
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCIDNP as a tool to unveil the reaction mechanism: interaction of mixed phosphonium–iodonium ylide with p-methoxyphenylacetylene†
Irina I. Levinaa,
Valery F. Tarasovb,
Tatyana A. Podruginaac and
Tatiana D. Nekipelova *a
*a
aEmanuel Institute of Biochemical Physics RAS, Kosygin St. 4, Moscow, 119334, Russia. E-mail: nekip@sky.chph.ras.ru
bSemenov Institute of Chemical Physics RAS, Kosygin St. 4, Moscow, 119991, Russia
cDepartment of Chemistry, Lomonosov Moscow State University, Lenin Hills 1, Moscow, 119991, Russia
First published on 21st August 2019
Abstract
In situ acquisition of the reaction between benzoyl phosphonium–iodonium ylide and p-methoxyphenylacetylene in an NMR spectrometer reveals the CIDNP effect in 31P and 1H NMR spectra of major products, substituted furan (emission) and phosphonium salt (enhanced absorption). The mechanism of products formation via radical pairs is discussed.
The synthesis of mixed phosphonium–iodonium ylides was described for the first time by Moriarty et al. in 1984.1 These compounds have attracted considerable attention in the last decade because of their high potential in the synthesis of different important and novel heterocycles.2 The interaction between mixed phosphonium–iodonium ylides and acetylenes allows for the synthesis of substituted phosphorus-containing heterocycles such as λ5-phosphinolines (3) and substituted furans (4) in a simple, one-pot, metal-free reaction at room temperature. Scheme 1 is given for a particular case of mixed benzoyl ylide 1 used in this study, in the following schemes counterion BF4− is omitted for simplicity, because no products containing B or F were identified. For some ylide–acetylene pairs the reaction proceeds only under irradiation, whereas for the others, as we have shown here, no irradiation is required. Reaction of ylide 1 with acetylenes 2 is characterized by remarkable features: (i) it proceeds only in dichloromethane (DCM) (ii) at high ylide concentrations (>0.01 M) (iii) with induction time,3 (iv) is catalyzed by acids4 and (v) relatively stable free radicals are observed by EPR in the reaction mixture.5
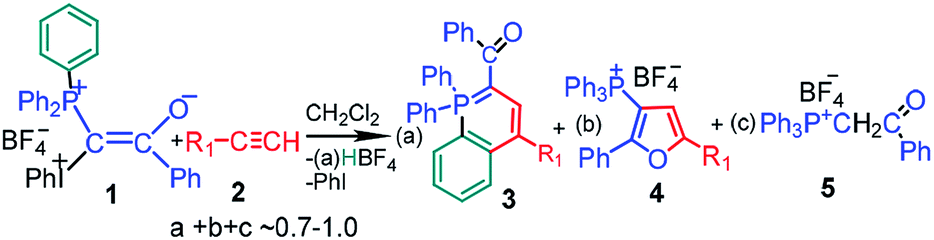 | ||
| Scheme 1 Principal scheme of the reaction between mixed phosphonium–iodonium ylide 1 and acetylenes. | ||
If the critical concentration dependence for the reaction with acetylenes is accounted for by the specific microheterogeneoos structure of the ylide solutions in DCM,6 the unusual features of the reaction, such as induction time, acid catalysis and presence of radicals, are inconsistent with the primarily suggested mechanism of product formation.7 This mechanism involves heterolytic scission of the C–I+Ph bond with elimination of PhI under irradiation and formation of λ5-phosphinoline 3 and furan derivative 4 from an intermediate carbocation (or a carbene) in parallel reactions. However the participation of carbene is unlikely because of the absence of cyclopropane derivative in the ylide reaction with styrene neither in thermolysis nor under irradiation.2a
For a given ylide the relative yields of heterocycles 3 and 4 depend on the acetylene structure: the lower ionization potential of acetylene, the higher yield of furan derivative 4. Phosphonium salt 5, the major product in the photolysis of the ylide alone and under storage of the solution in the dark, is formed in the mixture with acetylenes as a minor product.2a The presence of the iodonium group in the mixed ylides allowed us to propose the involvement of radicals in the formation of phosphonium salt 5,3,8 because radical decomposition of iodonium salts under irradiation is well-known.9 It has been shown that under irradiation the phosphonium salt is formed as a result of homolytic scission of the C–I+Ph bond with elimination of a radical cation (PhI)+˙. The generated radical 6 is protonated and then abstracts the hydrogen atom from DCM (Scheme 2).5a In the dark the decomposition of ylide 1 is accelerated by the traces of an acid due to the formation of dication 8, which decomposes to give radical cations easier than the parent ylide.4b,6 The acid is formed in the reaction of the radical cation (PhI)+˙ with DCM providing the acid autocatalysis.9
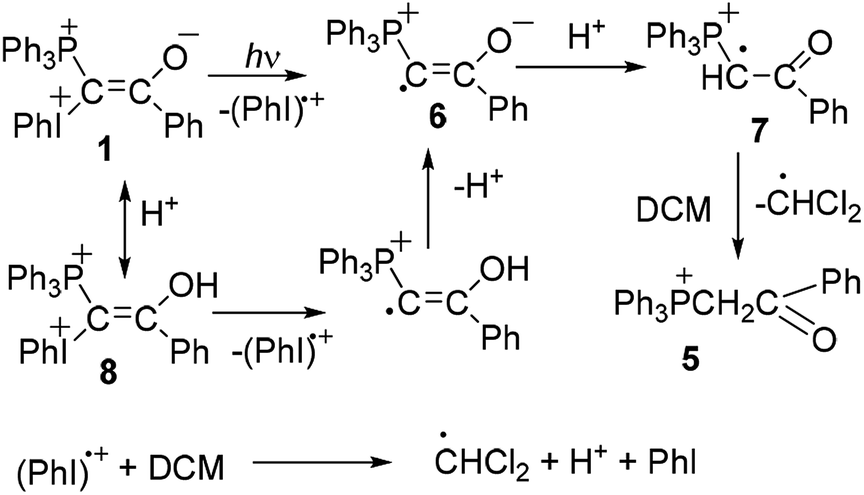 | ||
| Scheme 2 Mechanism of decomposition of ylide 1 and formation of salt 5. | ||
This route for the formation of salt 5 was confirmed by the experimental observation of a radical with the EPR spectrum corresponding to the spectrum of radical cation 7.5a However the question about the mechanism of the formation of heterocycles 3 and 4 still remains, although relatively stable radical cations were observed not only in the photolysis of ylide 1 alone, but in its mixtures with phenylacetylene, p-methoxyphenylacetylene, and 9-ethynylphenanthrene.5 For the two latter acetylenes the radical cations were observed even without irradiation after mixing the reagents.
In order to gain insight into the reaction mechanism, here we have studied the kinetics of the reaction between ylide 1 and p-methoxyphenylacetylene 2 by 31P NMR spectroscopy because the ylide itself and products 3–5 contain a phosphorus atom. The appeal of this system lies in the fact that not only radicals, but also heterocycles 3 and 4 are formed with comparable yields (30–60%) without irradiation at room temperature for a reasonable time (∼30–150 min). The comparison of the reaction between ylide 1 and acetylene 2 under irradiation and in the dark has shown the insignificant difference: the product yields are slightly, but not crucially different, and under irradiation the induction time decreases. The most impressive result of our experimental research is the detection of Chemically Induced Dynamic Nuclear Polarization (CIDNP) following the chemical transformations in the system (Fig. 1).
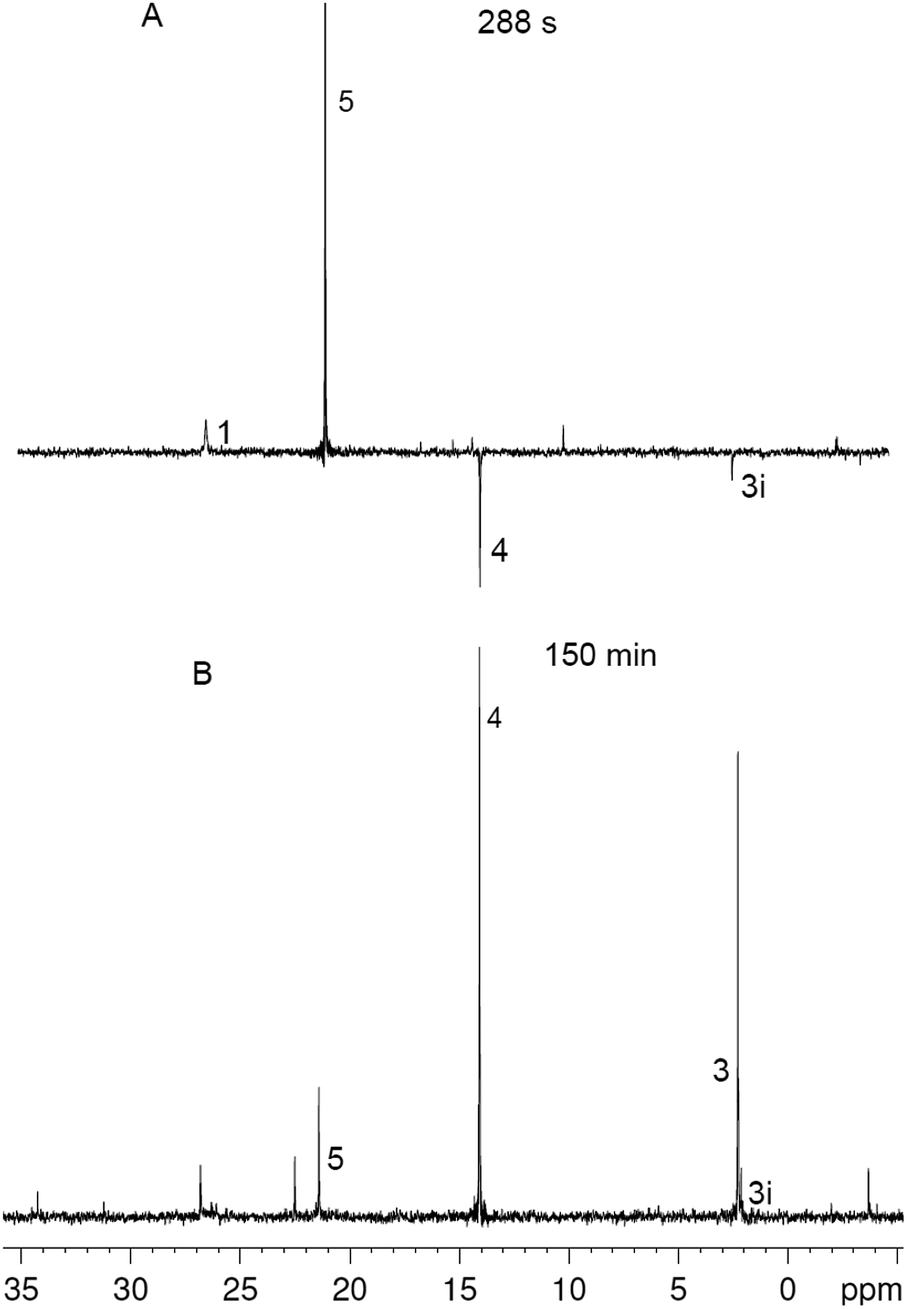 | ||
| Fig. 1 Fragment of 31P NMR spectra in the course of the reaction between 1 and 2 in DCM at different times, numbers correspond to the numbers of reagents and products in Scheme 1, for 3i see the text. | ||
The reaction between 1 (0.05 M) and 2 (0.15 M) in DCM was studied directly in the cavity of NMR spectrometer. The acquisition started in 1.5 min after mixing and was carried out continuously with four scans (∼6 s) per spectrum during 10 min, then with 32 scans (∼40 s) per spectrum during next 20 min and with 128 scans (∼1 min) per spectrum till the reaction accomplishment. Under these experimental conditions a first product is observed already in 2 min after mixing, and the ylide is consumed completely for 7 min (Fig. 2). During this time the 31P NMR spectra demonstrate the appearance of the signals with enhanced absorption and emission (Fig. 1A). The spectra at the most significant times of changes and their description are given in ESI (Fig. S1–S11†).
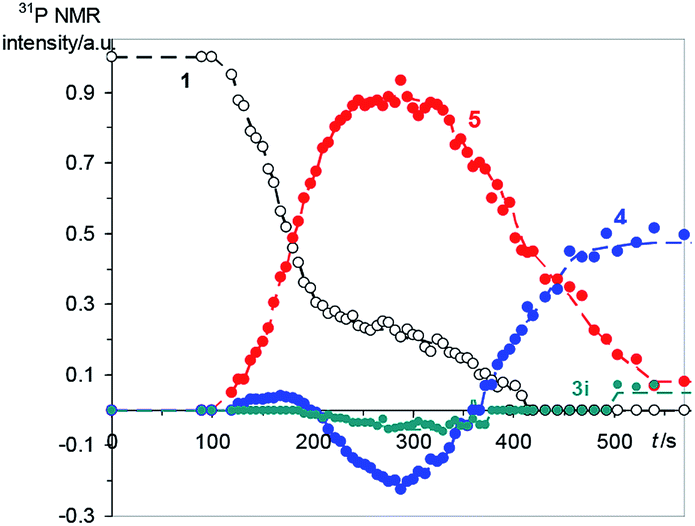 | ||
| Fig. 2 Relative integrated yields of the main products during first 10 min of the reaction, numbers correspond to the numbers of reagents and products in Scheme 1, for 3i see the text. | ||
In that what will follow further, such concepts as the sign of polarization and its intensity will refer to phosphorus atoms, unless otherwise indicated. Positively polarized phosphonium salt 5 (δ 21.4 ppm), negatively polarized furan 4 (δ 14.4 ppm) and a series of negatively polarized minor signals are observed (Fig. 1A). The maximum enhanced absorptive signal of 5 and the emissive signal of 4 are attained at the same time (288 s). All minor emission signals disappear by 360 s and the products with signals at 22.3, 2.2 and −3.9 ppm are minor products in the final mixture (Fig. 1B). We tentatively assign the 31P NMR signal at 2.2 ppm (final yield 3%), which demonstrates emission (Fig. 1, ESI Fig. S5–S7†), to λ5-phosphinoline 3i (Scheme 3), a regioisomer of 3 (Scheme 1, 2.3 ppm, final yield ∼30%). Since the yield of 3 is high, 3 was isolated and the position of the p-methoxyphenyl fragment in the heterocycle was determined in 3 by physicochemical methods before.2a Unlike the signals of 3i, 4 and 5, which achieve their final values practically at the time of the ylide consumption (7 min), the signal of λ5-phosphinoline 3 (2.3 ppm) appears when the ylide is almost consumed. However positive signals at 16.9, 15.4, −2.75 and −2.8 ppm appearing at 4.5 min and attaining their maximum values by 7 min are likely the precursors of 3, because the sum of concentrations of 3 and these intermediates.
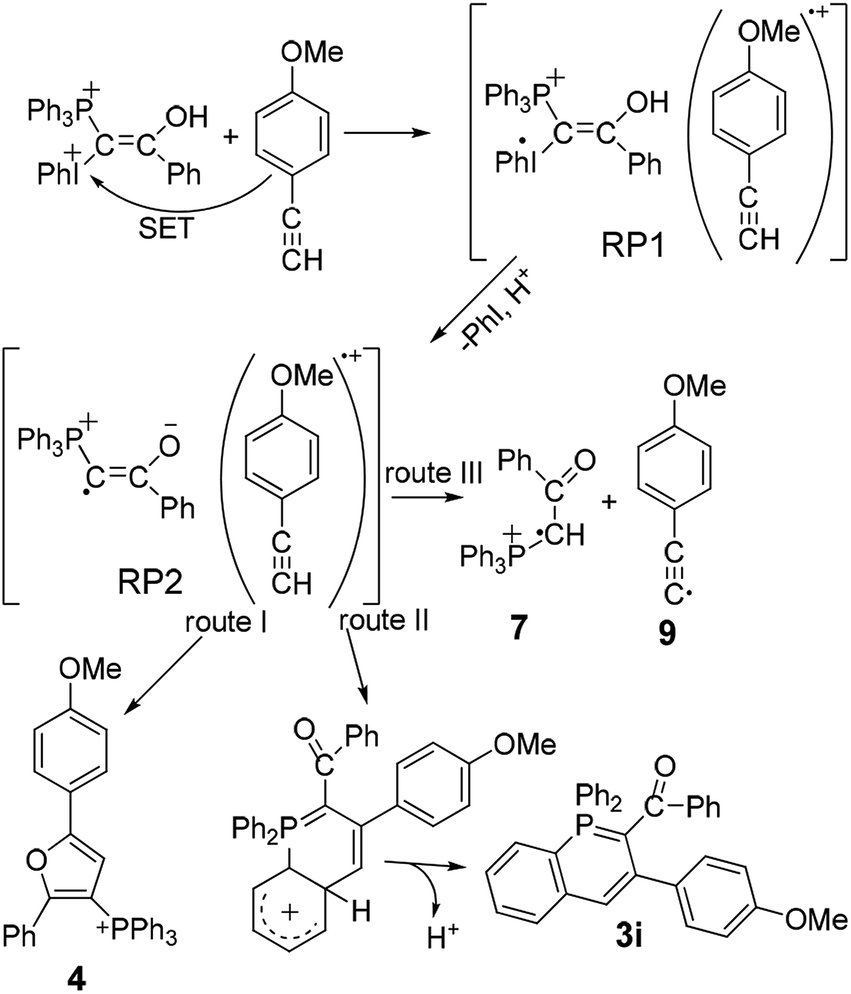 | ||
| Scheme 3 Formation of the products in the radical pairs. | ||
Due to the complexity and overlapping of the 1H NMR spectra of the initial reagents and products in the low field, it is difficult to process the 1H NMR spectra. However, emissive signal of the H(4) proton of 4 (δ ∼6.5 ppm), which does not overlap with the signals of 1, 2, 3 and 5, has been observed in DCM and deuterated DCM (DCMd2) (Fig. 3, ESI, Fig. S13–S18†). The CIDNP for the proton H(4) of furan 4 in DCMd2 indicates that this originates from 2 rather than from the solvent. In the 31P NMR spectra for the reaction in deuterated DCMd2 salt 5 appears as two signals slightly shifted to the high field (21.5 and 21.45 ppm, final yields 8 and 1%, respectively) in comparison to the protonated 5 (21.65 ppm in DCMd2). Both signals show enhanced absorption in the 31P NMR spectra in the course of the reaction. From the comparison with the spectrum of protonated form of 5 in DCMd2, the observed signals were attributed to the deuterated forms of salt 5 (Ph3P+–CHD–C(O)Ph)BF4− (5-HD) and (Ph3P+–CD2–C(O)Ph)BF4− (5-2D), respectively.
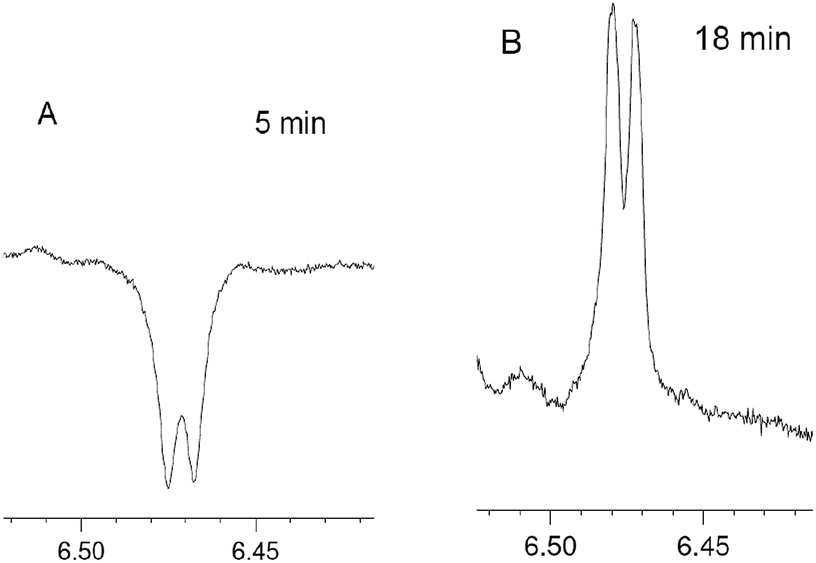 | ||
| Fig. 3 Fragments corresponding to H(4) protons of 4 in the 1H NMR spectrum in the course of the reaction between 1 and 2 in DCM at different times. | ||
According to Radical Pair Mechanism the CIDNP can be developed, if there is at least one transient state of the reaction which is comprised of a pair of radicals,10 and this pair should be spin-correlated, by another word, at definite time of its existence it is in a definite spin state which can evolve from one spin state to the other. In all the schemes for the reaction between ylides and acetylenes proposed before there was no place for radical pairs.2,7 Thus, the observation of CIDNP does not agree with any of these schemes.
The CIDNP effect was observed in the reduction of iodonium salts by amines,11 for P-containing compounds in their thermal reactions with peroxides12a–c and in the photolysis of benzoylphosphonate.12d In essence, all these examples are specially selected reactions to study the CIDNP phenomenon. The work presented here is a rather rare example of the use of CIDNP as a tool for studying the reaction mechanism. In other words, we have to propose a reaction mechanism for the interaction of ylide 1 with acetylene 2, which, on the way to the formation of the final products, would include spin-selective intermediate reactions occurring in the corresponding spin-correlated radical pairs. To begin with, we assume that single electron transfer (SET) from 2 to the protonated form of ylide 1, dication 8, which is present in the solution to the end of induction time in sufficient amounts due to acid catalysis, results in a geminate ion-radical pair RP1 (Scheme 3). This is a rather unusual assumption for reactions of mixed ylides with acetylenes. But it is supported by the dependence of the product yields on the ionization potential of acetylenes.7
The protonation of ylide 1 not only accelerates the rate of generation of RP1, but also the rate of homolytic dissociation of the dication 8, followed by deprotonation and the formation of the ion radical pair RP2. The radicals generated in the RP2 can either interact in or escape from the GP. The escaped radicals can recombine in an encounter pair or enter into reactions with non-radical initial compounds, solvent or intermediates. Phosphinoline 3 originates from intermediates formed with participation of the escaped radicals or radicals generated in the system independently.
Three routes for the interaction of the radicals in the RP2 are considered (Scheme 3): the major route I is the formation of furan 4 (∼80% of the products formed in the RP2) from the radical 6 E-isomer and acetylene radical cation, route II gives λ5-phosphinoline 3i from the radical 6 Z-isomer. Route III suggests the formation of radical cation 7 as a result of proton transfer (PT) from acetylene radical cation to radical 6. The escaped 7 forms salt 5 in the reaction with the solvent (Scheme 2). Salt 5 formed from radical 7 generated in the RP2 should show enhanced absorption. The final concentration of salt 5 is almost fivefold lower, than the final concentration of furan 4 confirming that the major reaction in the RP2 is the formation of furan. The formation of 5 in two forms in DCMd2, with 5-HD prevailing over 5-2D and the protonated form 5 almost being absent, does mean that the main source of radical 7 is the PT reaction in the RP2 (Scheme 3).
In summary, the CIDNP effect in the reaction between ylide 1 and p-methoxyphenylacetylene clearly shows the participation of radical pairs in the formation of furan derivative 4 and phosphonium salt 5. The presence of the induction time means that small amounts of H+ should be generated in the decomposition of the ylide (Scheme 2), and dication 8 should form. This initiates SET from 2 to dication 8. It should be emphasized that SET between 8 and 2 is promoted by the microheterogeneity of the ylide solution in DCM, which provides the close proximity of the reagents.6 Recently, we have shown complete inhibition of the formation of all phosphorus-containing products upon addition of TEMPOL (2 mol%) into the reaction mixture. The usual development of the reaction after TEMPOL consumption counts in favor of the assumption that radicals participate in the formation of all products in one or another degree.5b
In the case of phenylacetylene, the reaction proceeds under irradiation with an induction time of several minutes, furan derivative is formed in trace amounts and λ5-phosphinoline is the major heterocyclic product. The difference in the reaction course for phenylacetylene and p-methoxyphenylacetylene is accounted for by the lower ionization energy for the latter by about 0.8 eV.13 This makes possible SET from p-methoxyphenylacetylene to iodonium group of the ylide initiating PhI splitting off and the formation of the RP, in which 4 is formed. For phenylacetylene this process practically does not take place, and the formation of λ5-phosphinoline occurs in the reactions of photogenerated radical 6 with acetylene via several intermediates similarly to the formation of 3 for p-methoxyphenylacetylene.
The spectra were recorded on an Avance 500 NMR spectrometer of the Core Facility of IBCP RAS “New Materials and Technologies”. The study was supported by the IBCP RAS RF State Program no. 1201253303.
Conflicts of interest
There are no conflicts to declare.Notes and references
- R. M. Moriarty, I. Prakash, O. Prakash and W. A. Freeman, J. Am. Chem. Soc., 1984, 106, 6082–6084 CrossRef CAS.
- (a) E. D. Matveeva, T. A. Podrugina, A. S. Pavlova, A. V. Mironov, A. A. Borisenko, R. Gleiter and N. S. Zefirov, J. Org. Chem., 2009, 74, 9428–9432 CrossRef CAS PubMed; (b) E. D. Matveeva, T. A. Podrugina, M. A. Taranova, A. M. Ivanova, R. Gleiter and N. S. Zefirov, J. Org. Chem., 2012, 77, 5770–5774 CrossRef CAS PubMed; (c) E. D. Matveeva, T. A. Podrugina, M. A. Taranova, E. Y. Melikhova, R. Gleiter and N. S. Zefirov, Tetrahedron, 2013, 69, 7395–7402 CrossRef CAS; (d) E. D. Matveeva, D. S. Vinogradov, T. A. Podrugina, T. D. Nekipelova, A. V. Mironov, R. Gleiter and N. S. Zefirov, Eur. J. Org. Chem., 2015, 7324–7333 CrossRef CAS.
- T. D. Nekipelova, M. A. Taranova, E. D. Matveeva, T. A. Podrugina, V. A. Kuzmin and N. S. Zefirov, Dokl. Chem., 2012, 447, 262–265 CrossRef CAS.
- (a) T. D. Nekipelova, M. A. Taranova, E. D. Matveeva, V. A. Kuz’min and N. S. Zefirov, Kinet. Catal., 2015, 56, 403–411 CrossRef CAS; (b) T. D. Nekipelova, T. A. Podrugina, D. S. Vinogradov, P. I. Dem’yanov and V. A. Kuzmin, Kinet. Catal., 2019, 60, 44–51 CrossRef CAS.
- (a) T. D. Nekipelova, V. V. Kasparov, A. L. Kovarskii, A. Kh. Vorobiev, T. A. Podrugina, D. S. Vinogradov, V. A. Kuzmin and N. S. Zefirov, Dokl. Phys. Chem., 2017, 474(2), 109–112 CrossRef CAS; (b) T. D. Nekipelova, M. V. Motyakin, V. V. Kasparov, Ye. N. Degtyarev, I. I. Levina, I. D. Potapov and T. A. Podrugina, Russ. J. Phys. Chem. B, 2019, 13 Search PubMed , accepted for publication..
- I. I. Levina, O. N. Klimovich, D. S. Vinogradov, T. A. Podrugina, D. S. Bormotov, A. S. Kononikhin, O. V. Dement’eva, I. N. Senchikhin, E. N. Nikolaev, V. A. Kuzmin and T. D. Nekipelova, J. Phys. Org. Chem., 2018, 31, e3844, DOI:10.1002/poc.3844.
- E. D. Matveeva, T. A. Podrugina, M. A. Taranova, A. A. Borisenko, A. V. Mironov, R. Gleiter and N. S. Zefirov, J. Org. Chem., 2011, 76, 566–572 CrossRef CAS PubMed.
- T. D. Nekipelova, V. A. Kuzmin, E. D. Matveeva, R. Gleiter and N. S. Zefirov, J. Phys. Org. Chem., 2013, 26, 137–143 CrossRef CAS.
- (a) J. L. Dektar and N. P. Hacker, J. Org. Chem., 1990, 55, 639–647 CrossRef CAS; (b) J. A. Kampmeief and T. W. Nalli, J. Org. Chem., 1994, 59, 1381–1388 CrossRef.
- (a) R. Kaptein, Adv. Free-Radical Chem., 1975, 5, 319–380 CAS; (b) K. M. Salikhov, Yu. N. Molin, R. Z. Sagdeev and A. L. Buchachenko, Spin Polarization and Magnetic Effects in Radical reactions, ed. Yu. N. Molin, Elsevier, Amsterdam, Akadémiai Kiadó, Budapest, 1984 Search PubMed.
- (a) I. P. Gragerov, A. F. Levit, L. O. A. Kiprianova and A. L. Buchachenko, Org. Magn. Reson., 1973, 5, 445–448 CrossRef CAS; (b) L. K. Skrunts, A. F. Levit, L. A. Kiprianova and I. P. Gragerov, Zh. Org. Khim., 1980, 16, 604–608 CAS.
- (a) Ya. A. Levin, Dokl. Akad. Nauk SSSR, 1976, 230, 358–361 CAS; (b) Ya. A. Levin, A. V. Il’yasov, E. I. Goldfarb and E. I. Vorkunova, Org. Magn. Reson., 1973, 5, 487–496 CrossRef CAS; (c) D. G. Pobedimsky, P. A. Kirpichnikov, Yu. Yu. Samitov and E. I. Goldfarb, Org. Magn. Reson., 1973, 5, 503–506 CrossRef CAS; (d) G. S. Ananchenko, P. A. Purtov, E. G. Bagryanskaya and R. Z. Sagdeev, J. Phys. Chem. A, 1999, 103, 3430–3437 CrossRef CAS.
- There is no data for the ionization potential (IP) of p-methoxyphenylacetylene (IP = 8.8 eV for phenylacetylene, B. West, A. Sit, A. Bodi, P. Hemberger and P. M. Mayer, J. Phys. Chem. A, 2014, 118, 11226–11234), but the IP of 9.2 and 8.4 eV for benzene and anisole, respectively, allows us to assume that IP for p-methoxyphenylacetylene is near 8.0 eV..
Footnote |
| † Electronic supplementary information (ESI) available: 31P and 1H NMR spectra. See DOI: 10.1039/c9ra05697a |
| This journal is © The Royal Society of Chemistry 2019 |