
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComparison of nano-structured transition metal modified tri-metal MgMAl–LDHs (M = Fe, Zn, Cu, Ni, Co) prepared using co-precipitation†
Bianca R. Gevers *a,
Sajid Naseemb,
Andreas Leuteritzb and
Frederick J. W. J. Labuschagnéa
*a,
Sajid Naseemb,
Andreas Leuteritzb and
Frederick J. W. J. Labuschagnéa
aUniversity of Pretoria, Lynnwood Road, 0002, Pretoria, South Africa. E-mail: bianca.gevers@tuks.co.za
bLeibniz-Institut für Polymerforschung Dresden e. V., Hohe Straße 6, 01069, Dresden, Germany
First published on 9th September 2019
Abstract
Comparison of layered double hydroxides (LDHs) synthesised using different methods, conditions and post-treatment is difficult to achieve because these greatly modify their material properties. This paper aims to provide a comparison of material properties for modified quintinite, where all LDHs were synthesised at the same conditions – thus allowing for direct comparison of the material properties obtained. Nano-structured materials were formed in all cases. The nano-structured transition metal (TM) MgMAl–LDHs were synthesised using constant pH co-precipitation. Five TMs (M = Fe, Co, Ni, Cu, Zn) were included in the LDH layers with molar substitutions of 0.5%, 1%, 5%, 10%, and 25% based on Mg-replacement for divalent TM cations and Al-replacement for trivalent TM cations. The materials were characterised using powder X-ray diffraction (XRD), X-ray fluorescence spectroscopy (XRF), scanning electron microscopy (SEM), attenuated total reflectance Fourier transform infrared analysis (ATR-FTIR), thermogravimetric analysis (TGA) and particle size analysis (PSA). The modified LDHs were synthesised free of major by-products and with similar morphologies. It could be shown that the crystallite dimensions varied between the different TM substitutions, that morphological changes were visible for some of the TMs used, that the processability depended on the TMs substituted, and that the substitution of TMs influenced the thermal stability of the LDHs.
1 Introduction
LDHs are anionic clays with the general formula| [MII1−xMIIIx(OH)2][Xq−x/q·nH2O] |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) describes the composition of the LDH layers consisting of trivalent (MIII) and divalent (MII) metal cations, while [Xq−x/q·nH2O] represents the composition of the anionic interlayer. Further, x is the molar fraction of trivalent cations to total metal cations in the layer structure and is typically limited to 0.1 ≤ x ≤ 0.33. X represents the interlayer anion with charge q.1 LDHs have received increasing research attention due to their simple adaptability and tailoring options with respect to metal composition, anion usage and morphology.1,2
describes the composition of the LDH layers consisting of trivalent (MIII) and divalent (MII) metal cations, while [Xq−x/q·nH2O] represents the composition of the anionic interlayer. Further, x is the molar fraction of trivalent cations to total metal cations in the layer structure and is typically limited to 0.1 ≤ x ≤ 0.33. X represents the interlayer anion with charge q.1 LDHs have received increasing research attention due to their simple adaptability and tailoring options with respect to metal composition, anion usage and morphology.1,2
LDHs are most frequently synthesised using co-precipitation due to the ease of preparation for most compositions and the purity of the LDHs formed. Other options include urea hydrolysis, sol–gel synthesis and hydrothermal synthesis.1,3
One of the most common LDHs is hydrotalcite, which naturally occurs as Mg6Al2(OH)16CO3·4H2O and can, with carbonate anions in the interlayer space, be synthetically produced with ratios of divalent to trivalent metal cations of 1.0–3.0.1 Hydrotalcite can be modified to include other metals in its layer structure and is thus frequently investigated as a support structure for TMs. A variety of such substitutions have been performed in literature. In fact, publications exist with many different combinations of metals in the layer, and almost all metals have successfully been included in LDHs' layers.4
A subdivision of the hydrotalcite supergroup comprises the quintinite-group which consists of LDHs where the ratio MII+/MII+ is 2:1 and typical anions (CO2−3 and Cl−) and water are present in the interlayer.5 Quintinite has the chemical formula Mg4Al2(OH)12CO3·3H2O. The use of modified quintinite is widespread (albeit the LDHs are typically named hydrotalcite-like materials).
As mentioned by Mills et al. (2012), the predominant mentions of divalent metals in the layers of LDHs within the hydrotalcite supergroup are Mg, Ca, Mn, Fe, Ni, Cu and Zn, while the predominant trivalent metals are Al, Mn, Fe, Co and Ni.5 Common interlayer anions include CO2−3, Cl−, SO2−4, OH−, S2− and [Sb(OH)6]−, the most common one being CO2−3.5 For this study, the divalent metals Co, Ni, Cu and Zn, and trivalent metal Fe were chosen to modify the layer composition of standard quintinite. All LDHs were prepared with CO2−3 in the interlayer, as this was found to be the most common anion used in modified quintinite. The TMs for substitution were chosen for their variety of applications across catalysis, renewable energy generation, environmental remediation, drug delivery and polymer stabilisation – all big or upcoming fields of application for LDHs.1,6–8
Some examples of quintinite modification within these fields include: catalysis (ethylbenzene dehydrogenation,9 hydrocarbon oxidation,10 CO2 reforming of methane,11 dry reforming of methane12), drug delivery (NSAID release13), renewable energy (H2 generation,14 transesterification of soybean oil15), environmental remediation (fluoride absorption,16 wet air oxidation of Basic Yellow 11,17 N2O decomposition,18 methyl orange adsorption19) and polymer stabilisation/functional additives (PVC stabilisation,20 HCl scavenging in PVC,21 UV degradation stabiliser22).
Some researchers have performed investigations into the influence of the type and amount of TM substituted into LDH layers. A comprehensive search was conducted to find literature relevant to the synthesis and characterisation of modified quintinite. The results of the search are listed in Table 1.
| Anion | Mg:Al ratio |
Subs. | Syn. pH | SynM | Ref. |
|---|---|---|---|---|---|
| a SynM: synthesis method, CP: co-precipitation, hls CP: high and low saturation CP, v CP: variable CP, m CP: microwave CP, UH: urea hydrolysis, sa CP: simultaneous addition CP, C + S: CO2−3 + SO2−4. | |||||
| MgFeAl–LDH | |||||
| CO2−3 | 5.66, 2.33, 1 & 0.43:1 |
33.33, 16.66, 10, 7 | 9.6–9.9 | hls CP | 23,24 |
| CO2−3 | 3:2 |
25, 50, 75 | 9 | CP | 25 |
| C + S | 2:1 |
50 | 2.4–13 | v CP | 26 |
| CO2−3 | 2:1 |
4, 8, 12, 16, 20, 40, 60, 80 | 9.5 | CP | 14 |
| CO2−3 | 2:1 |
16.66, 33.33, 50, 66.66, 83.33 | — | sa CP | 16 |
| NO−3 | 3:1 |
5 | 10 | CP | 27 |
| CO2−3 | 2:1 |
5, 10 | — | UH | 28 |
|
|||||
| MgCoAl–LDH | |||||
| CO2−3 | 3.5:1 |
12.5 | 10 | CP | 29 |
| CO2−3 | 2:1 |
3, 5 | — | UH | 30 |
| C8H4O4 | 2.33, 1:1 |
20, 40, 60, 80 | 6.5 | CP | 31 |
| CO2−3 | 9, 10:1 |
11.11, 20 | 9 | CP | 32 |
| NO−3 | 3:1 |
5 | 10 | CP | 27 |
| CO2−3 | 2:1 |
5, 10 | — | UH | 28 |
|
|||||
| MgNiAl–LDH | |||||
| CO2−3 | 3.5:1 |
12.5 | 10 | CP | 29 |
| NO−3 | 3:1 |
5 | 10 | CP | 27 |
| NO−3 | 2, 3, 4:1 |
50 | 13 | m CP | 33 |
| CO2−3 | 7, 3:1 |
33.33, 66.67, 85.7 | 10 | CP | 34 |
| CO2−3 | 5, 3, 1, 0.33, 0.2:1 |
8 | 10.5 | CP | 35 |
| CO2−3 | 2:1 |
5, 10 | — | UH | 28 |
|
|||||
| MgCuAl–LDH | |||||
| CO2−3 | 3.5:1 |
12.5 | 10 | CP | 29 |
| CO2−3 | 1–5:1 |
1, 3, 5, 7, 10, 20 | 10 | CP | 36 |
| NO−3 | 3:1 |
5 | 10 | CP | 27 |
| CO2−3 | 2:1 |
5, 10 | — | UH | 28 |
|
|||||
| MgZnAl–LDH | |||||
| CO2−3 | 5.67 | 25, 50, 75 | — | UH | 37 |
| NO−3 | 3:1 |
5 | 10 | CP | 27 |
| NO−3 | 2, 3, 4:1 |
50 | 13 | m CP | 33 |
| CO2−3 | 2:1 |
12.5, 50, 80 | — | UH | 19 |
| CO2−3 | 2:1 |
5, 10 | — | UH | 28 |
As clearly visible from the table, papers that focus on the synthesis and characterisation of TM-modified quintinite and compare a variety of TMs are quite rare. Of the papers listed in Table 1, Rivera et al. (2006), Pavel et al. (2012) and Tsyganok et al. (2006) provided a comparison between different substituted TMs at the same substitution percentage.27,29,33
Some authors have investigated the effect of different amounts of TMs substituted into quintinite layers. Examples of these are the varying substitution of Fe,14,16,17 Co,30,31, Ni,15,17,18 and Zn,19,20,22 into quintinite. However, no papers (other than our previous paper on the urea hydrolysis synthesis of TM-modified LDHs,28 comparing the effects of two substitutions) could be found that focused on the synthesis, characterisation and comparison of varying amounts and types of TM substituted into quintinite specifically and using co-precipitation. This could be a result of the thermodynamic stability of the LDH phase depending on the synthesis pH, thus favouring the precipitation of divalent metal hydroxides if an unsuitable synthesis pH was chosen and producing impure LDH phases difficult to compare. Based on the solubility products of di-metal quintinite ([M4Al2(OH)12]CO3·nH2O), the formation of divalent hydroxides is favoured above pH ≈ 10 for M = Zn, pH ≈ 9 for M = Co, pH ≈ 8 for M = Ni and pH ≈ 12 for M = Mg.3
What is also evident from Table 1, is that the synthesis conditions and methods varied in most studies. As the product LDH phases' morphology and physical characteristics are heavily influenced by the synthesis method and conditions used, it renders the research that has already been performed difficult if not impossible to compare. Because of the difficulty of comparison between the different data sets, this paper aims to give the reader information on the effect of the amount and type of TM-substituted into the LDH layers with focus on purity, structural differences, morphological differences and thermal stability differences for LDHs all synthesised at the same conditions.
2 Experimental
2.1 Materials
Chemically pure (CP), reagent grade (RG) or analytical grade (AR) reactants were used for all experiments. AlCl3·6H2O (CP), MgCl2·6H2O (CP), ZnCl2 (CP), FeCl3·6H2O (CP), CoCl2·6H2O (AR), NaOH (AR) and Na2CO3 (AR) were sourced from ACE Chemicals. NiCl2·6H2O (AR) and CuCl2·2H2O (AR) were obtained from Merck, and FeCl3 (RG) from Sigma Aldrich. Deionised water was used for all experiments.2.2 Material preparation
All LDHs were synthesised by making use of the constant pH co-precipitation method. The stoichiometric amounts of metal salts were dissolved in deionised water to form a 2.5 M solution (based on cations). Na2CO3 was used in excess to ensure availability of carbonate anions in solution. The salt solution was added into a 0.4 M Na2CO3 solution in a dropwise fashion while maintaining a pH of 11 ± 0.3 in all experiments by adding 10 M NaOH solution as required. The synthesis pH was specifically chosen to be higher than the optimum/frequently chosen synthesis pH for hydrotalcite synthesis (pH = 10)38 so that all metal species are kept well above their theoretical hydroxide formation limit.3,39 Further, the precipitation was carried out at high supersaturation to limit the formation of the hydroxides. The resulting slurry was stirred vigorously at 500 rpm throughout the reaction. The precipitated LDH was filtered off using vacuum filtration, washed with 5 L of deionised water, and dried at 60 °C overnight (18 h).MgAl–LDHs with five TM substitutions (Fe, Co, Ni, Cu and Zn) were prepared with substitution percentages (s) of 0.5%, 1%, 5%, 10% and 25%. Hereby, Mg was substituted with the divalent metal cations of Co, Ni, Cu, and Zn and Al was substituted with trivalent Fe cations. The stoichiometrically correct amounts of salts for the divalent metal (Co, Ni, Cu and Zn) substituted LDHs were calculated using
| (2 − x)MgCl2·6H2O + xMCl2·yH2O + 1AlCl3·6H2O + 0.5Na2CO3 → Mg(2−x)MxAl1(OH)6(CO3)0.5·zH2O |
| 2MgCl2·6H2O + xFeCl3·6H2O + (1 − x)AlCl3·6H2O + 0.5Na2CO3 → Mg2MxAl(1−x)(OH)6(CO3)0.5·zH2O |
2.3 Materials characterisation
X-ray diffraction measurements were performed on a Panalytical X'Pert PRO X-ray diffractometer in θ–θ configuration, equipped with Fe-filtered Co-Kα radiation (1.789 Å) and an X'Celerator detector and variable-divergence- and fixed receiving slits. Samples were prepared according the standardised Panalytical backloading system, which facilitates the nearly random distribution of the particles. Data were collected in the angular range 5° ≤ 2θ ≤ 80°, with a step size of 0.008° 2θ and a 13 s scan step time. The phases were identified using X'Pert HighScore Plus software. The spectra were converted from variable slit to fixed slit prior to phase identification. The detection limit for phases was 2%.X-ray fluorescence (XRF) analysis was performed using a Thermo Fisher ARL Perform'X Sequential XRF instrument. The samples were dried at 100 °C and roasted at 1000 °C to determine loss on ignition (LOI). 1 g roasted sample (where available, if not, maximum grams remaining after roasting) was then placed together with 6 g of Li2B4O7 into a Pt/Au crucible and fused. Analyses were executed using the corresponding Quantas software.
Scanning electron microscopy (SEM) images were taken with a Zeiss Gemini Ultra Plus FEG SEM, and a Zeiss Gemini 2 Crossbeam 540 FEG SEM. The samples were prepared by evenly distributing the samples on carbon tape on an aluminium stub and coating it with two layers of carbon (angles: 0°, −45° and +45°) using a sputter coater. The micrographs were taken at 1 keV.
Thermogravimetric analysis (TGA) was performed with a heating rate of 10 °C min−1 using a TGA Q5000 from TA instruments in an inert nitrogen atmosphere.
ATR-FTIR spectra were obtained using a PerkinElmer 100 Spectrophotometer. Samples were pressed in place with a force arm. Spectra were obtained in the range of 550–4000 cm−1 each with 32 scans at a resolution of 2 cm−1.
The particle size analysis (PSA) was carried out using a Malvern Mastersizer 3000. Each sample was sieved to remove large agglomerates and diluted with distilled and deionised water. The sample was dispersed using tetrasodium pyrophosphate, blended, filtered and the filter cake re-dispersed in distilled and deionised water by shaking.
3 Results
3.1 Structure, composition and purity
The XRD patterns obtained for the synthesised samples are shown in Fig. 1 and a summary of the data obtained from XRD analysis (angular reflection positions, d-spacing, crystal parameters and crystallite sizes) is given in the ESI.† All samples were observed to have very similar patterns and clear (003), (006), (009), (110) and (115) reflections, as expected for an LDH in the rhombohedral R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group. The observed patterns show the expected reflections for synthetic quintinite in the rhombohedral (3R) stacking sequence prepared at near-ambient conditions.40 Broad reflections on XRD patterns for LDHs can indicate the formation of small crystallites and stacking faults.41 From comparison of our data to previous data obtained from samples prepared using urea hydrolysis,28 it could be concluded that the broad peaks observed in this study are indicative of the formation of small crystallites.41
m space group. The observed patterns show the expected reflections for synthetic quintinite in the rhombohedral (3R) stacking sequence prepared at near-ambient conditions.40 Broad reflections on XRD patterns for LDHs can indicate the formation of small crystallites and stacking faults.41 From comparison of our data to previous data obtained from samples prepared using urea hydrolysis,28 it could be concluded that the broad peaks observed in this study are indicative of the formation of small crystallites.41
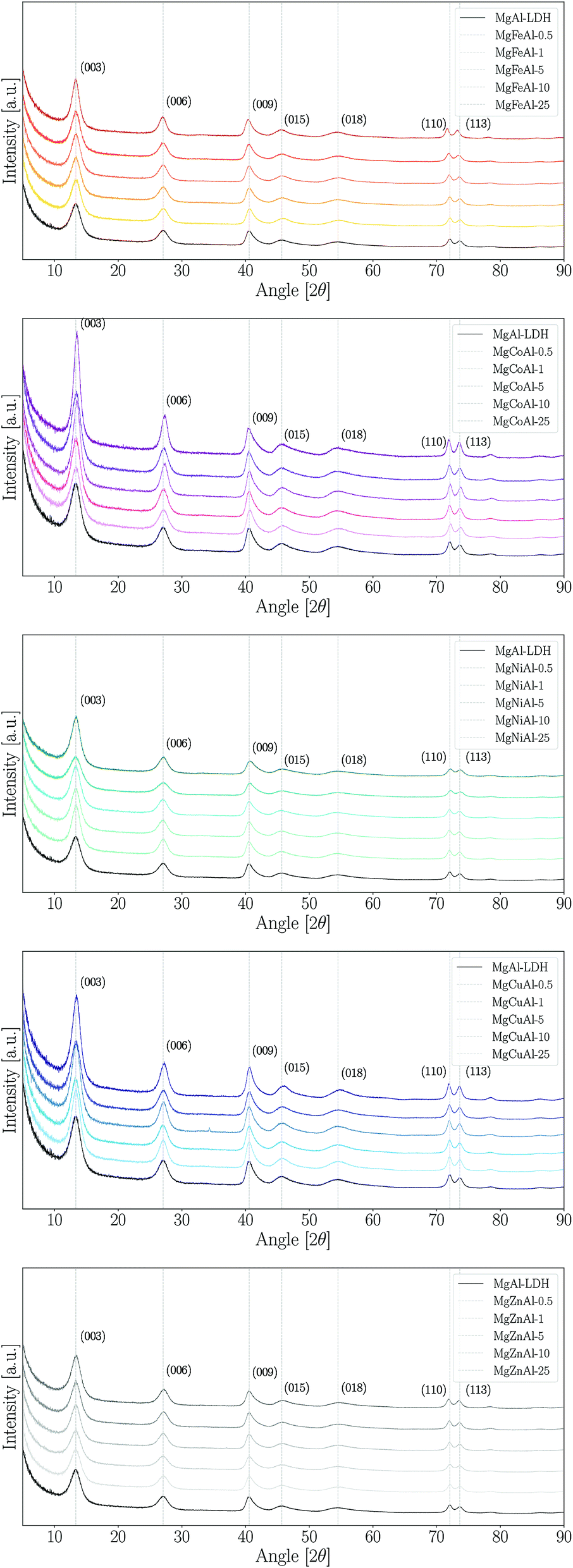 | ||
| Fig. 1 XRD patterns of the TM-substituted MgMAl–LDHs synthesised. | ||
The materials were prepared with great purity. No immediately visible reflections indicating an impurity phase were found for any LDH other than MgCuAl-5. However, three other LDHs were found to contain very small amounts of an additional phase. The impurity phases identified through XRD analysis showed that jamborite (Ni(OH)2·NiOOH), calumetite (Cu(OH,Cl)2·2H2O) and caresite (Fe4Al2(OH)12(CO3)·3H2O) were present in very small amounts in MgNiAl-0.5, MgNiAl-5, MgCuAl-5 and MgFeAl-10, respectively. Halo-formation, if present, was found to be small in the samples prepared, thus indicating that the LDHs were mostly synthesised free of appreciable amounts of amorphous material. However, it could not be excluded that some of the LDH phases were formed with low crystallinity, thus closely resembling amorphous material.
Further, the LDHs showed very similar peak definitions and intensities. For all but four LDHs, the intensity of the main reflection was higher compared to that of unmodified MgAl–LDH (Fig. 2). The Fe-, Co- and Zn-substituted LDHs all showed a definite increase in primary reflection intensity (PRI) with an increase in TM substitution. The PRIs of the Ni- and Cu-substituted LDHs varied upon an increase in TM substitution. The PRIs of the Ni-substituted LDHs were found to increase, except for a dip at 10% substitution. The net change in PRI of the Cu-substituted LDHs was positive; a zig-zag upwards trend seemed to exist. It was concluded that the general trend in PRI increase with an increase in TM substitution was significant enough in the Fe, Co, Zn and Cu-substituted LDHs to potentially relate these to increases in crystallinity rather than constructive interference due to preferential platelet orientation upon powder pressing during the XRD sample preparation – a phenomenon commonly observed in the XRD analysis of clays.42 This could be further substantiated by the increased narrowness of the PRI with an increase in TM substitution. However, because multiple factors influence the reflection intensity and width (e.g. crystallite size and crystallinity) no definite conclusion could be drawn on the increase in the crystallinity of the LDHs with TM substitution.
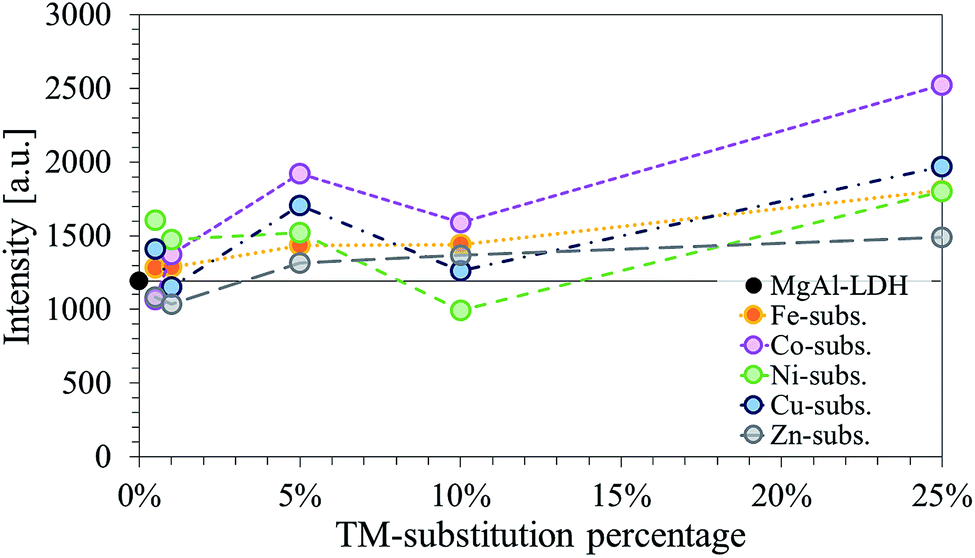 | ||
| Fig. 2 Comparison of the background adjusted intensities of the primary (003) reflection shown as a function of the TM-substitution percentage used to modify the MgMAl–LDHs with M = Fe, Co, Ni, Cu and Zn. | ||
Considering the XRD patterns shown in Fig. 1, it can be observed that some deviations in the position of the (most significantly) (003) and (110) reflections existed. Crystal parameters c and a were thus calculated in order to quantify structural changes in the LDHs. Changes in the c- and a-parameter with TM substitution are shown in Fig. 3. Bragg's law (nλ = 2dsinθ) was used to calculate the layer spacings d00l and d110 of the LDH. An average parameter c was calculated as shown by dos Reis et al. (2004)43 and using the relation that d003 = 2d006 = 3d009 for LDHs of rhombohedral symmetry2 so that c = 3(1/3(d003 + 2d006 + 3d009)) = d003 + 2d006 + 3d009. Parameter a was calculated using a = 2d110.2 It could be shown that the a-parameter increased linearly for the Fe-substituted LDHs and almost linearly with Cu- and Zn-substitution. The most significant increase in a-parameter was observed for the Fe-substituted LDHs. Only small increases in the a-parameter were observed for Cu- and Zn-substitution. The a-parameter varied for Co-substitution, albeit an increased a-parameter was observed for the 1%, 10% and 25% substituted LDHs. A decline in the a-parameter was observed upon Ni-substitution.
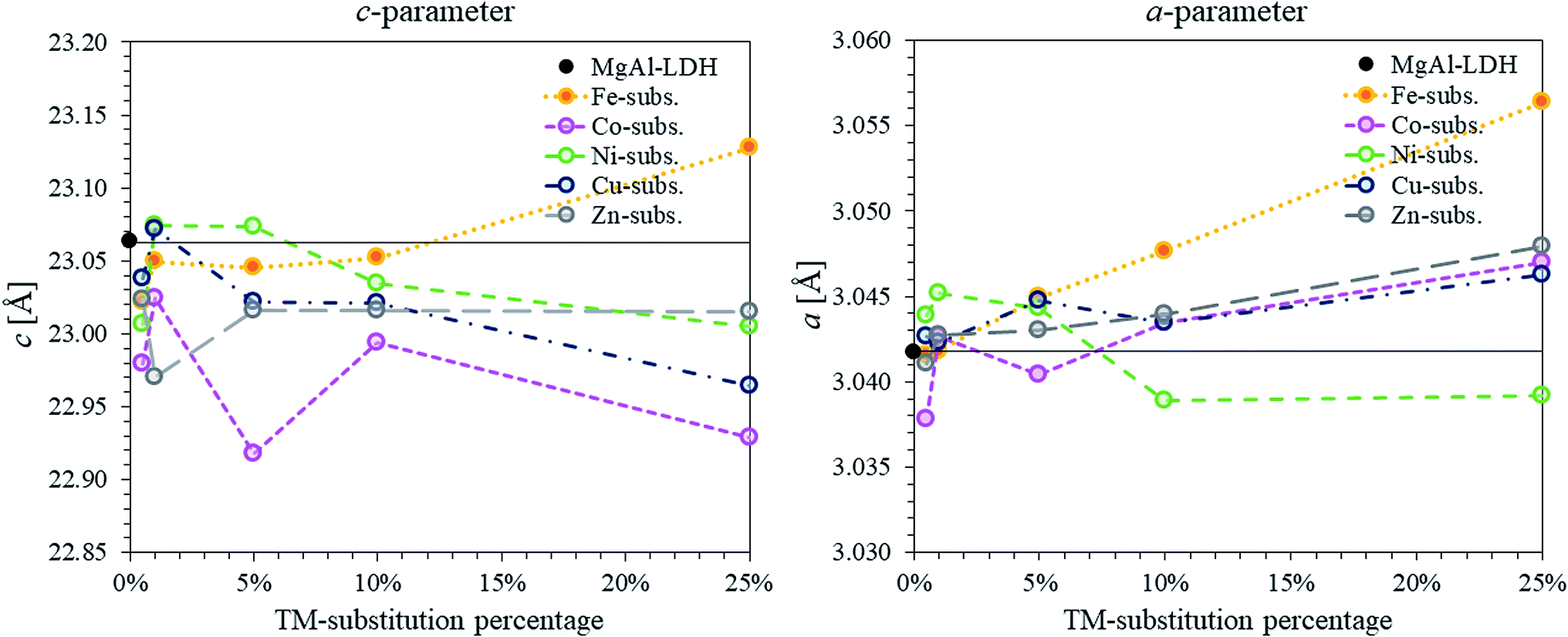 | ||
| Fig. 3 Comparison of c- and a-parameters calculated for the series of TM-substituted MgMAl–LDHs with M = Fe, Co, Ni, Cu and Zn. The parameters were calculated from the XRD data shown in Fig. 1. | ||
Changes in the a-parameter are a function of changes in the layer structure of LDHs. Being independent of the interlayer composition, the a-parameter can be used to determine successful layer composition modification.2 Considering the atomic radii of the VI coordinated cations in the crystal lattice (Mg2+ = 0.72 Å, Co2+ = 0.745 Å, Ni2+ = 0.69 Å, Cu2+ = 0.73 Å, Zn2+ = 0.74 Å, Al3+ = 0.535 Å and Fe3+ = 0.645 Å)44 the results correspond very well to the expected results for the modification of the LDH layer composition. We believe that the fluctuations in a-parameter observed for the Co-substituted samples occurred due to a change in valence state of the Co2+ cation to Co3+ (r(Co3+ = 0.545 − 0.61 Å)).
The results of the c-parameter calculations showed that some deviation existed in the layer distance between all groups of TM-substituted LDHs and between the different substitution percentages. Changes in the c-parameter are a function of the cation radius of the metals in the layer, as well as the number of water molecules and orientation of the anions (including coordination of the water and anions) within the interlayer.2 The differences seen here were found to match the change in interlayer water present in the LDHs (see TGA discussion) taking into account the layer thickness (a-parameter).
XRF analysis was used to ascertain the layer composition of the LDHs formed. Table 2 shows that the M2+/M3+ ratio was close to 2:1 for all synthesised LDHs. The desired TM substitution was closely matched for all but the Co-substituted LDHs.
| Name | MgO | Al2O3 | TM-oxide | Ratio | Subs. |
|---|---|---|---|---|---|
| MgAl | 0.811 | 0.192 | 0.000 | 2.11 | 0 |
| MgNiAl-0.5 | 0.831 | 0.186 | 0.005 | 2.24 | 0.58% |
| MgNiAl-1 | 0.846 | 0.176 | 0.007 | 2.43 | 0.80% |
| MgNiAl-5 | 0.772 | 0.180 | 0.045 | 2.26 | 5.51% |
| MgNiAl-10 | 0.682 | 0.182 | 0.086 | 2.11 | 11.22% |
| MgNiAl-25 | 0.581 | 0.162 | 0.193 | 2.39 | 24.93% |
| MgCuAl-0.5 | 0.814 | 0.182 | 0.004 | 2.24 | 0.53% |
| MgCuAl-1 | 0.799 | 0.189 | 0.009 | 2.13 | 1.13% |
| MgCuAl-5 | 0.779 | 0.184 | 0.043 | 2.23 | 5.24% |
| MgCuAl-10 | 0.712 | 0.180 | 0.081 | 2.20 | 10.24% |
| MgCuAl-25 | 0.546 | 0.170 | 0.194 | 2.18 | 26.18% |
| MgFeAl-0.5 | 0.796 | 0.189 | 0.001 | 2.09 | 0.61% |
| MgFeAl-1 | 0.824 | 0.189 | 0.002 | 2.15 | 1.15% |
| MgFeAl-5 | 0.821 | 0.177 | 0.010 | 2.20 | 5.37% |
| MgFeAl-10 | 0.801 | 0.170 | 0.020 | 2.11 | 10.44% |
| MgFeAl-25 | 0.739 | 0.129 | 0.044 | 2.13 | 25.32% |
| MgZnAl-0.5 | 0.814 | 0.190 | 0.005 | 2.15 | 0.57% |
| MgZnAl-1 | 0.804 | 0.193 | 0.009 | 2.10 | 1.15% |
| MgZnAl-5 | 0.762 | 0.189 | 0.043 | 2.12 | 5.32% |
| MgZnAl-10 | 0.710 | 0.181 | 0.081 | 2.18 | 10.26% |
| MgZnAl-25 | 0.571 | 0.176 | 0.189 | 2.16 | 24.90% |
| MgCoAl-0.5 | 0.615 | 0.146 | 0.002 | 2.12 | 0.86% |
| MgCoAl-1 | 0.799 | 0.192 | 0.006 | 2.12 | 2.07% |
| MgCoAl-5 | 0.744 | 0.201 | 0.025 | 2.04 | 9.22% |
| MgCoAl-10 | 0.697 | 0.190 | 0.044 | 2.18 | 15.85% |
| MgCoAl-25 | 0.568 | 0.172 | 0.107 | 2.59 | 36.19% |
XRF analysis revealed that other impurity cations were present in the samples, stemming from impurities in the chemicals used for synthesis. The Ni-, Zn- and Co-substituted samples contained impurities of Si and Ca, the Fe-substituted samples impurities of Si, Ca and S, and the Cu-substituted samples impurities of Si, Ca, S, Ni and Fe.† The absence of Na contamination confirmed sufficient washing of the LDHs after synthesis. None of the impurities listed were significant enough to greatly affect the Co2+/(Mg2+ + Co2+) ratio.
We believe that the increased presence of Co in the LDHs occurred as a result of the formation of a small, structurally similar and most likely amorphous Co side-species or a partially oxidised MgCoAl–LDH phase. This could be α-Co(OH)2, which is structurally similar to the MgCoAl–LDH prepared and simulated by Rajamathi, Kamath and Seshadri (2002) in terms of its XRD pattern.45 The formation of a partially oxidised MgCoAl–LDH phase of the form MgCo2+Co3+Al–CO2−3–LDH instead of its un-oxidised counterpart could also lie at the root of this problem. So too could the formation of numerous contaminant phases such as CoAl–LDH, MgCo2+Co3+–CO32−–LDH similar to the NO3− intercalated form prepared by Xu and Zeng (2000),46 or Co2+Co3+–CO32−–LDH similar to that of Ma et al. (2008)47 prepared with intercalated NO3−, be at the root of the problem.
Because of changes in oxidation state, bonding of the cations in the layer and interaction with the interlayer, these changes should be detectable using infrared spectroscopy (even for amorphous material). However, FTIR analysis (as discussed below) showed that it is unlikely that a large contaminant Co-containing phase was formed and the analysis could not give any clear indication of the phase formed. It could also be shown that the formation of an LDH species without Al is unlikely, considering the consistent presence of strong Al–OH stretching vibrations on the FTIR spectra discussed below.
FTIR spectra were obtained for all samples and are shown in Fig. 4 for the Co-substituted LDHs and in Fig. 5 for all other MgMAl–LDHs. The spectra obtained for the Fe-, Ni-, Cu- and Zn-substituted LDHs matched the MgAl–LDH well, while a lessening intensity of vibrational bands was observed for an increase in s in the MgCoAl–LDHs.
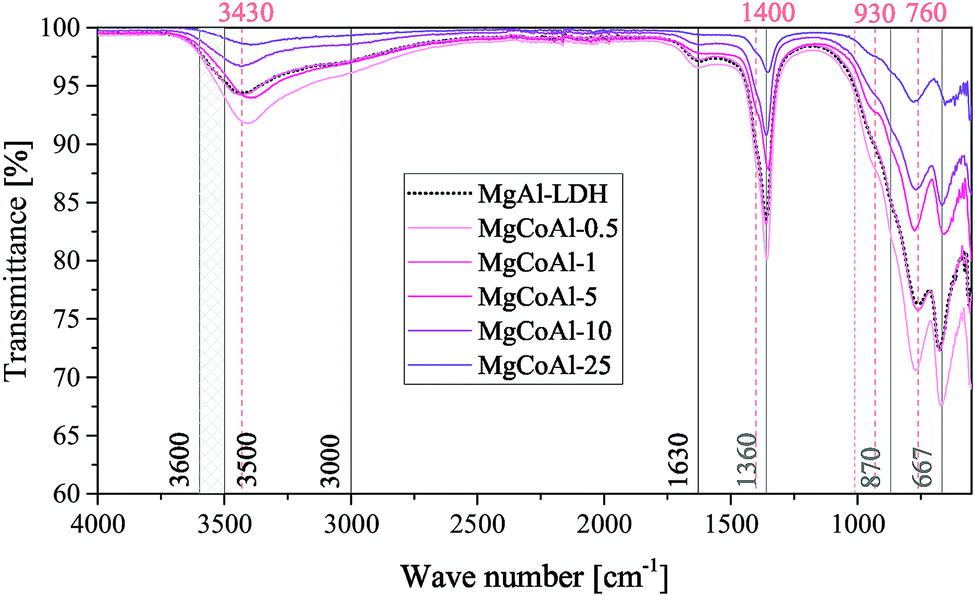 | ||
| Fig. 4 FTIR results of the MgCoAl–LDH samples (s = 0.05%, 1%, 5%, 10% and 25%) with indication of the vibrations for the intercalated CO32− species (grey), the typical vibrations for an LDH phase (black) and other important markers (apricot). | ||
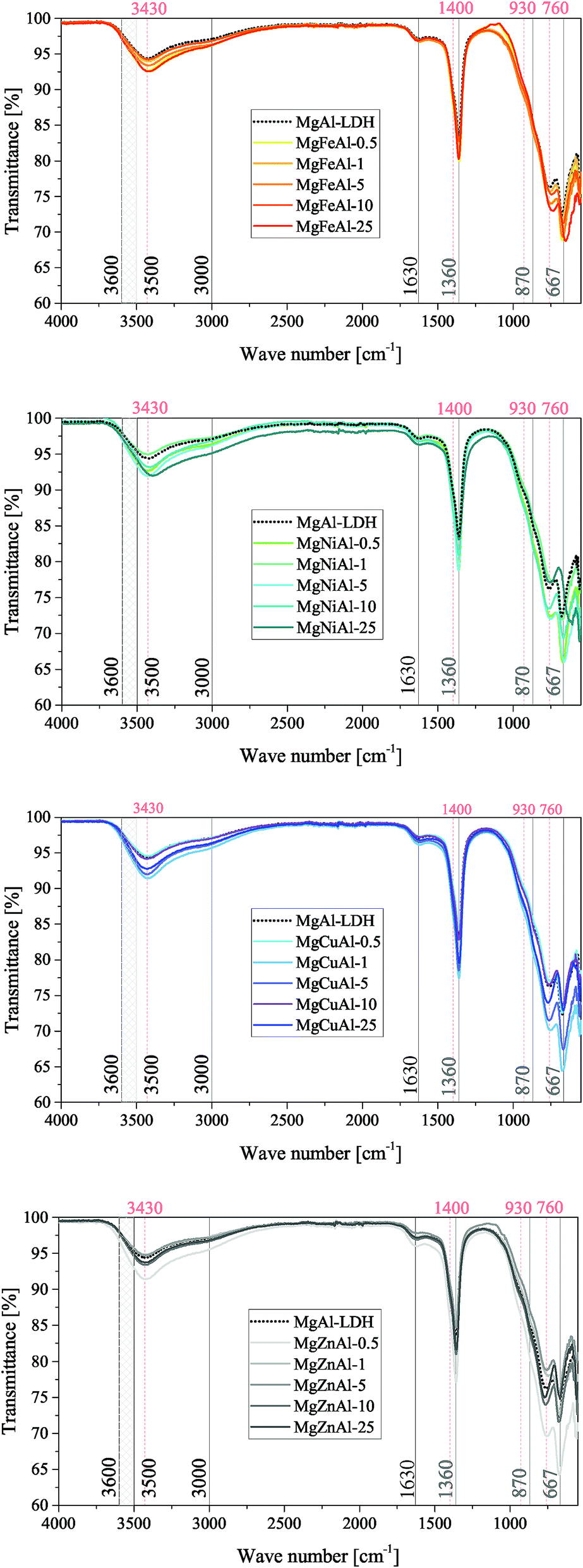 | ||
| Fig. 5 FTIR results of the MgMAl–LDH samples (M = Fe, Ni, Cu and Zn) with vibrations for the intercalated CO32− species (grey), the typical vibrations for an LDH phase (black) and other important markers (apricot) indicated. | ||
Typically, H-bonding stretching vibrations of the brucite-like layer OH-groups (M–OH bonds) in hydrotalcite are found between 3500 cm−1–3600 cm−1, but are dependent on the M3+/(M2+ + M3+) ratio.2 A spectroscopic study of natural quintinite showed that three broad infrared bands exist between 3000 cm−1 and 4000 cm−1, which correspond to the OH-stretching vibration (3388 cm−1) and stretching vibrations of water units (3029 cm−1 and 3148 cm−1).48 Another study was more specific on the assignment of these three vibrations. At similar vibrational band positions to those discussed by Theiss et al. (2015), Kloprogge and Frost (1999) assigned the three bands to an M–OH stretching mode (3467 cm−1), H-bonding of H2O to interlayer anions (3297 cm−1), and to CO2–H2O bridging (2972 cm−1).49 The same paper also found that the position of the bands is dependent on the TM used in the layer of the LDH.
The three bands mentioned above were observed in all prepared LDHs (albeit slightly shifted). The assignment of the peaks discussed by Theiss et al. (2015) fit the bands of this study better than those discussed by Kloprogge and Frost (1999) which can be explained by the use of a M2+/M3+ ratio of 3:1 by Kloprogge and Frost (1999).48,49 The band assigned to H-bonding of H2O to interlayer anions (≈3148 cm−1) was not as prominent for the LDHs prepared in this study compared to the intensity of the band in Theiss et al. (2015).48
In the mid-range of the FTIR spectra, a vibrational band at 1600 cm−1 that typically corresponds to water-bending2 of molecules associated to the interlayer anion was observed at 1630 cm−1.
On the lower end of the spectrum, Theiss et al. (2015) showed that vibrational bands at 776, 841, 866 and 949 cm−1 are linked to water librational modes.48 Further, they assigned vibrational bands at 1350 cm−1 and 1407 cm−1 to CO32− v3 antisymmetric stretching modes.48 Other authors linked vibrations at 1012, 870, 1365/1400 and 667 to the v1, v2, v3 and v4 vibrational modes of interlayer CO32− respectively.50 In addition to this, Kloprogge and Frost (1999) assigned a vibrations at 553, 597, 630, 759 and 939/1012 cm−1 to Al–OH translation, TM–O translation (Ni and Co), Mg–OH translation, Al–OH translation and a doublet Al–OH deformation respectively.
The spectra obtained for the MgMAl–LDHs exhibited vibrational bands at 1630, 1400, 1360, 930, 870, 760, 667 and ≈550 cm−1. The presence of the v1 vibrational mode of interlayer CO32− was not observed intensely. However, all other CO32− vibrational modes described by Kloprogge et al. (2002) were clearly visible on the spectra.50 Further, the CO32− antisymmetric stretching modes at 1360 cm−1 and 1400 cm−1 were especially prominent. It was thus concluded that CO32− was successfully intercalated into all LDHs. The remaining vibrations were assigned to Al–OH translation (≈550 cm−1 and ≈ 759 cm−1), and Al–OH deformation (≈930 cm−1). The Mg–OH translation band (≈630 cm−1), was not very intense. Furthermore, slight shifts in the position of the ≈ 760 cm−1 vibration were observed and associated to an increasing amount of TM–O bonds and thus TM–O stretching vibrations.51
3.2 Morphology, surface area and platelet characteristics
A similar morphology was obtained for all LDHs. The micrographs in Fig. 6 show that the LDHs were synthesised as globular assemblies of nano-sized platelets (full size images are available in the ESI†). This morphology can frequently be observed for unaged, co-precipitated LDHs. However, the platelet size varied with the type and amount of TM-modification. Some samples showed the formation of tiny particles covering the globular platelet assemblies. This typically led to complications in filtering and increased difficulty in the post-processing of the LDHs in comparison to that of the globularly assembled LDHs. The morphology shown on the micrographs in Fig. 6 were also observed in other spots of the samples. | ||
| Fig. 6 SEM micrographs (at 1 keV) of the TM-substituted MgMAl–LDHs synthesised. | ||
The following was observed as trends in the change of the morphology with an increase in TM substitution:
• MgFeAl–LDH: increase in platelet size and space between platelets with an increase in Fe-substitution.
• MgCoAl–LDH: first an increase in platelet size and space between the platelets, then a decrease in both.
• MgNiAl–LDH: first an increase in platelet size and space between the platelets, then a decrease in both.
• MgCuAl–LDH: slow increase in platelet size and space between the platelets.
• MgZnAl–LDH: increase in platelet size and increase in the space between the platelets.
Because the globular platelet assemblies achieved in this study were observed to be around 200 nm in size on the SEM micrographs, it was possible to calculate the crystallite sizes of the LDHs using the Scherrer equation
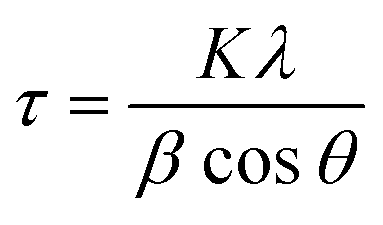
Two crystallite dimensions were calculated: the crystallite size parallel to the LDH layers and the crystallite size perpendicular to the LDH layers. The FWHM and Bragg angle of reflection (003) was used for the perpendicular dimension and the FWHM and Bragg angle of reflection (110) was used for the parallel dimension. As the calculation of crystallite size typically requires subtraction of instrumental line broadening for the determination of an accurate FWHM, the crystallite sizes given here are only estimates.
All results are shown in Fig. 7. It was found that the observations from SEM regarding the changes in platelet size and the pore structure correlated extremely well to the crystallite sizes calculated. Overall, the smallest crystallites were obtained for the Ni-substituted LDHs (s > 1%). Overall, the largest crystallites were obtained for MgFeAl-25. The crystallite sizes in direction parallel to the LDH layers were found not to follow a trend throughout the range of TM substitution. In terms of the crystallite thickness, however, a stable trend across the TM substitutions could be observed. The crystallite thickness increased with an increase in TM substitution (exception MgNiAl-10) and followed the general trend MgNiAl- ≈ MgZnAl- < MgCuAl- ≈ MgFeAl- < MgCoAl–LDH.
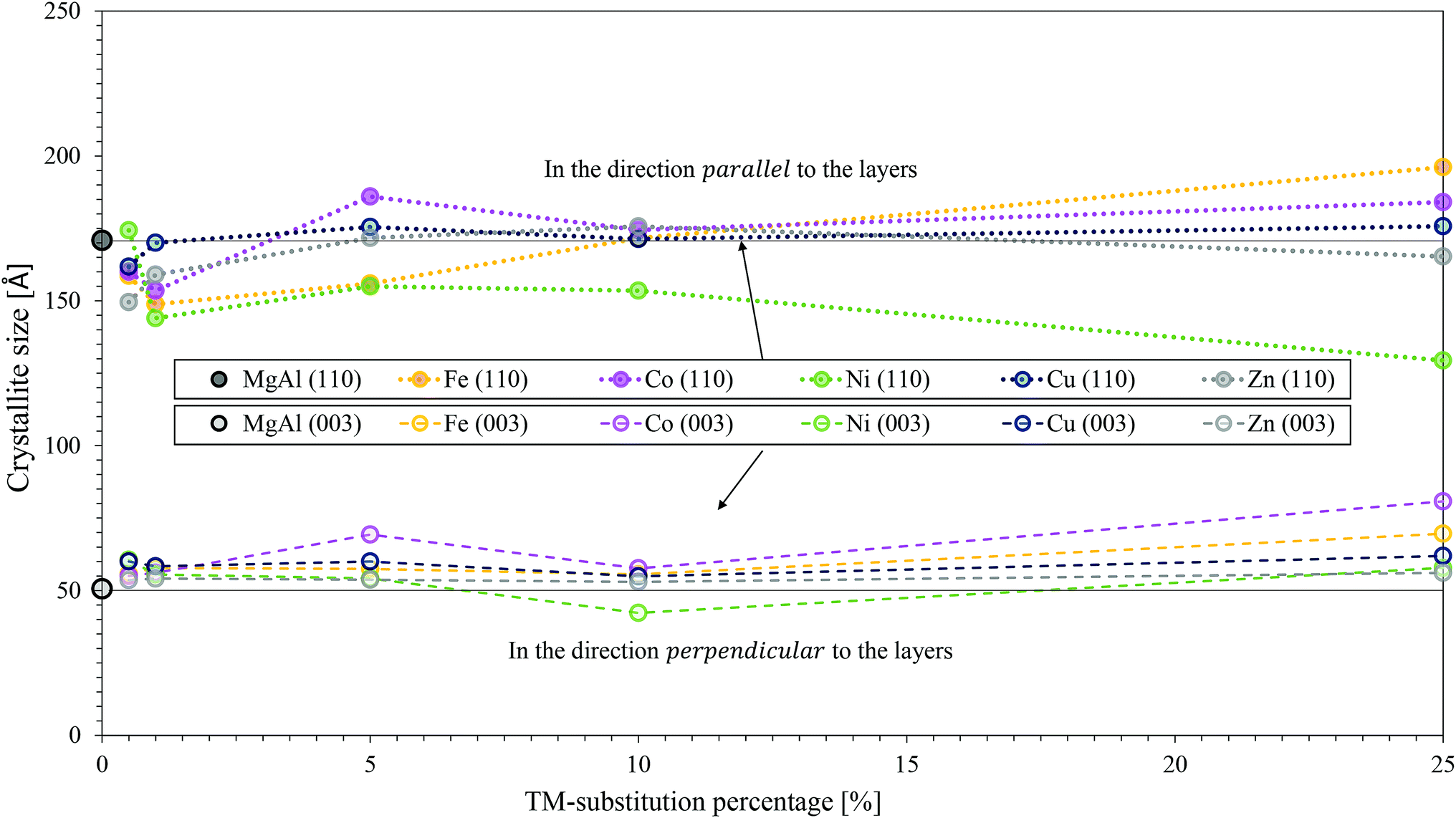 | ||
| Fig. 7 Comparison of crystallite sizes calculated from XRD data of the TM-substituted MgMAl–LDHs with M = Fe, Co, Ni, Cu and Zn. Crystallite sizes in the parallel and perpendicular direction are shown. | ||
In conclusion, TM-modified LDHs prepared in this study showed more complex structures than reported by other authors. Parida et al. (2012) formed smaller but better separated platelets upon Fe-substitution.14 Pu et al. (2013) formed particle agglomerates of 50–100 nm for Fe-substitution.16 The particles were not as intertwined as in this study. Much larger, but similarly intertwined structures were prepared by Vulić et al. (2008) for Fe-substitution and size variations were observed with a change in the percentage of Fe-substitution.23 Materials of similar morphology to those produced were prepared by Wang et al. (2011) (Ni-substituted),15 Chagas et al. (2015) (Co-substituted),30 and Zeng et al. (2016) (Cu-substituted).54 Zheng et al. (2012) formed very-well-defined globular platelet assemblies (microspheres) but of much larger size (approx. 2 μm) for Zn-substituted LDHs.19 Wang et al. (2013) and Wang et al. (2014) prepared similarly sized but well-separated platelets (Zn-substitution).20,22 All materials mentioned in this comparison were post-treated (unlike those prepared in this study). However, different synthesis methods and conditions were used for most studies, especially in terms of the synthesis pH, post-treatment conditions and the concentration of the salt and base solutions used for co-precipitation. Because the synthesis conditions affect the morphology of the material greatly, obtaining differing morphologies between the different studies thus comes at no surprise. A thorough comparison between the data produced in this study to others could not be made, as the morphology of LDHs prepared, or the influence of metal composition on morphological changes, is frequently left unreported.
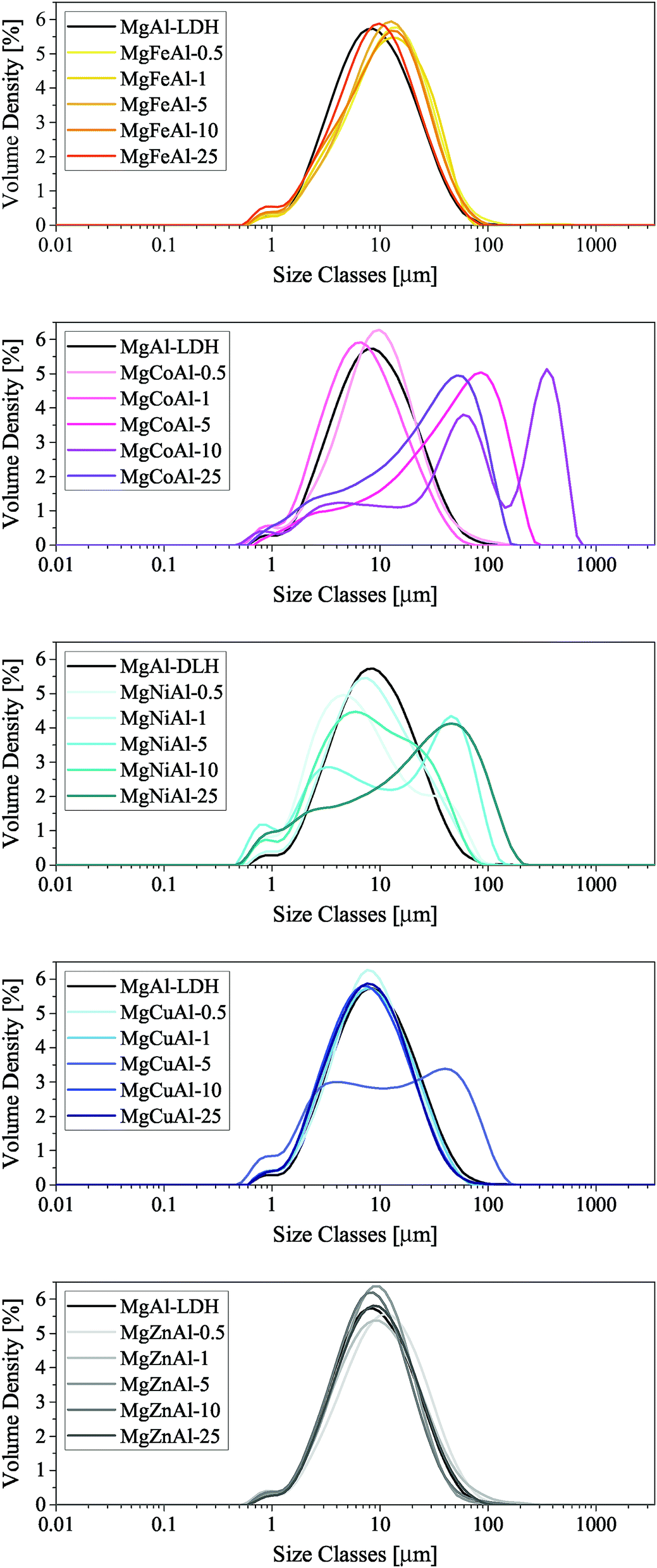 | ||
| Fig. 8 PSA of the TM-substituted MgMAl–LDHs synthesised (NOTE: the x-axis label of these figures is not compiling correctly). | ||
3.3 Thermal stability
The TGA results of the TM-substituted LDHs are shown in the ESI.† Fig. 9 was constructed as a summary of the results for all samples and to show changes in the thermal degradation behaviour of the materials. TGA is a useful characterisation technique to determine the stability of LDHs in desired applications and is used to find optimum thermal post-treatment temperatures (calcination temperatures). The calcination of an LDH leads first to the loss of adsorbed surface species, then a loss of crystal water and volatile interlayer species, and, finally, conversion of the hydroxide layers to oxide layers (layered double oxide formation (LDO)).1,2,55 To obtain this information, the LDH is typically heated from room temperature to the highest temperature of interest. For this study, 1000 °C was chosen – a temperature frequently applied to investigate the thermal degradation products of LDHs up to the irreversible formation of spinel- and mixed oxide phases.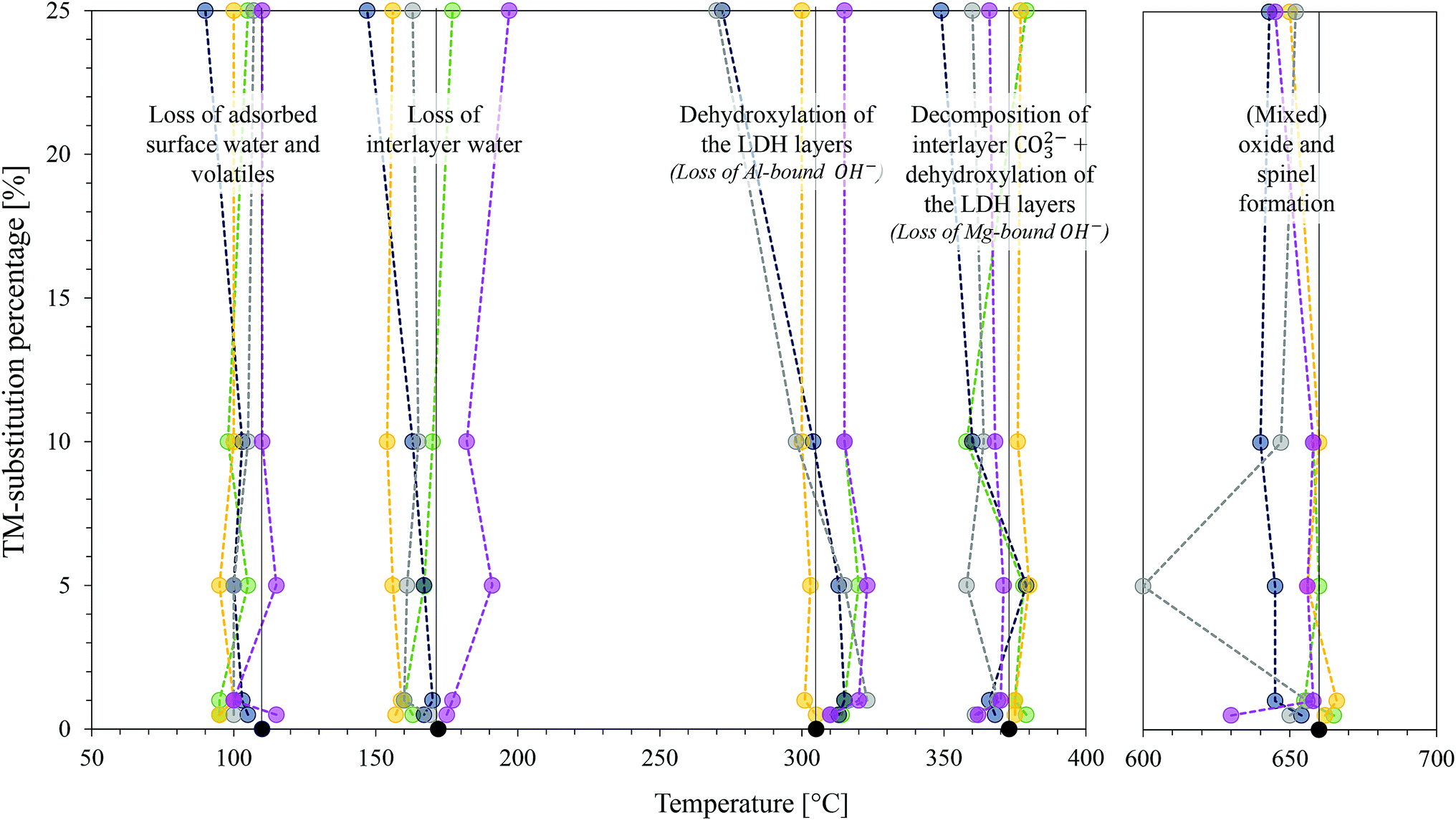 | ||
| Fig. 9 Change in position of Tmax of the derivative curve of the TGA profile with a change in TM-substitution percentage for all TM-substituted MgMAl–LDHs with M = Fe, Co, Ni, Cu and Zn. Decomposition stages are indicated. The usual colour spectrum applies: MgAl–LDH = black, Fe-subs. = yellow, Co-subs. = pink, Ni-subs. = green, Cu-subs. = blue and Zn-subs. = grey. | ||
Thermal decomposition of carbonate intercalated LDHs occurs as follows: at temperatures below and around 100 °C, adsorbed surface species are lost.55 Then there typically exist two decomposition stages. The first stage is associated with the loss of interlayer water, the second with the dehydroxylation of the layers and loss of interlayer anions.1 Hereby, the second stage can split into two distinct stages, the first being associated with the loss of OH− bound to Al cations in the layer and the second to the loss of OH− bound to Mg cations in the layer as well as the loss of interlayer carbonate anions. The loss of surface hydroxyl groups is associated with the conversion of the hydroxide- to oxide species. At this point, the LDH structure is retained as a layered double oxide species and rehydration is possible. Increasing the temperature further leads to the formation of amorphous metal oxide and mixed metal oxide phases that ultimately convert into better crystallised (mixed) metal oxide and spinel phases. These products cannot be rehydrated to form LDH structures.1,2
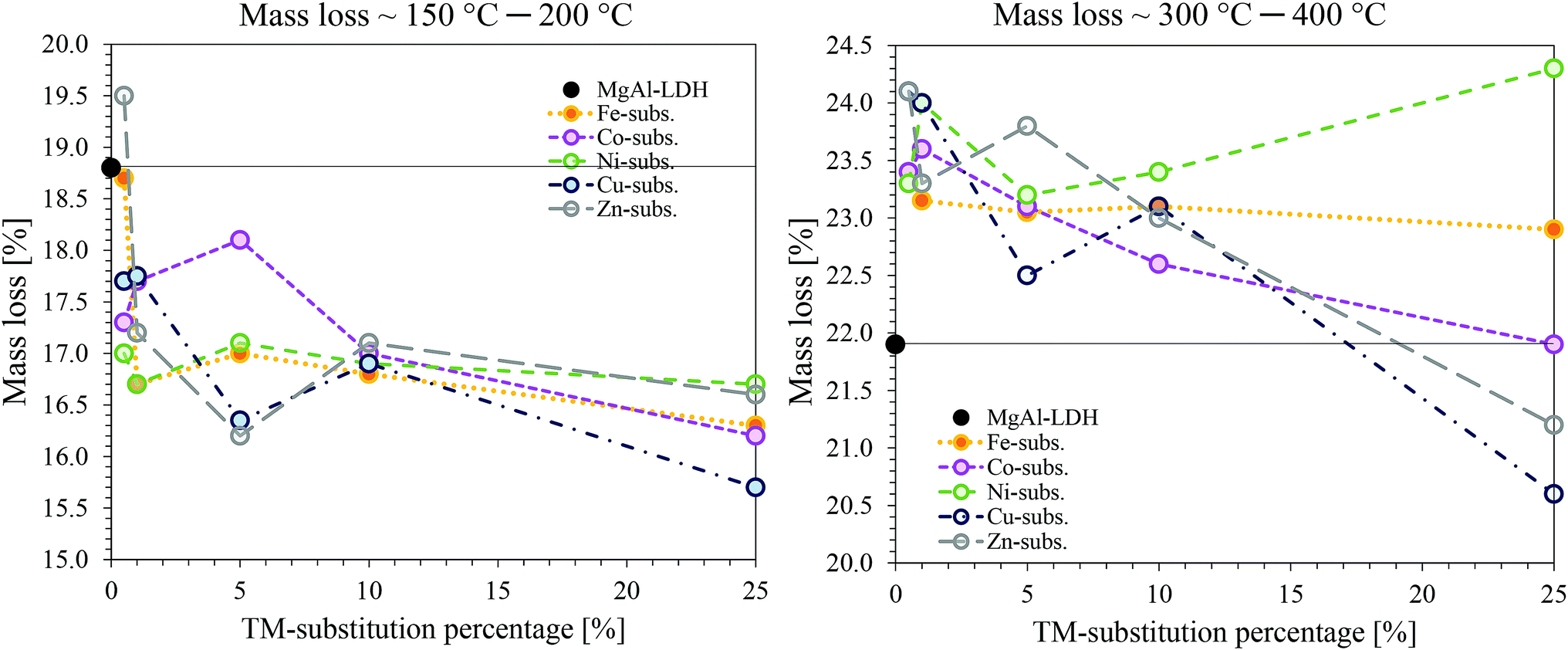 | ||
| Fig. 10 Comparison of mass loss with the different TMs and TM substitutions. On the right: mass loss of the combined effect of adsorbed surface species and interlayer water loss (≈100 °C to 200 °C). On the left: Mass loss of the combined effect of dehydroxylation and decarbonation (≈300 °C to 400 °C). | ||
A general trend could be observed at this stage, as the crystallisation effect was mostly unaffected by the TM-substitution percentage. The mixed oxides resulting from MgCuAl–LDH were found to be the least stable and underwent crystallisation to spinel phases at the lowest temperatures. This was followed very closely by MgZnAl–LDH, MgCoAl–LDH, MgFeAl–LDH and MgNiAl–LDH – the last to start crystallisation being MgAl–LDH.
MgNiAl-25 did not show any decomposition peak at T > 400 °C. MgZnAl-5, MgCuAl-5, MgFeAl-25 and MgCuAl-25 had additional, very small mass loss peaks at 879 °C, 663 °C, 790 °C and 800 °C, respectively.
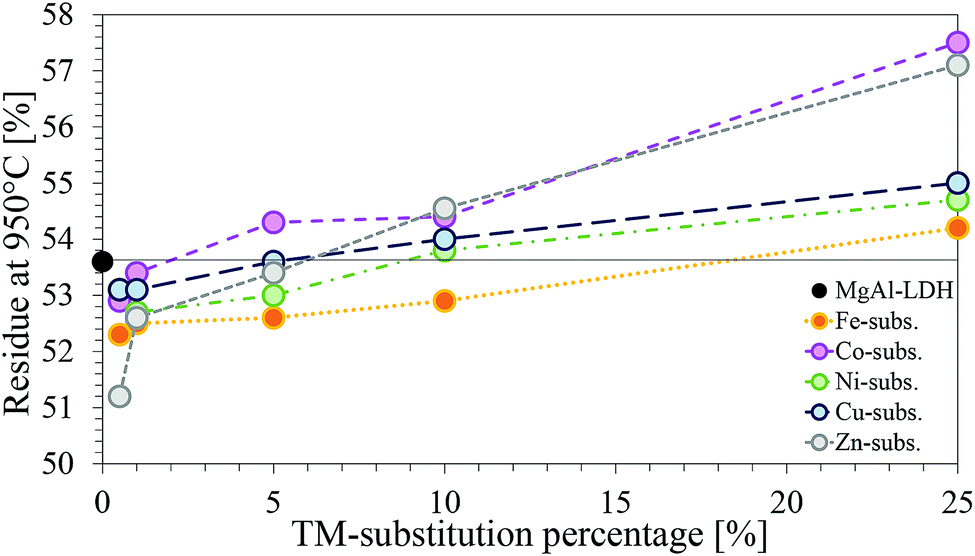 | ||
| Fig. 11 Comparison of the residue remaining at 950 °C for each of the TM-substituted MgMAl–LDHs with M = Fe, Co Ni, Cu and Zn. | ||
Considering the molar masses of each of the metals, an increase in mass of residue is logical. Interesting was, however, that, although every substitution percentage should have led to an increase in residue-mass, this increase was only observed for substitutions ≥5%.
Negative deviation from the percentage residue of MgAl–LDH can indicate the presence of impurities. Based on the chemicals used for synthesis, possible impurities could be oxides, hydroxides, carbonates, chlorides or combinations thereof, of the metals present in the LDH. For the impurity to lose more mass than the LDH form, however, the metal would have to be coordinated to more volatile species than the LDH.
It has already been established through XRD analysis that all but four samples contained no crystalline impurity phases in amounts larger than 2%. Considering the TGA curves shown in the ESI† and the percentage residue remaining, it could be confirmed once more that impurities present are very small and amorphous. However, other than additional decomposition peaks at temperatures >600 °C for samples MgCuAl-5 (calumetite contamination confirmed with XRD), MgZnAl-5, MgFeAl-25 and MgCuAl-25, which could have been caused by impurities, no indication towards the nature of the impurity phases could be found through any of the characterisation techniques used in this study. Further, no comparable references could be found indicating decomposition temperatures of the impurities found through XRD analysis.
Finally, a note on the residue-mass retained of the TM-substituted LDHs. According to the difference in weight caused through substitution of higher weight cations into the LDH layers, the percentage residue remaining should have followed the trend Zn > Cu > Ni > Co > Fe. The observed trend, however, was Co > Zn > Cu > Ni > Fe. This matches with the XRF results showing a larger amount of Co present in the samples. Combined with the TGA results which showed no additional mass loss peaks and very similar results for all substitutions, this serves as an indication towards the formation of an LDH phase with increased Co-content or a structurally similar contaminant Co-phase, seeing that this would decompose in the regions of LDH decomposition and thus remain undetected.57
4 Conclusion
While the synthesis of LDHs and also quintinite is studied well, this study, for the first time, provides a comprehensive comparison between the materials obtained with commonly substituted TMs in the layer (Fe, Co, Ni, Cu and Zn) and varying substitutions (up to 25%).The synthesis conditions used to prepare the LDHs led to the successful formation of all desired phases other than the Co-substituted LDHs. Considering all characterisation techniques used in this study, the LDHs could be synthesised with great purity and the desired composition (apart from Co-substituted LDHs). Further study of the Co-containing phase is advised to determine whether an impurity Co-phase or a partially oxidised MgCoII+CoIII+Al–LDH phase was formed.
It was found that the amount and type of TM substituted influenced the a- and c-parameter, platelet dimensions, post-processing method required for comparable results, and thermal stability.
It was shown through calculation of crystallite sizes from XRD data using the Scherrer equation and corroborated with SEM observations, that the largest and thickest platelets overall were produced with Co-substitution, while the thinnest and smallest platelets were produced with Ni-substitution. While the platelet dimensions varied with TM substitution, the morphology of the materials remained remarkably similar in the form of nanostructured globular platelet assemblies.
Analysis of the TGA data showed that the interlayer water retention is weakest overall in Fe-substituted LDHs and strongest in Co-substituted LDHs. Decomposition of the layers (dehydroxylation and decarbonation) followed in a two-step process (first a loss of Al-bound OH−, then a loss of Mg-bound OH− and CO32−). These two decomposition steps were dependent on the type and amount of TM substituted, and TM substitution influenced the amount of interlayer water present and the mass loss upon dehydroxylation and decarbonation.
Because the type and amount of TM substituted influenced the material properties with respect to size and thermal stability quite significantly, these differences should be taken into account when authors prepare materials to be compared against each other during application. Additionally, to obtain samples with comparable particle agglomerates, the frequently used mortar-and-pestle-grinding technique should be substituted with another sample preparation method.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by Techsparks (Pty) Ltd and the Technology and Human Resources for Industry Programme (THRIP) administered by the Department of Trade and Industry, South Africa, (grant number THRIP/133/31/03/2016). The authors would like to thank the Leibniz-Institut für Polymerforschung Dresden e. V., Germany for providing access to their TGA-DTA facilities for this research. Further, the authors would like to thank Mrs Jeanette Dykstra (X-Ray fluorescence analyst at the Department of Geology, University of Pretoria, South Africa) for analysing the samples with XRF and particularly thank Mrs Wiebke Gröte (X-Ray diffraction analyst at the Department of Geology, University of Pretoria, South Africa) for the analysis of the samples with XRD and her advice on the analysis of the XRD results. Sajid Naseem also thanks the Higher Education Commission (HEC), Pakistan and the German Academic Exchange Service (DAAD) for providing his scholarship. Finally, the authors are thankful for the ongoing support of the Leibniz-Institut für Polymerforschung and Techsparks (Pty) Ltd for collaborative work between the two groups.References
- C. Forano, T. Hibino, F. Leroux and C. Taviot-Guého, Handbook of Clay Science, Elsevier, 2006, vol. 1, pp. 1021–1095 Search PubMed
.
- F. Cavani, F. Trifirò and A. Vaccari, Catal. Today, 1991, 11, 173–301 CrossRef CAS
- X. Duan and D. G. Evans, Layered Double Hydroxides, Springer-Verlag Berlin Heidelberg, 2006, vol. 119, pp. 1–120 Search PubMed
- J. J. Bravo-Suárez, E. A. Páez-Mozo and S. T. Oyama, Quim. Nova, 2004, 27, 601–614 Search PubMed
- S. J. Mills, A. G. Christy, J.-M. R. Génin, T. Kameda and F. Colombo, Mineral. Mag., 2012, 76, 1289–1336 CrossRef CAS
- D. G. Evans and X. Duan, Chem. Commun., 2006, 485–496 RSC
- Y. Zhao, X. Jia, G. I. Waterhouse, L.-Z. Wu, C.-H. Tung, D. O'Hare and T. Zhang, Adv. Energy Mater., 2016, 6, 1501974 CrossRef
- A. Gomes, D. Cocke, D. Tran and A. Baksi, in Layered Double Hydroxides in Energy Research: Advantages and Challenges, ed. A. Jha, C. Wang, N. R. Neelameggham, D. P. Guillen, L. Li, C. K. Belt, R. Kirchain, J. S. Spangenberger, F. Johnson, A. J. Gomes, A. Pandey and P. Hosemann, Springer International Publishing, Cham, 2016, pp. 309–316 Search PubMed
- Y. Ohishi, T. Kawabata, T. Shishido, K. Takaki, Q. Zhang, Y. Wang, K. Nomura and K. Takehira, Appl. Catal., A, 2005, 288, 220–231 CrossRef CAS
- S. Tanasoi, G. Mitran, N. Tanchoux, T. Cacciaguerra, F. Fajula, I. Săndulescu, D. Tichit and I.-C. Marcu, Appl. Catal., A, 2011, 395, 78–86 CrossRef CAS
- L. Jin, B. Ma, S. Zhao, X. He, Y. Li, H. Hu and Z. Lei, Int. J. Hydrogen Energy, 2018, 43, 2689–2698 CrossRef CAS
- H. Düdder, K. Kähler, B. Krause, K. Mette, S. Kühl, M. Behrens, V. Scherer and M. Muhler, Catal. Sci. Technol., 2014, 4, 3317–3328 RSC
- M. del Arco, A. Fernández, C. Martín and V. Rives, Appl. Clay Sci., 2009, 42, 538–544 CrossRef CAS
- K. Parida, M. Satpathy and L. Mohapatra, J. Mater. Chem., 2012, 22, 7350–7357 RSC
- Y.-B. Wang and J.-M. Jehng, Chem. Eng. J., 2011, 175, 548–554 CrossRef CAS
- Y. B. Pu, J. R. Wang, H. Zheng, P. Cai and S. Y. Wu, Research Efforts in Material Science and Mechanics Engineering, 2013, pp. 21–25 Search PubMed
- G. Ovejero, A. Rodríguez, A. Vallet, P. Gómez and J. García, Water Sci. Technol., 2011, 63, 2381–2387 CrossRef CAS
- L. Obalová, K. Jirátová, F. Kovanda, M. Valášková, J. Balabánová and K. Pacultová, J. Mol. Catal. A: Chem., 2006, 248, 210–219 CrossRef
- Y.-M. Zheng, N. Li and W.-D. Zhang, Colloids Surf., A, 2012, 415, 195–201 CrossRef CAS
- G. Wang, M. Yang, Z. Li, K. Lin, Q. Jin, C. Xing, Z. Hu and D. Wang, J. Nanopart. Res., 2013, 15, 1882 CrossRef
- F. J. W. J. Labuschagné, D. M. Molefe, W. W. Focke, I. Van Der Westhuizen, H. C. Wright and M. D. Royeppen, Polym. Degrad. Stab., 2015, 113, 46–54 CrossRef
- G. Wang, D. Rao, K. Li and Y. Lin, Ind. Eng. Chem. Res., 2014, 53, 4165–4172 CrossRef CAS
- T. Vulić, M. Hadnadjev and R. Marinković-Nedučin, J. Microsc., 2008, 232, 634–638 CrossRef PubMed
- T. Vulić, A. Reitzmann, J. Ranogajec and R. Marinković-Nedučin, J. Therm. Anal. Calorim., 2012, 110, 227–233 CrossRef
- A. C. Heredia, M. I. Oliva, U. Agú, C. I. Zandalazini, S. G. Marchetti, E. R. Herrero and M. E. Crivello, J. Magn. Magn. Mater., 2013, 342, 38–46 CrossRef CAS
- S. Paikaray, J. Essilfie-Dughan and M. J. Hendry, Geochim. Cosmochim. Acta, 2018, 220, 217–234 CrossRef CAS
- O. D. Pavel, D. Tichit and I.-C. Marcu, Appl. Clay Sci., 2012, 61, 52–58 CrossRef CAS
- S. Naseem, B. Gevers, R. Boldt, F. J. W. J. Labuschagné and A. Leuteritz, RSC Adv., 2019, 9, 3030–3040 RSC
- A. Tsyganok and A. Sayari, J. Solid State Chem., 2006, 179, 1830–1841 CrossRef CAS
- L. Chagas, G. D. Carvalho, W. D. Carmo, R. S. Gil, S. Chiaro, A. L. ao, R. Diniz, L. D. Sena and C. Achete, Mater. Res. Bull., 2015, 64, 207–215 CrossRef CAS
- T. Coelho, R. Micha, S. Arias, Y. E. Licea, L. A. Palacio and A. C. Faro, Catal. Today, 2015, 250, 87–94 CrossRef CAS
- K. Shekoohi, F. S. Hosseini, A. H. Haghighi and A. Sahrayian, MethodsX, 2017, 4, 86–94 CrossRef PubMed
- J. Rivera, G. Fetter, Y. Jiménez, M. Xochipa and P. Bosch, Appl. Catal., A, 2007, 316, 207–211 CrossRef CAS
- U. Sikander, S. Sufian and M. A. Salam, Procedia Eng., 2016, 148, 261–267 CrossRef CAS
- Y. Zhu, S. Zhang, B. Chen, Z. Zhang and C. Shi, Catal. Today, 2016, 264, 163–170 CrossRef CAS
- I.-C. Marcu, D. Tichit, F. Fajula and N. Tanchoux, Catal. Today, 2009, 147, 231–238 CrossRef CAS
- A.-E. Sakr, T. Zaki, O. Elgabry, M. Ebiad, S. El-Sabagh and M. Emara, Appl. Clay Sci., 2018, 160, 263–269 CrossRef CAS
- S. Miyata, Clays Clay Miner., 1980, 28, 50–56 CrossRef CAS
- F. Scholz and H. Kahlert, ChemTexts, 2015, 1, 7 CrossRef
- V. Rives, Layered Double Hydroxides: Present and Future, Nova Science Publishers, 2001, pp. 39–92 Search PubMed
- S. Britto, S. Joseph and P. Vishnu Kamath, J. Chem. Sci., 2010, 122, 751–756 CrossRef CAS
- R. Dohrmann, K. B. Rüping, M. Kleber, K. Ufer and R. Jahn, Clays Clay Miner., 2009, 57, 686–694 CrossRef CAS
- M. J. dos Reis, F. Silvério, J. Tronto and J. B. Valim, J. Phys. Chem. Solids, 2004, 65, 487–492 CrossRef
- R. D. Shannon, Acta Crystallogr. A, 1976, 32, 751–767 CrossRef
- M. Rajamathi, P. Kamath and R. Seshadri, Mater. Res. Bull., 2000, 35, 271–278 CrossRef CAS
- Z. P. Xu and H. C. Zeng, Chem. Mater., 2000, 12, 2597–2603 CrossRef CAS
- R. Ma, K. Takada, K. Fukuda, N. Iyi, Y. Bando and T. Sasaki, Angew. Chem., Int. Ed., 2008, 47, 86–89 CrossRef CAS PubMed
- F. Theiss, A. López, R. L. Frost and R. Scholz, Spectrochim. Acta, Part A, 2015, 150, 758–764 CrossRef CAS PubMed
- J. Kloprogge and R. L. Frost, J. Solid State Chem., 1999, 146, 506–515 CrossRef CAS
- J. T. Kloprogge, D. Wharton, L. Hickey and R. L. Frost, J. Solid State Chem., 2002, 87, 623–629 CAS
- X. Wang and L. Andrews, J. Phys. Chem. A, 2006, 110, 10035–10045 CrossRef CAS PubMed
- A. L. Patterson, Phys. Rev., 1939, 56, 978–982 CrossRef CAS
- Y. Zhao, F. Li, R. Zhang, D. G. Evans and X. Duan, Chem. Mater., 2002, 14, 4286–4291 CrossRef CAS
- Y. Zeng, T. Zhang, Y. Xu, T. Ye, R. Wang, Z. Yang, Z. Jia and S. Ju, Appl. Clay Sci., 2016, 126, 207–214 CrossRef CAS
- V. Rives, Mater. Chem. Phys., 2002, 75, 19–25 CrossRef CAS
- J. Valente, G. Rodriguez-Gattorno, M. Valle-Orta and E. Torres-Garcia, Mater. Chem. Phys., 2012, 133, 621–629 CrossRef CAS
- Z. P. Xu and H. C. Zeng, J. Mater. Chem., 1998, 8, 2499–2506 RSC
Footnotes |
| † Electronic supplementary information (ESI) available: Table containing results from XRD analysis and crystal data calculations, full mass-based XRF results table, full-sized SEM micrographs, and figures showing profiles obtained through thermogravimetric analysis. See DOI: 10.1039/c9ra05452a |
| ‡ It is believed that the multipeak PSA curve of MgCuAl-5 resulted through an error in PSA sample preparation. |
| This journal is © The Royal Society of Chemistry 2019 |