J = 7.7 Hz, 2H), 7.04 (brs, 2H), 6.95 (dd, J = 7.8, 1.3 Hz, 2H), 6.69 (ddd, J = 7.8, 2.1, 1.0 Hz, 2H), 5.20 (brs, 4H), 3.64–3.21 (m, 4H). 13C-NMR (50 MHz, DMSO-d6): δ 167.0, 148.4, 135.1, 128.3, 116.1, 114.0, 112.6, 37.5. MS (FAB+) m/z: 299 [M + H]+. HRMS (FAB+) calculated for C16H19N4O2: 299.1508; found: 299.1509.
N,N'-(Propane-1,3-diyl)bis(3-aminobenzamide) (7b). Brown solid, 97.8% yield. Mp 98–100 °C. FTIR: 3324, 3226, 3063, 2931, 1622, 1579, 1528, 1487 cm−1. 1H-NMR (200 MHz, DMSO-d6): δ 8.29 (t, J = 5.7 Hz, 2H), 7.08 (t, J = 7.7 Hz, 2H), 7.05 (s, 2H), 6.96 (d, J = 7.7 Hz, 2H), 6.69 (ddd, J = 7.7, 2.4, 1.0 Hz, 2H), 5.22 (brs, 4H), 3.35–3.20 (m, 4H). 13C-NMR (50 MHz, DMSO-d6): δ 167.5, 149.1, 136.0, 129.1, 116.8, 114.6, 113.2, 37.2, 29.8. MS (FAB+) m/z: 313 [M + H]+. HRMS (FAB+) calculated for C17H21N4O2: 313.1665; found: 312.1699.
N,N'-(Butane-1,4-diyl)bis(3-aminobenzamide) (7c). Orange solid, 69.2% yield. Mp 120–122 °C. FTIR: 3433, 3332, 3222, 3057, 2943, 2866, 1647, 1583, 1547, 1520 cm−1. 1H-NMR (200 MHz, DMSO-d6): δ 8.23 (t, J = 5.5 Hz, 2H), 7.04 (t, J = 7.5 Hz, 2H), 7.01 (s, 2H), 6.94 (ddd, J = 7.5, 2.7, 1.1 Hz, 2H), 6.67 (ddd, J = 7.5, 2.4, 1.3 Hz, 2H), 5.21 (brs, 4H), 3.32–3.12 (m, 4H), 1.52 (brs, 4H). MS (CI+) m/z: 326 [M + H]+. HRMS (FAB+) calculated for C18H23N4O2: 327.1821; found: 327.1820.
N,N'-(Hexane-1,6-diyl)bis(3-aminobenzamide) (7d). Melon color solid, 80.3% yield. Mp 100–102 °C. FTIR: 3390, 3324, 3218, 3054, 2928, 2854, 1625, 1582, 1533, 1489 cm−1. 1H-NMR (200 MHz, DMSO-d6): δ 8.18 (t, J = 5.6 Hz, 2H), 7.00 (t, J = 7.6 Hz, 2H), 7.01 (s, 2H), 6.92 (td, J = 7.6, 0.5 Hz, 2H), 6.66 (ddd, J = 7.6, 2.0, 1.0 Hz, 2H), 5.19 (s, 4H), 3.31–3.08 (m, 4H), 1.63–1.40 (m, 4H), 1.31 (brs, 4H). 13C-NMR (50 MHz, DMSO-d6): δ 166.9, 148.6, 135.8, 128.5, 116.2, 114.2, 112.8, 39.0, 29.2, 26.3.
N,N'-(Octane-1,8-diyl)bis(3-aminobenzamide) (7e). Beige solid, 337 mg, 0.88 mmol, 90.4% yield. Mp 120–122 °C. FTIR: 3393, 3349, 3208, 3054, 2926, 2853, 1625, 1583, 1538, 1490 cm−1. 1H-NMR (200 MHz, DMSO-d6): δ 8.17 (t, J = 5.5 Hz, 2H), 7.15–6.85 (d, J = 7.8 Hz, 2H), 7.01 (s, 2H), 6.92 (dd, J = 7.8, 1.2 Hz, 2H), 6.66 (td, J = 7.8, 1.1 Hz, 1H), 5.19 (brs 4H), 3.19 (q, J = 6.5 Hz, 4H), 1.58–1.39 (m, 4H), 1.28 (s, 8H). 13C-NMR (50 MHz, DMSO-d6): δ 166.9, 148.6, 135.8, 128.5, 116.2, 114.2, 112.9, 29.2, 28.8, 26.5. MS (FAB+) m/z: 383 [M + H]+. HRMS (FAB+) calculated for C22H31N4O2: 383.2447; found: 383.2469.
N,N'-(Propane-1,3-diyl)bis(3-(3-(4-nitrophenyl)ureyl)benzamide) (8b). Brown solid, 51.7% yield. Mp 184–186 °C. FTIR: 3288, 3088, 1638, 1559 cm−1. 1H-NMR (400 MHz, DMSO-d6): δ 9.47 (s, 2H), 9.08 (s, 2H), 8.49 (t, J = 5.6 Hz, 2H), 8.20 (d, J = 9.3 Hz, 4H), 7.94 (t, J = 1.7 Hz, 1H), 7.71 (d, J = 9.3 Hz, 4H), 7.64 (ddd, J = 7.9, 2.2, 1.2 Hz, 2H), 7.50 (td, J = 7.9, 1.2 Hz, 2H), 7.40 (t, J = 7.9 Hz, 2H), 3.59–3.03 (m, 4H), 1.79 (qnt, J = 6.8 Hz, 2H). 13C-NMR (101 MHz, DMSO-d6): δ 166.1, 151.9, 146.2, 141.0, 139.0, 135.4, 128.7, 125.0, 121.1, 120.1, 117.7, 117.5, 37.0, 29.2. MS (FAB+) m/z: 641 [M + H]+. HRMS (FAB+) calculated for C31H29N8O8: 641.2108; found: 641.2143.
N,N'-(Buthane-1,4-diyl)bis(3-(3-(4-nitrophenyl)ureyl)benzamide) (8c). Pink solid, 77.0% yield. Mp 262–264 °C. FTIR: 3256, 3058, 1633, 1535 cm−1. 1H-NMR (400 MHz, DMSO-d6): δ 9.45 (s, 2H), 9.06 (s, 2H), 8.46 (t, J = 5.5 Hz, 2H), 8.20 (d, J = 9.4 Hz, 4H), 7.91 (t, J = 1.8 Hz, 2H), 7.71 (d, J = 9.4 Hz, 4H), 7.63 (ddd, J = 8.0, 2.2, 1.2 Hz, 2H), 7.49 (td, J = 8.0, 1.1 Hz, 2H), 7.39 (t, J = 8.0 Hz, 2H), 3.32–3.21 (m, 4H), 1.64–1.51 (m, 4H). 13C-NMR (101 MHz, DMSO-d6): δ 166.0, 151.9, 146.2, 141.0, 139.0, 135.5, 128.6, 125.0, 121.0, 120.9, 117.8, 117.5, 38.9, 26.7. MS (FAB+) m/z: 655 [M + H]+. HRMS (FAB+) calculated for C32H31N8O8: 655.2265; found: 656.2221.
N,N'-(Hexane-1,6-diyl)bis(3-(3-(4-nitrophenyl)ureyl)benzamide) (8d). Yellow solid, 70.8% yield. Mp 241–243 °C. FTIR: 3328, 3093, 1624, 1550 cm−1. 1H-NMR (400 MHz, DMSO-d6): δ 9.45 (s, 2H), 9.05 (s, 2H), 8.42 (t, J = 5.1 Hz, 2H), 8.19 (d, J = 9.0 Hz, 4H), 7.90 (s, 2H), 7.70 (d, J = 9.0 Hz, 4H), 7.62 (d, J = 8.1 Hz, 2H), 7.47 (d, J = 7.6 Hz, 2H), 7.38 (t, J = 7.8 Hz, 2H), 3.29–3.15 (m, 4H), 1.61–1.45 (m, 4H), 1.42–1.27 (m, 4H). MS (FAB+) m/z: 683 [M + H]+. HRMS (FAB+) calculated for C34H35N8O8: 683.2578; found: 683.2586.
N,N'-(Octane-1,8-diyl)bis(3-(3-(4-nitrophenyl)ureyl)benzamide) (8e). Yellow solid, 97% yield. Mp 266–268 °C. FTIR: 3278, 3082, 2930, 2855, 1706, 1628, 1585, 1543 cm−1. 1H-NMR (400 MHz, DMSO-d6): δ 9.46 (s, 2H), 9.06 (s, 2H), 8.41 (t, J = 5.5 Hz, 2H), 8.20 (d, J = 9.3 Hz, 4H), 7.90 (t, J = 1.8 Hz, 2H), 7.71 (d, J = 9.3 Hz, 4H), 7.63 (ddd, J = 8.0, 2.2, 1.0 Hz, 2H), 7.47 (td, J = 8.0, 1.5 Hz, 2H), 7.38 (t, J = 8.0 Hz, 2H), 3.29–3.19 (m, 4H), 1.61–1.45 (m, 4H), 1.32 (s, 8H). 13C-NMR (101 MHz, DMSO-d6): δ 165.8, 151.8, 146.1, 140.8, 138.8, 135.4, 128.5, 124.9, 120.7, 117.6, 117.3, 39.0, 28.9, 28.6, 26.3.
Molecular modeling
The molecular optimization of the geometries for the proposed complexes was carried out by DFT method, using B3LYP and the 6-31G(d) base in the Gaussian 09 computational program.24Results and discussion
Synthesis and characterization
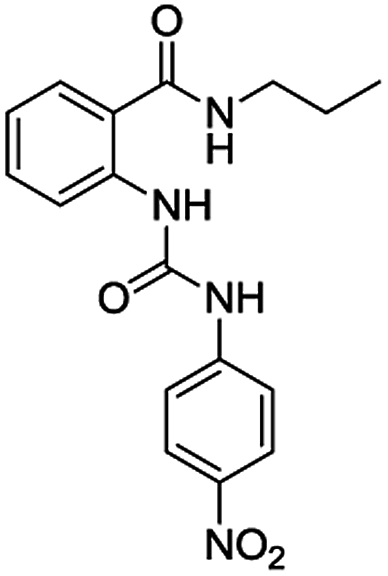
The synthesis of bis(ureas) 4 (ortho receptors) started with the reaction of isatoic anhydride (1) and an alkyldiamine (a: ethane-1,2-diamine, b: propane-1,3-diamine, c: buthane-1,4-diamine, d: hexane-1,6-diamine, e: octane-1,8-diamine) in THF at reflux temperature to obtain the corresponding oligomethylene bis(2-aminobenzamides) 2 with 81%, 41%, 72%, 55%, and 36% yields, respectively. The intermediates 2 were directly reacted with 4-nitrophenyl isocyanate in THF to obtain receptors 4 in 93%, 85%, 82%, 92%, and 96% yields, respectively (Scheme 1). An ortho mono-ureylbenzamide was synthetized under the same experimental conditions using propylamine in a 36% global yield.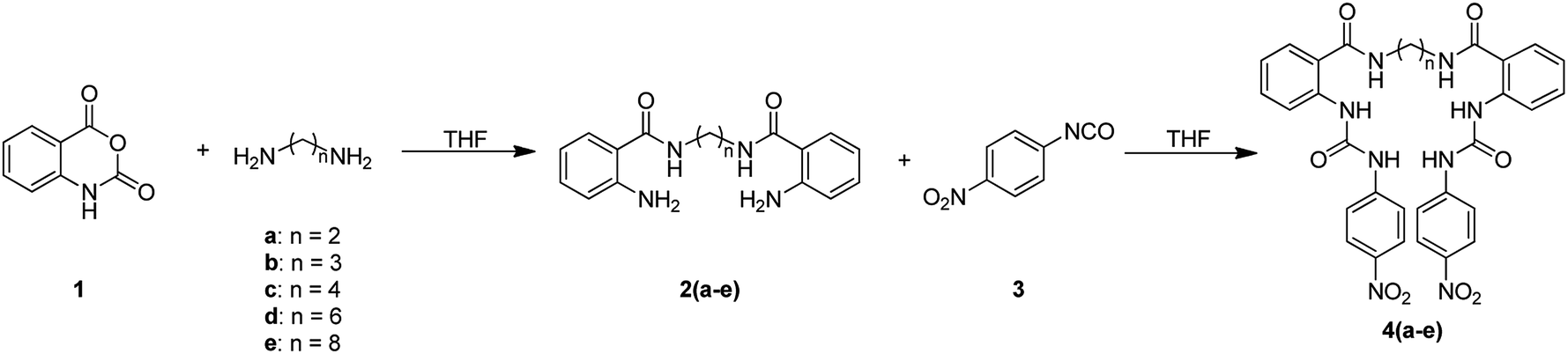 | ||
| Scheme 1 | ||
In the FTIR spectra, stretching N–H vibrations for amide and urea groups were observed in the range of 3154 to 3290 cm−1. The vibrations for urea carbonyls were found at 1705 to 1720 cm−1 and the amide carbonyls at 1688 to 1705 cm−1. The 1H NMR spectra display the typical ABCD pattern for the ortho-disubstituted benzamide and the A2B2 pattern for the nitrophenyl group. Both urea hydrogens are shifted downfield in the range of 10.40 to 10.62 ppm and the amide hydrogen at 8.70 to 8.85 ppm. The signals for the aliphatic hydrogens appear upfield (Fig. S1–S6†).
For the synthesis of bis(ureas) 8 (meta receptors), 3-nitrobenzoyl chloride (5) was reacted with a dialkylamine in the presence of TEA at RT in anhydrous THF. The bis-(3-nitrobenzamide) intermediates 6 were obtained with 88%, 90%, 80%, 59%, and 25% yields, respectively. Then, the nitro groups were reduced to amino using an H2 atmosphere and Pd (10%)/C at RT in dry MeOH. The bis-(3-aminobenzamide) intermediates 7 were obtained with 42%, 98%, 69%, 80%, and 90% yields. Compounds 7 were reacted with 4-nitrophenyl isocyanate in THF to obtain the bis-urea receptors 8 in 52%, 77%, 71%, and 97% yields (Scheme 2). A meta mono-ureylbenzamide was synthetized under the same experimental conditions using propylamine in a 20% global yield.
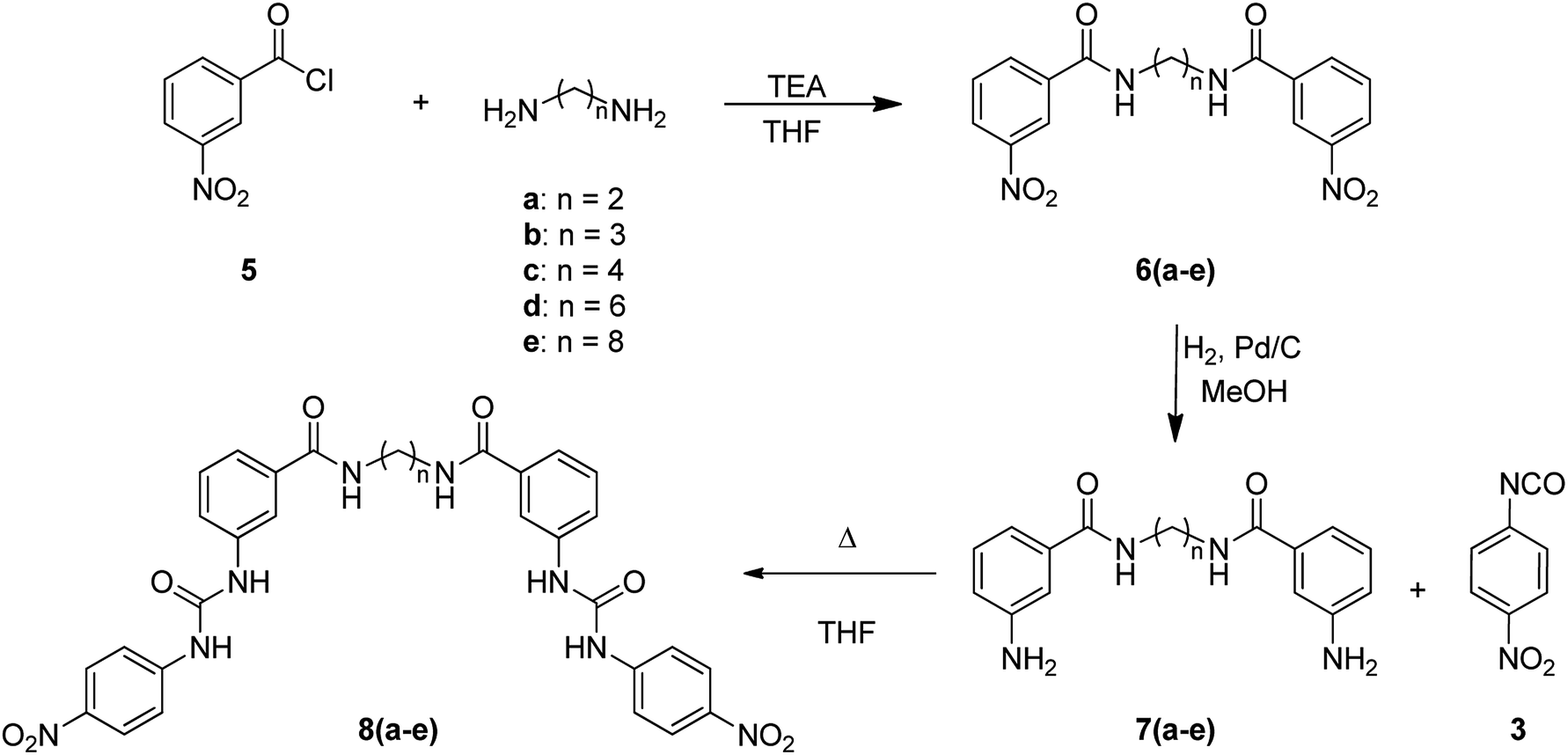 | ||
| Scheme 2 | ||
In the FTIR spectra, stretching N–H vibrations for amide and urea groups were observed in the range of 3221 to 3331 cm−1. The vibrations for urea carbonyls were found in the range 1706–1714 cm−1 and the amide carbonyls in the range 1625–1640 cm−1. In the 1H NMR spectra, the typical ABCX pattern for the meta-disubstituted benzamide and the A2B2 pattern for the nitrophenyl group were observed. Both urea hydrogens were shifted downfield in the range of 9.05 to 9.47 ppm and the amide hydrogen at 8.41 to 8.49 ppm. The signals for the aliphatic hydrogens appeared upfield (Fig. S7–S11†).
The chemical shift of urea and amide hydrogens in receptors 4 and 8 was directly related with the relative position of both functional groups, which was the result of a combination of resonance and inductive effects. The effect of the two electron-withdrawing groups, i.e., the nitrophenyl and benzamide, over these hydrogens was dramatically enhanced when they were in the ortho position. The acidity and the spatial availability of these hydrogens depends on the position and may result in different interaction strength with anions.
Study of anion recognition by UV-vis
The ortho and meta N-propyl ureabenzamides and receptors 4 and 8 presented an absorption band at 330–340 nm due to the nitrophenyl chromophore. The changes in this band were monitored by titration with different TBAX salts (X = AcO− (A), BzO− (B), F− (F), H2PO4− (HP), and HP2O73− (HPP)) in acetonitrile. All the receptors showed spectral changes in the titration depending on the receptor structure and the anion. Fig. 2 shows the spectra obtained in the titration of receptors 4d and 8d with TBAA in acetonitrile. There were bathochromic and hypochromic shifts in the absorption of receptor 4d at 338 nm during the titration, indicating the interaction with the anion, while there were bathochromic and hyperchromic shifts in the absorption band at 336 nm in receptor 8d. A new maximum absorbance band was observed at 362 nm. As can be seen, the changes in 8d were more evident than in 4d. In general, there were more evident spectroscopic changes in receptors 8 than in receptors 4, which was directly related to the relative position of the urea and benzamide groups in these receptors (Fig. S12–S23†). All the receptors showed a spectroscopic response toward the studied anions. However, the TBHPP induced the greatest changes in the absorption bands when lower molar equivalents were added. This was probably due to the higher number negative charges and the geometry of this anion. The TBAHPP salt induced a partial deprotonation at the ortho N-propyl ureabenzamide, which was evidenced by the presence of a band at 450 nm (Fig. S12i†). However, this new band was not observed in the titrations of meta N-propyl ureabenzamide and receptors 4 and 8. | ||
| Fig. 2 UV-vis spectra and absorbance profiles obtained in the titration of 4d (a and b) and 8d (c and d) with TBAA in acetonitrile [4d] and [8d] = 1 × 10−5 M. | ||
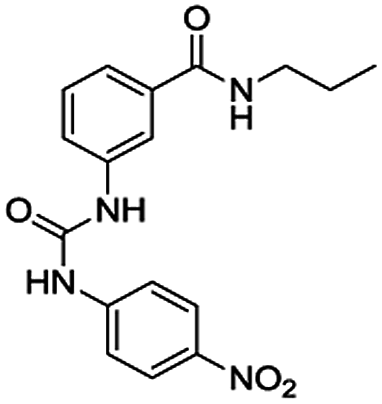
Table 1 lists the association constants (logK) for the complexes of receptors 4 calculated from the absorbance data using the HypSpec program,21–23 and are compared with those obtained with the ortho mono-ureylbenzamide receptor. Interestingly, the N-propyl mono-ureylbenzamide formed complexes with a 2:1 (receptor:anion) stoichiometry with TBAA, TBAB, and TBAHPP, which agreed with the possible cooperativity of both ureylbenzamide units in receptors 4 with the shorter oligomethylene spacer chains, and 1:1 complexes with TBAF and TBAHP. This behavior was not observed with the previously reported N-benzyl ortho mono-ureylbenzamide,20 where all the complexes had a 1:1 stoichiometry and the logK values ranged from 3.85 to 4.35. Thus, the stoichiometry and affinity toward the anions were improved with the N-propyl substituent. All receptors 4 formed exclusively a 1:1 complex with TBAA and TBAB and there was a tendency for the logK11 to decrease as the oligomethylene chain length increased. This behavior suggests the cooperativity of both ureylbenzamide units interacting with the carboxylate unit in these complexes. Two complexes with 1:1 and 1:2 (receptor:anion) stoichiometry were observed with TBAF and TBAHP; in particular, receptor 4e formed only the 1:2 complex. Noteworthy, two complexes with 2:1 and 1:1 stoichiometry were detected with TBAHPP and the values of logK11 and logK21 increased as the spacer length increased. This unusual behavior with pyrophosphate was due to its differences in size, shape, and charge. Also, the logK11 values for fluoride and phosphate complexes with receptors 4 were one or two units higher compared to the mono ureylbenzamide, which may indicate a cooperative effect of both interaction sites.
K)a for receptors 4 with TBAX salts determined by UV-vis in CH3CN
| Receptor | X | logK11 |
logK12 |
logK21 |
|---|---|---|---|---|
| a Standard deviation of the fit is given in parenthesis. | ||||
 |
AcO− | 10.11(2) | ||
| BzO− | 10.92(2) | |||
| F− | 5.62(1) | |||
| H2PO4− | 5.09(2) | |||
| 4a | AcO− | 6.51(2) | ||
| BzO− | 5.322(4) | |||
| F− | 6.64(6) | 12.87(5) | ||
| H2PO4− | 6.3(1) | 11.9(1) | ||
| HP2O73− | 5.55(4) | 10.75(7) | ||
| 4b | AcO− | 5.689(4) | ||
| BzO− | 4.661(4) | |||
| F− | 6.08(5) | 11.41(4) | ||
| H2PO4− | 5.2(1) | 10.64(6) | ||
| HP2O73− | 4.86(6) | 10.38(5) | ||
| 4c | AcO− | 6.012(8) | ||
| BzO− | 4.941(2) | |||
| F− | 6.34(7) | 11.98(7) | ||
| H2PO4− | 7.54(6) | 12.50(8) | ||
| HP2O73− | 5.73(5) | 11.27(9) | ||
| 4d | AcO− | 5.321(2) | ||
| BzO− | 4.806(3) | |||
| F− | 6.07(4) | 11.50(3) | ||
| H2PO4− | 6.55(2) | |||
| HP2O73− | 7.4(4) | 13.9(9) | ||
| 4e | AcO− | 4.997(3) | ||
| BzO− | 4.716(3) | |||
| F− | 9.14(2) | |||
| H2PO4− | 6.85(2) | 12.24(7) | ||
| HP2O73− | 6.9(1) | 13.3(3) | ||
Table 2 lists the association constants (logK) for the complexes of receptors 8 calculated from the absorbance data using the HypSpec program21–23 and these are compared with those obtained with the corresponding meta mono-ureylbenzamide. The mono-ureylbenzamide mainly formed complexes of a 1:1 receptor–anion stoichiometry with all the TBAX salts, and formed 2:1 complexes with acetate and fluoride, indicating a different behavior with its ortho analogue. The logK values were one to three units higher than those obtained with the N-benzyl ortho mono-ureylbenzamide previously reported,20 evidencing the positive effect of the N-propyl substituent in the affinity toward anions. Also, there were difference in the complex stoichiometries with TBAA and TBAF. In general, receptors 8 formed complexes with both 1:1 and 1:2 stoichiometries with the TBAX salts, and a 2:1 complex with TBAHPP. This anion may serve as a template for the self-assembly of two receptor molecules at low concentration, and there was a tendency for logK21 to increase as the oligomethylene chain length increased, indicating a higher affinity. The receptor 8a (with the shorter spacer chain) formed a 1:1 complex with TBAA, TBAB, and TBAF and a 1:2 complex with TBAHP, while 8c did this with TBAA and TBAB.
K)a for receptors 8 with TBAX salts determined by UV-vis in CH3CN
| Receptor | X | logK11 |
logK12 |
logK21 |
|---|---|---|---|---|
| a Standard deviation of the fit is given in parenthesis. | ||||
 |
AcO− | 7.5(2) | 13.7(4) | |
| BzO− | 5.931(4) | |||
| F− | 8.1(2) | 13.5(2) | ||
| H2PO4− | 6.31(1) | 12.1(1) | ||
| HP2O73− | 6.31(1) | |||
| 8a | AcO− | 6.84(2) | ||
| BzO− | 6.007(6) | |||
| F− | 5.050(4) | |||
| H2PO4− | 5.76(5) | 11.17(4) | ||
| HP2O73− | 10.93(2) | |||
| 8b | AcO− | 5.90(5) | 10.0(2) | |
| BzO− | 5.83(4) | 9.83(9) | ||
| F− | 8.4(3) | 13.9(3) | ||
| H2PO4− | 5.932(8) | 11.29(1) | ||
| HP2O73− | 10.94(1) | |||
| 8c | AcO− | 5.502(6) | ||
| BzO− | 5.160(5) | |||
| F− | 8.6(3) | 14.7(3) | ||
| H2PO4− | 5.12(3) | 10.51(3) | ||
| HP2O73− | 7.02(7) | 14.3(1) | ||
| 8d | AcO− | 6.01(5) | 10.0(2) | |
| BzO− | 5.80(3) | 10.25(5) | ||
| F− | 5.20(2) | |||
| H2PO4− | 7.74(7) | 13.43(7) | ||
| HP2O73− | 6.80(9) | 14.5(1) | ||
| 8e | AcO− | 5.89(6) | 9.8(5) | |
| BzO− | 5.55(5) | 9.2(4) | ||
| F− | ||||
| H2PO4− | 6.07(3) | 11.88(4) | ||
| HP2O73− | 11.90(3) | |||
Study of anion recognition by 1H NMR
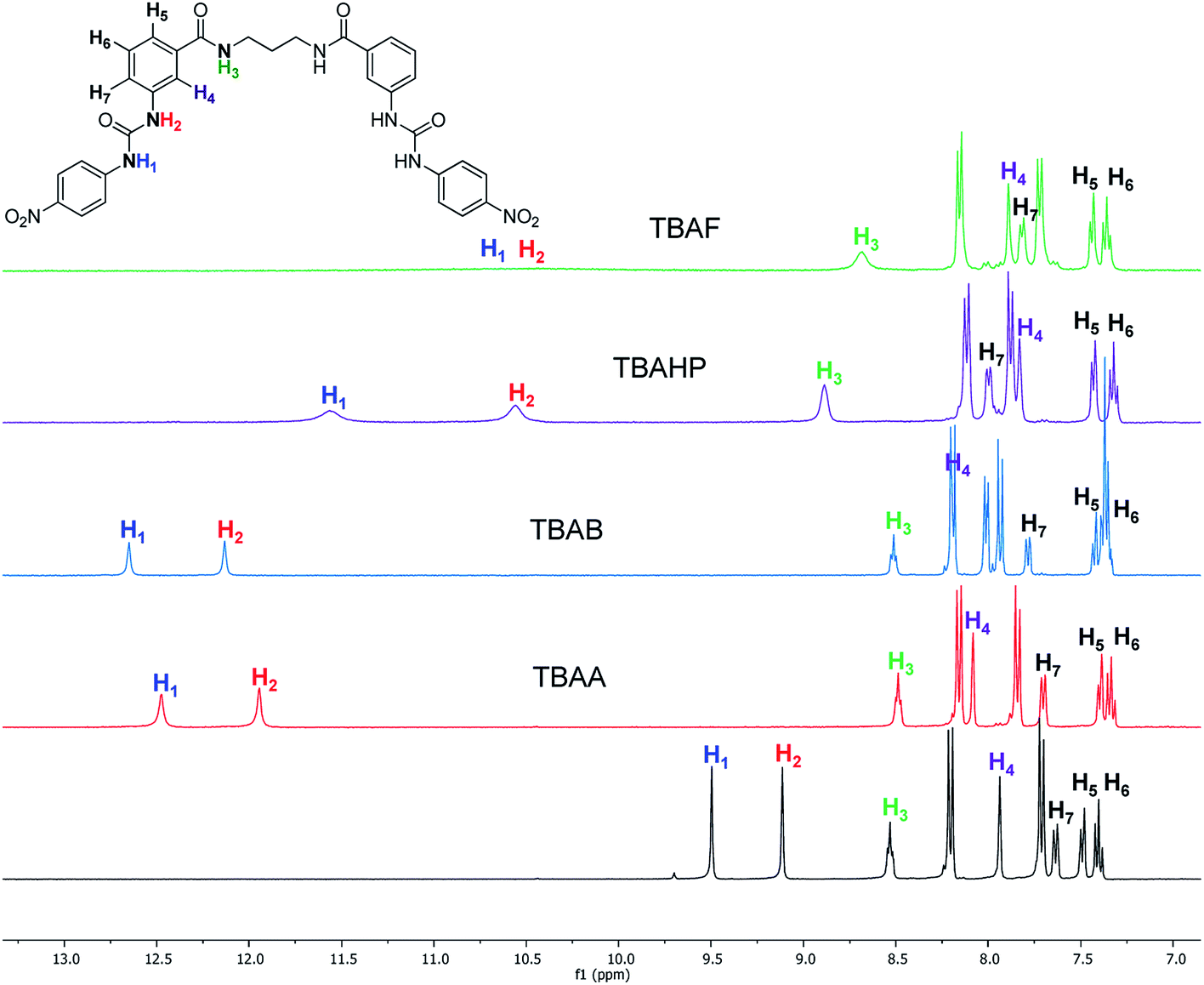
The interaction of the ortho monorureylbenzamide, 4b, 4c, and 4d with TBAA, TBAB, TBAF, TBAHP, and TBAHPP was analyzed by 1H NMR in DMSO-d6 due to the solubility of these receptors (Fig. S24–S43†). As depicted in Fig. 3, all the anions promoted different changes in the NMR signals of receptor 4b once the complex was formed in solution because of their differences in size, shape, and basicity. There was a non-equivalent downfield shift of the urea signals with TBAA and TBAB, indicating that both hydrogens H1 and H2 interacted with the anion, but not with the same strength. The spectra obtained during the titration (Fig. 4) indicated that H2 was more affected than H1. Also, it is interesting that the amide hydrogen (H3) signal was broadened, which means this hydrogen was also involved in the complex, and the H4 hydrogen at the ortho position to the amide carbonyl shifted to upfield when the interaction with the anion occurred. The same spectral changes were observed in the other the receptors with these anions.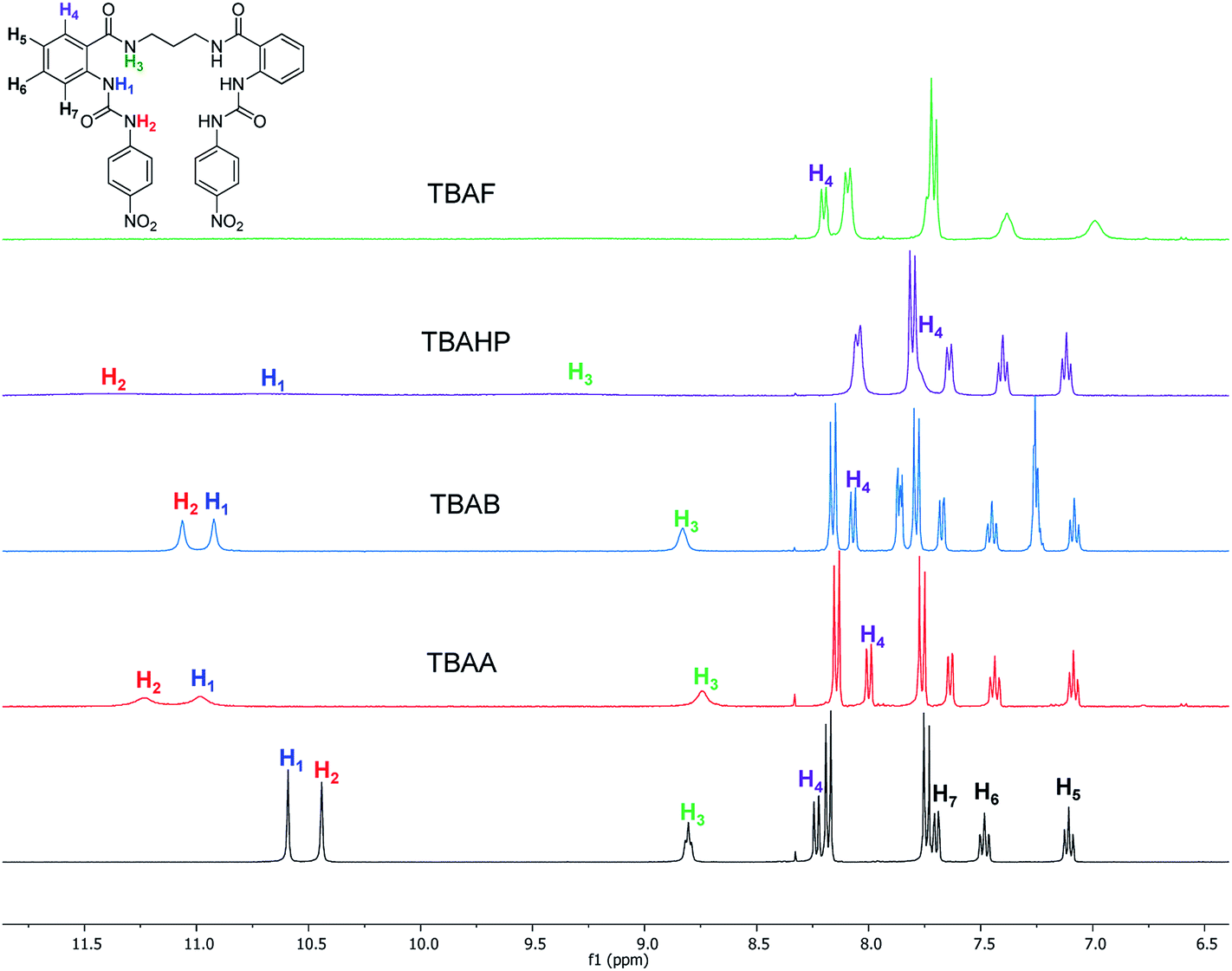 | ||
| Fig. 3 Partial 1H-NMR spectra of free 4b and with two molar equivalents of TBAA, TBAB, TBAHP, and TBAF in DMSO-d6. | ||
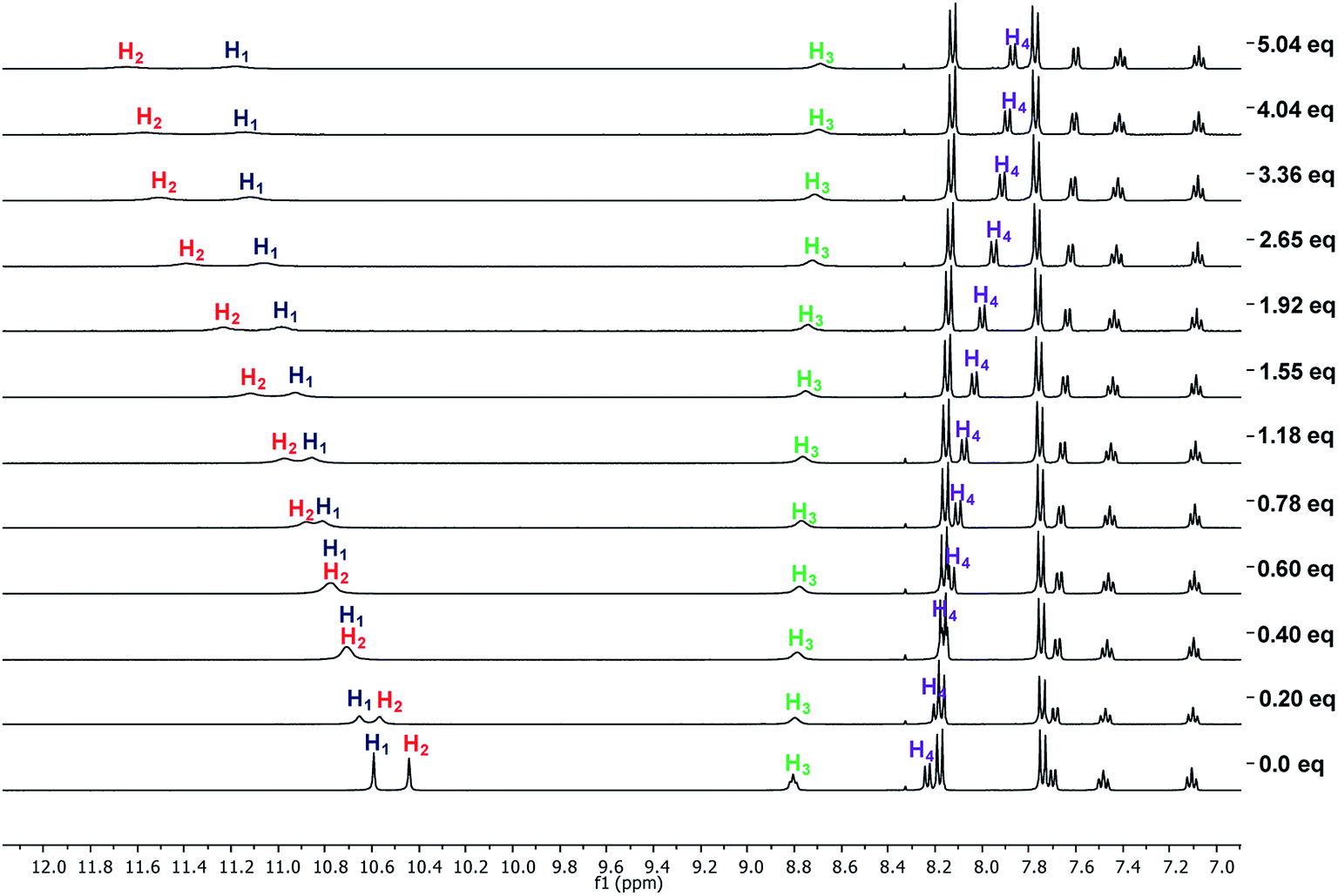 | ||
| Fig. 4 Partial 1H-NMR spectra obtained in the titration of 4b (6 mM) with TBAA in DMSO-d6. | ||
There was a downfield shift and broadening of the H1, H2, and H3 signals, indicating a strong participation of the three hydrogens in the supramolecular complex with TBAHP. Again, H2 was more affected than H1 and H4, being considerably shifted upfield, probably due to a conformational change in the amide group, where the “ortho effect” over H4 was avoided. In the titration with TBAF, all the NH hydrogens disappeared, indicating an acid–base reaction; a change of color also occurred in the solution. The titration with TBAHPP showed the deprotonation of urea hydrogens since the first anion aliquot due to an acid–base reaction. Also, the signal of the amide hydrogens H3 underwent a downfield shift and widening, indicating a strong interaction with this hydrogen. The signal for H4 was considerably shifted to upfield and became broad. Noteworthy, there were exceptional changes in the NMR spectra when more than two equivalents of TBAHPP were added to the solution. The signals of the A2B2 system for the nitrophenyl group were collapsed in one signal, and the rest of the signals showed significant changes in their chemical shifts and become broad. The same patterns were observed with the rest of the studied receptors (Fig. S28, S33, S38 and S43†).
The association constants for the complexes of ortho mono-ureylbenzamide, 4b, 4c, and 4d receptors were calculated from the chemical shifts data using the HypNMR program21–23 and are reported in Table 3. As expected, the ortho monoureylbenzamide receptor formed complexes with a 1:1 stoichiometry with all the studied anions. The chemical shift profiles obtained with TBAA and TBAB fitted the 1:1 stoichiometry model, and the values of logK11 decreased as the oligomethylene chain length increased in receptors 4. These association constants are comparable to those reported in the literature for other receptors,19 even though receptors 4 had a flexible interconnection chain. The chemical shift profiles in the titration with TBAHP fitted 1:1 and 1:2 stoichiometries and the values of logK11 were higher than those obtained with TBAA and TBAB. Both logK11 and logK12 values depended on the oligomethylene chain length decreasing as the oligomethylene chain increased. The values of logK with TBAF and TBAHPP could not be calculated due to the deprotonation reaction. The stoichiometry of the supramolecular complexes was in agreement with those found by UV-vis for these receptors.
K)a for the complexes of receptors 4 with different TBAX salts determined by 1H-NMR
The interaction of the receptors meta mono-ureylbenzamide, 8b, 8c, and 8d with TBAA, TBAB, TBAF, TBAHP, and TBAHPP was analyzed by 1H NMR in DMSO-d6 (Fig. S44–S63†). As seen in Fig. 5, all the TBAX salts promoted different changes in the signals of receptor 8b once the complex was formed in solution. Here, an equivalent downfield shift of the urea signals with TBAA and TBAB was observed, indicating that both hydrogens interacted with the anion with the same strength. The spectra obtained during the titration with TBAA (Fig. 6) indicated that both H2 and H1 were gradually shifted downfield when up to two equivalents of TBAA were added. The amide hydrogen (H3) slightly shifted to upfield and interestingly, the hydrogen at the amide carbonyl and urea ortho position (H4) shifted to downfield, suggesting a bond polarization due to a C–H⋯O interaction in the complex. This effect over the H4 signal was stronger with TBAB than with TBAA, and it was partially lost by increasing the oligomethylene chain length in receptors 8c and 8d.
 | ||
| Fig. 5 Partial 1H-NMR spectra of free 8b and with two molar equivalents of TBAA, TBAB, TBAHP, and TBAF in DMSO-d6. | ||
 | ||
| Fig. 6 1H NMR spectra obtained in the titration of 8b with TBAA in DMSO. | ||
The successive formation of three different complexes was detected in the titration of 8b with TBAHP (Fig. 7). The two urea signals were shifted downfield with the same magnitude when one equivalent of TBAHP was added. Then, the chemical shift of H1 was higher than H2 when from one to three equivalents of anion was added, but beyond this concentration it remained constant. The signal of H2 was gradually shifted downfield when up to two equivalents of anion were added. The amide signal became wide when one equivalent of anion was added, and was then shifted downfield with the addition from one to three equivalents and then remained constant. Interestingly, the signal of H4 was shifted to upfield and the signal of H7 was shifted to downfield, indicating a significant change in the chemical environment of these hydrogens. Considering the changes of the signals, the urea hydrogens as well as the amide hydrogen equally participated in the 1:1 complex (see Fig. S66†), while the hydrogens H1 and H3 were more affected in the 1:2 complex. The chemical shifts of H4 and H7 indicated a conformational change in the receptor, allowing the interaction with a third anion unit, where the H1 and H3 hydrogens were the most affected. Receptors 8c and 8d showed a similar signal pattern with TBAHP, although the H3 signal was less affected as the oligomethylene chain increased (Fig. S57 and S62†).
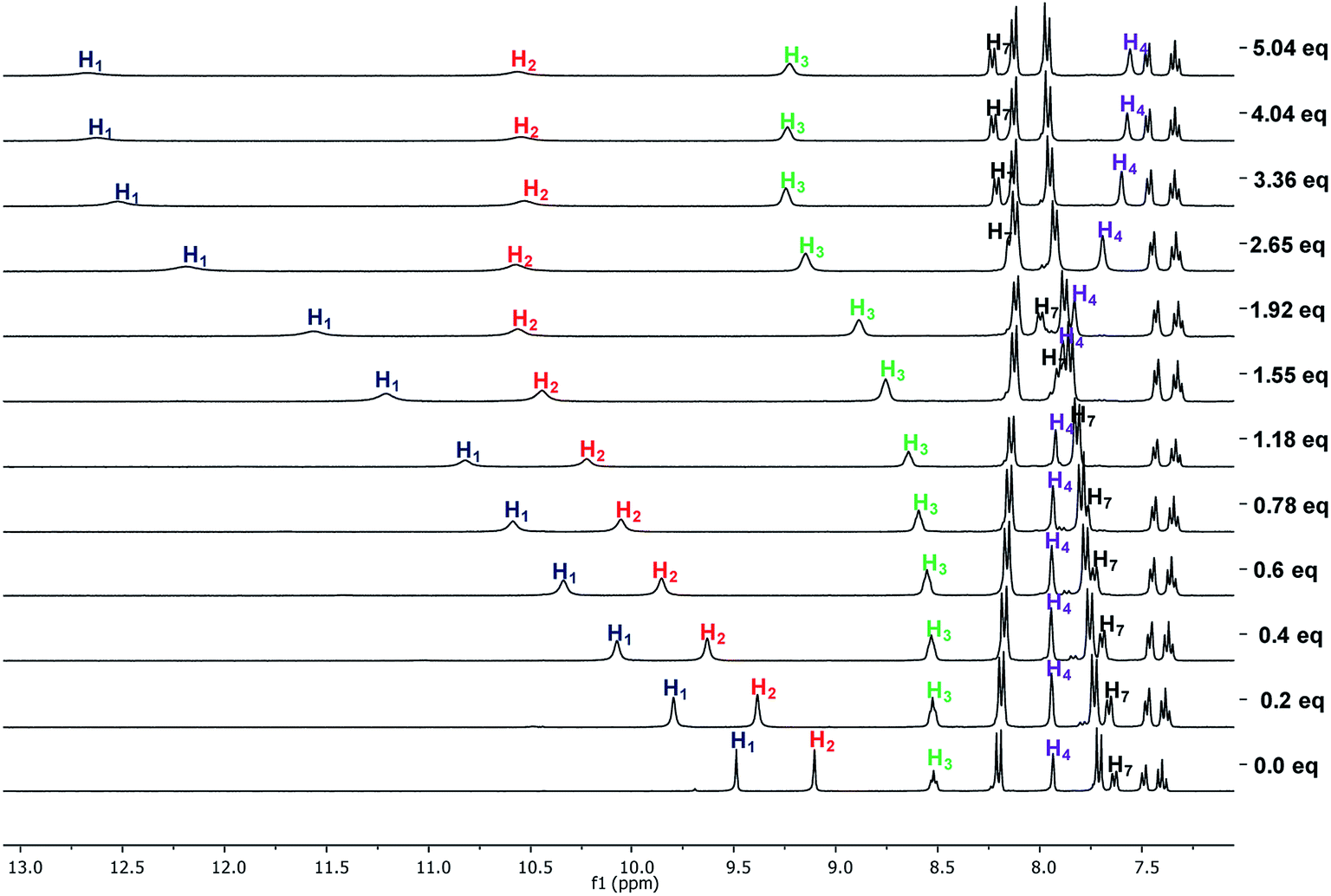 | ||
| Fig. 7 1H NMR spectra obtained in the titration of 8b with TBAHP in DMSO-d6. | ||
The titrations of 8b, 8c, and 8d with TBAHPP showed deprotonation of the urea groups since the first aliquot was added. Interestingly, all the signals become broad and shifted in different ways, indicating three clear stages during the course of the titration. All the signals showed the maximum broadening between 0.4 and 0.6 molar equivalents of TBAHPP added, suggesting a 2:1 (receptor:anion) complex. The same behavior was also found with the UV titrations. Then, all the signals recovered their multiplicity when 1.2 equivalents were added, and the H3 signal was shifted 1.8 ppm downfield, indicating the interaction with the anion. These changes showed the formation of a 1:1 complex. Interestingly, an additional signal shifted about 0.5 ppm appeared for H3, and the two signal of the nitrophenyl group collapsed into one at 2.4 equivalents of TBAHPP added, showing changes in the complex. All these changes in the NMR spectra indicated an acid–base reaction, but also the interaction with the anion in different forms depending on the anion concentration.
The association constants (logK) for the complexes of receptors meta mono-ureylbenzamide, 8b, 8c, and 8d were calculated from the chemical shifts and are reported in Table 4. The meta mono-ureylbenzamide formed 1:1 complexes with TBAA and TBAB salts, and the data fitting showed the presence of additional complexes with TBAHP. The chemical shift profiles obtained with TBAA and TBAB fitted with a 1:1 and 1:2 stoichiometry model, where the values of logK decreased as the oligomethylene chain length increased in receptors 8. The values of logK were considerably superior compared to those obtained with receptors 4.
K)a for the complexes of receptor 8 with different TBAX salts determined by 1H-NMR in DMSO-d6
| Receptor | X | logK11 |
logK12 |
logK13 |
|---|---|---|---|---|
| a Standard deviation of the fit is given in parenthesis.b Excessive standard deviation. | ||||
 |
AcO− | 3.61(6) | 8.2 | 11.0 |
| BzO− | 3.25(3) | |||
| H2PO4−b | 5.0 | |||
| 8b | AcO− | 4.1(1) | 7.6(2) | 12.6 |
| BzO− | 5.3(4) | 8.9(7) | ||
| H2PO4−b | 5.1 | 9.4 | ||
| 8c | AcO− | 5.46(8) | 11.3 | |
| BzO− | 5.33(1) | |||
| H2PO4−b | 4.9 | 9.0 | ||
| 8d | AcO− | 5.29(1) | ||
| BzO− | 3.0(6) | 5.91(9) | ||
| H2PO4−b | 2.50 | 4.47 | ||
The results obtained by UV-vis and 1H NMR demonstrated that receptors 4 and 8 established interactions with all the anions (TBAX salts) used in the study. However, there were significant differences regarding the urea and amide position, the oligomethylene chain length, and the geometry and charge of the anion. Receptors 8 showed more evident spectroscopic changes due to the interaction with the anion compared with the observed response of receptors 4, which was attributed to the different position of the urea and amide groups in the receptors. The values of the association constants obtained by UV-vis were in the same orders for both types of receptors (logK11 = 4–8, logK12 = 10–15, logK21 = 10–15). This similarity in the affinity was related to the strength of the interaction with the anions, considering that in receptors 4, three hydrogen bonding per each ureabenzamide unit may participate in the supramolecular complex, as was observed in the NMR analysis. The fitting of the absorbance data revealed the formation of 2:1 (receptor:anion) complexes with TBAA, TBAB, and TBAHPP with the ortho mono-ureylbenzamide. This behavior was consistent with the formation of 1:1 complexes with these anions in receptors 4, where the two ureylbenzamide units participated in the interaction with one unit of the anion. Also the increase of the association constants in the receptors with the shorter oligomethylene chains (4a and 4b) confirmed a major cooperativity between the two units due to their proximity. Instead, there was an opposite tendency with TBAHPP in both 1:1 and 2:1 (receptor:anion) complexes, probably due to the different geometry and higher number of negative charges. The successive formation of 1:1 and 1:2 complexes with TBAF and TBAHP indicated a low stability of the 1:1 complexes, probably due to size and geometry of these anions compared with the carboxylates and pyrophosphate.
The successive formation of 1:1 and 1:2 with TBAA, TBAB, TBAF, and TBAHP, and only 2:1 complexes with TBAHPP at low concentration occurred in receptors 8, where this latter case is an interesting peculiarity. A significant difference in receptors 8 was the strong equivalent interaction of the urea hydrogens and the apparent C–H⋯O interaction with carboxylates. Interestingly, the tetrahedral geometry of dihydrogen phosphate induces an interaction with the receptor with different stoichiometries, where even the amide hydrogen is involved in the 1:2 and 1:3 complexes (receptor:anion). The formation of higher ratio complexes with TBAHP even with the meta N-propyl ureabenzamide up to 1:3 was rather unexpected. The fitting quality with this anion was also poor, but all attempts to exclude the higher ratio complexes failed; fitting to a simpler model always was divergent and did not allow any estimate of the binding constants. We believe that these complications resulted from the recently established strong auto-association of dihydrogen phosphate anion, particularly complicated by a possible contribution of free phosphoric acid, which may be generated by water traces in the solvent employed.25 Moreover, the UV-vis and NMR studies with TBAHPP demonstrated the formation of supramolecular complexes of different stoichiometries with all receptors at high concentration, which was consistent with the tendency observed in the UV-vis studies at low concentration.
Theoretical calculations of the supramolecular complexes
In order to support these experimental findings, the geometries of the supramolecular complexes were optimized by theoretical calculations using DFT with a B3LYP 6-31G(d) basis set.24 First, the mono-ureylbenzamide complexes with AcO− were calculated and interesting differences were observed (Fig. 8). The distance between the urea hydrogens H1 and H2 and the carboxylate was not equivalent, indicating a different strength in the ortho complex (structure I), and a strong interaction with amide hydrogen. The inequivalent chemical shift of the urea hydrogen signals and broadening of the amide hydrogen in the NMR spectra is now evident (Fig. S24†). Also, the amide carbonyl lay out of the plane with the aromatic ring, which is consistent with the upfield shift of H4 signal in the NMR spectra, indicating the loss of the “ortho effect” over this hydrogen by the carbonyl. In the meta complex (structure II), the H1, H2, H3, and H4 hydrogens were oriented toward the carboxylate; the interaction of both urea hydrogens was equivalent and an interaction with amide hydrogen was present, but the distance between the carboxylate and the hydrogen was longer than that observed for the ortho complex. In this complex the distance of the C–H4 with the carboxylate was about 2.2 Å, indicating a C–H⋯O interaction which was consistent with the downfield of the H4 signal during the titration. The interactions in this model are in agreement with the results obtained in the NMR experiment (Fig. S44†).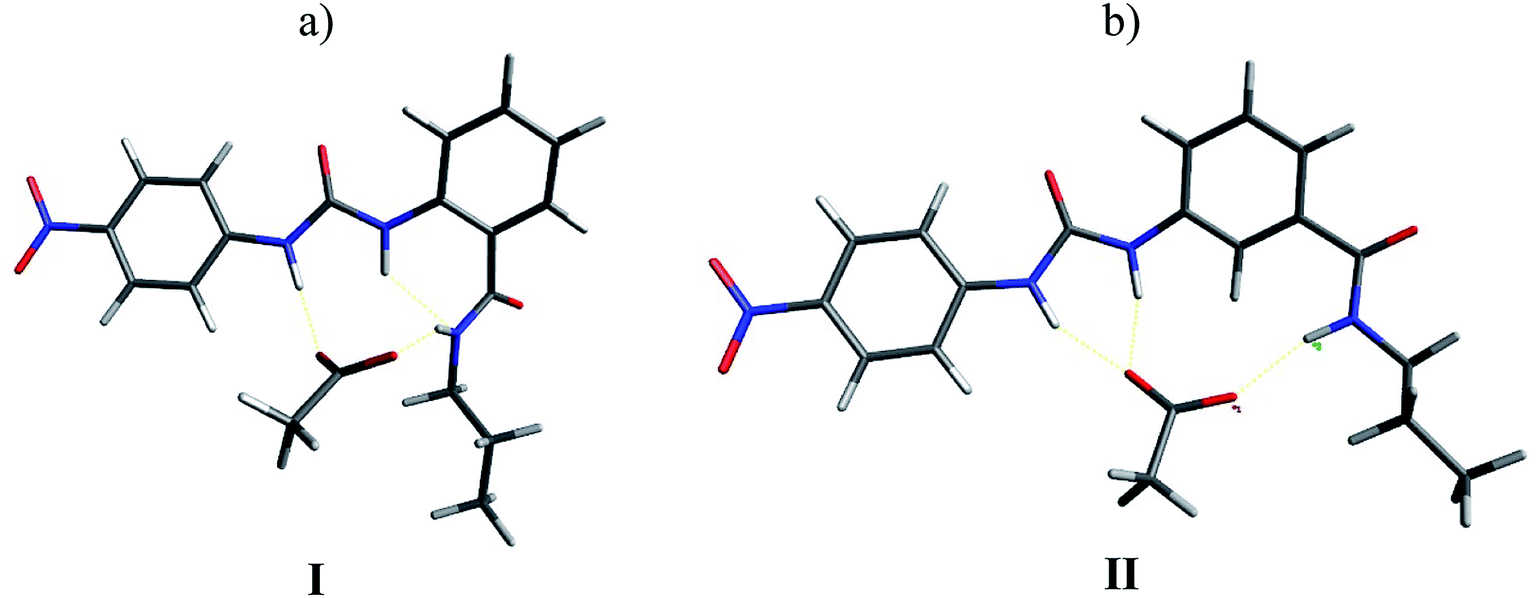 | ||
| Fig. 8 Optimized structures of monoureabenzamide-TBAA complexes I (a) and II (b) by DFT. | ||
Then, the interaction of 4b with AcO− was analyzed, optimizing a complex in which the AcO− interacts only with one ureylbenzamide unit (structure III) and another structure where both units participate in the complexation (structure IV). This calculation was done in order to elucidate which complex would be favored based on their total energy. The results indicated that structure IV was 9.76 kcal mol−1 more stable than structure III (Fig. 9). As can be seen in Fig. 9a, complex IV showed more hydrogen bond interactions between the receptor and the AcO− compared with complex III and the nitrophenyl groups appear face to face, indicating a possible additional interaction. This structure agrees with the chemical shifts and the signals affected during the titration with TBAA by NMR, which are very similar to those observed with the mono-ureylbenzamide (Fig. 4).
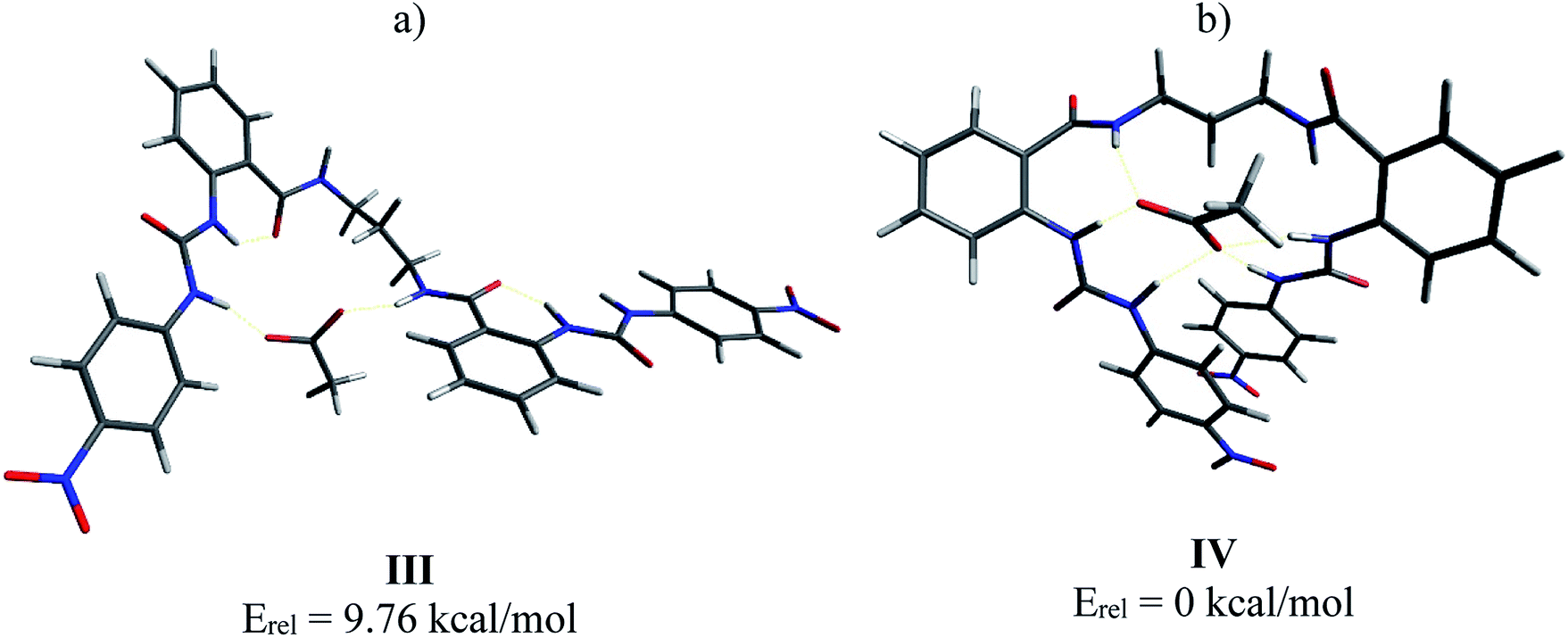 | ||
| Fig. 9 Optimized structures III (a) and IV (b) for complex 4b-AcO− by DFT. | ||
The complexes with H2PO4− and HP2O73− were also optimized (Fig. 10). In the 1:1 4b-H2PO4− complex (structure V), both urea units interacted with the anion through the N–H2, leaving the N–H1 free, while only one of the N–H3 interacted with the anion. This means that three sites were available to interact with a second anion. In fact, the NMR experiment showed a wide signal for H1 with the TBAHP addition but no shift to downfield was observed (see Fig. S32†). This result was consistent with the experimental observed tendency of these receptors to form multi-equivalent complexes with H2PO4−. The complex with HP2O73− showed deprotonation of the N–H2 in one urea unit. This acid–base reaction was evident in the NMR titrations, and a change of color in the solution was also observed. Almost all the hydrogen bonding sites interacted with the anion in the complex.
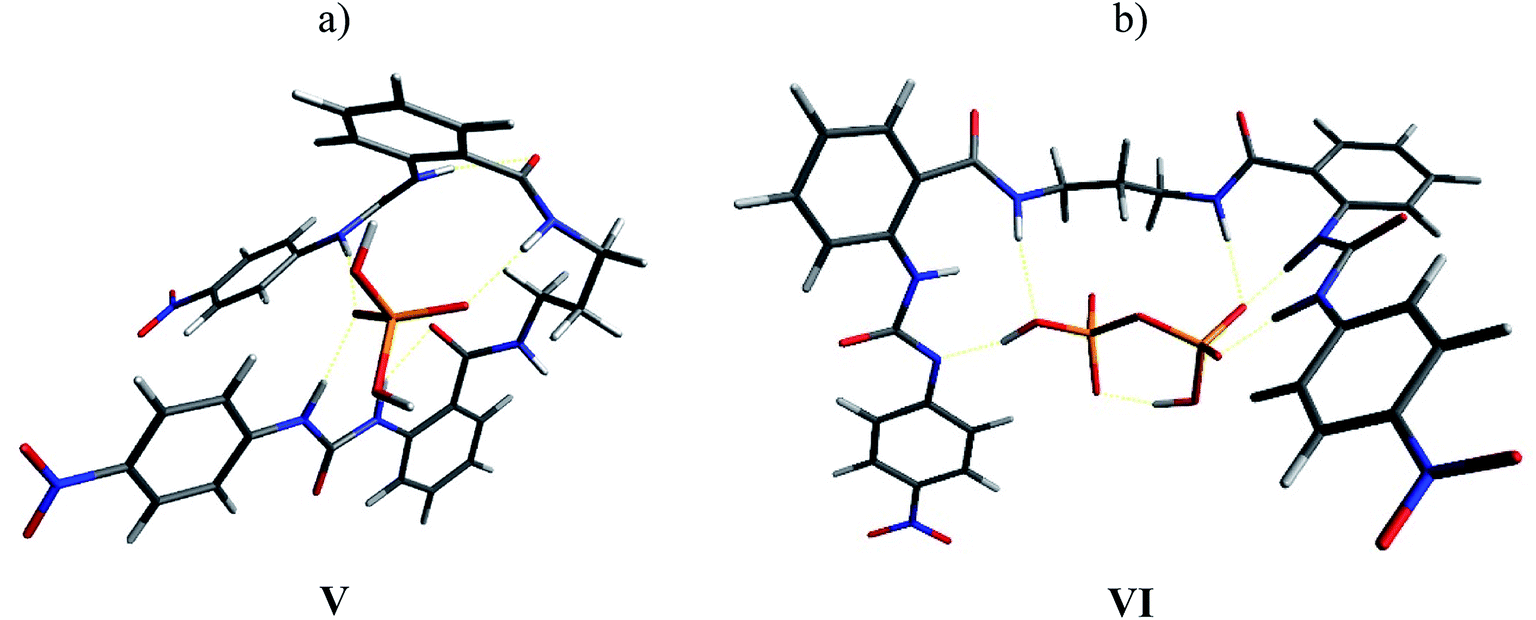 | ||
| Fig. 10 Optimized structures V (a) and VI (b) for the complexes 4b-H2PO4− and 4b-HP2O73− by DFT. | ||
It was noteworthy that the complexes of 4c and 4d with AcO− showed a helical conformation, where both ureylbenzamide units had a coplanar and antiparallel position allowing the interaction with the anion through five hydrogen bonds (Fig. 11). The enhancement of the spacer chain from four to six methylenes favored the coplanarity between the two units. The complex with BnO− showed a similar helical conformation (Fig. S64†). This singular conformation with the carboxylate anions may induce the exclusive formation of 1:1 complexes, as identified by the UV-vis and NMR techniques. The helical conformation of receptor 4d was distorted in the complexes with H2PO4− and HP2O73− due to the differences in the sizes and geometry of these anions (Fig. S64†).
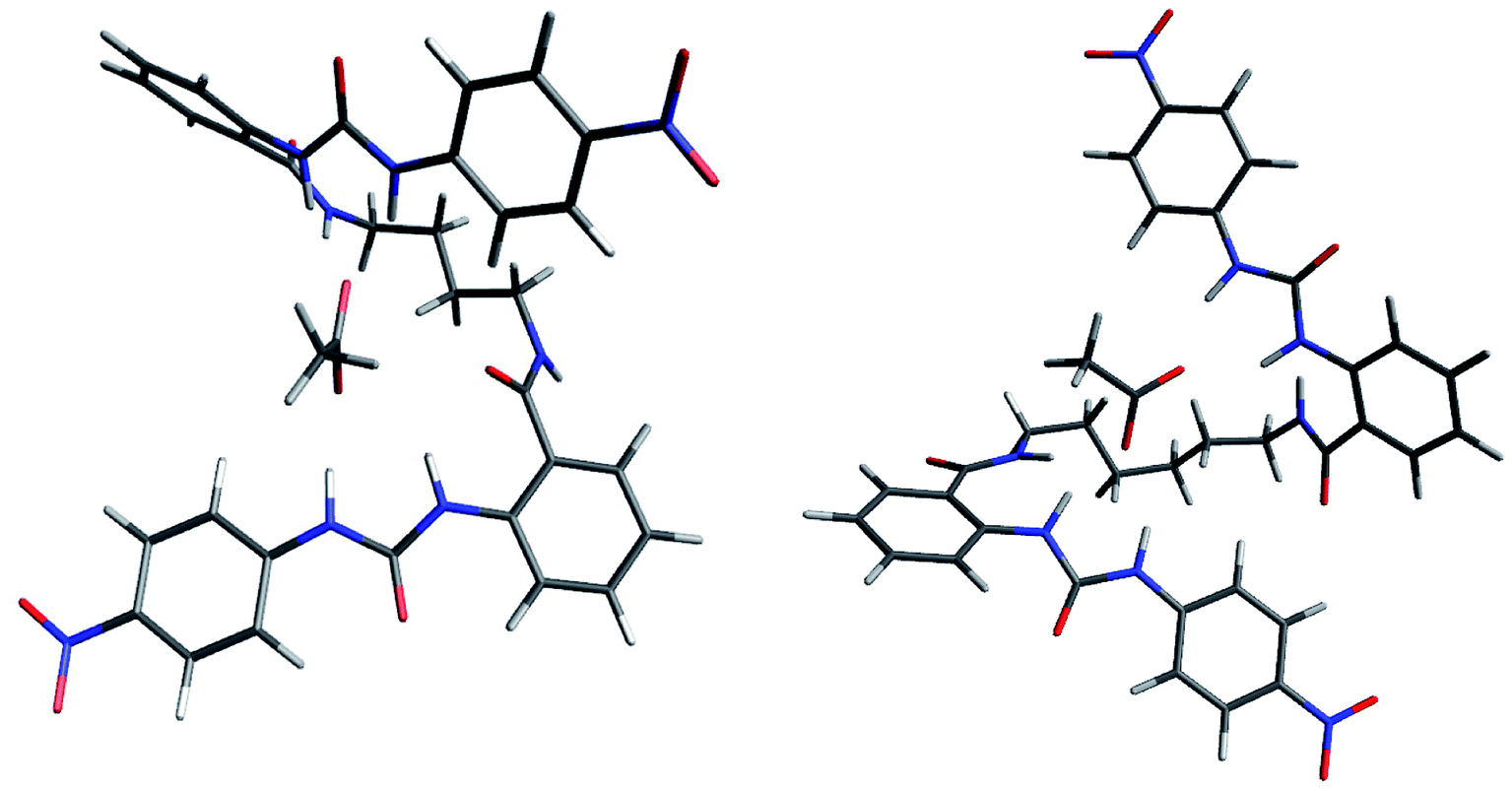 | ||
| Fig. 11 Optimized structures for the complexes 4c-AcO− and 4d-AcO− by DFT. | ||
The 1:1 complex of receptor 8b with acetate was optimized at the same level of theory. The “saddle” shape of the receptor in the complex allowed the H1, H2, H3, and H4 hydrogens of both ureylbenzamide units to be oriented in the direction of the anion (Fig. 12a and b). The distance between the anion and the interacting groups were minimal and the hydrogen bonding was established in the cavity of the receptor in this arrangement. The ureylbenzamide units adopted a perpendicular position to form a compact cavity, where H1, H2, H3, and H4 were oriented to the anion in the complex of 8d with acetate (Fig. 12c and d). The flexibility of the oligomethylene spacer chain allowed the necessary conformational changes to form a 1:1 complex despite the distance between both ureylbenzamide in these receptors (Fig. S65†).
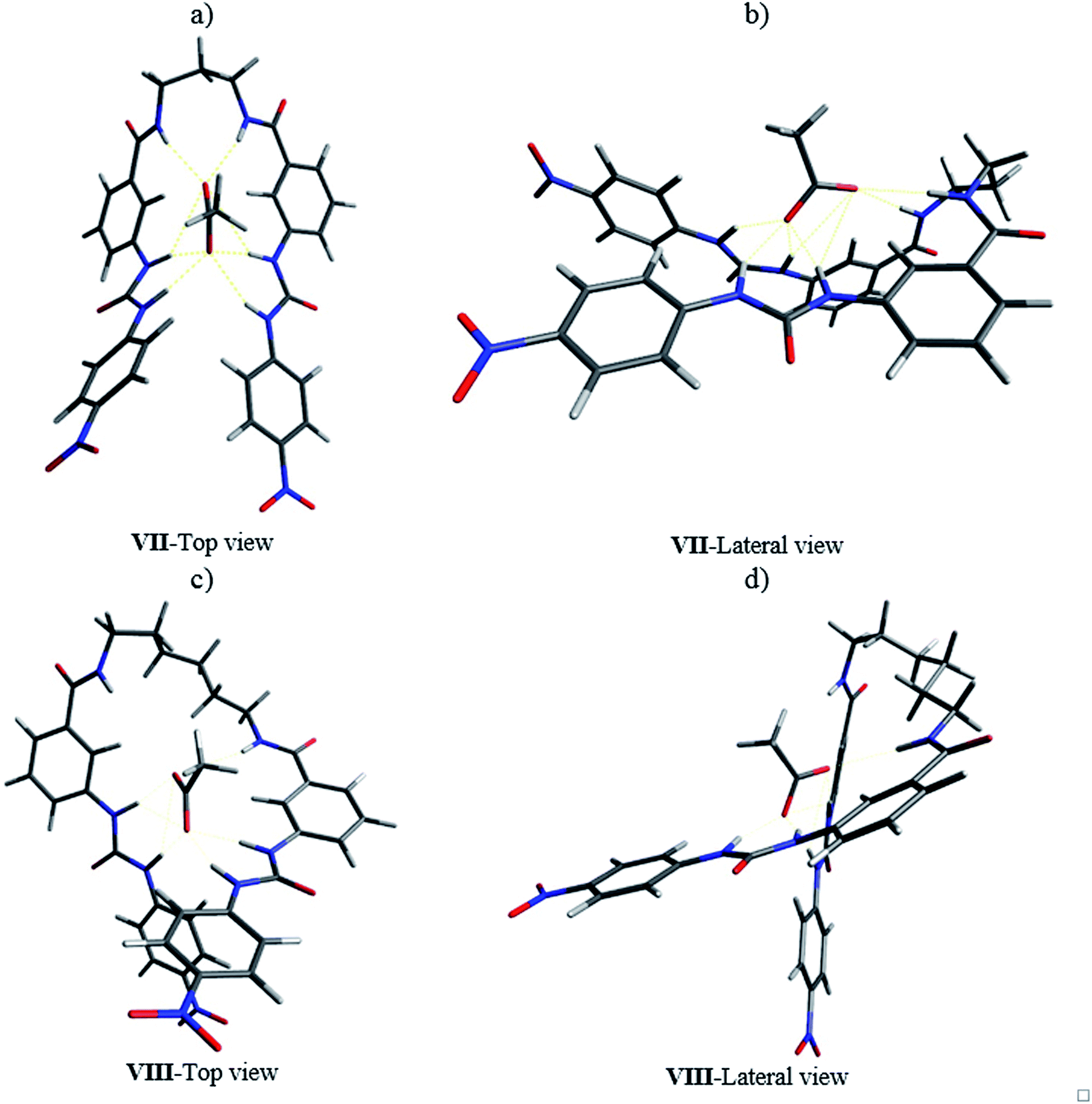 | ||
| Fig. 12 Optimized structures for the complexes 8b-AcO− (a and b) and 8d-AcO− (c and d) by DFT. | ||
The optimization of the complexes with phosphate and pyrophosphate showed configurational changes in the oligomethylene chain induced by the size and geometry of the anion in order to minimize the distance between the interaction sites and the anions (Fig. S66 and S67†). Interestingly, the complexes with 8b showed both anions located in a superficial position on the cavity formed by the two ureylbenzamide units. The distances allowed the interaction of the anions with the H1, H2, H3, and H4 hydrogens. The receptors 8d and 8e with a longer oligomethylene spacer formed a big cavity and the anions occupied an internal or equatorial position. These observations partially explain or justify the high values of logK for 1:1 complexes in receptors with long spacer chain.
Conclusions
The results obtained by UV-vis and 1H NMR as well as the values of logK and the theoretical analysis revealed a strong interaction of receptors 4 and 8 with the anions due to their bifunctionality. The flexible oligomethylene chain in these receptors enhance the possibility to adopt different conformational arrangement to form 1:1 complexes regardless of differences in the size and shape of the anion. Receptors 4 formed 1:1 complexes with carboxylates due to the singular helical conformation of the receptors, which is an obvious consequence of the relative ortho position of the urea and amide groups. The anions with different sizes and geometries also formed 1:1 complexes, but the helical conformation was distorted to maximize the interactions. These results also revealed the formation of complexes with variable stoichiometry with dihydrogen phosphate and pyrophosphate due to their geometry, charge number, and autoassociation process.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors thank Consejo Nacional de Ciencia y Tecnología (CONACyT Mexico) (Grant No. CB-2014-239581) for financial support for this project. José García-Elías thanks to CONACyT for the graduate fellowship. The authors acknowledge grateful support from the Supramolecular Chemistry Thematic Network (CONACyT Grant No. 294810). Also, thank CONACyT for the ITT NMR facilities (Grant INFR-201103-173395).References
- X. Ma and Y. Zhao, Biomedical Applications of Supramolecular Systems Based on Host-Guest Interactions, Chem. Rev., 2015, 115, 7794–7839 CrossRef CAS PubMed.
- S. N. Berry, V. Soto-Cerrato, E. N. W. Howe, H. J. Clarke, I. Mistry, A. Tavassoli, Y.-T. Chang, R. Pérez-Tomás and P. A. Gale, Fluorescent Transmembrane Anion Transporters: Shedding Light on Anionophoric Activity in Cells, Chem. Sci., 2016, 7, 5069–5077 RSC.
- P. A. Gale, E. N. W. Howe and X. Wu, Anion Receptor Chemistry, Chem, 2016, 1, 351–422 CAS.
- P. A. Gale, Anion Receptor Chemistry: Highlights from 1999, Coord. Chem. Rev., 2001, 213, 79–128 CrossRef CAS.
- M. Boiocchi, L. Del Boca, D. E. Gómez, L. Fabbrizzi, M. Licchelli and E. Monzani, Nature of Urea–Fluoride Interaction: Incipient and Definitive Proton Transfer, J. Am. Chem. Soc., 2004, 126, 16507–16514 CrossRef CAS PubMed.
- D. A. Jose, D. K. Kumar, P. Kar, S. Verma, A. Ghosh, B. Ganguly, H. N. Ghosh and A. Das, Role of Positional Isomers on Receptor–anion Binding and Evidence for Resonance Energy Transfer, Tetrahedron, 2007, 63, 12007–12014 CrossRef CAS.
- T. H. Russ, A. Pramanik, M. E. Khansari, B. M. Wong and M. A. Hossain, A Quinoline Based Bis-Urea Receptor for Anions: A Selective Receptor for Hydrogen Sulfate, Nat. Prod. Commun., 2012, 7, 301–304 CrossRef CAS PubMed.
- V. Amendola, D. Esteban-Gómez, L. Fabbrizzi and M. Licchelli, What Anions Do to N–H-Containing Receptors, Acc. Chem. Res., 2006, 39, 343–353 CrossRef CAS PubMed.
- C. M. G. dos Santos, T. McCabe, G. W. Watson, P. E. Kruger and T. Gunnlaugsson, The Recognition and Sensing of Anions through “Positive Allosteric Effects” Using Simple Urea–Amide Receptors, J. Org. Chem., 2008, 73, 9235–9244 CrossRef CAS PubMed.
- P. Comba, The Relation between Ligand Structures, Coordination Stereochemistry, and Electronic and Thermodynamic Properties, Coord. Chem. Rev., 1993, 123, 1–48 CrossRef CAS.
- V. S. Bryantsev and B. P. Hay, De Novo Structure-Based Design of Bisurea Hosts for Tetrahedral Oxoanion Guests, J. Am. Chem. Soc., 2006, 128, 2035–2042 CrossRef CAS PubMed.
- B. P. Hay, T. K. Firman and B. A. Moyer, Structural Design Criteria for Anion Hosts: Strategies for Achieving Anion Shape Recognition through the Complementary Placement of Urea Donor Groups, J. Am. Chem. Soc., 2005, 127, 1810–1819 CrossRef CAS PubMed.
- B. Garg, T. Bisht and S. M. S. Chauhan, 2,2′-Diaminoazo-Benzene, a Potential Scaffold for the Synthesis of Bis-Ureas and Thioureas: Solution Phase Anion Sensing and Binding Studies, Sens. Actuators, B, 2012, 168, 318–328 CrossRef CAS.
- S. Nishizawa, P. Bühlmann, M. Iwao and Y. Umezawa, Anion Recognition by Urea and Thiourea Groups: Remarkably Simple Neutral Receptors for Dihydrogenphosphate, Tetrahedron Lett., 1995, 36, 6483–6486 CrossRef CAS.
- P. Bühlmann, S. Nishizawa, K. P. Xiao and Y. Umezawa, Strong Hydrogen Bond-Mediated Complexation of H2PO4− by Neutral Bis-Thiourea Hosts, Tetrahedron, 1997, 53, 1647–1654 CrossRef.
- S. Nishizawa, P. Bühlmann, K. P. Xiao and Y. Umezawa, Application of a Bis-Thiourea Ionophore for an Anion Selective Electrode with a Remarkable Sulfate Selectivity, Anal. Chim. Acta, 1998, 358, 35–44 CrossRef CAS.
- A. N. Leung, D. A. Degenhardt and P. Bühlmann, Effect of Spacer Geometry on Oxoanion Binding by Bis- and Tetrakis-Thiourea Hosts, Tetrahedron, 2008, 64, 2530–2536 CrossRef CAS.
- Y.-J. Kim, H. Kwak, S. J. Lee, J. S. Lee, H. J. Kwon, S. H. Nam, K. Lee and C. Kim, Urea/thiourea-Based Colorimetric Chemosensors for the Biologically Important Ions: Efficient and Simple Sensors, Tetrahedron, 2006, 62, 9635–9640 CrossRef CAS.
- C. Caltagirone, C. Bazzicalupi, F. Isaia, M. E. Light, V. Lippolis, R. Montis, S. Murgia, M. Olivari and G. Picci, A New Family of Bis-Ureidic Receptors for Pyrophosphate Optical Sensing, Org. Biomol. Chem., 2013, 11, 2445–2451 RSC.
- B. Moreno-Valle, M. Aguilar-Martínez, A. Ochoa-Terán, M. Martínez-Quiroz, V. Miranda-Soto, J. García-Elías, K. Ochoa-Lara, V. Labastida-Galván and M. Ordoñez, Synthesis and Anion Recognition Studies of New Ureylbenzamide-Based Receptors, Supramol. Chem., 2018, 30, 9–19 CrossRef CAS.
- P. Gans, A. Sabatini and A. Vacca, Talanta, 1996, 43, 1739–1753 CrossRef CAS PubMed.
- P. Gans, A. Sabatini and A. Vacca, Anal. Chim. Acta, 1999, 89, 45–49 CAS.
- A. Shokrollahi, F. Zarghampour, S. Akbari and A. Salehi, Anal. Methods, 2015, 7, 3551–3558 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, K. N. Brothers V. N. Kudin R. Staroverov J. Kobayashi K. Normand E. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision E.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- D. Barisic, V. Tomisic and N. Bregovic, Anal. Chim. Acta, 2019, 1046, 77–92 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra05783h |
| This journal is © The Royal Society of Chemistry 2019 |